

ABSTRACT
Raynaud's phenomenon (RP) is a common vasospastic condition which affects ~5% of the general population. The majority of individuals have primary RP; however, Raynaud's can also occur secondary to a broad range of underlying medical conditions and drug therapies. RP is a cardinal feature in patients with systemic sclerosis and is often the earliest symptom of the disease. Unlike primary RP, patients with secondary RP can develop persistent digital ischaemia, including ulcers and gangrene. Patients require a comprehensive clinical assessment and investigation, in particular, the detection of autoantibodies and nailfold capillaroscopic abnormalities. Non-pharmacological management is indicated in all patients. There are a wide range of available drug therapies to treat RP, including when complicated by digital ulceration, and surgical intervention is sometimes required. Future research is needed to understand the complex pathogenesis of RP and to measure the impact and severity of RP to develop optimised approaches to management.
KEYWORDS: Raynaud's phenomenon, systemic sclerosis, digital ulcers
Key points
Raynaud's phenomenon (RP) is common and should be considered as a ‘symptom complex’ and always requires explanation.
The majority (80–90%) of individuals have primary (‘idiopathic’) RP.
RP can occur secondary to a broad range of underlying medical conditions and drug causes.
RP occurs in the vast majority (>95%) of patients with systemic sclerosis and is often the earliest symptom of the disease.
Unlike patients with primary RP, persistent digital ischaemia, ulceration and gangrene can occur in patients with secondary RP.
Patients with RP require a comprehensive clinical assessment and investigation.
Key investigations in RP include testing for autoantibodies and performing nailfold capillaroscopy.
Non-pharmacological management (eg keeping warm) is indicated in all individuals with RP.
There are a wide range of available drug therapies to treat RP. First-line treatment is usually with calcium channel blockers.
Clinicians are increasingly using phosphodiesterase-type 5 inhibitors earlier to treat RP, particularly when secondary to connective tissue disease.
Prostanoid (eg iloprost) can be used in the setting of refractory RP and for ischaemic digital complications, and sometimes surgical intervention is required
Introduction
Raynaud's phenomenon (RP) is an episodic, vasospastic disorder typically characterised by digital vascular compromise following exposure to cold and/or emotional stressors (Fig 1). The majority (80–90%) of individuals have primary (‘idiopathic’) RP (PRP), whereas secondary RP (SRP) can occur due to a broad range of medical conditions and drug-related causes. Irrespective of the underlying cause, RP has a significant negative impact on individuals’ quality of life and can result in persistent digital ischaemia including ulceration and gangrene in secondary forms of RP (Fig 2).1–4 The purpose of this review is to describe the relevant epidemiology and pathogenesis, clinical features including complications, and provide a practical approach to the investigation and multi-faceted management of RP.
Fig 1.
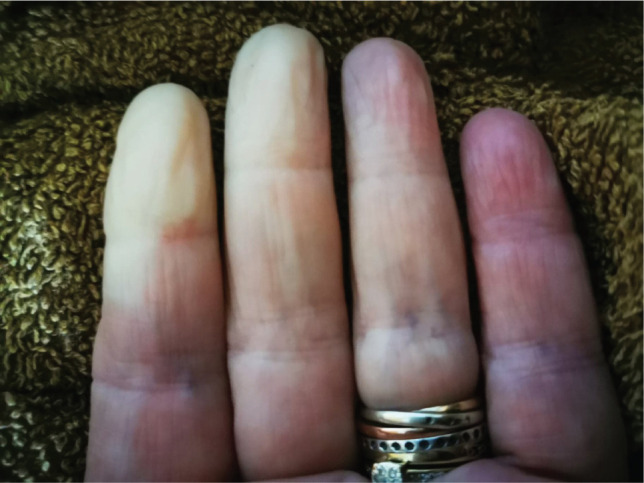
Photograph of a hand with an example of digital vascular compromise from Raynaud's phenomenon.
Fig 2.
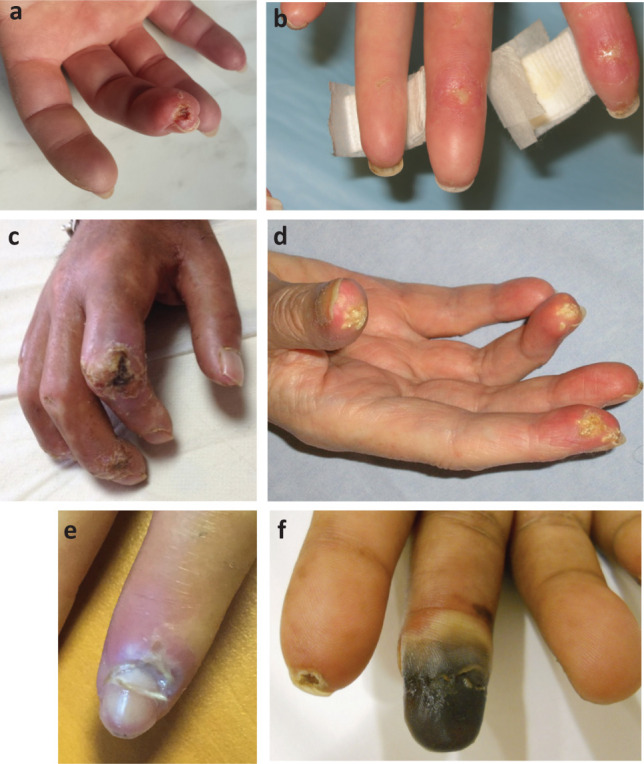
Photographs of hands with persistent digital ischaemia including ulceration and gangrene in secondary forms of Raynaud's phenomenon. Reproduced with permission from Hughes M, Allanore Y, Chung L, Pauling JD, Denton CP, Matucci-Cerinic M. Raynaud's phenomenon and digital ulcers in systemic sclerosis. Nat Rev Rheumatol 2020;4:208–21.
