

Abstract
Background
The number of patients living with co-existing diseases is growing. This study aimed to assess the extent of multimorbidity, medication use, and drug- and gene-based interactions in patients following a non-ST elevation acute coronary syndrome (NSTE-ACS).
Methods
In 1456 patients discharged from hospital for a NSTE-ACS, comorbidities and multimorbidity (≥ 2 chronic conditions) were assessed. Of these, 698 had complete drug use recorded at discharge, and 652 (the ‘interaction’ cohort) had drug use and actionable genotypes available for CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A5, DPYD, F5, SLCO1B1, TPMT, UGT1A1, and VKORC1. The following drug interactions were investigated: pharmacokinetic drug-drug (DDIs) involving CYPs (CYPs above, plus CYP1A2, CYP2C8, CYP3A4), SLCO1B1, and P-glycoprotein; drug-gene (DGIs); drug-drug-gene (DDGIs); and drug-gene-gene (DGGIs). Interactions predicted to be ‘substantial’ were defined as follows: DDIs due to strong inhibitors/inducers, DGIs due to variant homozygous/compound heterozygous genotypes, and DDGIs/DGGIs where the constituent DDI/DGI(s) both influenced the victim drug in the same direction.
Results
In the whole cohort, 727 (49.9%) patients had multimorbidity. Non-linear relationships between age and increasing comorbidities and decreasing coronary intervention were observed. There were 98.1% and 39.8% patients on ≥ 5 and ≥ 10 drugs, respectively (from n = 698); women received more non-cardiovascular drugs than men (median (IQR) 3 (1–5) vs 2 (1–4), p = 0.014). Overall, 98.7% patients had at least one actionable genotype. Within the interaction cohort, 882 interactions were identified in 503 patients (77.1%), of which 346 in 252 patients (38.7%) were substantial: 59.2%, 11.6%, 26.3%, and 2.9% substantial interactions were DDIs, DGIs, DDGIs, and DGGIs, respectively. CYP2C19 (49.5% of all interactions) and SLCO1B1 (18.4%) were involved in the largest number of interactions. Multimorbidity (p = 0.019) and number of drugs (p = 9.8 × 10−10) were both associated with patients having ≥ 1 substantial interaction. Multimorbidity (HR 1.76, 95% CI 1.10–2.82, p = 0.019), number of drugs (HR 1.10, 95% CI 1.04–1.16, p = 1.2 × 10−3), and age (HR 1.05, 95% CI 1.03–1.07, p = 8.9 × 10−7), but not drug interactions, were associated with increased subsequent major adverse cardiovascular events.
Conclusions
Multimorbidity, polypharmacy, and drug interactions are common after a NSTE-ACS. Replication of results is required; however, the high prevalence of DDGIs suggests integrating co-medications with genetic data will improve medicines optimisation.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12916-020-01827-z.
Keywords: Multimorbidity, Polypharmacy, Interactions, Pharmacogenomics, Acute coronary syndrome
Background
Coronary heart disease (CHD) is a leading cause of mortality worldwide [1]. The age-adjusted CHD death rate is falling in the developed world [2, 3] leading to a higher proportion of individuals living with CHD; approximately half of this decrease is attributable to improvements in interventional and pharmacological strategies, and the other half to attenuation of risk factors [2]. Additionally, the management of multiple other conditions has improved. Thus, the number of CHD patients with multimorbidity, often defined as the co-existence of two or more chronic diseases [4], is increasing [5].
Multimorbidity increases the prevalence of polypharmacy, which some have defined as five or more medications daily [6]. Polypharmacy however can be appropriate; for instance, after acute coronary syndrome (ACS), guidelines generally recommend dual antiplatelet therapy (aspirin and a P2Y12 inhibitor), a high intensity statin, an angiotensin-converting enzyme inhibitor (ACEI) or angiotensin II receptor blocker (ARB), and a beta blocker [7]. However, multimorbidity often further increases the number of concomitant drugs due to treatment of related conditions, such as ACS risk factors (e.g. hypertension) and sequelae (e.g. heart failure), and treatment of other co-existing diseases. As the number of co-prescribed medications rises, the risk of drug-drug interactions (DDIs) [8, 9] and adverse drug reactions [10] increases.
Pharmacogenomics is the study and application of the genomic determinants of drug response. Therapeutic recommendations (e.g. dose or drug selection) for ‘actionable’ drug-gene interactions (DGIs) involving germline genotypes have been developed by international guideline committees for over 75 drugs, which include cardiovascular drugs and many other commonly prescribed medications [11]. Some drugs, such as warfarin, have recommendations involving more than one gene leading to drug-gene-gene interactions (DGGIs). Several large pharmacogenomics clinical implementation programmes are underway [12]; for example, the Ubiquitous Pharmacogenomics implementation study has recruited ⁓ 7000 patients from seven European sites [12]. Therefore, although pharmacogenomics is not yet commonplace, it is expected to become increasingly available in clinical practice in the coming years.
Interactions can primarily affect a drug’s pharmacokinetics or pharmacodynamics. Many pharmacokinetic-based interactions arise through the altered function of drug-metabolising enzymes (e.g. phase I cytochrome P450 enzymes (CYPs)) and transporters (e.g. P-glycoprotein (P-gp) encoded by ABCB1, and organic anion-transporting polypeptide 1B1 (OATP1B1) encoded by SLCO1B1). Interestingly, around half of the 200 most commonly used drugs undergo CYP-mediated metabolism [13] with CYP3A4/5, CYP2D6, CYP2C9, CYP2B6, and CYP2C19 responsible for most of this xenobiotic metabolism [14]. Importantly, the function of these enzymes and transporters can be significantly influenced by both interacting drugs [15, 16] and common genetic variants [11]. Failure to consider both factors, independently and combined, can lead to imprecise predictions regarding the function of drug-metabolising enzymes, transporters, and pharmacodynamic targets [17]. This may be one reason why it is currently challenging to predict which patients will be clinically affected by a DDI and its severity. Therefore, assessment of how DDIs are affected by genetic variation in one or more metabolic pathways (Fig. 1) is an important area to investigate to a have fuller understanding of potential adverse clinical consequences. However, drug- and gene-based interactions have only been studied together in real-world patients in a handful of studies [18–20], and no studies have focused on patients at the point of hospital discharge nor following a non-ST elevation ACS (NSTE-ACS).
Fig. 1.
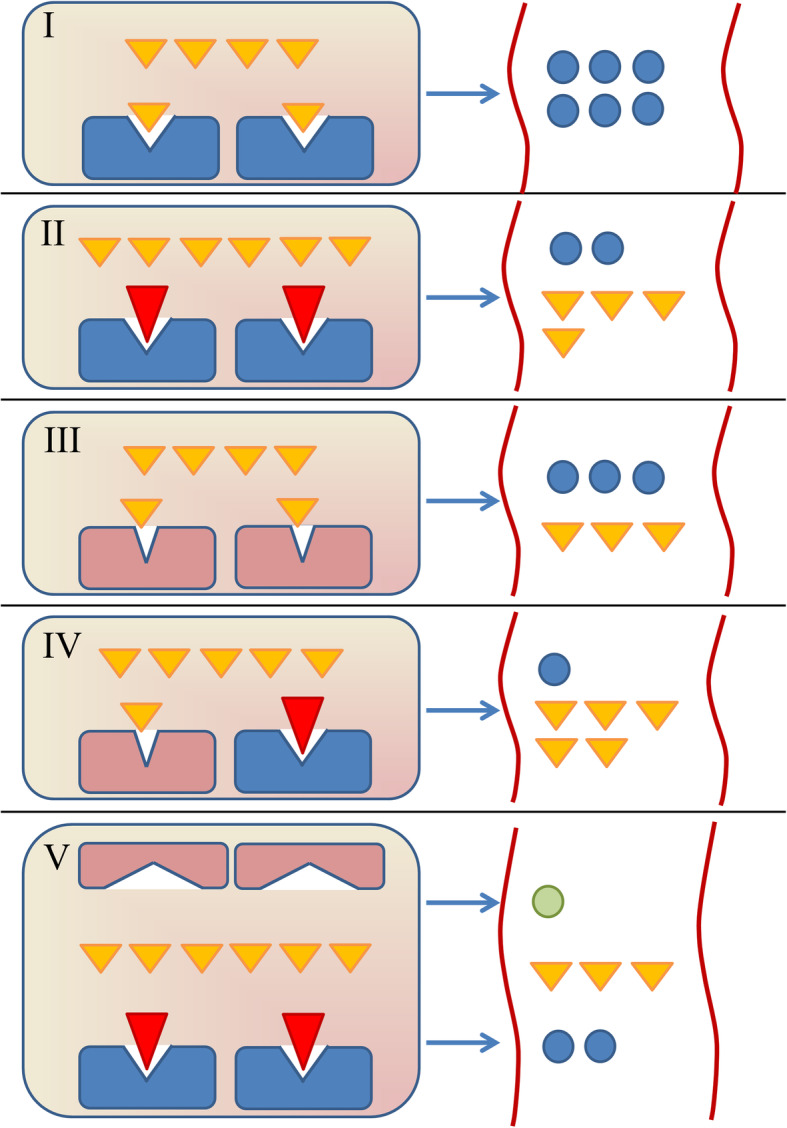
Drug-drug, drug-gene, and drug-drug-gene interactions. The hepatic extensive (normal) metabolism of small molecule drug A (yellow triangles) requires functional drug-metabolising enzyme Z (DME-Z, blue rectangles) for metabolism to metabolite A (blue circles), as shown in I. In II, DME-Z is inhibited by perpetrator drug B (red triangle) leading to reduced drug A metabolism, and hence a drug-drug interaction (DDI). Similarly in III, the function of DME-Z is reduced by genetic variation (red rectangles) conferring a poor metaboliser (homozygous) genotype, leading to reduced drug A metabolism, and hence a drug-gene interaction (DGI). In IV, a drug-drug-gene interaction (DDGI) is depicted whereby both genetic variation (e.g. a heterozygous intermediate metaboliser genotype) and perpetrator drug B conceivably act together to collectively reduce DME-Z function, strongly attenuating drug A metabolism. In V, drug A is primarily metabolised by DME-Z, but also a second enzyme, DME-Y, (flattened red rectangles) involved to a lesser extent is shown. If perpetrator drug B inhibits DME-Z, less drug A is metabolised by DME-Z and so drug A metabolism is more reliant on DME-Y conversion to metabolite B (green circle). However, if the genotype of DME-Y confers a reduced function (poor metaboliser DME-Y phenotype depicted in V), then the overall metabolism of drug A will be greatly reduced, constituting a DDGI. Similarly, if the functions of both DME-Z and DME-Y were affected by genetic variants, a drug-gene-gene interaction (DGGI) would manifest. The result of all of these interactions is less metabolism and so increased systemic exposure to drug A. These interactions equally apply to perpetrator drug inducers and genetic variation conferring rapid/ultra-rapid metaboliser predicted phenotypes. Similarly, these interactions apply to both deactivating metabolism of active drugs and bioactivation of prodrugs. Beyond enzymes, pharmacokinetic interactions can also be mediated by drug- and/or gene-based alterations to drug transporters
Therefore, the aim of this work was to determine the extent of multimorbidity, medication use, and drug- and gene-based mainly pharmacokinetic interactions in patients discharged after a NSTE-ACS, to explore the influence of patient age and sex on these factors, and to investigate the impact of these factors on subsequent clinical events.
