

Abstract
Among the diverse development of stimuli-responsive assemblies, plasmonic nanoparticle (NP) assemblies functionalized with responsive molecules are of a major interest. In this review, we outline a comprehensive and up-to-date overview of recently reported studies on in vitro and in vivo assembly/disassembly and biomedical applications of plasmonic NPs, wherein stimuli such as enzymes, light, pH, redox potential, temperature, metal ions, magnetic or electric field, and/or multi-stimuli were involved. Stimuli-responsive assemblies have been applied in various biomedical fields including biosensors, surfaced-enhanced Raman scattering (SERS), photoacoustic (PA) imaging, multimodal imaging, photo-activated therapy, enhanced X-ray therapy, drug release, stimuli-responsive aggregation-induced cancer therapy, and so on. The perspectives on the use of stimuli-responsive plasmonic assemblies are discussed by addressing future scientific challenges involving assembly/disassembly strategies and applications.
Keywords: plasmonic assemblies, stimuli-responsive, assembly/disassembly, theranostics, applications, gold nanoparticle
1. Introduction
Properties of plasmonic nanomaterials are associated with support surface plasmon polariton modes wherein electromagnetic excitations are coupled with collective oscillations of free charges in a conductive medium.[1] The main constituents of plasmonic nanomaterials are metals, e.g., gold (Au), silver (Ag), and copper (Cu).[2–8] The surface plasmon polariton modes are mainly determined by plasmon structure, size, composition, shape, and local dielectric environment, allowing nanostructures to focus and direct light down to the nanoscale. Nanoplasmonics is a field of study concerned with the investigation of the ability of plasmonic nanostructures to interact strongly with light at wavelengths that significantly exceed their dimensions.[9–11] Therefore, latest methods for the preparation and assembly of plasmonic nanostructures for precisely manipulating light have opened new doors for the applications that were previously considered impossible.
Assembly is a technique for the formation of ordered structures by basic structural units such as polymers and biomolecules.[12–17] The basic structural units spontaneously organize or aggregate into a stable structure with a certain geometric appearance under noncovalent bond-based interactions during the processes of assembly.[18–25] Practical requirements for disease diagnosis, detection, and therapy put forward the expectation for nanoassembly technology to obtain advanced drug delivery systems and sensors for early disease diagnosis. The combination and mutual penetration of nanoassemblies and biomedicine provide great innovation opportunities and attractive market prospects in the biomedical field.
The preparation of novel and efficient nanoassemblies is an important guide for advanced biomedical applications. The application of nanoassembly technology in biomedical research has undergone three developmental stages. In the first stage, the in vitro assembly of nanoparticles (NPs) is directly applied to the biomedical field.[26–34] Here, stimuli-responsive in vitro assembly is the focus of attention. Wherein plasmonic NPs functionalized with responsive molecules cross-link and aggregate and even undergo reversible triggered transitions upon physical or chemical triggers such as light, temperature, magnetic or electric field, pH, metal ions, enzymes, and redox agents.[35–38] Nanoassemblies have been widely applied to biosensors and theranostics.[39–43] However, their disadvantages are poor specificity and sensitivity when applied to living organisms, which can also cause damage to the surrounding normal tissues while killing cancer cells; in addition, the tumor matrix prevents large-size nanoassemblies from entering cancer cells, resulting in low utilization. In the second stage, targeted nanoassemblies containing targeting molecules can carry drugs into the tumor region, effectively improving their specificity and utilization.[44–49] The two types of in vitro nanoassemblies mentioned above have common disadvantages during in vivo applications: increased environmental interference by living organisms, requirement of high doses, and short intratumoral retention time, among others. To circumvent the limitations of in vitro nanoassemblies, there is an increasing interest in the development of stimuli-responsive in vivo reversible or irreversible assemblies as they would effectively reduce the doses and prolong the retention time of NPs, increase imaging signal at lesions and improve therapeutic efficacy.[50–53] The size and morphology of NPs will change on encountering different stimuli, such as light, ultrasound (US), abnormal tumor microenvironment including acidic tumor microenvironment, overexpressed enzymes, abnormal metal ions enrichment; as well as high levels of reduced glutathione (GSH) and reactive oxygen species (ROS) resulting from rapid metabolism and proliferation.