Search strategy
Our search strategy was developed in accordance with published recommendations for the writing of narrative reviews to inform the stated purpose of the review.5 The following search criteria were applied within the National Institutes of Health's National Library of Medicine (PubMed) with a focus on recent publications within the past 5 years (1,074 results identified on 18 August 2020): (Raynaud*) AND ((assessment) OR (classification) OR (clinical trial) OR (pathogenesis) OR (management) OR (treatment) OR (treatment)).
The results of this search formed the majority of the major subheadings of the review. In addition, a grey search of the manuscripts cited within these articles was undertaken, and with the inclusion of key legacy papers.
Epidemiology
In general, the prevalence of RP within the general population has been reported to be approximately 5%.6 The authors of a recent systematic review and meta-analysis estimated that the prevalence and incidence of PRP was 4.85% (95% confidence interval (CI) 2.08–8.71) and 0.25% (95% CI 0.19–0.32), respectively.7 However, other studies have reported that the prevalence of PRP may be higher, affecting 2–20% of women and 1–12% men.6 Around half of patients with PRP have a family history of RP, particularly in females and those with early-onset RP.6 As a generalisation, PRP tends to occur before the age of 30; whereas, Raynaud's that develops over the age of 40 is more likely to be SRP. In general, RP (and particularly PRP) is more common in women compared with men.8 The prevalence of RP increases with age in men and is more likely to be due to occupational exposures (eg vibratory tool use) or peripheral vascular disease (from atherosclerosis).6,9
Secondary causes of RP
The causes of SRP are presented in Box 1.10 A comprehensive clinical assessment including investigations is required to identify any potential causes of SRP, considering the broad ranging pathology (described more later). This is of crucial importance as these may require a significantly different approach to treatment (eg treating severe hypothyroidism or management of paraproteinaemia). RP is a cardinal feature in patients with systemic sclerosis (SSc) and is often the first symptom of the condition, particularly in patients with the limited subset of the disease, potentially occurring decades before the onset of skin thickening.11,12
Box 1.
Proposed criteria for primary Raynaud's phenomenon. Adapted from LeRoy EC, Medsger TA. Raynaud's phenomenon: a proposal for classification. Clin Exp Rheumatol 1992;10:485-8
| Episodic attacks of acral pallor or cyanosis |
| Peripheral pulses should be strong and symmetrical |
| No evidence of digital pitting, ulceration or gangrene |
| Normal nailfold capillaries |
| Negative antinuclear antibody |
| Normal erythrocyte sedimentation rate |
Pathogenesis of RP
The pathogenesis of RP involves a complex interplay between genetic, neural, vascular and intravascular factors. Although a detailed review of the pathogenesis of RP is beyond the scope of this review, an overview of the key pathogenetic mechanisms informs a systematic approach to both the assessment and management of RP. We will highlight the main pathogenic mechanisms that have been implicated in PRP and/or SSc-RP to date.
Genetic
Genetic factors appear to play an important role in the development of RP as demonstrated in familial and twin studies.13 Indeed, previous work has demonstrated half of patients with PRP have positive family history with first degree relatives having RP.6 Of mechanistic and therapeutic interest, Munir et al reported that RP is associated with a polymorphism in the NOS1 gene.14
Vascular
Increased vascular tone including vasospasm including the digital arteries is central to the pathogenesis of RP.15,16 In SSc, there is endothelial dysfunction and overproduction of vasoconstrictors (eg endothelin-1 and angiotensin II) and impaired vasodilation with reduced production or reduced efficacy of vasodilators (eg nitric oxide and prostacyclin).17,18
Neural
The primary deficit in PRP is considered to be a ‘local fault’ in vascular function of thermoregulation.19 Central autonomous involvement has also been suggested to be important in the pathogenesis of connective tissue disease-associated RP.20 Cold stimuli causes relocation of alpha 2c-adrenergic receptors from the Golgi apparatus to the cell surface, through activation of the rho kinase, which then increases the sensitivity of contractile proteins and subsequent vasoconstriction.21 Increased alpha 2c-adrenergic expression in vascular smooth muscle cells has been reported in RP.22 Vasoconstriction compromises arteriovenous anastomoses and nutritional blood flow, which is further compounded in patients with SSc by vascular damage and occlusion.18,19 Abnormalities in vasoactive peptides (eg of calcitonin gene-related peptide vascular biology) have also been implicated in the pathogenesis of RP.23
Intravascular factors
Intravascular factors are also likely to play an important part in the pathogenesis of RP through increased viscosity (for example in certain haematological disorders). Contributory mechanisms which have been implicated in the pathogenesis of RP include (but are not limited to) platelet and white cell activation, red cell structural deformity and defective fibrinolysis.24–26
Clinical features
Typical attacks of RP consist of episodic colour change of the hands and often the feet triggered by exposure to cold temperature and/or emotional stress. The classical description of RP is a triad of initially white/pallor from vasoconstriction, blue/cyanosis from sequestration of deoxygenated blood and finally redness from reperfusion and hyperaemia (Fig 1). It is important to highlight that patients do not need to have all three colour changes.27 Cyanosis (without blanching) has been reported to be more common in patients with SSc compared with primary RP, and reactive hyperaemia less common. The majority of classification systems have traditionally mandated the presence of at least two (biphasic) colour changes.4 Raynaud's can also affect other vascular beds including the lips, nose, ears and nipples.