Methods
Participants
This investigation utilised the Pharmacogenetics of Acute Coronary Syndrome (PhACS) study, which has been described previously [21]. Briefly, PhACS was a UK 16-site prospective observational study between 2008 and 2013 that recruited 1470 patients hospitalised with an NSTE-ACS. Standard data collection included medical history and cardiovascular medications at discharge; other medications were optionally recorded in an open-ended medication appendix. Participants were followed up for incident events at 1 and 12 months, and the study ended when all had received 12 months follow-up. However, whilst the study was running, participants recruited earlier were followed up annually after their 12-month visit. The protocol was approved by the Liverpool UK (adult) research Ethics Committee (07/H1005/117), site-specific approval was granted at all study sites, and written informed consent was ascertained from all study subjects in accordance with the Declaration of Helsinki.
In this present study, all PhACS patients alive at discharge from their index hospitalisation (n = 1456) were used to describe clinical characteristics and secondary prevention cardiovascular drug use, and all with available genetic data were used to assess the frequency of actionable genetic variants in pharmacogenes. Patients whose non-cardiovascular and cardiovascular drugs were both known at discharge were used to describe total drug use (n = 698), and of these patients, those who had quality-controlled (QC) genetic data were used to assess all drug and gene interactions (n = 652, the ‘interaction cohort’) (Fig. 2).
Fig. 2.
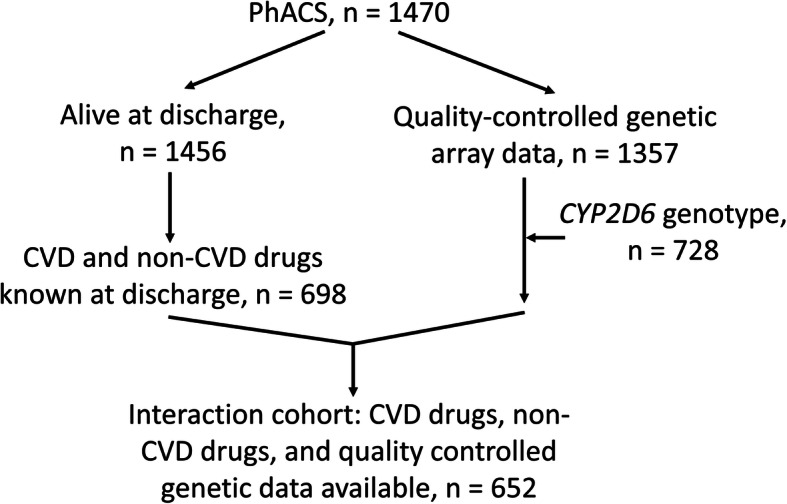
Study cohort selection process. There were 1470 patients included in the PhACS study, of whom 1456 were discharged alive from their index NSTE-ACS; 1357 had array genetic data available following standard quality control procedures; CYP2D6 genotype was available in 728 after determination of copy number variation in those on CYP2D6 substrates; 698 had non-cardiovascular disease (CVD) drugs at discharge recorded in their case report form (CRF) medication appendix, in addition to CVD drugs explicitly required in the CRF; and in 652 patients (the interaction cohort), CVD drugs, non-CVD drugs, and genetic data were available (although patients not on a CYP2D6 substrate did not require CYP2D6 genotype for inclusion in the interaction cohort)
Clinical characteristics, multimorbidity, and medication use
Within the whole cohort (n = 1456), sex, age, body mass index (BMI), smoking, pre-existing comorbidities, raised troponin at index NSTE-ACS, investigation of index NSTE-ACS by coronary catheterisation and treatment by percutaneous coronary intervention or coronary artery bypass grafting (PCI/CABG), and use of secondary prevention drugs (aspirin, P2Y12 inhibitor, ACEI/ARB, beta blocker, statin) at discharge were determined. Comorbidities were further subdivided into cardiovascular and non-cardiovascular conditions. Pre-existing cardiovascular multimorbidity was defined as ≥ 2 of the following: hypertension, prior myocardial infarction (MI, did not include index NSTE-ACS), stroke, transient ischaemic attack, subarachnoid haemorrhage, and peripheral artery disease. Pre-existing non-cardiovascular multimorbidity was ≥ 2 of the following: chronic pulmonary disease (chronic obstructive pulmonary disease, contemporary asthma, bronchiectasis, pulmonary fibrosis), diabetes, chronic kidney disease (CKD, serum creatinine > 150 μmol/L) or renal transplantation, peptic ulcer disease, chronic liver disease (alcoholic liver disease, cirrhosis, autoimmune hepatitis), osteoarthritis, connective tissue disease (e.g. rheumatoid arthritis, polymyalgia rheumatica), chronic neurological conditions (e.g. epilepsy, motor neuron disease, dementia, Parkinson’s disease), and any cancer. Only physical illnesses were recorded in PhACS; mental health disease data were not available. Overall multimorbidity was defined as ≥ 2 of any of the above cardiovascular and non-cardiovascular conditions, in keeping with the Academy of Medical Sciences definition [22].
The individually recorded discharge medications in those with complete drug data (n = 698) were grouped together according to the British National Formulary chapter-level categories [23]. However, drugs in the skin, eye, and ear/nose/oropharynx chapters were combined here due to being mainly non-oral. An ‘Other’ category was also constructed, consisting mainly of small numbers of drugs from the malignant disease/immunosuppression and gynaecology/urinary tract disorder chapters. A patient was counted for a category if they were on one or more drugs within that category.
Identification of actionable genetic variants
Participants were genotyped using the Illumina HumanOmniExpressExome-8 v1.0 BeadChip at Edinburgh Genomics (Roslin Institute, Scotland). The genetic QC and imputation for PhACS have been described previously, with 1357 patients passing all QC [24]. The following actionable variants were extracted: CYP2B6*6/*9 (rs3745274), CYP2C9*2 (rs1799853), *3 (rs1057910), CYP2C19*2 (rs4244285), *17 (rs12248560), CYP3A5*3 (rs776746), CYP2D6*3 (rs35742686), *4 (rs3892097), *9 (rs5030656), *10 (rs1065852), *41 (rs28371725), DPYD HapB3 (rs56038477), F5 (rs6025), SLCO1B1*5/*15 (rs4149056), TPMT*3B (rs1800460), *3C (rs1142345), *3A (rs1800460 with rs1142345), UGT1A1 -364C>T (rs887829), and VKORC1 1173C>T (rs9934438). The variant allele of UGT1A1 rs887829 is in linkage disequilibrium (r2 ≈ 0.99) with the tandem nucleotide repeat polymorphism (UGT1A1*28) [25]. Of these 18 variants, 10 contained imputed values (information scores all > 0.7, indicating good imputation quality). If required, imputed genotypes were categorised as wild-type (imputed score 0–0.3), heterozygous (0.7–1.3), and homozygous (1.7–2).
