

Abstract
Opioid drugs are a first line treatment for severe acute pain and other chronic pain conditions, but long-term opioid drug use produces opioid-induced hyperalgesia (OIH). Co-administration of cannabinoids with opioid receptor agonists produce anti-nociceptive synergy, but cannabinoid receptor agonists may also produce undesirable side effects. Therefore, positive allosteric modulators (PAM) of cannabinoid type-1 receptors (CB1R) may provide an option reducing pain and/or enhancing the anti-hyperalgesic effects of opioids without the side effects, tolerance and dependence observed with the use of ligands that target the orthosteric binding sites. This study tested GAT211, a PAM of cannabinoid type-1 receptors (CB1R) for its ability to enhance the anti-hyperalgesic effects of the mu-opioid receptor (MOR) agonist DAMGO in rats treated chronically with morphine (or saline) and tested during withdrawal. We tested the effects of intra-periaqueductal gray (PAG) injections of 1) DAMGO, 2) GAT211, or 3) DAMGO + GAT211 on thermal nociception in chronic morphine-treated rats that were hyperalgesic and also in saline-treated control rats. We used slice electrophysiology to test the effects of DAMGO/GAT211 bath application on synaptic transmission in the vlPAG. Intra-PAG DAMGO infusions dose-dependently reversed chronic morphine-induced hyperalgesia, but intra-PAG GAT211 did not alter nociception at the doses we tested. When co-administered into the PAG, GAT211 antagonized the anti-nociceptive effects of DAMGO in morphine-withdrawn rats. DAMGO suppressed synaptic inhibition in the vlPAG of brain slices taken from saline- and morphine-treated rats, and GAT211 attenuated DAMGO-induced suppression of synaptic inhibition in vlPAG neurons via actions at CB1R. These findings show that positive allosteric modulation of CB1R antagonizes the behavioral and cellular effects of a MOR agonist in the PAG of rats.
Keywords: Morphine, Opioids, Opiates, Pain, Hyperalgesia, PAG, Dose-response, Positive allosteric modulators
INTRODUCTION
Chronic opioid treatment for pain is associated with tolerance to the analgesic effects of opioids, and cessation of opioid use produces or exaggerates hyperalgesia (Lee, Silverman et al. 2011). Many patients abuse prescription opioids (Vowles, McEntee et al. 2015), and approximately 100 people die every day from opioid abuse (Rudd, Aleshire et al. 2016). Therefore, it is critical to identify ways to reduce reliance on opioids for pain management in the clinic.
Cannabinoids are a potential alternative or adjunct to opioids for the treatment of pain (Guindon and Hohmann 2009). Endogenous cannabinoid (eCB) and opioid systems in the CNS interact to modulate pain and nociception (Welch 2009). Opioid and cannabinoid receptor co-expression is observed in specific CNS sites important for mediating pain-related outcomes, including the periaqueductal gray (PAG) where they are abundantly co-expressed (Wilson-Poe, Morgan et al. 2012, Befort 2015). Cannabinoids are said to have opioid-sparing effects because co-administration of cannabinoids allows for reduction of opioid drug doses without loss of analgesic efficacy, and because co-administration of cannabinoid and opioid drugs produces additive or synergistic analgesic or anti-hyperalgesic effects (Cichewicz 2004, Welch 2009, Nielsen, Sabioni et al. 2017). That said, in both heterologous cells and rat brain tissue expressing mu-opioid receptor (MOR) and cannabinoid type-1 receptors (CB1R), co-application of MOR and CB1R agonists exert reciprocally antagonistic effects on G-protein signaling (Rios, Gomes et al. 2006). In agreement with this notion of functional antagonism, CB1R antagonism enhances MOR agonist-induced suppression of synaptic inhibition in the ventrolateral PAG (vlPAG) of morphine-treated animals (Wilson-Poe, Lau et al. 2015). In general, the mechanisms underlying the anti-nociceptive effects of cannabinoids are not well understood, nor are the mechanisms underlying opioid-cannabinoid interaction effects on pain-related behavior and synaptic transmission in PAG.
Physiological pain, pathological pain and opioid analgesia are mediated at multiple sites in the brain and spinal cord. The PAG receives ascending pain information from the spinal cord (Millan 2002, Tracey and Mantyh 2007) and modulates incoming pain signals via descending projections to the medulla (Hirakawa, Tershner et al. 1999). Chronic opioid drug treatement produces robust and persistent hyperalgesia in humans and rodents (Angst and Clark 2006), and is accompanied by altered synaptic transmission in the PAG (Bagley, Gerke et al. 2005, Wilson-Poe, Lau et al. 2015). Acute systemic treatment with morphine produces analgesia, chronic treatment produces tolerance to this analgesic effect, and both effects are blocked by infusion of a MOR antagonist into the vlPAG (Lane, Patel et al. 2005). Furthermore, injection of morphine into the vlPAG produces robust analgesia and chronic intra-vlPAG morphine injections produce tolerance to this analgesic effect (Morgan, Clayton et al. 2005). Opioid antinociception in the PAG is mediated by inhibition of GABA neurons and disinhibition of PAG output neurons (Moreau and Fields 1986, Bagley, Gerke et al. 2005). Finally, chronic continuous and chronic intermittent in vivo morphine treatment each produce anti-nociceptive tolerance but they may produce different types of plasticity in the PAG (Chieng and Christie 1996).
The use of cannabinoids in conjunction with opioids to treat pain or for the alleviation of opioid-induced withdrawal symptom has met with limited success (De Aquino and Ross 2018, Wiese and Wilson-Poe 2018). Similar to opioids, chronic exposure to cannabinoids leads to the development of tolerance, physical dependence, and withdrawal symptoms during abstinence (Wiese and Wilson-Poe 2018) Positive allosteric modulators (PAM) of CB1Rs enhance the functional effects of cannabinoid receptor activation, increase the efficacy of cannabinoid binding at traditional orthosteric binding sites, reduce dissociation rates of ligands from orthosteric binding sites, and amplify downstream G-protein coupled signaling pathways (Khurana, Mackie et al. 2017, Laprairie, Kulkarni et al. 2017, Alaverdashvili and Laprairie 2018). Systemic administration of a CB1R PAM reduces nociception, enhances the anti-nociceptive effects of eCBs, enhances the anti-nociceptive effects of exogenous cannabinoids, reduces tolerance to cannabinoid anti-nociceptive effects and reduces cannabinoid withdrawal effects in mice (Martini, Waldhoer et al. 2007, Ignatowska-Jankowska, Baillie et al. 2015, Slivicki, Xu et al. 2018). More recently, it was shown that systemic administration of the CB1R PAM GAT211 reduces paclitaxel-induced behavioral hypersensitivities to mechanical and cold stimulation in mice, and that co-administration of GAT211 and morphine results in anti-allodynic synergism, but that GAT211 does not alter somatic signs of morphine dependence or morphine reward in mice (Slivicki, Iyer et al. 2020).
The primary goals of these experiments were to test interaction effects of the CB1R PAM GAT211 and the MOR agonist DAMGO 1) in PAG on morphine withdrawal hyperalgesia and 2) on synaptic transmission in the PAG of rats treated chronically with morphine or saline. We hypothesized that intra-PAG infusions of the MOR agonist DAMGO and the CB1R PAM GAT211 would have additive anti-nociceptive effects in rats, that bath application of DAMGO and GAT211 would produce additive suppression of synaptic inhibition onto PAG neurons of rats, and that these additive behavioral and cellular effects would be enhanced in hyperalgesic morphine-treated rats tested or sacrificed during withdrawal.
METHODS
Animals
Adult male Wistar rats 2.5 months old weighing between 250–300 g were obtained from Charles River (RRID: RGD_13508588). Animals were pair-housed in standard cages with bedding in humidity- and temperature-controlled (22°C) vivarium on a 12 h light/dark cycle (lights off at 8:00 A.M.). Access to standard lab chow and water was provided at libitum. Following their arrival, rats were acclimated for 1 week prior to the start of any experiments. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the Louisiana State University Health Sciences Center and were in accordance with the National Institute of Health guidelines.