It is important to differentiate RP from conditions which can potentially mimic cutaneous features including (but not limited to) acrocyanosis and erythromelalgia. Acrocyanosis is seen in vaso-occlusive disorders (eg cold agglutinin disease) which is characterised by persistent symmetrical cyanosis of the extremities and is exacerbated by cold exposure (akin to RP). Erythromelalgia is characterised by erythema and pain of the extremities which is exacerbated by heat rather than cold temperatures (unlike RP). The key is eliciting a clear temporal relationship between the cutaneous features and the environmental (eg cold vs hot) triggers.
Clinical assessment
A comprehensive clinical assessment is required in all patients with RP, with particular reference to the secondary causes of RP (Table 1).28 This should seek to address two main areas of concern.
Does the patient have primary or secondary RP? In particular, does the patient have evidence of an autoimmune connective tissue disease?
How severe is the patient's RP including the burden of ischaemic digital complications (eg in SSc)? This will dictate management including the need for drug therapies and potentially surgical intervention.
Table 1.
Secondary causes of Raynaud's phenomenon. Reproduced from Devgire V, Hughes M. Raynaud's phenomenon. Br J Hosp Med (Lond) 2019;80:658–64.
| Vascular (usually proximal large vessel disease, often unilateral symptoms) | Compressive (eg cervical rib) Obstructive: non-inflammatory (ie atherosclerosis); inflammatory vascular disease (eg thromboangiitis obliterans (Buerger's disease)) |
| Occupational | Hand–arm-vibration syndrome (vibration white finger) |
| Autoimmune conditions | Systemic sclerosis Systemic lupus erythematosus Sjogren's syndrome Mixed connective tissue disease/overlap syndromes Undifferentiated connective tissue disease Idiopathic inflammatory myopathies |
| Drug-/chemical-related | Amphetamines Beta-blockers Bleomycin Cisplatin Clonidine Cyclosporine Interferons Methysergide Polyvinyl chloride |
| Conditions associated with increased plasma viscosity and reduced digital perfusion | Cryoglobulinaemia Cryofibrinogenaemia Paraproteinaemia Malignancy (including as a paraneoplastic phenomenon) |
| Other causes and associations | Carpal tunnel syndrome Frostbite Hypothyroidism |
History
As previously described, the age of onset of RP can often help to distinguish PRP from SRP. A detailed description of RP attacks should be elicited (eg the pattern of colour change and body areas affected). Thumb involvement is more likely to occur in patients with SRP compared to PRP.29 Asymmetrical RP attacks can indicate proximal (large) vessel disease. The impact of RP including the activities of daily living and occupation should be explored. Detailed past medical and drug histories (with reference to the causes of SRP) as well as an occupational history (eg the use of vibratory tools) should be obtained (Table 1).28 A family history of RP and any potentially associated autoimmune and/or rheumatological diseases (eg SSc) should be elicited.
Examination
A full physical examination should be performed with a particular focus on the extremities and signs of systemic diseases associated with SRP including established digital ischaemia (eg pitting scars and ulcers). Cutaneous manifestations of SSc should be actively sought such as skin thickening (scleroderma) including of the digits (sclerodactyly), calcinosis (subcutaneous calcium deposition) and telangiectases (dilated blood vessels). The peripheral pulses should be strong and symmetrical and abnormalities (eg low volume/absent) require urgent investigation (eg by arterial Doppler ultrasound).30,31 Cardiovascular and respiratory examinations should be undertaken including (but not limited to) elicit features of pulmonary fibrosis and hypertension.
Investigations
When the history and examination strongly suggest PRP, ‘minimal’ investigations include full blood count, inflammatory markers (erythrocyte sedimentation rate or C-reactive protein), antinuclear antibody and nailfold capillaroscopy (discussed later). These should be within the normal range or low titre (eg antinuclear antibody). Autoantibody testing is particularly useful to help identify those patients who are at the greatest risk of developing a definitive connective tissue disease, including SSc.32 Extractable nuclear antibody testing can determine the antigenic target of autoantibodies and are very helpful in identifying SSc (eg anticentromere and anti-Scl-70) and related connective tissue diseases (eg anti-Jo-1 in myositis). In addition to these minimal investigations, many clinicians also request routine biochemistry (eg renal and liver function tests), bone biochemistry, creatinine kinase, immunoglobulins with serum electrophoresis (if there is a clinical suspicion of paraproteinaemia), complements C3 and C4, fasting lipid profile (especially in older patients at high risk of cardiovascular disease), and a chest (or thoracic outlet) X-ray looking for a bony cervical rib.33 Where clinically appropriate (eg vascular thrombosis and/or pregnancy morbidity and mortality), testing for antiphospholipid syndrome (anticardiolipin and anti-b2-glycoprotein antibodies and lupus anticoagulant) can be performed. As earlier, if proximal vessel involvement is suspected, then urgent Doppler arterial ultrasound must be urgently performed to assess for proximal (large) vessel involvement, which may be amenable to therapeutic intervention.30–33
Specialist investigations
Specialist investigations include assessment of microvascular structure (as assessed by nailfold capillaroscopy) and functional vascular imaging (including thermography). Laser-derived techniques (eg full-field perfusion of the digits) show great promise as a surrogate for SSc-digital vasculopathy (including RP) particularly to support drug development programmes/clinical trials.34
Nailfold capillaroscopy
Nailfold capillaroscopy (Figs 3a and 3b) provides a unique non-invasive insight into the structure of the microcirculation. Capillaroscopy is performed at the base of the fingernails where the capillaries lie parallel to the surface of the skin. Capillaroscopy can be performed with either low-magnification examination (∼×10 magnification) using a variety of devices (dermatoscope, stereomicroscope or ophthalmoscope) or high-magnification examination (∼×200–600 magnification) using videocapillaroscopy.35,36 The former provides a wide-field examination of the nailfold; whereas, higher-magnification allows for a more detailed examination and quantification of microvascular structure.