CYP2D6 copy number variants (CNVs) were identified with the TaqMan™ copy number assay (Thermo Fisher Scientific) in those on CYP2D6 substrate drugs (n = 728). qPCR data were acquired using Quantstudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific) and analysed with CopyCaller v2.1 software (Life Technologies). CYP2D6 genotype to metaboliser phenotype translation was based on recent consensus recommendations from the Clinical Pharmacogenetics Implementation Consortium (CPIC) and Dutch Pharmacogenetics Working Group (DPWG) [26], determined from CYP2D6 activity score (AS) as follows: poor metaboliser (PM, AS = 0), intermediate metaboliser (IM, AS = 0.25–1), extensive metaboliser (EM, AS = 1.25–2.25), and ultra-rapid metaboliser (UM, AS > 2.25). The metaboliser phenotypes for the other CYPs were determined from CPIC standard diplotype-phenotype tables [11].
Drug substrates, inhibitors, and inducers for drug-drug interactions
Drug substrates, inhibitors, and inducers for CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5 are listed in Additional files 1, 2, and 3 [15, 16, 27] and were based on the US Food and Drug Administration (FDA) clinical tables [15] and the Indiana University cytochrome P450 drug interactions Flockhart Table™ [16] (both accessed January 2019), with specific input from the literature when required [27]. Tobacco smoke constituted a CYP1A2 inducer.
Substrates and inhibitors for drug transporters, P-gp and SLCO1B1, are in Additional file 4 [15, 28–30]. Transporter substrates/inhibitors were extracted from the FDA clinical tables [15] with supplementation of P-gp substrates and addition of P-gp inducers from Wessler et al. [28]; no SLCO1B1 inducers were listed. Assessment of the strength of transporter inhibitors/inducers was based on relevant literature [28–30].
A DDI was present if a patient was on a victim drug and perpetrator drug that influenced the victim drug through one or more investigated enzymes/transporters; autoinhibition/autoinduction was not considered interactions.
Drug substrates for drug-gene interactions
For DGIs, substrate drugs were limited to those with a pharmacogenomic clinical guideline available from CPIC/DPWG for the above genotyped pharmacogenes (Additional file 5) [11, 31–41]. For each substrate, only the genotypes/metaboliser statuses considered actionable for that drug contributed to a DGI; thus, for example, CYP2D6 IM was considered a DGI with amitriptyline but not codeine.
Assessment of drug and gene interactions
We considered pharmacokinetic drug- and gene-based interactions and two pharmacodynamic interactions: warfarin-VKORC1 rs9934438 (affects warfarin dose) and oestrogen-containing contraceptives-F5 rs6025 (augments the risk of venous thromboembolism).
First, we identified and counted the total number of unique DDIs, DGIs, drug-drug-gene interactions (DDGIs), and DGGIs of any strength (‘all’ interactions). Second, we identified those interactions predicted to have a ‘substantial’ effect, defined as follows:
-
i)
DDIs involving strong drug inhibitors/inducers. Strong CYP inhibitors increase the area under the concentration-time curve (AUC) of sensitive substrates ≥ 5-fold, strong CYP inducers decrease the AUC of sensitive substrates by ≥ 80%, and strong transporter inhibitors/inducers were determined from the literature [28–30].
-
ii)
DGIs due to variant homozygous/compound heterozygous genotypes.
-
iii)
DDGIs due to any strength perpetrator drug and actionable genotype provided the directions of effect of the constituent DDI and DGI on the victim drug were the same (i.e. all interactions would be expected to increase/decrease victim drug exposure/response). The same principle was applied to DGGIs.
The numbers of interactions per patient and gene were determined. Unless otherwise stated, each interaction in a given patient was only counted once. Thus, for example, a DDGI was counted as one DDGI, whilst its constituent DDI and DGI were not counted.
Clinical endpoints
The main endpoints of PhACS were major adverse cardiovascular events (MACE—a composite of cardiovascular death and non-fatal MI or ischaemic stroke) and all-cause mortality (ACM) [21]. It was expected that interactions influencing cardiovascular drugs would predominate given PhACS is a cardiovascular cohort. Thus, these available main endpoints were harnessed to explore whether interactions and other relevant factors (see below) might be associated with clinical sequelae. A calculation of power was not conducted as this assessment was exploratory using available events.
Statistical analysis
Categorical clinical variables are presented as group number (percentage), age as mean (standard deviation) as normally distributed, and BMI as median (interquartile range (IQR) as non-normally distributed; integer count variables are presented as median (IQR/range) for numbers of drugs and interactions, or number of all comorbidities (percentage).
To assess for baseline differences between the interaction cohort and the remainder of the whole cohort, the following were compared: clinical characteristics and secondary prevention drug use (Pearson’s chi-squared test), number of comorbidities and overall number of cardiovascular drugs (Fisher’s exact test), age (Student’s t test), and BMI (Mann-Whitney U test).
In the whole cohort, age was associated with patient sex (p = 2.0 × 10−8). Thus, the influence of sex and age on clinical variables/drug use was ascertained by their inclusion in appropriate regression (linear/logistic/Poisson/negative binomial) models. In all analyses involving age, when a quadratic relationship with an outcome was suspected from visual inspection, age and age squared were entered into the model. In this situation, age was mean centred before its squared term calculated. When a linear relationship was identified, uncentred age was used.
To investigate the association between multimorbidity and number of drugs, Poisson regression was used, whilst logistic regression assessed whether multimorbidity was associated with the number of patients that received all five secondary prevention cardiovascular drug classes (vs < 5); both analyses were adjusted for age.
Multicollinearity was tested between age (or with mean centred age and age squared), sex, multimorbidity, number of drugs, and all and substantial interactions and was not found (highest variance inflation factor < 1.73). Thus, age, sex, multimorbidity, and number of drugs underwent univariate and multivariable logistic regression with forward (likelihood ratio) variable selection to investigate associations with patients that have ≥ 1 interaction, or ≥ 1 substantial interaction (vs patients with < 1). Lastly, univariate and multivariable Cox’s proportional hazards regression tested whether age, sex, multimorbidity, number of drugs, and all or substantial interactions were associated with time to incident MACE, or ACM, during follow-up within the interaction cohort.
In sensitivity analyses, dichotomous multimorbidity was replaced by number of comorbidities and all relevant analyses repeated. p < 0.05 was taken as statistically significant as all analyses were exploratory. Statistical analysis was carried out in the R computing environment [42] version 3.5.1 or above and IBM SPSS version 24.0 (IBM Corp, Armonk, NY, USA).
Results
Baseline differences in cohorts
Table 1 reports baseline differences between the interaction cohort (n = 652) and other patients from the whole cohort (n = 804). The interaction cohort was marginally older (p = 0.012) and included more women (p = 0.040). The main difference was the higher prevalence of multimorbidity in the interaction cohort (59.0% vs 42.5%, p = 3.7 × 10−10).
Table 1.
Comparison of those included and not included in the interaction cohort
| Characteristic | Interaction cohort, n = 652 | Not in interaction cohort, n = 804 | Unadjusted p value |
|---|---|---|---|
| Demographics | |||
| Sex (male), n (%) | 457 (70.1) | 600 (74.6) | 0.040 |
| Age (years), mean (SD) | 66.0 (11.5) | 64.4 (11.8) | 0.012 |
| BMI, median (IQR) | 28.3 (25.3–31.8) | 27.9 (24.9–31.4) | 0.21 |
| Comorbidities | |||
| Smoking, n (%) | 185 (28.4) | 180 (22.4) | 0.011 |
| Cardiovascular multimorbidity | 218 (33.4) | 175 (21.8) | 6.1 × 10−7 |
| Non-cardiovascular multimorbidity | 167 (25.6) | 98 (12.2) | 4.1 × 10−11 |
| All multimorbidity | 385 (59.0) | 342 (42.5) | 3.7 × 10−10 |
| Number of comorbidities, n (%) | |||
| 0 | 116 (17.8) | 205 (25.5) | 5.0 × 10−4 |
| 1 | 151 (23.2) | 257 (32.0) | |
| 2 | 152 (23.3) | 174 (21.6) | |
| 3 | 107 (16.4) | 105 (13.1) | |
| 4–5 | 117 (17.9) | 55 (6.8) | |
| 6–7 | 9 (1.4) | 8 (1.0) | |
| Index NSTE-ACS | |||
| Raised troponin, n (%) | 623 (95.6) | 765 (95.1) | 0.45 |
| Coronary catheterisation, n (%) | 445 (68.3) | 529 (65.8) | 0.32 |
| PCI/CABG, n (%) | 283 (43.4) | 376 (46.8) | 0.20 |
| Cardiovascular drugs at discharge | |||
| Aspirin, n (%) | 612 (93.9) | 758 (94.3) | 0.52 |
| P2Y12 inhibitor, n (%) | 556 (85.3) | 680 (84.6) | 0.81 |
| ACEI/ARB, n (%) | 540 (82.8) | 651 (81.0) | 0.39 |
| Beta blocker, n (%) | 528 (81.0) | 664 (82.6) | 0.34 |
| Statin, n (%) | 609 (93.4) | 767 (95.4) | 0.026 |
| Number of patients on all five secondary prevention cardiovascular drugs, n (%)† | 373 (57.2) | 464 (57.7) | 0.84 |
| Number of cardiovascular drugs/patient, median (IQR, range)†† | 6 (5–7, 0–9) | 6 (5–6, 2–10) | 0.11 |
ACEI angiotensin-converting enzyme inhibitor, ARB angiotensin II receptor blocker, CABG coronary artery bypass graft surgery, IQR interquartile range, n (%) number (percent) of patients, PCI percutaneous coronary intervention, SD standard deviation
†Secondary prevention cardiovascular drugs were aspirin, P2Y12 inhibitor, ACEI/ARB, beta blocker, and a statin
††Includes secondary prevention and other cardiovascular drugs
Clinical variables, comorbidities, medication use, and influence of patient age and sex
Patient clinical characteristics and medication use are shown in Table 2. In the whole cohort (n = 1456), 72.6% were male, mean age was 65.1 years old (standard deviation 11.7, range 26.4–93.4), 26.9% had cardiovascular multimorbidity, 18.1% had non-cardiovascular multimorbidity (most commonly diabetes, then osteoarthritis and chronic pulmonary diseases), 49.9% had ≥ 2 any comorbidities, and 4.7% had ≥ 5 any comorbidities. The index NSTE-ACS was treated with PCI/CABG in 45.3% patients, and 57.5% received a drug from all five secondary prevention cardiovascular drug classes at discharge. In those with full drug data (n = 698), 98.1%, 39.8%, and 6.2% were on at least five, ten, and 15 different drugs, respectively; the median numbers of cardiovascular, non-cardiovascular, and total drugs per patient were 6 (IQR 5–7, range 0–9), 2 (IQR 1–4, range 0–16), and 9 (IQR 7–11, range 2–26), respectively.