Although many studies have been reported in the field of stimuli responsiveness, to the best of our knowledge, very few reviews have comprehensively summarized stimuli-responsive plasmonic assemblies especially in vivo stimuli-responsiveness and its applications in the biomedical field. This review mainly provides a comprehensive summary of representative stimuli-responsive plasmonic assemblies and their biomedical applications reported in recent years. Stimuli-responsive plasmonic assemblies are classified into two categories depending on the different responsive sites: (1) In vitro stimuli-responsive plasmonic assemblies triggered by enzymes, light, pH, temperature, metal ions, magnetic or electric field, and multi-stimuli responsiveness, and (2) In vivo stimuli-responsive plasmonic assemblies include external stimuli-induced in situ assembly and tumor microenvironment-induced in situ assembly. Stimuli-responsive plasmonic assemblies with high sensitivity and specificity have been applied to various fields; however, herein, we focus on their recent development in biomedical applications, including biosensors, molecular imaging, drug release, and theranostics (Scheme 1). Finally, the overview and future perspective for the use of such plasmonic assemblies based on stimuli-responsiveness are provided.
Scheme 1.
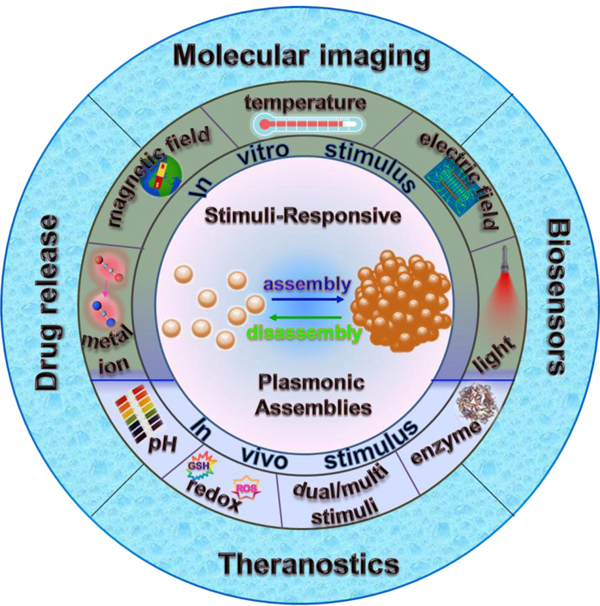
Schematic illustration of stimuli-responsive plasmonic assemblies and their biomedical applications.
2. In vitro stimuli-responsive plasmonic assemblies
2.1. Enzyme responsiveness
Enzymes are specific and efficient biocatalysts, and a vast majority of enzymes are proteins produced by living cells. The catalytic conditions of the enzymes are mild and enzymatic reactions can be conducted at room temperature and atmospheric pressure. Most chemical reactions in organisms are enzymatic reactions. Enzymes, as biocatalysts, not only have the characteristics of a general catalyst but also have characteristics different from a general catalyst when they catalyze a chemical reaction. The rate of an enzymatic reaction is more than 100 times faster than that of a corresponding non-enzymatic reaction.[54–56] Based on these advantages, enzyme reaction-induced assembly of NPs has been widely used in the biomedical field.[57–68] For example, He et al.[65] developed enzyme-responsive Ag nanoassemblies that were applied in high-performance targeted antimicrobial therapy for methicillin-resistant Staphylococcus aureus infections. Further, alkaline phosphatase (ALP), a common hydrolytic enzyme, can trigger the assembly of plasmonic NPs. Jiang et al.[69] designed a cascade reaction-induced magnetic relaxation switching sensor based on the assembly of magnetic NPs (MNP) and silver nanoparticles (AgNP). Ascorbic acid was formed by ALP-induced cascade reactions and then Ag+ was transformed into AgNP to enable the aggregation of dispersed MNP for generating MNP/Ag nanoassemblies. These nanoassemblies could dramatically increase the state change of MNP and greatly enhance the sensitivity of traditional magnetic relaxation switching sensors and were promising for sensing small molecules with high sensitivities. Besides AgNP, gold nanoparticles (AuNP) are also common plasmonic NPs because of their unique optical and electronic properties. Jiang et al.[70] designed a plasmonic nanosensor that used ALP-induced Cu(I)-catalyzed azide/alkyne cycloaddition (CuAAC) between azide/alkyne functionalized AuNP. As shown in Fig. 1A, a phosphate group was removed from ascorbic acid-phosphate via an ALP enzyme reaction, thus leading to a reduction of Cu(II) to Cu(I) via ascorbic acid, followed by CuAAC reactions between azide- and alkyne-terminated groups, resulting in the ALP-mediated aggregation of AuNP. This “dispersed to aggregated” state of AuNP was shown with a “red to blue” color change, attributing high sensitivity to the ALP-sensing naked-eye readout (Fig. 1B). With an increase in ALP concentration, the UV-Vis absorption gradually decreased at 530 nm and increased at 650 nm (Fig. 1C) because of the local surface plasmon resonance (LSPR) originating from the enzyme-triggered aggregation of AuNP. The CuAAC reactions led to the aggregation of AuNP (Fig. 1D); the hydrodynamic size of AuNP with ALP was much larger than that of AuNP without ALP (Fig. 1E). Plasmonic NPs, functionalized with enzyme-responsive groups such as polypeptides can aggregate when the corresponding enzymes are added; however, some plasmonic nanosystems are also linked with other luminescent molecules, thus preparing assemblies that have signal changes to enzymes for the development of different enzyme-detection systems. Kikuchi et al.