Drug Treatment
Morphine sulphate (a gift from National Institute of Drug Abuse) at a dose of 10 mg/Kg dissolved in physiologic saline was injected twice per day (9:00 a.m. and 4:00 p.m.) subcutaneously (26 G hypodermic needles) for 7 consecutive days. Control animals received daily injections of saline on a protocol identical to the morphine-treated individuals. Rats were always tested behaviorally at 8:00 a.m. prior to the day’s morphine/saline treatment. Rats were sacrificed for slice electrophysiology recordings at 9:00 a.m. DAMGO (Catalog No.: 1171, Tocris) and GAT211 (SML1926, Sigma-Aldrich) used for intra-PAG injections were dissolved in physiologic saline and DMSO, respctively.
Microinjections
DAMGO and GAT211 were administered through a 31-gauge injection cannula inserted into and extending 1 mm beyond the tip of the guide cannula. To reduce confounds caused by mechanical stimulation of neurons during testing, rats received a sham injection in which the injector was inserted into the guide cannula without drug administration before the Hargreaves Testing on Day 7, a day prior to the actual dose response experiment conducted on Day 8. Microinjections were administered (0.4 μL) over a period of 2 mins using a 1 μl syringe. To minimize backflow of the drug up the cannula track, the injection cannula was maintained in place for an additional 1 min. The doses, injection volumes` and inter-dose intervals used in this cumulative dosing procedure were adapted from Meyer et al., 2007 (Meyer, Fossum et al. 2007).
Behavioral Testing
Thermal nociception was measured in morphine-treated and saline-treated rats (n=18 morphine, 20 saline) using the Hargreaves plantar test (Hargreaves Apparatus, Model 309, IITC Life Sciences). Prior to testing, animals were always acclimatized to the testing room (for 1 hour) and to the testing apparatus (5 min), which consists of several 4 × 8 × 5 inch clear Plexiglas enclosures on top of a glass pane suspended 8 inches above the table top. Following the 5-min habituation period, baseline nociception was assessed using the Hargreaves test, which consists of measuring the latency for a rat to withdraw its hindpaw when stimulated by a halogen lamp heat source from underneath. The reaction time (paw withdrawal latency, PWL) to elicit a withdrawal response was recorded for each trial. The trial was terminated if the animal failed to respond within 20 sec to prevent potential burn injury. Four readings were obtained from each rat in one session, two from each hindpaw in an alternating manner with at least 1 min between the readings. The readings were averaged to determine a thermal nociception score for each rat for that session. Animals were assigned to morphine or saline treatment groups counterbalanced for baseline thermal nociception (i.e., PWL) values. Following the start of daily morphine/saline treatments, rats underwent two Hargreaves test sessions, once on Day 2 (after a single day of morphine treatment) and again on Day 7 (after 6 days of morphine treatment). All tests occurred 16 hours after the most recent morphine/saline injection.
Experiment 1: Dose response effects of DAMGO on morphine WD thermal hyperalgesia
Please refer to Figure 1A for the full experimental design regarding morphine treatment, behavioral test schedules and sacrifice for cellular recordings. This experiment tested the effect of intra-PAG injections of DAMGO on morphine withdrawal induced hyperalgesia in morphine-treated (n=6) and saline-treated (n=5) rats. Rats received DAMGO microinjections of 0.03, 0.07, 0.2, and 0.7 μg/0.4 μl into the PAG, resulting in cumulative half log doses (0.03, 0.1, 0.3, and 1 μg/0.4 μl). Injections were spaced 20 min apart, and nociception was assessed at baseline and 15 min after each injection. The doses and inter-dose intervals used in this cumulative dosing procedure were adapted from Meyer et al., 2007 (Meyer, Fossum et al. 2007).
Figure 1. Intra-PAG infusions of the MOR agonist DAMGO produce dose-dependent and time-dependent anti-hyperalgesia in rats treated with chronic morphine and this effect is attenuated by co-infusion of the CB1R PAM GAT211 into the PAG.
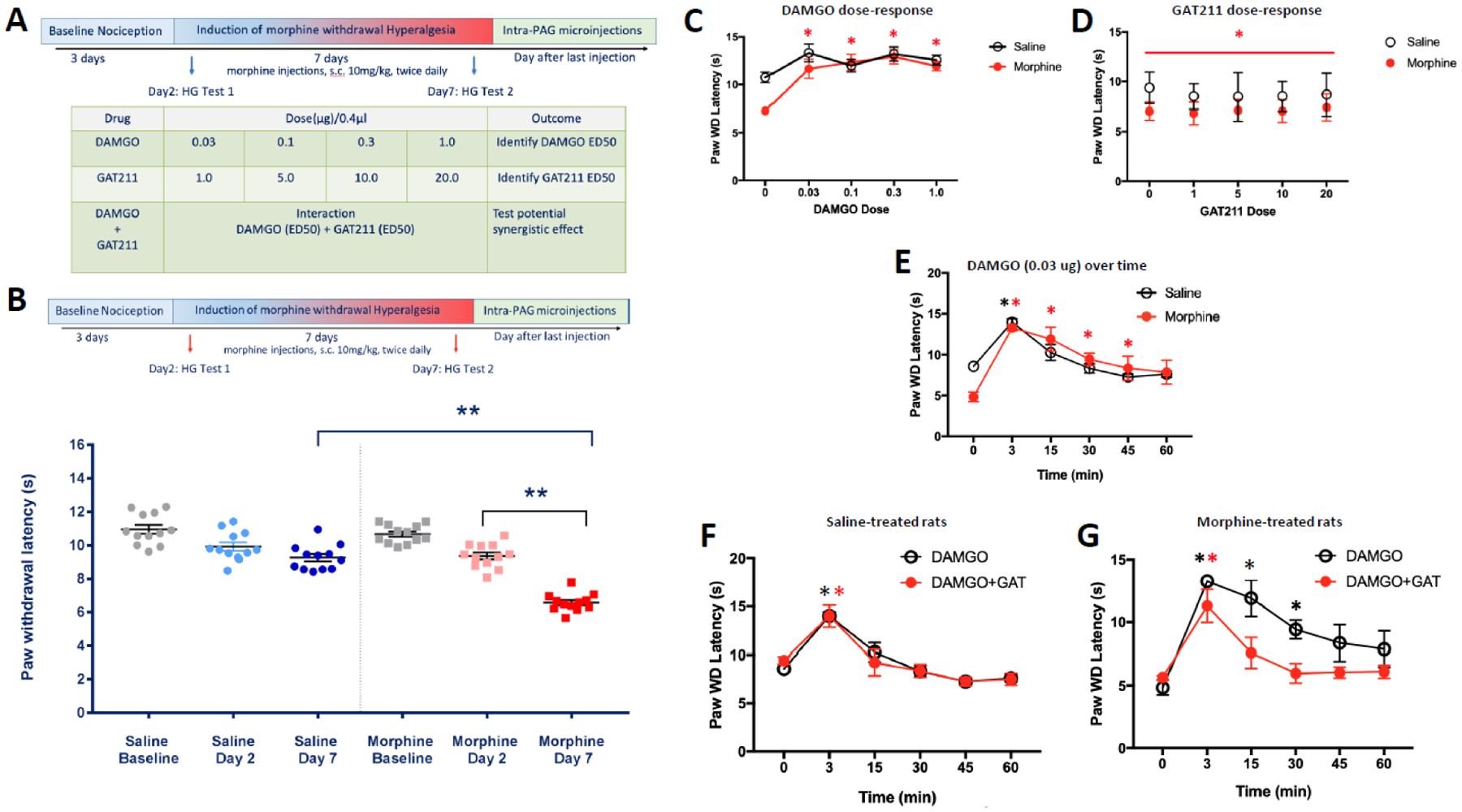
(A) Timecourse of chronic in vivo morphine treatment and intra-PAG infusions. (B to G) show mean ± SEM hindpaw withdrawal latencies in response to a noxious thermal stimulus using the Hargreaves test. Hindpaw withdrawal latency for each animal at each dose/timepoint is the average of two trials on the right hindpaw and two trials on the left hindpaw. (B) Rats treated with 20 mg/kg morphine per day for 6 days (two s.c. injections of 10 mg/kg/day at 9:00 a.m. & 4:00 p.m.) exhibited thermal hyperalgesia, as exhibited by shorter hindpaw withdrawal latencies, relative to the same rats treated with morphine for 1 day and also relative to a separate group of rats treated with saline for 6 days, **p<0.001; (C) dose-response effects of DAMGO infused into the PAG on hindpaw withdrawal latencies in morphine-treated (n=6) and saline-treated (n=5) rats; *p<0.001 DAMGO dose effect relative to the zero dose condition in morphine-treated rats; (D) dose-response effects of GAT211 infused into the PAG on hindpaw withdrawal latencies in morphine-treated (n=6) and saline-treated (n=6) rats; *p<0.05 main effect of morphine; (E) timecourse effects of DAMGO (0.03 ug) infused into the PAG on hindpaw withdrawal latencies in morphine-treated (n=3) and saline-treated (n=4) rats; red asterisk: p<0.02 DAMGO dose effect relative to zero minutes time point in morphine-treated rats, black asterisk: p<0.001 DAMGO dose effect relative to zero minutes time point in saline-treated rats; (F) timecourse effects of DAMGO (0.03 ug) and GAT211 (10 ug) co-infused into the PAG on hindpaw withdrawal latencies in saline-treated rats (n=4); black asterisk: p<0.01 DAMGO effect relative to zero minutes time point, red asterisk: p<0.01 DAMGO+GAT211 effect relative to zero minutes time point; (G) timecourse effects of DAMGO (0.03 ug) and GAT211 (10 ug) co-infused into the PAG on hindpaw withdrawal latencies in morphine-treated rats (n=6); black asterisk: p<0.01 DAMGO effect relative to zero minutes time point, red asterisk: p<0.01 DAMGO+GAT211 effect relative to zero minutes time point.