Fig 3.

Nailfold capillaroscopy. a) Normal capillaries with a uniform, regularly distributed and ‘hairpin’-like appearance. b) Abnormal capillaries of patient with systemic sclerosis showing progressive structural alterations.
Normal capillaries have a uniform, regularly distributed and ‘hairpin’-like appearance (Fig 3a). Whereas, in patients with SSc, there are progressive structural alterations (Fig 3b). Early abnormal findings include dilated capillaries and few haemorrhages. As these progress, there are more haemorrhages and frequent ‘giant’ capillaries. Late features include loss of capillaries (so-called ‘drop out’) with extensive avascular areas, and an aberrant (failed) attempt at neoangiogenesis.37 Similar nailfold capillaroscopic findings have been reported in mixed connective tissue disease including ‘ramified’ (or bushy) capillaries, which also share a number of similar clinical features of SSc (eg pulmonary hypertension) and systemic lupus erythematous. Progressive nailfold capillaroscopic abnormalities have been reported to be predictive of disease progression in SSc including internal organ involvement and digital ulceration.38,39 A simple ‘fast-track’ algorithm has been developed to differentiate a ‘non-scleroderma’ from a ‘scleroderma’ pattern on capillaroscopic images, with excellent reported reliability.40 Experts in the field of capillaroscopy have recently produced consensus guidance on standardisation of nailfold capillaroscopy in patients with RP and SSc.41
Infrared thermography
Infrared thermography uses a thermal imaging camera to measure the skin surface temperature which is an indirect of measure of tissue perfusion. Patients with RP often have cooler fingertips (compared with healthy individuals) and can be used to differentiate between PRP and SRP. However, at present, there is no internationally agreed standardised thermography protocol, and some include dynamic (eg a cold challenge) assessment to further help to differentiate PRP and SRP.42 A limiting factor is that thermography requires relatively expensive equipment and a temperature-controlled laboratory. However, the increasing availability of low-cost (eg mobile phone) technology could facilitate the wider adoption of thermal imaging into routine clinical practice.43
Management
The management of RP can be broadly considered under the two headings (which significantly overlap) of ‘uncomplicated RP’ and ‘complicated RP’, the latter of which involves progression to persistent tissue ischaemia (ie digital ulceration and/or gangrene; Fig 4).44
Fig 4.
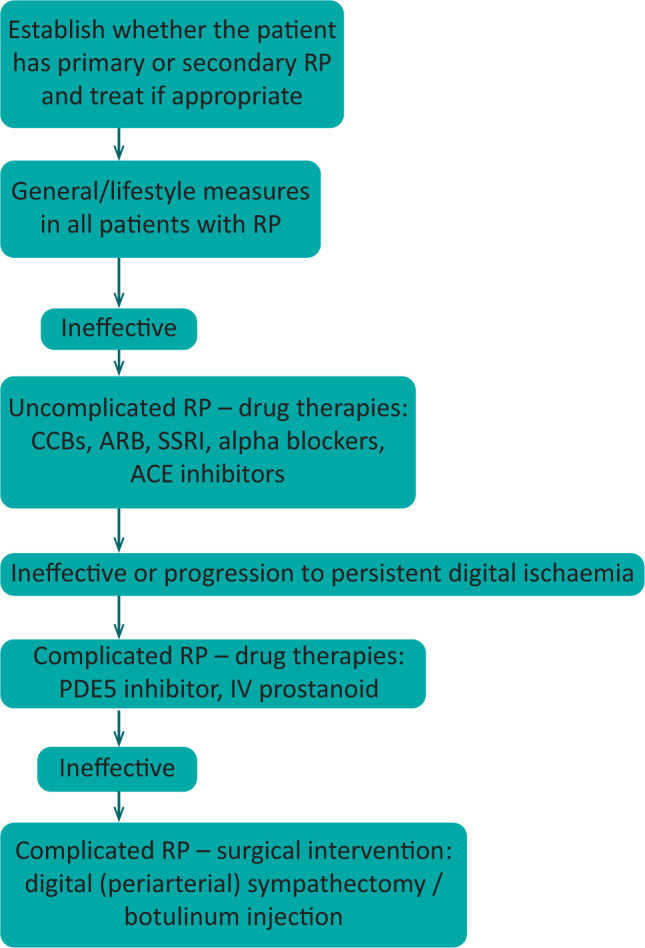
Flowchart for treatment of Raynaud's phenomenon. ACE = angiotensin-converting enzyme; ARB = angiotensin II receptor blockers; CCB = calcium-channel blockers; IV = intravenous; PDE5 = phosphodiesterase type 5; RP = Raynaud's phenomenon; SSRI = selective serotonin reuptake inhibitor.