[71] successfully prepared a β-galactosidase enzyme-responsive near-infrared (NIR) fluorescent sensor based on metal-enhanced fluorescence by modifying AuNP with enzyme-responsive self-assembly moieties and NIR fluorophores (Fig. 1F). Time-dependent LSPR absorption spectrum of functionalized-AuNP in the presence of β-galactosidase was achieved (Fig. 1G), and the aggregation of functionalized AuNP triggered by β-galactosidase enzymatic reaction was observed (Fig. 1H). The NIR fluorescence of functionalized AuNP was obviously increased because of improvements in both the radiative decay rate and the light scattering intensity of fluorophores, whereas the change in the non-radiative decay rate was relatively small. Therefore, the stimuli-responsive fluorescence enhancement possessed great potential for applications in bioimaging and diagnosis. The hybrid plasmonic assemblies realized the complementary advantages of different materials, thus broadening the scope of application and improving the detection sensitivity. Further, Ma et al. [72] designed a sensitive caspase-3 sensing system based on the inner-filter effect of AuNP on CdTe quantum dots. The caspase-3 enzyme could trigger the aggregation of AuNP and reduce their surface plasmonic absorption at 520 nm. Simultaneously, the fluorescence emission of CdTe quantum dots was significantly recovered, and a high sensitivity of caspase-3 enzyme activity was achieved with a detection limit of 18 pM.
Fig. 1.

(A) ALP enzyme-induced Cu(I)-catalyzed azide/alkyne cycloaddition (CuAAC) between alkyne- and azide-functionalized AuNP. (B) The plasmonic immunoassays based on ALP enzyme-induced CuAAC. (C) Absorption spectra of the AuNP solution after CuAAC under various ALP concentrations. (D) TEM images of AuNP before and after ALP-triggered CuAAC reaction. (E) DLS data of AuNP before and after ALP enzyme-induced CuAAC reaction. (F) Schematic illustration of the enzyme-responsive fluorescence enhancement by triggering aggregation of NGal-NIR-AuNP/CHO-NIR-AuNP. (G) Time-dependent UV-Vis spectra of NGal-NIR-AuNP/CHO-NIR-AuNP in the presence of β-gal. (H) Morphological variation of AuNP triggered by enzymatic reaction. (A-E) Reproduced with permission.[70] Copyright 2014, American Chemical Society. (F-H) Reproduced with permission.[71] Copyright 2015, The Royal Society of Chemistry.
2.2. Light responsiveness
Light is an external stimulus that can be instantly delivered to precise locations and closed systems. Further, light can be used in the form of various wavelengths, which may trigger a series of different reactions. For plasmonic NP surfaces functionalized with light-responsive molecules, light can serve as a trigger to mediate the assembly or disassembly of these plasmonic NPs.[73–82] In recent years, light-triggered reversible assembly technology has gained increasing attention because of its characteristics such as simplicity, cost effectiveness, and high efficiency in constructing new structures and materials; further, light may be used in many fields, such as switching sensors and controlled dye/drug release. For example, Qi et al.[81] realized light-triggered reversible self-assembly of Au nanorods (AuNRs) without surface functionalization by photo-exchange adsorption of azoNa from anionic azobenzene derivatives; it was expected to be used in photoswitch sensors. self-assembly of plasmonic NPs via photodimerization reactions can also be achieved. The [2+2] cycloaddition reaction of coumarin occurs with long wave (365 nm) UV irradiation, and this reaction can be reversed with a shorter wavelength (254 nm) UV irradiation.[83] For example, Zhan et al.[84] reported that facilitated by the coumarin functional group of colloidal coumarin-modified AuNP, light illumination at 365 nm induced the self-assembly of monodispersed AuNP, and the resulting nanoassemblies could be disassembled to the dispersed state with light illumination at a relatively shorter wavelength of 254 nm (Fig. 2A-D); this reversible cycle depending on light responsive photolysis of coumarin could be repeated multiple times (Fig. 2E). “Smart” polymers are a class of dynamic polymers that extend the dynamics of molecular scale to macroscopic polymer materials, and thus become an important means for constructing intelligent materials with responsive behavior.[85] Plasmonic NPs modified with a responsive polymer can effectively switch between assembly and disassembly using light to control the bonding strength of noncovalent polymer bonds for controlled dye/drug release. Chen et al.[76] developed an amphiphilic poly(acrylic acid)-block-poly(7-methylacryloyloxy-4-methylcoumarin) copolymer nanoreactor to prepare light-responsive poly(7-methylacryloyloxy-4-methylcoumarin) (PMAMC)-functionalized AuNP. The size of these AuNP and the length of the light-responsive PMAMC hair located on the AuNP surface could be readily adjusted by controlling the atom transfer radical polymerization time of the respective PMAMC and poly(acrylic acid) (PAA) blocks. The PMAMC functionalized-AuNP could be reversibly self-assembled and disassembled with different wavelengths of UV light (Fig. 2F). The absorption redshift upon irradiation with 365 nm light indicated the formation of Au nanoassemblies (Fig. 2G). The PMAMC-functionalized AuNPs were used to encapsulate Rhodamine B (RhB) dyes through the photodimerization of PMAMC blocks triggered by 365 nm light, and achieve light-responsive controlled dye release through photocleavage of PMAMC block triggered by 254 nm light (Fig. 2H, I).