Experiment 2: Dose-response effects of GAT211 on morphine WD thermal hyperalgesia
The effect of intra-PAG injections of GAT211 on morphine induced hyperalgesia was assessed in a similar manner as described for DAMGO microinjections. Rats (n=6 morphine-treated, n=6 saline-treated) received GAT211 microinjections of 1, 4, 5, and 10μg/0.4 μl dissolved in DMSO into the PAG, resulting in cumulative doses of 1, 5, 10, and 20 μg/0.4 μl. Injections were spaced 20 min apart, and nociception was assessed at baseline (following vehicle injection) and 15 min after each injection.
Experiment 3: Time course effects of DAMGO on morphine WD thermal hyperalgesia
The time course of anti-nociceptive effects of DAMGO was evaluated using a single 0.03ug/0.4uL, a dose at which a robust increase in PWL had previously been observed (n=3 morphine-treated, n=4 saline control). The effect of DAMGO microinjection into the PAG was evaluated over a 60 min period at multiple time points (3, 15, 30, 45 and 60 min) post infusion.
Experiment 4: Effects of GAT211 on DAMGO-induced attenuation of morphine WD thermal hyperalgesia
Synergistic or additive interactions between MOR agonist and CB1 agonist have been reported by several groups (Tham, Angus et al. 2005, Cox, Haller et al. 2007, Maguire, Yang et al. 2013). However, the effects appear to be compound specific and require correct pairing in order to be effective. The objective of this experiment was to determine the interactions between DAMGO, a μ- opioid agonist and GAT211, a positive allosteric modulator of CB1 receptor when administered into the vPAG. Interactions of drug combinations was evaluated by sequential administration of DAMGO and GAT211 in morphine-treated (n=6) and saline-treated (n=4) rats. First, DAMGO was infused into the PAG, then rats were tested for PWL in the Hargreaves test 3 minutes later. Finally, GAT211 (10 ug/0.4uL) was infused 10 minutes after DAMGO infusion and rats were tested for GAT211 modulation of DAMGO effect on PWL at 15, 30, 45 and 60-minute time points.
Experiment 5: DAMGO-GAT211 effects on synaptic transmission in PAG of morphine-withdrawn rats
After 7 days of morphine or saline treatment and Hargreaves testing, animals were sacrificed 16 hours following the most recent morphine/saline treatment (i.e., during withdrawal at the same time point hyperalgesia was observed) and brain slices were prepared for vlPAG recordings. Recordings were performed with blockers of synaptic excitation circulating in the bath, specifically, CNQX (10 μM, Tocris) for AMPA receptors and AP5 (50 μM , Tocris) for NMDA receptors. When whole-cell recording configuration was established, the voltage was clamped at −50 mV and 5 min of spontaneous postsynaptic current was recorded (n=30 neurons from morphine-treated rats and n=47 neurons from saline-treated rats). Inhibitory spontaneous postsynaptic events were detected in these recordings by thresholding rapid excursions in outward current; the average event amplitude (n=27 neurons from morphine-treated rats and n=47 neurons from saline-treated rats) and mean frequency (n=30 neurons from morphine-treated rats and n=47 neurons from saline-treated rats) over the 5-min recording period were quantified. Resting membrane potential (RMP) was estimated by recording in current clamp mode (with 0 pA injected current) and averaging the resting voltage over a 1.5 second period (n=25 neurons from morphine-treated rats and n=22 neurons from saline-treated rats). For neurons that were spontaneously active at rest, we estimated firing rate by dividing the number of spikes counted over a 4.5-sec period by the time period. For experiments that tested the effect of MOR and CB1R interaction on inhibitory synaptic transmission, we recorded from vlPAG neurons in voltage clamp mode (@ −50 mV) and electrically stimulated nearby (~150 – 300 μm) vlPAG with a bipolar chromium wire electrode to evoke inihibtory postsynaptic currents (eIPSCs). vlPAG was stimulated and eIPSCs were recorded every 20 seconds for 10 minutes to establish baseline amplitude. At this point, either DAMGO (100 nM) or GAT211 (0.1, 1 or 10 μM) was delivered to the bath and eIPSCs were recorded for 15 minutes. Mean eIPSC amplitude during this period was estimated using eIPSC responses between 5 minutes (or later if stable equilibrium was not reached at 5 minutes) and 15 minutes of drug application. When GAT211 was applied to the bath, DAMGO was then successively applied and eIPSCs were recorded for 15 more minutes, with the last 10 minutes of recordings being used to estimate mean eIPSC amplitude for this drug combination condition. In a subset of experiments, the interaction effects of GAT211 and DAMGO were tested in the presence of the CB1R inverse agonist rimonabant (RIM; 5 μM). In these experiments, RIM was circulating in the aCSF before targeting vlPAG neurons for whole cell recordings. After whole-cell configuration was established, a baseline synaptic amplitude was obtained by recording synaptic responses every 20 seconds for 10 minutes. Then GAT211 (1 μM) was added to the bath and 15 minutes of synaptic responses were recorded, at which point DAMGO (100 nM) was added to the bath synaptic responses were recorded for 15 more minutes.
Statistics
Group data are plotted as mean ± standard error of the mean (SEM). To detect morphine effects on thermal nociception, one-way analysis of variance (ANOVA) was used. To analyze single-drug (DAMGO or GAT211) dose-response effects in morphine- and saline-treated rats, a two-way repeated-measures (RM) ANOVA (within-subjects factor: dose) was used. To analyze timecourse effects of DAMGO and GAT211 alone or in combination, a two-way RM ANOVA (within-subjects factor: time) was used. Resting membrane potential (RMP), spontaneous firing rates, and spontaneous inhibitory postsynaptic currents (sIPSCs) in morphine- and saline-treated rats were compared using 2-sample t-tests. Relative supression of evoked IPSCs by DAMGO was analyzed using 2-way ANOVA with in vivo morphine treatment and bath-applied GAT211 concentration as between-subjects factors. Where appropriate, pairwise comparisons were completed using Sidak’s or Bonferroni post-hoc tests. Statistical significance was set at p<0.05.