Table 2.
Clinical characteristics, multimorbidity, and medication use
| Characteristic | Female, n = 396 | Male, n = 1057 | Adjusted p value† | All, n = 1456†† |
|---|---|---|---|---|
| Demographics | ||||
| Age (years), mean (SD) | 68.0 (11.6) | 64.1 (11.6) | 2.0 × 10−8 | 65.1 (11.7) |
| BMI, median (IQR) | 27.5 (24.2–32.5) | 28.3 (25.4–31.5) | 0.85 | 28.1 (25.0–31.6) |
| Comorbidities | ||||
| Smoking, n (%) | 98 (24.7) | 266 (25.2) | 0.060 | 365 (25.1) |
| Cardiovascular multimorbidity, n (%) | 104 (26.2) | 288 (27.2) | 0.12 | 392 (26.9) |
| Non-cardiovascular multimorbidity, n (%) | 96 (24.2) | 168 (15.9) | 8.0 × 10−3 | 264 (18.1) |
| All multimorbidity, n (%)††† | 217 (54.8) | 507 (48.0) | 0.39 | 727 (49.9) |
| Number of comorbidities, n (%)††† | ||||
| 0 | 77 (19.4) | 244 (23.1) | 0.60 | 321 (22.0) |
| 1 | 102 (25.8) | 306 (28.9) | 408 (28.0) | |
| 2 | 97 (24.5) | 227 (21.5) | 326 (22.4) | |
| 3 | 56 (14.1) | 155 (14.7) | 212 (14.6) | |
| 4–5 | 59 (14.9) | 113 (10.7) | 172 (11.8) | |
| 6–7 | 5 (1.3) | 12 (1.1) | 17 (1.2) | |
| Index NSTE-ACS | ||||
| Raised troponin, n (%) | 378 (95.5) | 1007 (95.3) | 0.91 | 1388 (95.3) |
| Coronary catheterisation, n (%) | 250 (63.1) | 723 (68.4) | 0.49 | 974 (66.9) |
| PCI/CABG, n (%) | 156 (39.4) | 502 (47.5) | 0.082 | 659 (45.3) |
| Cardiovascular drugs at discharge | ||||
| Aspirin, n (%) | 371 (93.7) | 996 (94.2) | 0.82 | 1370 (94.1) |
| P2Y12 inhibitor, n (%) | 334 (84.3) | 899 (85.1) | 0.89 | 1236 (84.9) |
| ACEI/ARB, n (%) | 318 (80.3) | 871 (82.4) | 0.61 | 1191 (81.8) |
| Beta blocker, n (%) | 300 (75.8) | 889 (84.1) | 1.8 × 10−3 | 1192 (81.9) |
| Statin, n (%) | 367 (92.7) | 1006 (95.2) | 0.14 | 1376 (94.5) |
| Patients on all five secondary prevention cardiovascular drugs, n (%)‡ | 205 (51.8) | 630 (59.6) | 0.081 | 837 (57.5) |
| Drug use by category at discharge | ||||
| N = 698‡‡ | n = 208 | n = 490 | n = 698 | |
| Gastro-intestinal, n (%) | 129 (62.0) | 267 (54.5) | 0.18 | 396 (56.7) |
| Cardiovascular, n (%) | 207 (99.5) | 490 (100.0) | 0.99 | 697 (99.9) |
| Respiratory, n (%) | 45 (21.6) | 99 (20.2) | 0.85 | 144 (20.6) |
| Central nervous system, n (%) | 85 (40.9) | 160 (32.7) | 0.026 | 245 (35.1) |
| Infections, n (%) | 17 (8.2) | 33 (6.7) | 0.65 | 50 (7.2) |
| Endocrine, n (%) | 83 (39.9) | 136 (27.8) | 0.012 | 219 (31.4) |
| Nutrition and blood, n (%) | 44 (21.2) | 63 (12.9) | 0.024 | 107 (15.3) |
| Musculoskeletal, n (%) | 36 (17.3) | 65 (13.3) | 0.37 | 101 (14.5) |
| Eye, ear, nose, oropharynx, and skin, n (%) | 10 (4.8) | 28 (5.7) | 0.33 | 38 (5.4) |
| Other, n (%) | 9 (4.3) | 41 (8.4) | 0.017 | 50 (7.2) |
| Numbers of drugs at discharge | ||||
| Number of cardiovascular drugs/patient, median (IQR, range) | 6 (5–6, 0–9) | 6 (5–7, 2–9) | 0.21 | 6 (5–7,0–9) |
| Number of non-cardiovascular drugs/patient, median (IQR, range) | 3 (1–5, 0–15) | 2 (1–4, 0–16) | 0.014 | 2 (1–4, 0–16) |
| Number of drugs/patient, median (IQR, range) | 9 (8–11, 2–22) | 9 (7–11, 3–26) | 0.20 | 9 (7–11, 2–26) |
ACEI angiotensin-converting enzyme inhibitor, ARB angiotensin II receptor blocker, CABG coronary artery bypass graft surgery, IQR interquartile range, n (%) number (percent) of patients, PCI percutaneous coronary intervention, SD standard deviation
†Adjusted for age, except when testing the association between sex and age itself
††The sex of three patients was missing from the n = 1456 cohort; thus, these patients were not included in the sex-stratified columns, but were counted in the All column
†††The age-adjusted p value for the association between sex and all comorbidities was 0.39 when comorbidities were dichotomised into multimorbidity (≥ 2 vs < 2 any cardiovascular or non-cardiovascular conditions) and p = 0.60 when number of comorbidities was treated as an integer count within negative binomial regression
‡Secondary prevention cardiovascular drugs were aspirin, P2Y12 inhibitor, ACEI/ARB, beta blocker, and a statin
‡‡For drug categories within the n = 698 cohort, a patient was counted if they were on one or more drugs within a given category
As expected, increasing age was strongly associated with increased multimorbidity (p = 1.8 × 10−22) and higher total drug use (p = 2.1 × 10−8). Increasing age was also associated with fewer patients currently smoking (p = 6.0 × 10−35) or receiving drugs from all five secondary prevention drug classes (p = 1.5 × 10−8) (see Additional file 6, which additionally includes results if age dichotomised at 65). Interestingly, non-linear relationships were observed between age and number of comorbidities (a decline in the rate of acquiring comorbidities at older age), and especially with both coronary catheterisation and PCI/CABG for the index NSTE-ACS (a decrease in interventions at older age, see Additional file 7). No other non-linear age relationships were detected (including when investigating interactions, MACE, and ACM).
Women were almost 4 years older on average in the whole cohort (Table 2). After adjustment for age, women had more non-cardiovascular multimorbidity than men (24.2% vs 15.9%, p = 0.008) but there were no differences between the sexes in cardiovascular multimorbidity (p = 0.12) or overall number of comorbidities (p = 0.60). Trends were observed for fewer women receiving PCI/CABG (39.4% vs 47.5%, p = 0.082) or drugs from all five cardiovascular secondary prevention drug classes (51.8% vs 59.6%, p = 0.081) compared to men. Notably, women were prescribed more non-cardiovascular drugs than men (median (IQR) 3 (1–5) vs 2 (1–4), p = 0.014). Specifically, a higher proportion of women received one or more drugs in the central nervous system category (40.9% vs 32.7%, p = 0.026, mainly antidepressants and analgesics), endocrine system category (39.9% vs 27.8%, p = 0.012, a combination including more insulin, levothyroxine, bisphosphonates, and oestrogens/progestrogens), and nutrition and blood category (21.2% vs 12.9%, p = 0.024; mainly iron supplementation and vitamin replacement) compared to men. Overall though, total drug use did not differ between women and men (median (IQR) of 9 (8–11) vs 9 (7–11), p = 0.20).