Uncomplicated RP
Non-pharmacological interventions
Non-pharmacological interventions are indicated in all patients with RP. Patient education is essential including lifestyle measures (eg cold avoidance and wearing multiple layers of clothing) and to emphasise the importance of smoking cessation (which promotes vasoconstriction). Patients should be provided with high-quality information about RP, including patient information booklets and they should be sign-posted to patient-led organisations for support and advice, which is increasingly being accessed online.45,46
First-line drug treatment
Drug treatment is indicated when non-pharmacological interventions are insufficient to manage RP and is often required in patients with SRP (especially SSc).47 Calcium channel blockers (eg nifedipine) are often used as first-line drug treatment for PRP and SRP.8,33,48,49 Other drug classes which are used for the treatment of RP include angiotensin receptor II antagonists (eg losartan), angiotensin-converting enzyme (ACE) inhibitors, and selective serotonin receptor antagonists (eg fluoxetine).33,49 The latter of which is particularly useful in patients who are prone to significant vasodilatory side effects.33 In general, drug therapies for RP are started at a low-dose and gradually increased based on the benefits of treatment vs dose-limiting side effects.8,33
Complicated RP
Drug therapy for refractory RP
Clinicians are increasingly using phosphodiesterase-type 5 inhibitors (particularly sildenafil) earlier in the pharmacological treatment of RP, particularly when complicated CTD-associated RP is complicated by digital ischaemia and/or gangrene.33,50 Prostanoid therapy (eg iloprost or epoprostenol) is administered (intravenously) in cases of refractory digital ulcers (to foster ulcer healing and reduce new ulceration) and is also used in patients with severe RP (despite the previously described interventions).51–53 The endothelin-receptor antagonist bosentan is licensed in Europe for the treatment of digital ulcers in SSc. Bosentan reduces the development of new ulcers but does not impact on the healing of existing ulcers.54 However, a similar effect was not observed with macitentan (another endothelin-receptor antagonist).55 Such drug therapies are often used in combination in the context of complicated RP.56 Of note, the therapeutic pathways which are targeted for refractory RP are also similarly targeted for the treatment of pulmonary hypertension.57
Surgical intervention
Surgery is reserved for severe (refractory) RP and/or persistent digital ischaemia (ie ulcers and gangrene). Indications for surgery include severe (and often nocturnal pain) which suggests underlying necrosis or abscess development, removal of infected tissue including deeper bony involvement (ie osteomyelitis), and debulking of subcutaneous calcinosis. There is increasing experience in digital (periarterial) sympathectomy for RP and this could also potentially be beneficial for early digital ischaemia.58 Similarly, there is increasing interest surrounding botulinum injections; however, the evidence base to support these interventions at present is limited and warrants further investigation.59,60
Unmet needs and challenges
Further research is required to understand the complex aetiopathogenesis of RP and how this differs between patient groups, including how to optimise approaches to treatment. Historically, assessment of treatment efficacy in clinical trials has relied upon patient-reported, diary-based methods, which experts in SSc and RP have expressed significant concerns relating to the conduct of drug development programmes.61 Ongoing work to understand the patient experience of RP (and digital ulcers) could lead to the development of novel patient reported outcome measures which would capture the multifaceted impact of RP and ischaemic digital complications.62,63 Another key consideration is that episodic (vasospastic) ischaemia from RP in SSc is complicated by progressive accumulation of structural microvascular damage, which is considered largely irreversible. The relative merits of sequential monotherapy vs initial combination therapy for RP (akin to the modern pharmacological treatment of pulmonary hypertension) has yet to be studied. A ‘unified’ vascular phenotype has been recently proposed in SSc. With this in mind, could a ‘treat-to-target’ approach, which is widely adopted in medicine, also be adopted for the treatment of RP?18,64 Furthermore, could such an approach to treatment also positively modify the other digital and visceral vascular complications of SSc?18 Topical approaches to treatment also merit revisiting, as such, a ‘local’ approach to treatment could be better tolerated compared with systemic therapies for RP (and also digital ischaemic complications).65–67
Conclusion
RP is a common condition which is associated with significant morbidity including pain and disability. A key feature is that secondary forms of SRP (especially in patients with SSc) can result in digital ulceration and gangrene. A comprehensive clinical assessment (history and physical examination) is required in all patients with RP and requesting investigations. Key investigations include the detection of autoantibodies and/or nailfold capillary abnormalities to identify those individuals who are at the highest risk of developing connective tissue diseases (eg SSc). Non-pharmacological intervention is indicated in all patients with RP. There are a wide range of oral drug therapies for RP including when complicated by digital ischaemia. Surgical intervention is sometimes required in severe (refractory) RP and/or persistent digital ischaemia. Future research is needed to understand the complex pathogenesis and lived experience of RP/outcome measures of treatment efficacy to develop optimised approaches to management including pharmacological therapies.