RESULTS
Experiment 1: Dose response effects of intra-PAG DAMGO on morphine WD thermal hyperalgesia
We tested the dose-response effects of intra-PAG DAMGO infusions on thermal nociception using a cumulative dosing procedure (Fig. 1A) and the Hargreaves test. Fig. 1B shows that 6 days of twice-daily morphine treatment produced significant thermal hyperalgesia when rats were tested 16 hours after the last treatment, as indicated by a significant main effect of morphine, F(1,72)=6.89, p<0.001, main effect of time, F(2,72)=45.33, p<0.001, and a significant interaction effect, F(2,72)=12.77, p<0.001. Post-hoc analysis revealed that 6 days of morphine injections produced thermal hyperalgesia relative to saline-treated controls, relative to morphine group baseline and relative to a single day of morphine treatment (p<0.001 in all cases). A 2-way (morphine treatment x DAMGO dose) RM ANOVA revealed a significant main effect of DAMGO dose, F(4,36)=13.64, p<0.001, and a significant treatment x DAMGO interaction effect, F(4,36)=3.01, p=0.03, on hindpaw withdrawal latencies (Fig. 1C). Post-hoc comparisons revealed that morphine-treated rats were hyperalgesic relative to saline-treated controls prior to DAMGO treatment, (p=0.007). Furthermore, all doses of DAMGO infused into the PAG produced anti-hyperalgesic effects (i.e., longer hindpaw withdrawal latencies) in morphine-treated rats relative to the zero dose condition (p<0.001 in all cases). The DAMGO dose-response effects suggest that rats treated chronically with morphine exhibit increased sensitivity to the anti-nociceptive effects of intra-PAG DAMGO treatment relative to saline-treated controls.
Experiment 2: Dose-response effects of intra-PAG GAT211 on morphine WD thermal hyperalgesia
We tested the dose-response effects of intra-PAG GAT211 infusions on thermal nociception using a cumulative dosing procedure and the Hargreaves test. A 2-way (morphine treatment x GAT211 dose) RM ANOVA revealed that chronic morphine treatment reduced hindpaw withdrawal latencies relative to saline-treated controls, F(1,10)=5.22, p=0.045 (Fig. 1D). There was not a main effect of GAT211 treatment nor was there a treatment x GAT211 interaction effect. These data suggest that intra-PAG treatment with GAT211 alone does not modulate nociception in morphine-withdrawn rats or controls.
Experiment 3: Time course effects of intra-PAG DAMGO on morphine WD thermal hyperalgesia
We tested the timecourse effects of intra-PAG infusion of a single DAMGO dose (0.3 ug) on thermal nociception over time using the Hargreaves test. We chose 0.3 ug DAMGO because it was the lowest effective dose in the dose-response experiment (Fig. 1C). A 2-way (morphine treatment x time) RM ANOVA revealed a significant main effect of time, F(5,25)=30.94, p<0.001, and a significant treatment x time interaction effect, F(5,25)=4.54, p=0.004, on hindpaw withdrawal latencies (Fig. 1E). Post-hoc comparisons revealed that 0.3 ug DAMGO produced a significant anti-hyperalgesic effect (i.e., longer hindpaw withdrawal latencies) in morphine-treated rats at 3, 15, 30 and 45 minutes post-treatment (p<0.02 in all cases) relative to the zero dose condition, but that this effect dissipated by 60 minutes post-treatment (p=0.055). Furthermore, 0.3 ug DAMGO produced a significant analgesic effect (i.e., longer hindpaw withdrawal latencies) in saline-treated controls at 3 minutes post-treatment (p<0.001) relative to the zero dose condition, but this effect dissipated by 15 minutes post-treatment (p>0.05 for all subsequent time points). The DAMGO timecourse effects suggest that rats treated chronically with morphine exhibit increased sensitivity to the anti-nociceptive effects of intra-PAG DAMGO treatment relative to saline-treated controls.
Experiment 4: Effects of intra-PAG DAMGO and GAT211 co-infusion on morphine withdrawal thermal hyperalgesia
We tested the effects of intra-PAG DAMGO (0.3 ug) and GAT211 (10 ug) co-infusion on thermal nociception over time using the Hargreaves test. Because no dose of GAT211 affected thermal nociception in Experiment 2, we chose a middle dose (10 ug) for this experiment. A 2-way (morphine treatment x time) RM ANOVA revealed a significant main effect of morphine treatment, F(1,9)=6.61, p=0.03, and a significant main effect of time, F(5,45)=23.66, p<0.001, on hindpaw withdrawal latencies. More specifically, morphine-treated rats exhibited thermal hyperalgesia (i.e., shorter hindpaw withdrawal latencies) relative to saline-treated controls across all time points (compare panels Fig. 1F & 1G).
To determine the effects of co-infused GAT211 on the anti-nociceptive effects of DAMGO in the PAG, we then analyzed DAMGO timecourse effects in rats that either were or were not co-infused with GAT211. In saline-treated rats (Fig. 1F), this 2-way (drug infusion x time) RM ANOVA revealed a main effect of time, F(5,35)=30.66, p<0.001, but no effect of drug treatment and no interaction effect on thermal nociception. In morphine-treated rats (Fig. 1G), this 2-way (drug infusion x time) RM ANOVA revealed a significant main effect of time, F(5,35)=20.82, p<0.001, and a significant drug treatment x time interaction effect, F(5,35)=2.63, p=0.04, on thermal nociception (i.e., hindpaw withdrawal latencies). Post-hoc comparisons revealed that DAMGO produced significant anti-hyperalgesic effects at 3, 15 and 30 minutes post-infusion (p<0.01 in all cases), similar to what was observed in Experiment 3 (DAMGO timecousre experiment; Fig. 1E), but that co-infusion of GAT211 with DAMGO produced significant anti-hyperalgesic effects at only the 3-minute timepoint (p<0.01). These results suggest that GAT211 blunts the anti-nociceptive effects of DAMGO in the PAG of morphine-withdrawn rats but not saline-treated controls.
Experiment 5: DAMGO-GAT211 effects on synaptic transmission in PAG of morphine-withdrawn rats
We treated rats with chronic morphine or saline to generate brain slices for electrophysiology recordings. Rats were tested for thermal nociception using the Hargreaves test on days 2 and 7 of the morphine treatment schedule (i.e., after 1 and 6 days of morphine treatment), then sacrificed for slice preparation on Day 8 (i.e., after 7 days of morphine treatment). A 2-way (morphine treatment x test day) RM ANOVA revealed a significant main effect of treatment, F(1,58)=29.66, p<0.001, a significant main effect of day, F(2,58)=20.61, p<0.001, and a significant treatment x day interaction effect, F(2,58)=32.64, p<0.001, on hindpaw withdrawal latencies (Fig. 2A). Post-hoc comparisons revealed that 6 days of morphine treatment produced thermal hyperalgesia (i.e., shorter hindpaw withdrawal latencies) in morphine-treated rats relative to baseline, relative to 1 day of morphine treatment, and relative to controls treated with saline for 6 days (p<0.001 in all cases).
Figure 2 – Chronic morphine hyperpolarizes PAG cells and reduces the number of spontaneously active cells in the PAG of hyperalgesic rats.