All multimorbidity (≥ 2 conditions vs < 2), adjusted for age, was strongly associated with both increased total number of drugs/patient (median (IQR) 10 (8–12) vs 8 (7–9), p < 2.0 × 10−16) and less patients being prescribed all five secondary prevention cardiovascular drug classes (51.4% vs 64.9%, respectively, p = 1.7 × 10−4). These associations persisted when multimorbidity was stratified into cardiovascular and non-cardiovascular multimorbidity, although non-cardiovascular multimorbidity was particularly strongly associated with fewer patients receiving all five secondary prevention drug classes (39.2% vs 62.4%, age-adjusted p = 5.8 × 10−9).
Patient genotypes
Figure 3 shows patient genotype-based metaboliser phenotypes (CYPs) and genotypes (non-CYPs) for actionable pharmacogenes. Importantly, 98.7% of patients with genotypes available for all analysed genes (n = 713 inclusive of CYP2D6 CNVs) had at least one actionable genotype (range 0–7 actionable genotypes from 11 genes).
Fig. 3.

Patient pharmacogene genotypes. Proportions of genotype-based CYP metaboliser phenotypes (a) and non-CYP pharmacogene genotypes (b) observed across the whole cohort with quality-controlled array genetic data available (n = 1357), except for CYP2B6 (n = 1347), F5 (n = 1346), and CYP2D6 (n = 728). The number of patients with CYP2B6, F5, and CYP2D6 available was less due to exclusion of patients with imputed genotypes outside the predefined imputation acceptance range, and determination of CYP2D6 copy number variation in patients on a CYP2D6 drug substrate (see the ‘Methods’ section). Overall, 713 patients had genotypes available for all analysed genes. EM, extensive (normal) metaboliser; IM, intermediate metaboliser; PM, poor metaboliser; RM, rapid metaboliser; UM, ultra-rapid metaboliser; WT, wild-type; HET, heterozygous; HOM, homozygous for the variant allele
Drug and gene interactions
In the interaction cohort (n = 652), the overall numbers of different drug substrates, inhibitors, and inducers considered were 199, 109, and 27, respectively. Twenty-four drugs with an associated pharmacogenomic guideline were prescribed in the interaction cohort, 17 of which contributed to ≥ 1 observed DGI (Additional file 5). The genes with DGIs were as follows: CYP2C9, CYP2C19, CYP2D6, CYP3A5, SLCO1B1, and VKORC1.
Overall, 882 interactions in 503 (77.1%) patients were identified, whilst 346 substantial interactions occurred in 252 (38.7%) patients (Fig. 4a). Almost half (45.0%) of all interactions were DDIs with 41.2% DGIs, 12.7% DDGIs, and 1.1% DGGIs. For substantial interactions, 59.2% were DDIs, 11.6% DGIs, 26.3% DDGIs, and 2.9% DGGIs. The observed DGGIs were due to either warfarin/CYP2C9/VKORC1 or amitriptyline/CYP2D6/CYP2C19 interactions. A complete breakdown of all and substantial interactions is in Additional files 8 and 9, respectively.
Fig. 4.
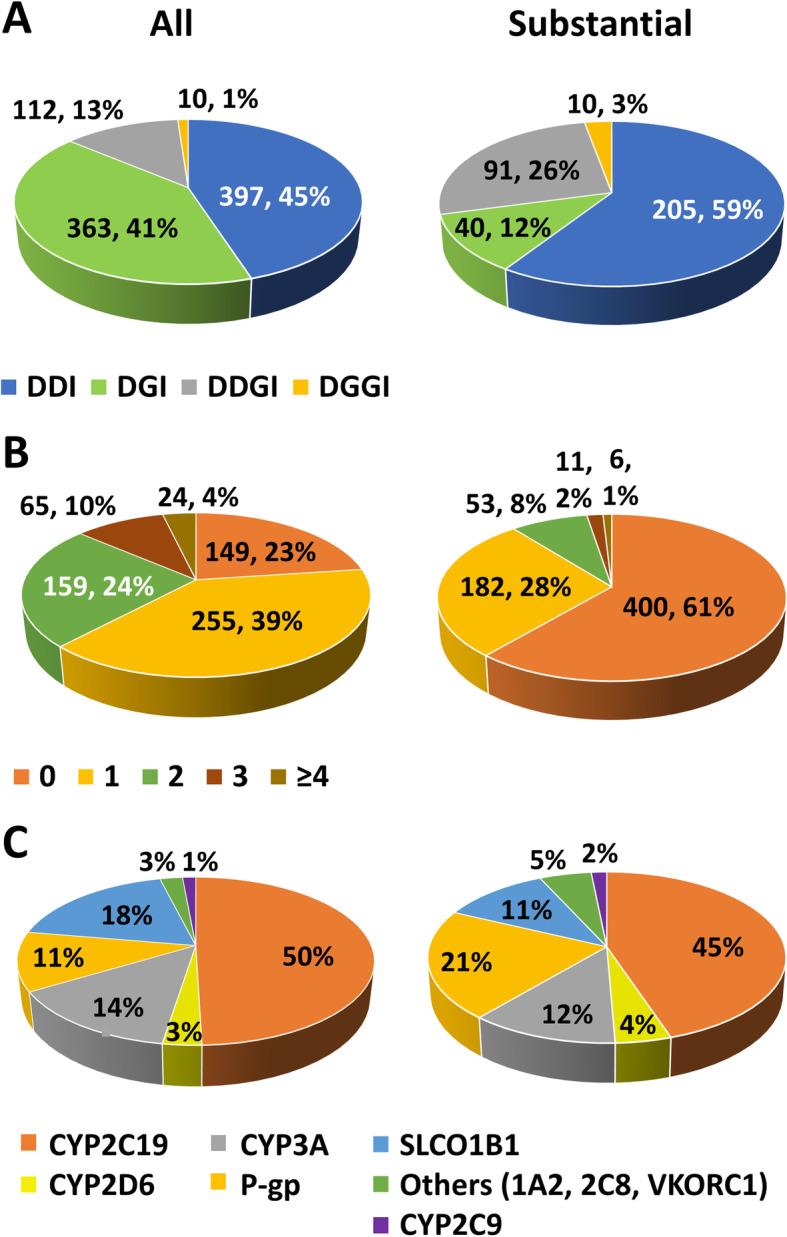
Drug- and gene-mediated interactions. This figure shows the total number (%) of interactions identified (a), the number (%) of interactions per patient (b), and the percentage of interactions mediated per enzyme/transporter (c) in 652 post-NSTE-ACS patients. The pie charts on the left display all interactions (of any or unconfirmed strength), whilst predicted substantial interactions only are presented in the charts on the right. In a and b, each interaction was counted only once. In c, some types of interaction were counted more than once in a given patient. These were as follows: DDIs where a perpetrator drug can influence the same victim drug through more than one enzyme/transporter (e.g. verapamil inhibiting atorvastatin at both CYP3A4/5 and P-gp), DDGIs involving more than one enzyme/transporter, and DGGIs. These interactions were included in the count of both involved enzymes/transporters to illustrate them clearly in c only
Of those with interactions, the median number (range) of all and substantial interactions per patient was 1 (1–9) and 1 (1–5), respectively (Fig. 4b). Interactions involving CYP2C19 were the most common (49.5% of all interactions), followed by SLCO1B1 (18.4%), CYP3A4/5 (13.8%), and P-gp (11.4%, Fig. 4c). Almost a third of all and substantial interactions involved transporters (P-gp, SLCO1B1, Fig. 4c). CYP2C9, CYP2C19, CYP2D6, and SLCO1B1 mediated both drug- and genotype-based interactions (Fig. 5). CYP2C19 interactions were the most common as 84.7% of patients were prescribed the CYP2C19 substrate, clopidogrel, and 44.2% a proton pump inhibitor (44.4% of these were (es)omeprazole, 52.8% lansoprazole, 2.8% other), which are CYP2C19 substrates and inhibitors. The majority of SLCO1B1 interactions were due to atorvastatin (79.9% of patients received) or, to a lesser extent, simvastatin (10.0% received). Thus, whilst DDIs made up 41.5% (202/487) of all CYP2C19-mediated interactions, 96.7% (175/181) of SLCO1B1-mediated interactions were dependent on SLCO1B1 rs4149056 (77.9% DGIs, 18.8% DDGIs).
Fig. 5.