References
- 1.Hughes M, Snapir A, Wilkinson J, et al. Prediction and impact of attacks of Raynaud's phenomenon, as judged by patient perception. Rheumatology (Oxford) 2015;54:1443–7. [DOI] [PubMed] [Google Scholar]
- 2.Bassel M, Hudson M, Taillefer SS, et al. Frequency and impact of symptoms experienced by patients with systemic sclerosis: results from a Canadian National Survey. Rheumatology (Oxford) 2011;50:762–7. [DOI] [PubMed] [Google Scholar]
- 3.Fábián B, Fábián AK, Bugán A, Csiki Z. Comparison of mental and physical health between patients with primary and secondary Raynaud's phenomenon Category: Article. J Psychosom Res 2019;116:6–9. [DOI] [PubMed] [Google Scholar]
- 4.Pauling JD, Hughes M, Pope JE. Raynaud's phenomenon - an update on diagnosis, classification and management. Clin Rheumatol 2019;38:3317–30. [DOI] [PubMed] [Google Scholar]
- 5.Gasparyan AY, Ayvazyan L, Blackmore H, Kitas GD. Writing a narrative biomedical review: considerations for authors, peer reviewers, and editors. Rheumatol Int 2011;31:1409–17. [DOI] [PubMed] [Google Scholar]
- 6.Maundrell A, Proudman SM. Epidemiology of Raynaud's phenomenon. In: Wigley FM, Herrick AL, Flavahan NA. (eds). Raynaud's phenomenon. New York: Springer, 2015:21–35. [Google Scholar]
- 7.Garner R, Kumari R, Lanyon P, Doherty M, Zhang W. Prevalence, risk factors and associations of primary Raynaud's phenomenon: Systematic review and meta-analysis of observational studies. BMJ Open 2015;5:6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belch J, Carlizza A, Carpentier P, et al. ESVM guidelines - the diagnosis and management of Raynaud's phenomenon. Vasa 2017;46:413–23. [DOI] [PubMed] [Google Scholar]
- 9.Cordeiro RA, de Andrade RM. Raynaud's phenomenon in the occupational context. Rev Assoc Med Bras 2019;65:1314–20. [DOI] [PubMed] [Google Scholar]
- 10.LeRoy EC, Medsger TA. Raynaud's phenomenon: a proposal for classification. Clin Exp Rheumatol 1992;10:485–8. [PubMed] [Google Scholar]
- 11.LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 1988;15:202–5. [PubMed] [Google Scholar]
- 12.Meier FMP, Frommer KW, Dinser R, et al. Update on the profile of the EUSTAR cohort: an analysis of the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis 2012;71:1355–60. [DOI] [PubMed] [Google Scholar]
- 13.Pistorius MA, Planchon B, Schott JJ, Lemarec H. Aspects héréditaires et génétiques de la maladie de Raynaud. J Mal Vasc 2006;31:10–5. [DOI] [PubMed] [Google Scholar]
- 14.Munir S, Freidin MB, Brain S, Williams FMK. Association of Raynaud's phenomenon with a polymorphism in the NOS1 gene. PLoS One 2018;13:e0196279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter SA, Dean E, Kroeger EA. Apparent finger systolic pressures during cooling in patients with Raynaud's syndrome. Circulation 1988;77:988–96. [DOI] [PubMed] [Google Scholar]
- 16.Rogers S, Hughes M. Digital artery vasospasm in primary Raynaud's phenomenon. Eur J Rheumatol 2020. [Epub Ahead of Print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrick AL. The pathogenesis, diagnosis and treatment of Raynaud phenomenon. Nat Rev Rheumatol 2012;8:469–79. [DOI] [PubMed] [Google Scholar]
- 18.Hughes M, Allanore Y, Chung L, Pauling JD, Denton CP, Matucci-Cerinic M. Raynaud's phenomenon and digital ulcers in systemic sclerosis. Nat Rev Rheumatol 2020;4:208–21. [DOI] [PubMed] [Google Scholar]
- 19.Herrick AL, Wigley FM. Raynaud's phenomenon. Best Pract Res Clin Rheumatol 2020;34:101474. [DOI] [PubMed] [Google Scholar]
- 20.Gosk-Bierska I, Misterska-Skóra M, Wasilewska M, et al. Analysis of peripheral nerve and autonomic nervous system function and the stage of microangiopathy in patients with secondary Raynaud's phenomenon in the course of connective tissue diseases. Adv Clin Exp Med 2018;27:1587–92. [DOI] [PubMed] [Google Scholar]
- 21.Bailey SR, Eid AH, Mitra S, Flavahan S, Flavahan NA. Rho kinase mediates cold-induced constriction of cutaneous arteries: Role of α2C-adrenoceptor translocation. Circ Res 2004;94:1367–74. [DOI] [PubMed] [Google Scholar]
- 22.Freedman RR, Baer RP, Mayes MD. Blockade of vasospastic attacks by alpha 2-adrenergic but not alpha 1-adrenergic antagonists in idiopathic Raynaud's disease. Circulation 1995;92:1448–51. [DOI] [PubMed] [Google Scholar]
- 23.Bunker CB, Dowd PM, Terenghi G, Springall DR, Polak JM. Deficiency of calcitonin gene-related peptide in Raynaud's phenomenon. Lancet 1990;336:1530–3. [DOI] [PubMed] [Google Scholar]
- 24.Ames PRJ, Lupoli S, Alves J, et al. The coagulation/fibrinolytic balance in systemic sclerosis: evidence for a haematological stress syndrome. Br J Rheumatol 1997;37:1045–50. [DOI] [PubMed] [Google Scholar]
- 25.Pamuk GE, Turgut B, Pamuk ON, et al. Increased circulating platelet-leucocyte complexes in patients with primary Raynaud's phenomenon and Raynaud's phenomenon secondary to systemic sclerosis: a comparative study. Blood Coagul Fibrinolysis 2007;18:297–302. [DOI] [PubMed] [Google Scholar]