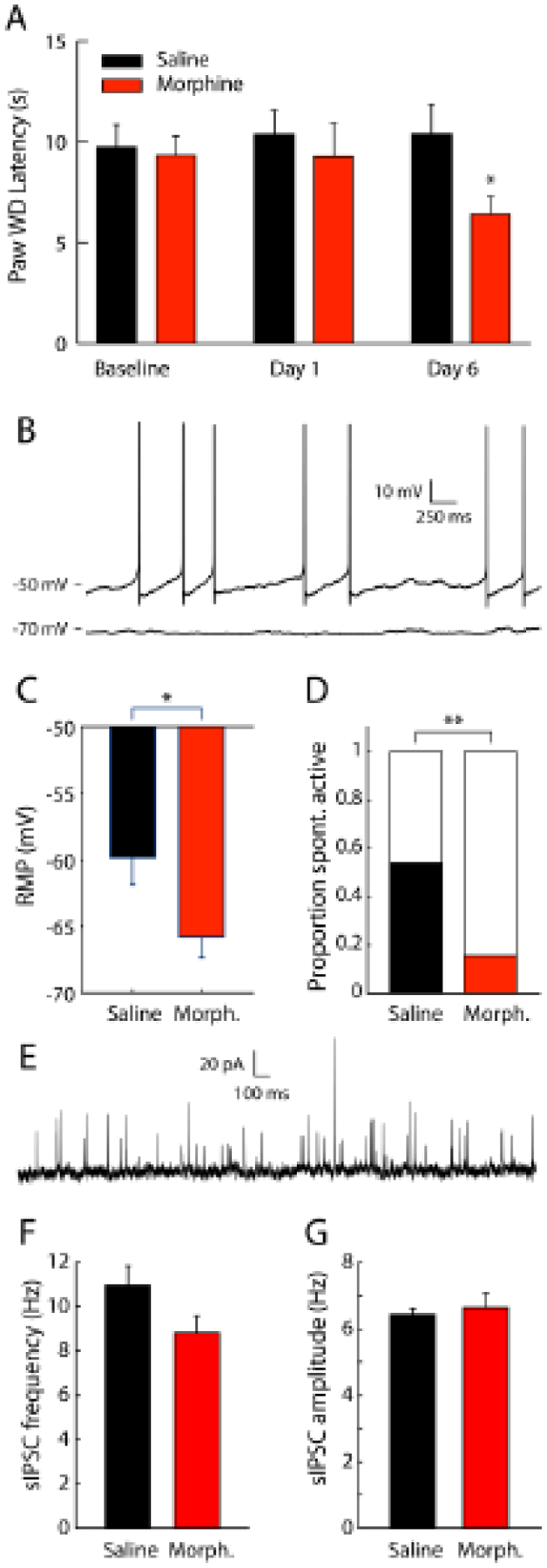
(A) Rats treated with 20 mg/kg morphine (n=15) per day for 6 days (two s.c. injections of 10 mg/kg/day at 9:00 a.m. & 4:00 p.m.) exhibited thermal hyperalgesia, as exhibited by shorter mean ± SEM hindpaw withdrawal latencies, relative to saline-treated control rats (n=16), *p<0.001; (B) Whole cell recordings in vlPAG neurons revealed some neurons that were spontaneously active (example trace above) and some neurons that had stable subthreshold resting membrane potentials (example recording below). (C) Resting membrane potential (RMP) from saline-treated control rats (n=22) was higher than morphine-treated rats (n=25), *p<0.05. (D) The proportion of spontaneously active neurons was higher for saline-treated rats relative to morphine-treated rats, **p<0.01. (E) Postsynaptic current was recorded at −50 mV and spontaneous inhibitory postsynaptic currents (sIPSCs) were detected. sIPSC frequency (F) and amplitude (G) were not different between neurons from saline-treated rats and morphine-treated rats (n=47 neurons from saline-treated rats and n=30 neurons from morphine-treated rats).
During electrophysiological recording sessions, we recorded membrane voltage from vlPAG neurons in current clamp mode with 0 pA injected current. Some neurons were spontaneously active while some had stable subthreshold resting voltages (Fig. 2B). Resting membrane potential (RMP) was estimated from neurons exhibiting stable shubtrehsold membrane voltage. RMP of saline rats (−59.8 ± 2.0 mV) was higher than that of morphine treated rats (−65.8 ± 1.5 mV, p < 0.05, Fig. 2C). The proportion of neurons from saline rats that were spontaneously active (0.54) is higher than that of neurons from morphine rats (0.16, p < 0.01, Fig. 2D). We also recorded postsynaptic current while clamping the membrane voltage at −50 mV to detect spontaneous inhibitory postsynaptic current (sIPSC) events (Fig. 2E). The mean sIPSC frequency of saline neurons (11.0 ± 0.9 Hz) and morphine neurons (8.8 ± 0.8 Hz) were not significantly different (Fig. 2F). The mean sIPSC amplitudes of saline (6.2 ± 0.2 pA) and morphine (6.6 ± 0.4 pA) neuorns are not significantly different (Fig. 2G).
To test the effect of MOR agonism and allosteric modulation of CB1R we recorded vlPAG neurons in voltage clamp mode at −50 mV and electrically stimulated nearby vlPAG tissue to evoked IPSCs (eIPSCs). eIPSC responses were recorded and amplitudes were calculated in the aCSF bath, followed by bath application of GAT211 (0.1, 1 or 10 μM) and then DAMGO (100 nM) (Fig. 3A). eIPSC responses were typically attenuated after the application of DAMGO (Fig. 3B). Morphine treatment history did not alter bath applied pharmacology effects on synaptic responses (F1,56 = 2.31, p = 0.1341), therefore we collapsed data collected in morphine and saline animals (Fig. 3C & 3D). We found that the amount of eIPSC amplitude suppression by DAMGO was affected by GAT211 concentration (1-way ANOVA, F3,59 = 2.91, p < 0.042, Fig. 3C). However, posthoc comparisons revealed no single pairwise differences between individual group means. To explore if this effect was contingent on direct CB1R activation we perform a subset of experiments with the same design as before except with the CB1R antagonist rimonabant (RIM, 5 μM) circulating in the bath. We found a main effect of bath condition (i.e. aCSF vs. GAT211 (1 μM) vs. RIM (5 μM) + GAT211 (1 μM)) on eIPSC suppression by DAMGO (F2,38 = 4.71, p = 0.015, Fig. 3D) with significant pairwise differences between aCSF and GAT211 (p < 0.05) and GAT211 and RIM+GAT211 (p < 0.05).
Figure 3 – Positive allosteric modulation of CB1Rs by GAT211 functionally antagonizes MOR function.