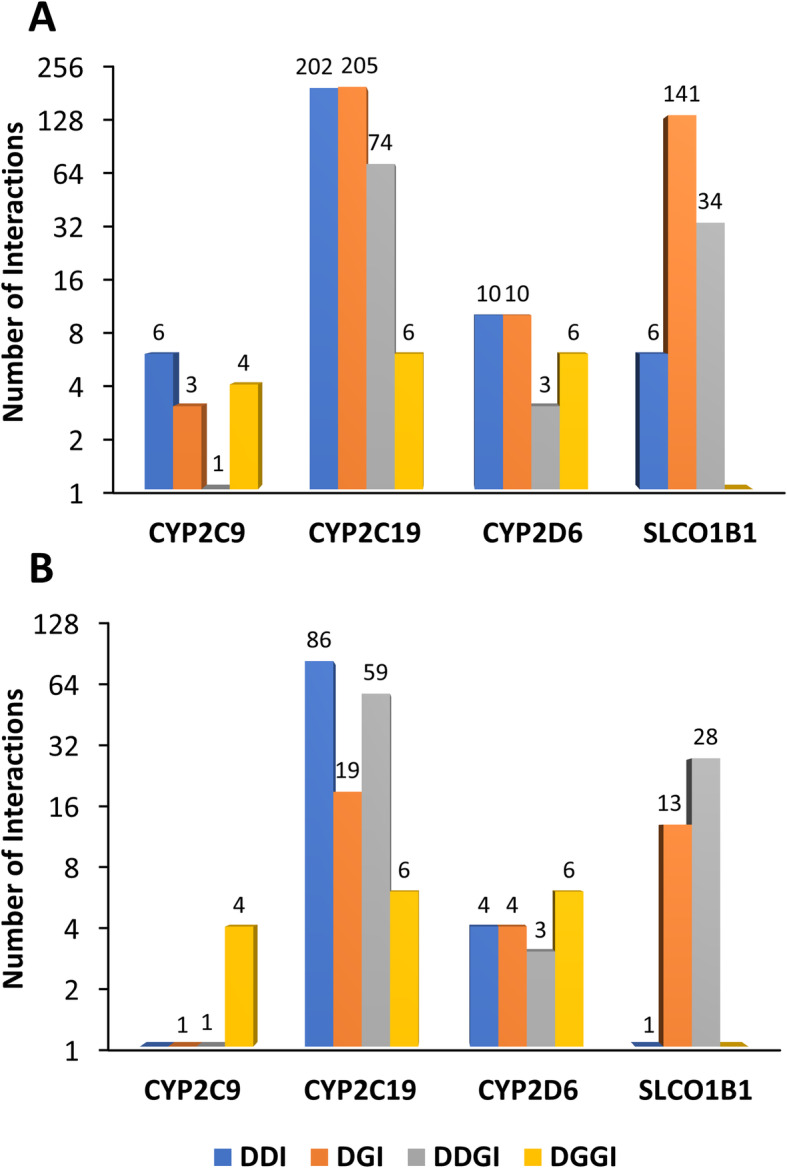
Interactions mediated by CYP2C9, CYP2C19, CYP2D6, and SLCO1B1. This figure displays the number of identified all (a) and substantial (b) interactions mediated through the three CYP enzymes (2C9, 2C19, 2D6) and transporter (SLCO1B1) that were directly influenced by both drug- and gene-based interactions, in 652 post-NSTE-ACS patients. In this patient cohort, CYP2C19-mediated interactions were the most common due to the high frequency of clopidogrel prescribing and, to a lesser extent, proton pump inhibitors, although many SLCO1B1-based interactions were also found due to atorvastatin and simvastatin
The median number (IQR, range) of interactions in those with and without multimorbidity was 1 (1–2, 0–9) and 1 (0–2, 0–4), respectively. Multimorbidity (p = 0.019) and number of drugs (p = 9.8 × 10−10) were both associated with patients that have ≥ 1 substantial interaction, and number of drugs (p = 3.8 × 10−8) with ≥ 1 all interaction, in multivariable analysis. Neither patient sex nor age was associated with all/substantial interactions (Additional file 10).
Survival analysis
There were 114 MACE and 74 ACM during a median follow-up of 19 months from hospital discharge within the interaction cohort. In multivariable survival analysis, age (p = 8.9 × 10−7), multimorbidity (p = 0.019), and number of drugs (p = 1.2 × 10−3) were associated with an increased risk of MACE, and age (p = 1.9 × 10−12) and number of drugs (p = 4.0 × 10−4) with increased ACM (Table 3). Whilst number of substantial interactions was borderline associated with MACE in univariate survival analysis (p = 0.053), it was not associated when added to the selected multivariable model (p = 0.89).
Table 3.
Associations with time to major adverse cardiovascular events and all-cause mortality
| MACE | ACM | |||
|---|---|---|---|---|
| HR (95% CI) | p value | HR (95% CI) | p value | |
| Univariate analysis | ||||
| Sex (F vs M) | 1.49 (1.02–2.18) | 0.038 | 1.40 (0.88–2.24) | 0.16 |
| Age | 1.05 (1.04–1.07) | 2.0 × 10−9 | 1.09 (1.06–1.11) | 1.9 × 10−12 |
| All multimorbidity | 2.69 (1.73–4.20) | 1.2 × 10−5 | 2.75 (1.56–4.84) | 4.8 × 10−4 |
| Number of drugs | 1.15 (1.10–1.21) | 2.0 × 10−8 | 1.14 (1.08–1.21) | 3.0 × 10−6 |
| Number of all interactions | 1.16 (0.995–1.34) | 0.058 | 1.01 (0.83–1.23) | 0.93 |
| Number of substantial interactions | 1.23 (0.997–1.51) | 0.053 | 1.11 (0.84–1.46) | 0.47 |
| Multivariable analysis | ||||
| Age | 1.05 (1.03–1.07) | 8.9 × 10−7 | 1.08 (1.06–1.11) | 1.9 × 10−12 |
| All multimorbidity | 1.76 (1.10–2.82) | 0.019 | – | – |
| Number of drugs | 1.10 (1.04–1.16) | 1.2 × 10−3 | 1.12 (1.05–1.19) | 4.0 × 10−4 |
ACM all-cause mortality, CI confidence interval, HR hazard ratio, MACE major adverse cardiovascular events
All types of interaction (drug-drug, drug-gene, drug-drug-gene, and drug-gene-gene) were counted to determine numbers of all and numbers of predicted substantial interactions per patient
Sensitivity analysis
When multimorbidity was replaced by number of comorbidities, all results remained equivalent, although the association between comorbidities and patients with ≥ 1 substantial interaction, adjusted for number of drugs, was reduced to borderline significance (p = 0.071, Additional file 11).
Discussion
The main findings of this study are: multimorbidity, polypharmacy, and drug interactions are common post-NSTE-ACS; drug- and gene-mediated perpetrators are involved in 85.5% (59.2% DDIs, 26.3% DDGIs) and 40.8% (11.6% DGIs, 26.3% DDGIs, 2.9% DGGIs) of identified substantial interactions, respectively; and both multimorbidity and number of drugs increase the risk of substantial interactions. Importantly, older age, multimorbidity, and number of drugs were associated with increased MACE, but despite trends, no statistically significant associations were found between all/substantial interactions and MACE or ACM. Lastly, differences in drug use were observed based on patient age and sex, and increasing age was associated with reduced coronary intervention.
The prevalence of multimorbidity is increasing [5] and so recognition of its clinical impact is important. We found multimorbidity post-NSTE-ACS to be associated with increased drug use, yet a lower likelihood of being prescribed secondary prevention medications, in keeping with other studies [43, 44]. Furthermore, multimorbidity and number of drugs were both associated with an increased likelihood of predicted substantial interactions. Of note, the interaction cohort had a higher proportion of patients with multimorbidity relative to the whole cohort, likely from exclusion of patients with no drug entries in their medication appendix due to being unable to ascertain whether this was accurate or represented missing data. Nevertheless, as clopidogrel and atorvastatin were the two most common victim drugs and were prescribed to similar extents across the whole cohort, the main interaction findings remain plausibly applicable to other cardiovascular patients, although further investigation in separate cohorts to determine generalisability is warranted. Still, the interaction cohort was likely enriched for patients with extreme polypharmacy and interaction burden; conceivably, it is these patients that may benefit the most from comprehensive medicines optimisation incorporating pharmacogenomics.
Importantly, the interactions assessed here included both genetic risk variants and drug perpetrators. In fact, 98.7% of patients had at least one actionable pharmacogene. In practice, only a small proportion of identified DDIs lead to clinically significant events [45]. Thus, 5–15% of older patients might suffer clinically significant adverse reactions involving drug interactions, whereas 35–60% have an identified DDI [45]. The reasons for this are complex [46], but genetic factors likely play a role, and therefore, identification of DDGIs is plausibly important, although these remain currently a novel and under-investigated subset of interactions. Still, the proportion of substantial interactions identified as DDGIs here (26.3%) was similar to estimates (19–22%) from the two previous studies in clinically heterogeneous patient populations [18, 19]. The reason(s) for the slightly higher DDGI estimate here likely pertains to considering SLCO1B1 as well as CYPs, and assessing NSTE-ACS patients enriched for multimorbidity mostly taking clopidogrel that requires CYP2C19 bioactivation.
One example of a cardiovascular DDGI is between warfarin and simvastatin, whereby simvastatin reduces warfarin dose requirements by 25% and 43% in CYP2C9*3 heterozygous and homozygous patients, respectively [47]. Nevertheless, a quantitative estimate of the impact of most DDGIs on victim drug systemic exposure is currently unavailable. A general principle appears to be that drug inhibitors decrease the metabolism of victim drugs to a greater extent in CYP EMs and IMs compared to PMs [48]. Nevertheless, this is not always the case; for example, the CYP2D6 inhibitor, fluoxetine, increased risperidone exposure in CYP2D6 PMs as well EMs, possibly due to the effects of its metabolite (norfluoxetine) on a secondary pathway of risperidone metabolism (CYP3A4) [48, 49].