- 26.Herrick AL. Pathogenesis of Raynaud's phenomenon. Rheumatology (Oxford) 2005;44:587–96. [DOI] [PubMed] [Google Scholar]
- 27.Maverakis E, Patel F, Kronenberg DG, et al. International consensus criteria for the diagnosis of Raynaud's phenomenon. J Autoimmun 2014;48–49:60–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Devgire V, Hughes M. Raynaud's phenomenon. Br J Hosp Med (Lond) 2019;80:658–64. [DOI] [PubMed] [Google Scholar]
- 29.Chikura B, Moore T, Manning J, Vail A, Herrick AL. Thumb involvement in Raynaud's phenomenon as an indicator of underlying connective tissue disease. J Rheumatol 2010;37:783–6. [DOI] [PubMed] [Google Scholar]
- 30.Sharp C, Akram Q, Hughes M, Muir L, Herrick AL. Differential diagnosis of critical digital ischemia in systemic sclerosis: Report of five cases and review of the literature. Semin Arthritis Rheum 2016;46:209–16. [DOI] [PubMed] [Google Scholar]
- 31.Haque A, Cleveland T, Powell L, et al. Large vessel disease as a potentially treatable cause of devastating critical digital ischaemia in systemic sclerosis. Clin Rheumatol 2020;39:2823–4. [DOI] [PubMed] [Google Scholar]
- 32.Koenig M, Joyal F, Fritzler MJ, et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud's phenomenon to systemic sclerosis: A twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum 2008;58:3902–12. [DOI] [PubMed] [Google Scholar]
- 33.Hughes M, Ong VH, Anderson ME, et al. Consensus best practice pathway of the UK Scleroderma Study Group: digital vasculopathy in systemic sclerosis. Rheumatology (Oxford) 2015;54:2015–24. [DOI] [PubMed] [Google Scholar]
- 34.Pauling JD, Hackett N, Guida A, Merkel PA. Performance of laser-derived imaging for assessing digital perfusion in clinical trials of systemic sclerosis-related digital vasculopathy: A systematic literature review. Semin Arthritis Rheum 2020. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 35.Baron M, Bell M, Bookman A, et al. Office capillaroscopy in systemic sclerosis. Clin Rheumatol 2007;26:1268–74. [DOI] [PubMed] [Google Scholar]
- 36.Hughes M, Moore T, O’Leary N, et al. A study comparing videocapillaroscopy and dermoscopy in the assessment of nailfold capillaries in patients with systemic sclerosis-spectrum disorders. Rheumatology (Oxford) 2015;54:1435–42. [DOI] [PubMed] [Google Scholar]
- 37.Maricq HR, Carwile LeRoy E. Patterns of finger capillary abnormalities in connective tissue disease by “wide-field” microscopy. Arthritis Rheum 1973;16:619–28. [DOI] [PubMed] [Google Scholar]
- 38.Smith V, Riccieri V, Pizzorni C, et al. Nailfold capillaroscopy for prediction of novel future severe organ involvement in systemic sclerosis. J Rheumatol 2013;40:2023–8. [DOI] [PubMed] [Google Scholar]
- 39.Cutolo M, Herrick AL, Distler O, et al. Nailfold videocapillaroscopic features and other clinical risk factors for digital ulcers in systemic sclerosis: a multicenter, prospective cohort study. Arthritis Rheumatol 2016;68:2527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith V, Vanhaecke A, Herrick AL, et al. Fast track algorithm: How to differentiate a “scleroderma pattern” from a “non-scleroderma pattern”. Autoimmun Rev 2019;18:102394. [DOI] [PubMed] [Google Scholar]
- 41.Smith V, Herrick AL, Ingegnoli F, et al. Standardisation of nailfold capillaroscopy for the assessment of patients with Raynaud's phenomenon and systemic sclerosis. Autoimmun Rev 2020;19:102458. [DOI] [PubMed] [Google Scholar]
- 42.Anderson ME, Moore TL, Lunt M, Herrick AL. The “distal-dorsal difference”: a thermographic parameter by which to differentiate between primary and secondary Raynaud's phenomenon. Rheumatology (Oxford) 2007;46:533–8. [DOI] [PubMed] [Google Scholar]
- 43.Wilkinson JD, Leggett SA, Marjanovic EJ, et al. A multicenter study of the validity and reliability of responses to hand cold challenge as measured by laser speckle contrast imaging and thermography: outcome measures for systemic sclerosis-related Raynaud's phenomenon. Arthritis Rheumatol 2018;70:903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hughes M, Herrick AL. Assessment and management of Raynaud's phenomenon. Prescriber 2017;28:11–6. [Google Scholar]
- 45.Hughes M, Baker A, Farrington S, Pauling JD. Patient organisation-led initiatives can play an important role in raising awareness about Raynaud's phenomenon and encourage earlier healthcare utilisation for high-risk groups. Ann Rheum Dis 2019;78:439–41. [DOI] [PubMed] [Google Scholar]
- 46.Devgire V, Martin AF, McKenzie L, Sandler RD, Hughes M. A systematic review of internet-based information for individuals with Raynaud's phenomenon and patients with systemic sclerosis. Clin Rheumatol 2020;39:2363–7. [DOI] [PubMed] [Google Scholar]