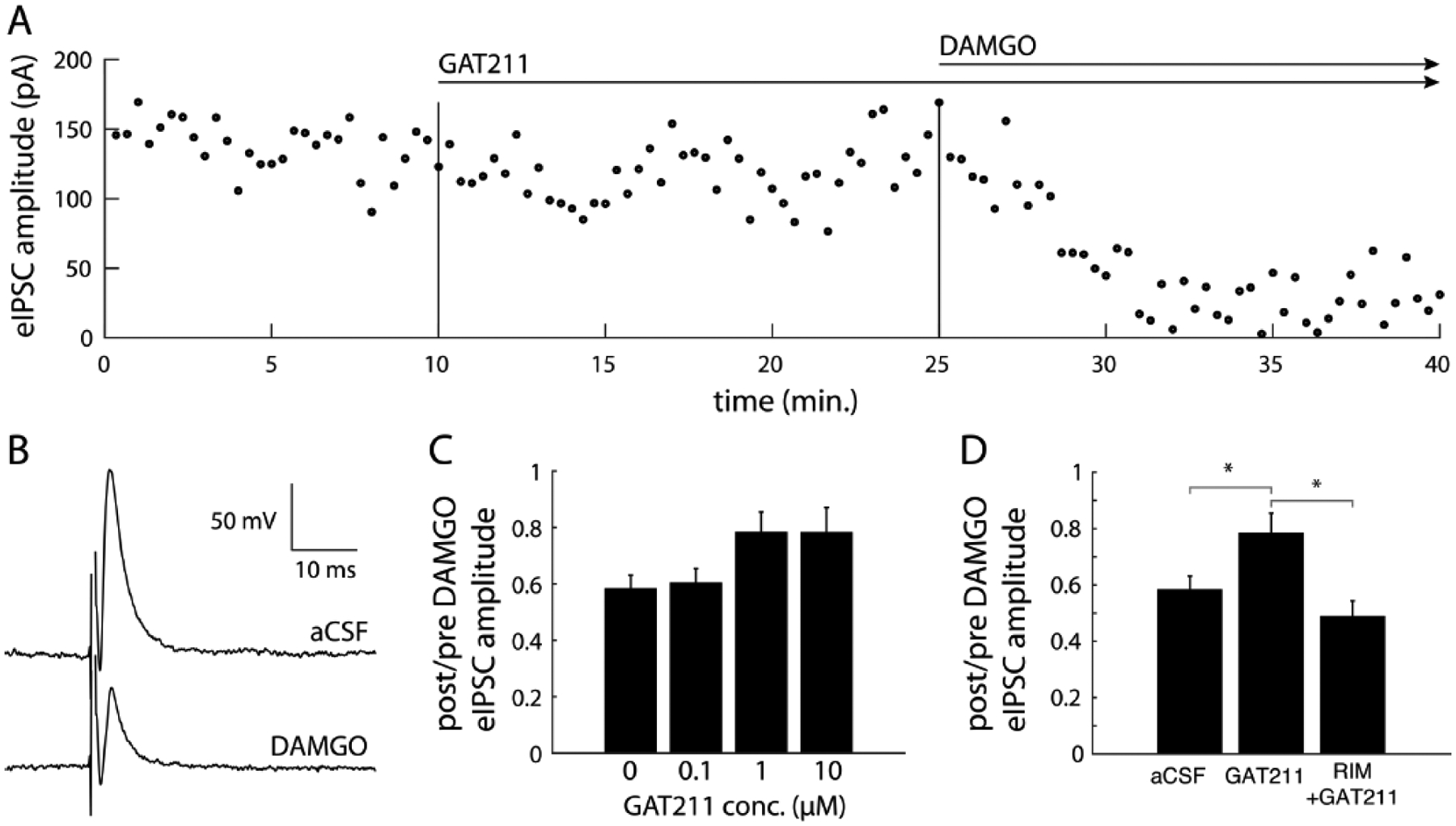
(A) Electrically evoked inhibitory postsynaptic currents (eIPSCs) were recorded periodically every 20 sec for 40 mins. Baseline eIPSCs were recorded for 10 mins after which GAT211 was applied to the bath and 15 mins after this DAMGO was applied. (B) Example traces show that DAMGO significantly attenuated eIPSC amplitude. (C) There was a main effect of GAT211 concentration on the suppression of eIPSC amplitude by DAMGO (p < 0.05), although posthoc comparisons with multiple comparison correction showed no significant differences. (D) GAT211 (1 μM) attenuates DAMGO-induced suppression of synaptic inhibition, an effect that is not observed in the presence of the CB1R antagonist rimonabant (RIM), *indicates p<0.05 difference between the two bracketed conditions.
DISCUSSION
We tested the hypothesis that a MOR agonist and CB1R PAM in the PAG would have additive anti-nociceptive effects and additive suppressive effects on synaptic inhibition, and that these additive effects would be enhanced in morphine-withdrawn rats. As expected, rats treated chronically with morphine exhibited robust thermal hyperalgesia during withdrawal. Rats treated chronically with morphine exhibited increased sensitivity to the anti-nociceptive effects of intra-PAG DAMGO treatment relative to saline-treated controls. Intra-PAG GAT211 treatment alone did not alter nociception in morphine-withdrawn rats or saline-treated controls. Contrary to our hypothesis, intra-PAG GAT211 treatment diminished the anti-nociceptive effects of DAMGO in the PAG of morphine-withdrawn rats but had no such effect in saline-treated controls. In vlPAG neurons, DAMGO suppressed synaptic inhibition similarly in saline-treated and morphine-withdrawn rats, whereas GAT211 did not affect synaptic inhibition in vlPAG. Again contrary to our hypothesis, GAT211 attenuated DAMGO-induced suppression of synaptic inhibition in vlPAG neurons of saline-treated and morphine-withdrawn rats and, because this effect was blocked by rimonabant, the GAT211 attenuation of DAMGO effects may be mediated by CB1R.
Despite their efficacy, the use of opioids for pain treatment is limited by side effects, including analgesic tolerance and opioid-induced hyperalgesia (OIH) (Trang, Al-Hasani et al. 2015). MORs are the primary target for opiates and mediates both analgesic and hyperalgesic effects of these compounds (Al-Hasani and Bruchas 2011, Lee, Silverman et al. 2011, Roeckel, Le Coz et al. 2016). The ability of opioids to activate MORs in either a Gi/Go-coupled mode or a Gs-coupled mode, depending on the dose of the agonist and the exposure regimen (acute versus chronic) results in reduced versus enhanced neuronal excitability. The Gs state of MOR predominates under chronic treatment condition and induces hyperalgesia through adenylyl cyclase activation (Crain and Shen 1990, Crain and Shen 1992). Additionally, internalization of MORs as a compensatory changes caused by sustained opioid receptor activation, has also been associated with OIH (Williams, Ingram et al. 2013).
In the PAG, MOR agonists inhibit GABAergic inhibition of output projection neurons, thereby facilitating analgesic or anti-hyperalgesic effects of those drugs (Christie, Connor et al. 2000). More specifically, pre-synaptic MOR activation reduces GABA release in the PAG by enhancing pre-synaptic voltage-gated potassium conductance, thereby disinhibiting neurons that project to the rostral ventral medulla (RVM) (Vaughan, Ingram et al. 1997, Ingram, Vaughan et al. 1998). The ventrolateral PAG contains high concentrations of MORs and CB1Rs that are co-localized in neurons (Wilson-Poe, Morgan et al. 2012). CB1R activation also reduces pre-synaptic GABA release, but does so via downregulation of voltage-gated calcium channels (Wilson and Nicoll 2001). CB1R PAMs (including GAT211) enhance the functional effects of cannabinoid receptor activation (Khurana, Mackie et al. 2017, Laprairie, Kulkarni et al. 2017, Alaverdashvili and Laprairie 2018) and systemic treatment with CB1R PAMs (GAT211 and others) reduces nociception in mice with chronic neuropathic or inflammatory pain at doses that are not inherently rewarding or aversive (Ignatowska-Jankowska, Baillie et al. 2015, Slivicki, Xu et al. 2018). Here, we showed that 1) CB1R PAM GAT211 antagonizes the anti-nociceptive effects of MOR activation in the PAG of morphine-withdrawn rats at GAT211 doses that had no effect on their own, and 2) attenuates MOR agonist-induced suppression of synaptic inhibition in the vlPAG at concentrations that have no effect on their own. These results are consistent with prior work showing that MOR and CB1R have functionally antagonistic effects on G-protein signaling (Rios, Gomes et al. 2006), and that CB1R antagonism enhances MOR agonist-induced suppression of synaptic inhibition in the vlPAG (Wilson-Poe, Lau et al. 2015), suggesting a functionally antagonistic interaction between MORs and CB1Rs at the cellular level.
A recent study examined the effect of systemic GAT211 injection on pain-related behaviors (mechanical nociception using Von Frey and sensitivity to cold using acetone) in adult male mice with neuropathic (paclitaxel) or inflammatory (Complete Freund’s Adjuvant [CFA]) pain (Slivicki, Xu et al. 2018). In that study, systemic GAT211 treatment attenuated mechanical allodynia induced by paclitaxel and CFA in a CB1R-dependent manner, and systemic GAT211 produced synergistic anti-allodynic effects with eCB degradative enzyme inhibitors in mice with neuropathic pain, suggesting that GAT211 effects are stronger when eCB levels are higher. Systemic GAT211 treatment did not produce a conditioned place preference or aversion, did not lose efficacy over 19 days of chronic treatment, and did not produce withdrawal after treatment with a precipitating dose of the CB1R antagonist rimonabant (Slivicki, Xu et al. 2018). An even more recent follow-up study (Slivicki, Iyer et al. 2020) showed that systemic administration of the CB1R PAM GAT211 reduces paclitaxel-induced allodynia in mice, that co-administration of GAT211 and morphine produces anti-allodynic synergism, but that GAT211 does not alter somatic signs of morphine dependence (unlike what has been observed for THC (Cichewicz and Welch 2003)) or morphine reward. These data suggest that the pain state (injured versus uninjured) of the animal, and perhaps the resultant effect of that pain state on eCB tone, may dictate GAT211 effects alone and in combination with MOR agonists (e.g., morphine or DAMGO). In our study, intra-PAG GAT211 infusions did not rescue thermal hyperalgesia in rats chronically treated with morphine and tested during withdrawal, nor did it exert analgesic effects in saline-treated animals. There are several possible explanations for these seemingly discrepant sets of findings: for example, it is possible that anti-hyperalgesic effects of systemic GAT211 are not mediated in the PAG, GAT211 effects differ for hyperalgesia induced by neuropathic/inflammatory pain and that induced by chronic intermittent morphine treatment and withdrawal, GAT211 effects differ in mouse vs. rat and/or GAT211 effects differ for thermal vs. mechanical stimuli. We may also have not selected the optimal dose range for testing GAT211 effects on nociception or we may have needed to boost eCB levels using an eCB degradative enzyme inhibitor to unmask GAT211 effects. That said, our electrophysiology data do agree with the Slivicki et al. (2018, 2020) finding that GAT211 effects are dependent on CB1R function. Our data, collected in uninjured rats, suggest that GAT211 actions in the PAG do not rescue opioid-induced hyperalgesia nor do they potentiate DAMGO effects on nociception or synaptic transmission.