Moving forward, it will be important to characterise DDGIs and understand their clinical sequelae in larger patient datasets. In this study, despite a univariate signal, no association between substantial interactions and MACE was observed. Thus, larger patient cohorts and additional use of other endpoints potentially more directly related to interactions, such as (causality-assessed) adverse drug reactions and all-cause hospitalisation, should be utilised. Irrespective, as pharmacogenomics becomes more available in clinical practice, prescribers will be faced with combining genetic, co-medication, and comorbidity (e.g. renal/hepatic impairment) information together when making prescribing decisions. This is not trivial, and prescribers should ideally be supported with electronic clinical decision support systems, which in turn will often require increasing interoperability between different healthcare information systems (e.g. between primary/secondary care, and blood test results/medication data) alongside national and international structures to review and implement evolving evidence. Increasing the clinical evidence base of specific DDGIs will help prioritise DDGIs for incremental implementation.
This study assessed multimorbidity, polypharmacy, and drug- and gene-based interactions within a UK-based NSTE-ACS cohort. However, it does have limitations. Beyond the limited patient sample size, mental illness diagnoses could not be included as comorbidities, no clinical frailty scale was recorded, and non-cardiovascular drug data were only established in a subset of patients.
Conclusions
This study demonstrated that multimorbidity, polypharmacy, and drug interactions are common after a NSTE-ACS, with CYP2C19 and SLCO1B1 being involved in the largest number of identified interactions. Multimorbidity and drug use were linked to interactions and clinical events. Differences in prescribing patterns between patients based on age and gender were identified. The high prevalence of DDGIs emphasises the need for further DDGI research and for healthcare systems to consider integration of medication information with genetic data and other clinical factors to enhance medicines optimisation.
Supplementary Information
Additional file 1. Table of drug-metabolising CYP substrates.
Additional file 2. Table of drug-metabolising CYP inhibitors.
Additional file 3. Table of drug-metabolising CYP inducers.
Additional file 4. Table of transporter substrates, inhibitors and inducers.
Additional file 5. Table of genotypes considered and actionable genotype-based metaboliser phenotypes for each drug.
Additional file 6. Table of clinical characteristics, multimorbidity and medication use by age.
Additional file 7. Visualisation of non-linear relationships with age.
Additional file 8. Table of all identified interactions.
Additional file 9. Table of identified substantial interactions.
Additional file 10. Table of associations with patients that have at least one identified drug interaction.
Additional file 11. Sensitivity analysis after substitution of dichotomous multimorbidity (≥2 comorbidities) with number of comorbidities.
Acknowledgements
EMK studied at the Department of Clinical Pharmacy & Toxicology, Leiden University Medical Center, Leiden, the Netherlands, during part of this work.
Abbreviations
- ACEI
Angiotensin-converting enzyme inhibitor
- ACM
All-cause mortality
- ARB
Angiotensin II receptor blocker
- AUC
Area under the concentration-time curve
- BMI
Body mass index
- CABG
Coronary artery bypass graft surgery
- CHD
Coronary heart disease
- CI
Confidence interval
- CNV
Copy number variant
- CPIC
Clinical Pharmacogenetics Implementation Consortium
- CYP
Cytochrome P450 enzyme
- DDI
Drug-drug interaction
- DDGI
Drug-drug-gene interaction
- DGI
Drug-gene interaction
- DGGI
Drug-gene-gene interaction
- DPWG
Dutch Pharmacogenetics Working Group
- EM
Extensive metaboliser
- FDA
US Food and Drug Administration
- HET
Heterozygous
- HOM
Homozygous for the variant allele
- HR
Hazard ratio
- IM
Intermediate metaboliser
- IQR
Interquartile range
- MACE
Major adverse cardiovascular events
- MI
Myocardial infarction
- NSTE-ACS
Non-ST elevation acute coronary syndrome
- PCI
Percutaneous coronary intervention
- PhACS
Pharmacogenetics of Acute Coronary Syndrome study
- PM
Poor metaboliser
- QC
Quality control
- RM
Rapid metaboliser
- UM
Ultra-rapid metaboliser
Authors’ contributions
RMT conceived the study; RMT, EMK, VF, and AT designed the study and carried out the data analysis; MP oversaw the study; all authors were involved in writing the manuscript and approved the final version.
Funding
RMT was supported by a Postdoctoral Research Fellowship from Health Education England Genomics Education Programme (HEE GEP). The views expressed in this publication are those of the authors and not necessarily those of HEE GEP. VF was supported by the Newton Fund, the Academy of Medical Sciences, and the Medical Research Council. The PhACS prospective clinical study was funded as part of the National Health Service NHS Chair of Pharmacogenetics, Department of Health. MP is Emeritus NIHR Senior Investigator.
Availability of data and materials
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
The protocol was approved by the Liverpool UK (adult) research Ethics Committee (07/H1005/117), site-specific approval was granted at all study sites, and written informed consent was ascertained from all study subjects in accordance with the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
MP receives other research funding from various organisations including the EU Commission and NIHR. He has also received partnership funding for the following: MRC Clinical Pharmacology Training Scheme (co-funded by MRC and Roche, UCB, Eli Lilly and Novartis); a PhD studentship jointly funded by EPSRC and Astra Zeneca; and grant funding from Vistagen Therapeutics. He has also unrestricted educational grant support for the UKP harmacogenetics and Stratified Medicine Network from Bristol-Myers Squibb and UCB. He has developed an HLA genotyping panel with MC Diagnostics, but does not benefit financially from this. None of these of additional funding sources have been used for the current paper. The other authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ford ES, Ajani UA, Croft JB, Critchley JA, Labarthe DR, Kottke TE, et al. Explaining the decrease in U.S. deaths from coronary disease, 1980-2000. N Engl J Med. 2007;356(23):2388–2398. doi: 10.1056/NEJMsa053935. [DOI] [PubMed] [Google Scholar]
- 3.Levi F, Lucchini F, Negri E, La Vecchia C. Trends in mortality from cardiovascular and cerebrovascular diseases in Europe and other areas of the world. Heart. 2002;88(2):119–124. doi: 10.1136/heart.88.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diederichs C, Berger K, Bartels DB. The measurement of multiple chronic diseases--a systematic review on existing multimorbidity indices. J Gerontol A Biol Sci Med Sci. 2011;66(3):301–311. doi: 10.1093/gerona/glq208. [DOI] [PubMed] [Google Scholar]
- 5.Uijen AA, van de Lisdonk EH. Multimorbidity in primary care: prevalence and trend over the last 20 years. Eur J Gen Pract. 2008;14(Suppl 1):28–32. doi: 10.1080/13814780802436093. [DOI] [PubMed] [Google Scholar]
- 6.Masnoon N, Shakib S, Kalisch-Ellett L, Caughey GE. What is polypharmacy? A systematic review of definitions. BMC Geriatr. 2017;17:230. doi: 10.1186/s12877-017-0621-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.National Institute for Health and Care Excellence (NICE) Myocardial infarction: cardiac rehabilitation and prevention of further cardiovascular disease (Clinical guideline CG172) 2013. [PubMed] [Google Scholar]
- 8.Salazar JA, Poon I, Nair M. Clinical consequences of polypharmacy in elderly: expect the unexpected, think the unthinkable. Expert Opin Drug Saf. 2007;6(6):695–704. doi: 10.1517/14740338.6.6.695. [DOI] [PubMed] [Google Scholar]
- 9.Shetty V, Chowta MN, Chowta KN, Shenoy A, Kamath A, Kamath P. Evaluation of potential drug-drug interactions with medications prescribed to geriatric patients in a tertiary care hospital. J Aging Res. 2018;2018:5728957. doi: 10.1155/2018/5728957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dequito AB, Mol PG, van Doormaal JE, Zaal RJ, van den Bemt PM, Haaijer-Ruskamp FM, et al. Preventable and non-preventable adverse drug events in hospitalized patients: a prospective chart review in the Netherlands. Drug Saf. 2011;34(11):1089–1100. doi: 10.2165/11592030-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 11.PharmGKB . Clinical Guideline Annotations. 2019. [Google Scholar]
- 12.van der Wouden CH, Cambon-Thomsen A, Cecchin E, Cheung KC, Davila-Fajardo CL, Deneer VH, et al. Implementing pharmacogenomics in Europe: design and implementation strategy of the ubiquitous pharmacogenomics consortium. Clin Pharmacol Ther. 2017;101(3):341–358. doi: 10.1002/cpt.602. [DOI] [PubMed] [Google Scholar]
- 13.Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32(11):1201–1208. doi: 10.1124/dmd.104.000794. [DOI] [PubMed] [Google Scholar]
- 14.Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138(1):103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Food and Drug Administration . Drug development and drug interactions: table of substrates, inhibitors and inducers. 2017. [Google Scholar]
- 16.Flockhart DA. Drug interactions: cytochrome P450 drug interaction table: Indiana University School of Medicine; 2007. https://drug-interactions.medicine.iu.edu/MainTable.aspx. Accessed 18 Nov 2019.