- 47.Lis-S´wie˛ty A. Recent advances in the workup and management of Raynaud phenomenon. Polish Arch Intern Med 2019;129:798–808. [DOI] [PubMed] [Google Scholar]
- 48.Denton C, Hughes M, Gak N, et al. BSR and BHPR guideline for the treatment of systemic sclerosis. Rheumatology (Oxford) 2016;55:1906–10. [DOI] [PubMed] [Google Scholar]
- 49.Fernández-Codina A, Walker KM, Pope JE, Scleroderma Algorithm Group Treatment algorithms for systemic sclerosis according to experts. Arthritis Rheumatol 2018;70:1820–8. [DOI] [PubMed] [Google Scholar]
- 50.Hachulla E, Hatron PY, Carpentier P, et al. Efficacy of sildenafil on ischaemic digital ulcer healing in systemic sclerosis: the placebo-controlled SEDUCE study. Ann Rheum Dis 2016;75:1009–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Negrini S, Magnani O, Matucci-Cerinic M, et al. Iloprost use and medical management of systemic sclerosis-related vasculopathy in Italian tertiary referral centers: results from the PROSIT study. Clin Exp Med 2019;19:357–66. [DOI] [PubMed] [Google Scholar]
- 52.Cruz JE, Ward A, Anthony S, et al. Evidence for the use of epoprostenol to treat Raynaud's phenomenon with or without digital ulcers. Ann Pharmacother 2016;50:1060–7. [DOI] [PubMed] [Google Scholar]
- 53.Hinze AM, Wigley FM. Pharmacotherapy options in the management of Raynaud's phenomenon. Curr Treat Options Rheumatol 2018;4:235–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matucci-Cerinic M, Denton CP, Furst DE, et al. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis 2011;70:32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khanna D, Denton CP, Merkel PA, et al. Effect of Macitentan on the development of new ischemic digital ulcers in patients with systemic sclerosis: DUAL-1 and DUAL-2 randomized clinical trials. JAMA 2016;315:1975–88. [DOI] [PubMed] [Google Scholar]
- 56.Moinzadeh P, Hunzelmann N, Krieg T. Combination therapy with an endothelin-1 receptor antagonist (bosentan) and a phosphodiesterase V inhibitor (sildenafil) for the management of severe digital ulcerations in systemic sclerosis. J Am Acad Dermatol 2011;65:e102–4. [DOI] [PubMed] [Google Scholar]
- 57.Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ 2018;360:j5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chiou G, Crowe C, Suarez P, et al. Digital sympathectomy in patients with scleroderma: an overview of the practice and referral patterns and perceptions of rheumatologists. Ann Plast Surg 2015;75:637– 43 [DOI] [PubMed] [Google Scholar]
- 59.Bello RJ, Cooney CM, Melamed E, et al. The therapeutic efficacy of botulinum toxin in treating scleroderma-associated Raynaud's phenomenon: a randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol 2017;69:1661–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dhaliwal K, Griffin MF, Salinas S, et al. Optimisation of botulinum toxin type a treatment for the management of Raynaud's phenomenon using a dorsal approach: a prospective case series. Clin Rheumatol 2019;38:3669–76. [DOI] [PubMed] [Google Scholar]
- 61.Pauling JD, Frech TM, Hughes M, et al. Patient-reported outcome instruments for assessing Raynaud's phenomenon in systemic sclerosis: A SCTC vascular working group report. J Scleroderma Relat Disord 2018;3:249–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pauling JD, Domsic RT, Saketkoo LA, et al. Multinational qualitative research study exploring the patient experience of Raynaud's phenomenon in systemic sclerosis. Arthritis Care Res (Hoboken) 2018;70:1373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hughes M, Pauling JD, Jones J, Denton CP, Domsic RT, Frech TM, et al. Multicenter qualitative study exploring the patient experience of digital ulcers in systemic sclerosis. Arthritis Care Res (Hoboken) 2020;72:723–33. [DOI] [PubMed] [Google Scholar]
- 64.Hughes M, Khanna DK, Pauling JD. Drug initiation and escalation strategies of vasodilator therapies for Raynaud's phenomenon: can we treat to target? Rheumatology (Oxford) 2020;59:464–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hughes M, Moore T, Manning J, et al. Reduced perfusion in systemic sclerosis digital ulcers (both fingertip and extensor) can be increased by topical application of glyceryl trinitrate. Microvasc Res 2017;111:32–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Curtiss P, Schwager Z, Cobos G, Lo Sicco K, Franks AG. A systematic review and meta-analysis of the effects of topical nitrates in the treatment of primary and secondary Raynaud's phenomenon. J Am Acad Dermatol 2018;78:1110–8.e3. [DOI] [PubMed] [Google Scholar]
- 67.Wasan EK, Zhao J, Poteet J, Mohammed MA, et al. Development of a UV-stabilized topical formulation of nifedipine for the treatment of Raynaud phenomenon and chilblains. Pharmaceutics 2019;11:594. [DOI] [PMC free article] [PubMed] [Google Scholar]