We showed here that chronic morphine treatment resulted in more hyperpolarized membrane potentials and fewer spontaneously active neurons in vlPAG of rats. Another study showed that proportion and mean rate of spontaneously active neurons in vlPAG was similar in saline- and morphine-treated animals (Chieng and Christie 1996). However, in that study, recordings from morphine-treated mice were performed in the presence of bath-applied morphine, and those mice were treated and sacrificed during adolescence (4–6 weeks of age). We also report that relative suppression of eIPSC amplitude by DAMGO is not significantly different between saline- and morphine-treated animals. This lack of morphine effect on DAMGO sensitivity differs from past work showing that the synaptic effects of MOR agonism were either sensitized (Ingram, Vaughan et al. 1998, Ingram, Macey et al. 2008) or desensitized (Wilson-Poe, Lau et al. 2015, Wilson-Poe, Jeong et al. 2017) after chronic morphine treatment. These discrepancies may in part be attributable to morphine dose and schedule (10 mg/kg twice daily for 7 days in current study, 5 mg/kg injection twice daily for 2 days in (Ingram, Macey et al. 2008, Wilson-Poe, Lau et al. 2015, Wilson-Poe, Jeong et al. 2017), 2 mg/kg injection twice daily for 5 days in (Ingram, Macey et al. 2008), or 75 mg subcutaneous pellet for 72 hours in [(Ingram, Macey et al. 2008)), time of recording after final morphine treatment (17 hours in current study, 18–24 hrs in (Wilson-Poe, Jeong et al. 2017), 1–2 days in (Ingram, Vaughan et al. 1998, Wilson-Poe, Jeong et al. 2017), or 1–4 days in (Ingram, Macey et al. 2008)), rat strain (Wistar in current study, Sprague-Dawley in all prior studies) and/or age of the animals (adult rats in current study, adolescent rats in all prior studies). Chronic morphine reduces the sensitivity of presynaptic reduction of ICa and upregulation of GIRKs (Bagley, Gerke et al. 2005), but we are unable to attribute our observed morphine-induced decreased in RMP and proportion of spontaneously active neurons to modification of a specific ionic conductance.
Relatively few studies have examined functional interactions of CBRs and MORs at the cellular and synaptic levels using electrophysiological approaches. One study demonstrated that DAMGO-induced suppression of inhibitory current in vlPAG neurons was weaker after chronic morphine treatment, and that antagonism of CB1Rs enhanced the relative suppressive effect of DAMGO in morphine-treated but not saline-treated rats (Wilson-Poe, Lau et al. 2015). Our findings seem to agree with the latter finding, in that GAT211, perhaps through actions at CB1R, attenuates the suppressive effect of DAMGO on synaptic inhibition in vlPAG; but we did not observe differences in bath-applied DAMGO sensitivity between saline- and morphine-treated animals. One potentially important point to note is that electrophysiological recordings were conducted in the presence of GAT211 bath application followed by DAMGO bath application, whereas in vivo nociception testing was conducted after intra-PAG infusions of DAMGO followed by GAT211.
Collectively, our results show that intra-PAG treatment with the CB1R PAM GAT211 antagonizes intra-PAG DAMGO-induced rescue of thermal hyperalgesia in rats treated chronically with morphine and tested during withdrawal. Our results also show that GAT211 antagonized DAMGO-induced suppression of inhibitory transmission in the vlPAG in a CB1R-dependent manner. These results are consistent with prior work showing that a CB1R agonist inhibits the anti-nociceptive effects of morphine when delivered concurrently but not when drug deliveries are staggered in time (Wilson, Maher et al. 2008). Future work will determine generalizability of these results across pain models, pain-related behaviors, CNS sites, species, and sex, and will also determine cell-type specific effects of GAT211 and/or opiates in the PAG.
Funding and Disclosures
This work was supported in part by Merit Review Award #I01 BX003451 from the United States (U.S.) Department of Veterans Affairs, Biomedical Laboratory Research and Development Service. This work was also supported by NIH grant AA023305 (NWG) and an internal pilot grant from the LSUHSC Alcohol & Drug Abuse Center of Excellence (JWM). NWG is a consultant for Glauser Life Sciences, Inc. All other authors report no biomedical financial interests or potential conflicts of interest.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
REFERENCES
- Al-Hasani R and Bruchas MR (2011). “Molecular mechanisms of opioid receptor-dependent signaling and behavior.” Anesthesiology 115(6): 1363–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaverdashvili M and Laprairie RB (2018). “The future of type 1 cannabinoid receptor allosteric ligands.” Drug Metab Rev 50(1): 14–25. [DOI] [PubMed] [Google Scholar]
- Angst MS and Clark JD (2006). “Opioid-induced hyperalgesia: a qualitative systematic review.” Anesthesiology 104(3): 570–587. [DOI] [PubMed] [Google Scholar]
- Bagley EE, Gerke MB, Vaughan CW, Hack SP and Christie MJ (2005). “GABA transporter currents activated by protein kinase A excite midbrain neurons during opioid withdrawal.” Neuron 45(3): 433–445. [DOI] [PubMed] [Google Scholar]
- Befort K (2015). “Interactions of the opioid and cannabinoid systems in reward: Insights from knockout studies.” Front Pharmacol 6: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng B and Christie MJ (1996). “Local Opioid Withdrawal in Rat Single Periaqueductal Gray NeuronsIn Vitro.” The Journal of Neuroscience 16(22): 7128–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ, Connor M, Vaughan CW, Ingram SL and Bagley EE (2000). “Cellular actions of opioids and other analgesics: implications for synergism in pain relief.” Clin Exp Pharmacol Physiol 27(7): 520–523. [DOI] [PubMed] [Google Scholar]
- Cichewicz DL (2004). “Synergistic interactions between cannabinoid and opioid analgesics.” Life Sciences 74(11): 1317–1324. [DOI] [PubMed] [Google Scholar]
- Cichewicz DL and Welch SP (2003). “Modulation of oral morphine antinociceptive tolerance and naloxone-precipitated withdrawal signs by oral Delta 9-tetrahydrocannabinol.” J Pharmacol Exp Ther 305(3): 812–817. [DOI] [PubMed] [Google Scholar]
- Cox ML, Haller VL and Welch SP (2007). “Synergy between delta9-tetrahydrocannabinol and morphine in the arthritic rat.” Eur J Pharmacol 567(1–2): 125–130. [DOI] [PubMed] [Google Scholar]
- Crain SM and Shen KF (1990). “Opioids can evoke direct receptor-mediated excitatory effects on sensory neurons.” Trends Pharmacol Sci 11(2): 77–81. [DOI] [PubMed] [Google Scholar]
- Crain SM and Shen KF (1992). “After chronic opioid exposure sensory neurons become supersensitive to the excitatory effects of opioid agonists and antagonists as occurs after acute elevation of GM1 ganglioside.” Brain Res 575(1): 13–24. [DOI] [PubMed] [Google Scholar]