- 17.Shah RR, Smith RL. Addressing phenoconversion: the Achilles’ heel of personalized medicine. Br J Clin Pharmacol. 2015;79(2):222–240. doi: 10.1111/bcp.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verbeurgt P, Mamiya T, Oesterheld J. How common are drug and gene interactions? Prevalence in a sample of 1143 patients with CYP2C9, CYP2C19 and CYP2D6 genotyping. Pharmacogenomics. 2014;15(5):655–665. doi: 10.2217/pgs.14.6. [DOI] [PubMed] [Google Scholar]
- 19.Hocum BT, White JR, Jr, Heck JW, Thirumaran RK, Moyer N, Newman R, et al. Cytochrome P-450 gene and drug interaction analysis in patients referred for pharmacogenetic testing. Am J Health Syst Pharm. 2016;73(2):61–67. doi: 10.2146/ajhp150273. [DOI] [PubMed] [Google Scholar]
- 20.Mostafa S, Kirkpatrick CMJ, Byron K, Sheffield L. An analysis of allele, genotype and phenotype frequencies, actionable pharmacogenomic (PGx) variants and phenoconversion in 5408 Australian patients genotyped for CYP2D6, CYP2C19, CYP2C9 and VKORC1 genes. J Neural Transm (Vienna) 2019;126(1):5–18. doi: 10.1007/s00702-018-1922-0. [DOI] [PubMed] [Google Scholar]
- 21.Turner RM, Yin P, Hanson A, FitzGerald R, Morris AP, Stables RH, et al. Investigating the prevalence, predictors, and prognosis of suboptimal statin use early after a non-ST elevation acute coronary syndrome. J Clin Lipidol. 2017;11(1):204–214. doi: 10.1016/j.jacl.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The Academy of Medical Sciences . Multimorbidity: a priority for global health research. 2018. [Google Scholar]
- 23.Joint Formulary Committee . British National Formulary. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turner RM, Fontana V, Zhang JE, Carr D, Yin P, Fitz Gerald R, et al. A genome-wide association study of circulating levels of atorvastatin and its major metabolites. Clin Pharmacol Ther. 2020;108(2):287-97. [DOI] [PubMed]
- 25.Gammal RS, Court MH, Haidar CE, Iwuchukwu OF, Gaur AH, Alvarellos M, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for UGT1A1 and atazanavir prescribing. Clin Pharmacol Ther. 2016;99(4):363–369. doi: 10.1002/cpt.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caudle KE, Sangkuhl K, Whirl-Carrillo M, Swen JJ, Haidar CE, Klein TE, et al. Standardizing CYP2D6 genotype to phenotype translation: consensus recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin Transl Sci. 2020;13(1):116–124. doi: 10.1111/cts.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Modak AS, Klyarytska I, Kriviy V, Tsapyak T, Rabotyagova Y. The effect of proton pump inhibitors on the CYP2C19 enzyme activity evaluated by the pantoprazole-(13) C breath test in GERD patients: clinical relevance for personalized medicine. J Breath Res. 2016;10(4):046017. doi: 10.1088/1752-7163/10/4/046017. [DOI] [PubMed] [Google Scholar]
- 28.Wessler JD, Grip LT, Mendell J, Giugliano RP. The P-glycoprotein transport system and cardiovascular drugs. J Am Coll Cardiol. 2013;61(25):2495–2502. doi: 10.1016/j.jacc.2013.02.058. [DOI] [PubMed] [Google Scholar]
- 29.BCGuidelines.ca . Use of non-vitamin K antagonist oral anticoagulants (NOAC) in non-valvular atrial fibrillation. Appendix A: potential NOAC drug interaction. 2015. [Google Scholar]
- 30.Karlgren M, Vildhede A, Norinder U, Wisniewski JR, Kimoto E, Lai Y, et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J Med Chem. 2012;55(10):4740–4763. doi: 10.1021/jm300212s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hicks JK, Sangkuhl K, Swen JJ, Ellingrod VL, Müller DJ, Shimoda K, et al. Clinical Pharmacogenetics Implementation Consortium guideline (CPIC) for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants: 2016 update. Clin Pharmacol Ther. 2017;102(1):37–44. doi: 10.1002/cpt.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutch Pharmacogenetics Working Group (DPWG) Pharmacogenetic recommendations. 2020. [Google Scholar]
- 33.Relling MV, Schwab M, Whirl-Carrillo M, Suarez-Kurtz G, Pui CH, Stein CM, et al. Clinical Pharmacogenetics Implementation Consortium guideline for thiopurine dosing based on TPMT and NUDT15 genotypes: 2018 update. Clin Pharmacol Ther. 2019;105(5):1095–1105. doi: 10.1002/cpt.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hicks JK, Bishop JR, Sangkuhl K, Müller DJ, Ji Y, Leckband SG, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98(2):127–134. doi: 10.1002/cpt.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott SA, Sangkuhl K, Stein CM, Hulot JS, Mega JL, Roden DM, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther. 2013;94(3):317–323. doi: 10.1038/clpt.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crews KR, Gaedigk A, Dunnenberger HM, Leeder JS, Klein TE, Caudle KE, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin Pharmacol Ther. 2014;95(4):376–382. doi: 10.1038/clpt.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caudle KE, Rettie AE, Whirl-Carrillo M, Smith LH, Mintzer S, Lee MTM, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C9 and HLA-B genotypes and phenytoin dosing. Clin Pharmacol Ther. 2014;96(5):542–548. doi: 10.1038/clpt.2014.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramsey LB, Johnson SG, Caudle KE, Haidar CE, Voora D, Wilke RA, et al. The Clinical Pharmacogenetics Implementation Consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin Pharmacol Ther. 2014;96(4):423–428. doi: 10.1038/clpt.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Birdwell KA, Decker B, Barbarino JM, Peterson JF, Stein CM, Sadee W, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin Pharmacol Ther. 2015;98(1):19–24. doi: 10.1002/cpt.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goetz MP, Sangkuhl K, Guchelaar HJ, Schwab M, Province M, Whirl-Carrillo M, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2D6 and tamoxifen therapy. Clin Pharmacol Ther. 2018;103(5):770–777. doi: 10.1002/cpt.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bristol-Myers Squibb Company . Highlights of prescribing information for coumadin (warfarin sodium) 2017. [Google Scholar]
- 42.R Core Team . R: a language and environment for statistical computing. 2016. [Google Scholar]
- 43.Rothnie KJ, Smeeth L, Herrett E, Pearce N, Hemingway H, Wedzicha J, et al. Closing the mortality gap after a myocardial infarction in people with and without chronic obstructive pulmonary disease. Heart. 2015;101(14):1103–1110. doi: 10.1136/heartjnl-2014-307251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith DJ, Martin D, McLean G, Langan J, Guthrie B, Mercer SW. Multimorbidity in bipolar disorder and undertreatment of cardiovascular disease: a cross sectional study. BMC Med. 2013;11(1):263. doi: 10.1186/1741-7015-11-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Magro L, Moretti U, Leone R. Epidemiology and characteristics of adverse drug reactions caused by drug-drug interactions. Expert Opin Drug Saf. 2012;11(1):83–94. doi: 10.1517/14740338.2012.631910. [DOI] [PubMed] [Google Scholar]
- 46.Pirmohamed M. Drug-drug interactions and adverse drug reactions: separating the wheat from the chaff. Wien Klin Wochenschr. 2010;122(3):62–64. doi: 10.1007/s00508-010-1309-1. [DOI] [PubMed] [Google Scholar]
- 47.Andersson ML, Eliasson E, Lindh JD. A clinically significant interaction between warfarin and simvastatin is unique to carriers of the CYP2C9*3 allele. Pharmacogenomics. 2012;13(7):757–762. doi: 10.2217/pgs.12.40. [DOI] [PubMed] [Google Scholar]
- 48.Bahar MA, Setiawan D, Hak E, Wilffert B. Pharmacogenetics of drug-drug interaction and drug-drug-gene interaction: a systematic review on CYP2C9, CYP2C19 and CYP2D6. Pharmacogenomics. 2017;18(7):701–739. doi: 10.2217/pgs-2017-0194. [DOI] [PubMed] [Google Scholar]
- 49.Bondolfi G, Eap CB, Bertschy G, Zullino D, Vermeulen A, Baumann P. The effect of fluoxetine on the pharmacokinetics and safety of risperidone in psychotic patients. Pharmacopsychiatry. 2002;35(2):50–56. doi: 10.1055/s-2002-25026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Table of drug-metabolising CYP substrates.
Additional file 2. Table of drug-metabolising CYP inhibitors.
Additional file 3. Table of drug-metabolising CYP inducers.
Additional file 4. Table of transporter substrates, inhibitors and inducers.
Additional file 5. Table of genotypes considered and actionable genotype-based metaboliser phenotypes for each drug.
Additional file 6. Table of clinical characteristics, multimorbidity and medication use by age.
Additional file 7. Visualisation of non-linear relationships with age.
Additional file 8. Table of all identified interactions.
Additional file 9. Table of identified substantial interactions.
Additional file 10. Table of associations with patients that have at least one identified drug interaction.
Additional file 11. Sensitivity analysis after substitution of dichotomous multimorbidity (≥2 comorbidities) with number of comorbidities.
Data Availability Statement
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.