- De Aquino JP and Ross DA (2018). “Cannabinoids and Pain: Weeding Out Undesired Effects With a Novel Approach to Analgesia.” Biological Psychiatry 84(10): e67–e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J and Hohmann AG (2009). “The endocannabinoid system and pain.” CNS Neurol Disord Drug Targets 8(6): 403–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa N, Tershner SA and Fields HL (1999). “Highly delta selective antagonists in the RVM attenuate the antinociceptive effect of PAG DAMGO.” Neuroreport 10(15): 3125–3129. [DOI] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Baillie GL, Kinsey S, Crowe M, Ghosh S, Owens RA, Damaj IM, Poklis J, Wiley JL, Zanda M, Zanato C, Greig IR, Lichtman AH and Ross RA (2015). “A Cannabinoid CB1 Receptor-Positive Allosteric Modulator Reduces Neuropathic Pain in the Mouse with No Psychoactive Effects.” Neuropsychopharmacology 40(13): 2948–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram SL, Macey TA, Fossum EN and Morgan MM (2008). “Tolerance to repeated morphine administration is associated with increased potency of opioid agonists.” Neuropsychopharmacology 33(10): 2494–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram SL, Vaughan CW, Bagley EE, Connor M and Christie MJ (1998). “Enhanced Opioid Efficacy in Opioid Dependence Is Caused by an Altered Signal Transduction Pathway.” The Journal of Neuroscience 18(24): 10269–10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana L, Mackie K, Piomelli D and Kendall DA (2017). “Modulation of CB1 cannabinoid receptor by allosteric ligands: Pharmacology and therapeutic opportunities.” Neuropharmacology 124: 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DA, Patel PA and Morgan MM (2005). “Evidence for an intrinsic mechanism of antinociceptive tolerance within the ventrolateral periaqueductal gray of rats.” Neuroscience 135(1): 227–234. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Kulkarni PM, Deschamps JR, Kelly MEM, Janero DR, Cascio MG, Stevenson LA, Pertwee RG, Kenakin TP, Denovan-Wright EM and Thakur GA (2017). “Enantiospecific Allosteric Modulation of Cannabinoid 1 Receptor.” ACS Chemical Neuroscience 8(6): 1188–1203. [DOI] [PubMed] [Google Scholar]
- Lee M, Silverman SM, Hansen H, Patel VB and Manchikanti L (2011). “A comprehensive review of opioid-induced hyperalgesia.” Pain Physician 14(2): 145–161. [PubMed] [Google Scholar]
- Maguire DR, Yang W and France CP (2013). “Interactions between mu-opioid receptor agonists and cannabinoid receptor agonists in rhesus monkeys: antinociception, drug discrimination, and drug self-administration.” J Pharmacol Exp Ther 345(3): 354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini L, Waldhoer M, Pusch M, Kharazia V, Fong J, Lee JH, Freissmuth C and Whistler JL (2007). “Ligand-induced down-regulation of the cannabinoid 1 receptor is mediated by the G-protein-coupled receptor-associated sorting protein GASP1.” FASEB J 21(3): 802–811. [DOI] [PubMed] [Google Scholar]
- Meyer PJ, Fossum EN, Ingram SL and Morgan MM (2007). “Analgesic tolerance to microinjection of the micro-opioid agonist DAMGO into the ventrolateral periaqueductal gray.” Neuropharmacology 52(8): 1580–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ (2002). “Descending control of pain.” Prog Neurobiol 66(6): 355–474. [DOI] [PubMed] [Google Scholar]
- Moreau JL and Fields HL (1986). “Evidence for GABA involvement in midbrain control of medullary neurons that modulate nociceptive transmission.” Brain Res 397(1): 37–46. [DOI] [PubMed] [Google Scholar]
- Morgan MM, Clayton CC and Boyer-Quick JS (2005). “Differential susceptibility of the PAG and RVM to tolerance to the antinociceptive effect of morphine in the rat.” Pain 113(1–2): 91–98. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Sabioni P, Trigo JM, Ware MA, Betz-Stablein BD, Murnion B, Lintzeris N, Khor KE, Farrell M, Smith A and Le Foll B (2017). “Opioid-Sparing Effect of Cannabinoids: A Systematic Review and Meta-Analysis.” Neuropsychopharmacology 42(9): 1752–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios C, Gomes I and Devi LA (2006). “mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis.” Br J Pharmacol 148(4): 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeckel LA, Le Coz GM, Gaveriaux-Ruff C and Simonin F (2016). “Opioid-induced hyperalgesia: Cellular and molecular mechanisms.” Neuroscience 338: 160–182. [DOI] [PubMed] [Google Scholar]
- Rudd RA, Aleshire N, Zibbell JE and Gladden RM (2016). “Increases in Drug and Opioid Overdose Deaths--United States, 2000–2014.” MMWR Morb Mortal Wkly Rep 64(50–51): 1378–1382. [DOI] [PubMed] [Google Scholar]
- Slivicki RA, Iyer V, Mali SS, Garai S, Thakur GA, Crystal JD and Hohmann AG (2020). “Positive Allosteric Modulation of CB1 Cannabinoid Receptor Signaling Enhances Morphine Antinociception and Attenuates Morphine Tolerance Without Enhancing Morphine- Induced Dependence or Reward.” Front Mol Neurosci 13: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slivicki RA, Xu Z, Kulkarni PM, Pertwee RG, Mackie K, Thakur GA and Hohmann AG (2018). “Positive Allosteric Modulation of Cannabinoid Receptor Type 1 Suppresses Pathological Pain Without Producing Tolerance or Dependence.” Biol Psychiatry 84(10): 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham SM, Angus JA, Tudor EM and Wright CE (2005). “Synergistic and additive interactions of the cannabinoid agonist CP55,940 with mu opioid receptor and alpha2-adrenoceptor agonists in acute pain models in mice.” Br J Pharmacol 144(6): 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey I and Mantyh PW (2007). “The cerebral signature for pain perception and its modulation.” Neuron 55(3): 377–391. [DOI] [PubMed] [Google Scholar]
- Trang T, Al-Hasani R, Salvemini D, Salter MW, Gutstein H and Cahill CM (2015). “Pain and Poppies: The Good, the Bad, and the Ugly of Opioid Analgesics.” J Neurosci 35(41): 13879–13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA and Christie MJ (1997). “How opioids inhibit GABA-mediated neurotransmission.” Nature 390(6660): 611–614. [DOI] [PubMed] [Google Scholar]
- Vowles KE, McEntee ML, Julnes PS, Frohe T, Ney JP and van der Goes DN (2015). “Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis.” Pain 156(4): 569–576. [DOI] [PubMed] [Google Scholar]
- Welch SP (2009). “Interaction of the cannabinoid and opioid systems in the modulation of nociception.” International Review of Psychiatry 21(2): 143–151. [DOI] [PubMed] [Google Scholar]
- Wiese B and Wilson-Poe AR (2018). “Emerging Evidence for Cannabis’ Role in Opioid Use Disorder.” Cannabis and Cannabinoid Research 3(1): 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ and Christie MJ (2013). “Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance.” Pharmacol Rev 65(1): 223–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Poe AR, Jeong HJ and Vaughan CW (2017). “Chronic morphine reduces the readily releasable pool of GABA, a presynaptic mechanism of opioid tolerance.” J Physiol 595(20): 6541–6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Poe AR, Lau BK and Vaughan CW (2015). “Repeated morphine treatment alters cannabinoid modulation of GABAergic synaptic transmission within the rat periaqueductal grey.” Br J Pharmacol 172(2): 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Poe AR, Morgan MM, Aicher SA and Hegarty DM (2012). “Distribution of CB1 cannabinoid receptors and their relationship with mu-opioid receptors in the rat periaqueductal gray.” Neuroscience 213: 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AR, Maher L and Morgan MM (2008). “Repeated cannabinoid injections into the rat periaqueductal gray enhance subsequent morphine antinociception.” Neuropharmacology 55(7): 1219–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI and Nicoll RA (2001). “Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses.” Nature 410(6828): 588–592. [DOI] [PubMed] [Google Scholar]