Fig. 2.
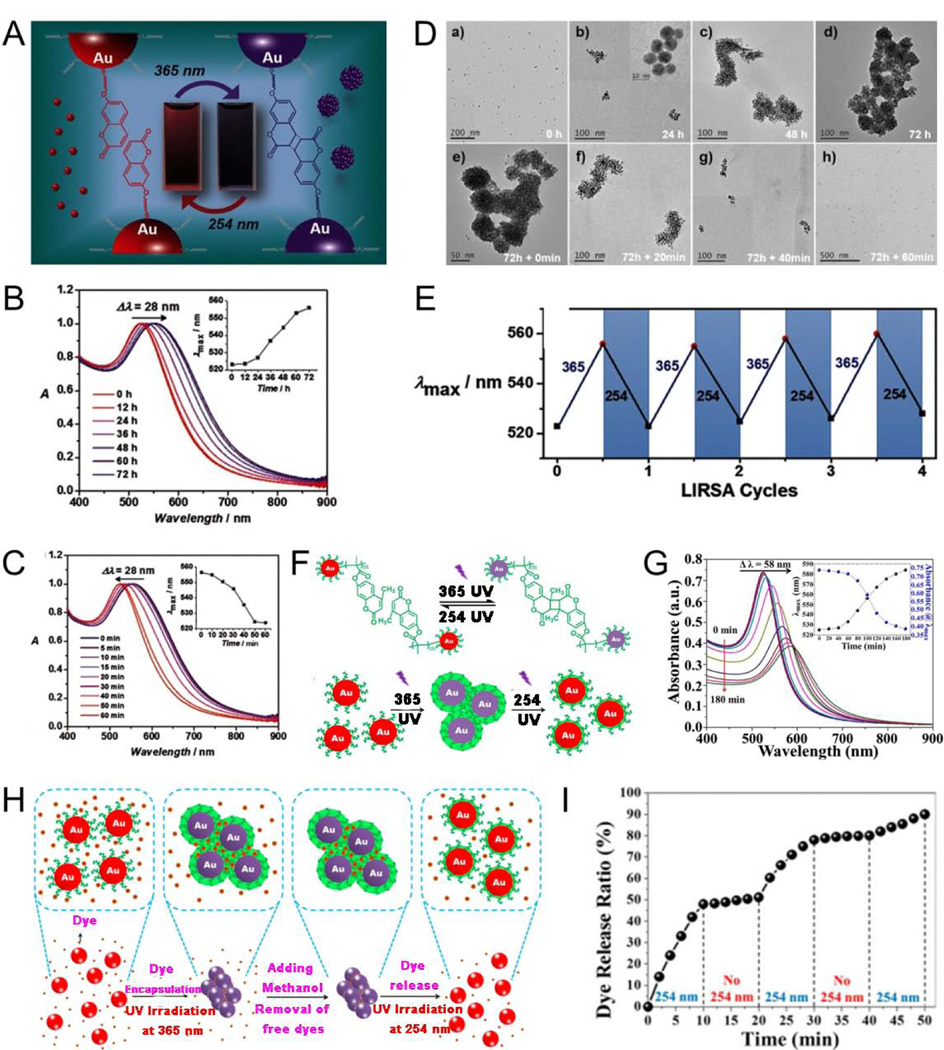
(A) The light-responsive behavior of AuNP. Normalized UV-Vis spectra of AuNP after light illumination at (B) 365 nm and (C) 254 nm. (D) TEM images of AuNP, a)–d) under illumination with 365-nm and e)–h) subsequent illumination with 254-nm light. (E) Reversible wavelength changes with light-responsive assembly/disassembly process. (F) Light-triggered reversible assembly of AuNP. (G) Absorption spectra of PMAMC-capped AuNP when irradiated with 365-nm light. (H) Encapsulation and release of a Rhodamine B dye using light-triggered reversible self-assembly of PMAMC-functionalized AuNP. (I) Controlled release of Rhodamine B. (A-E) Reproduced with permission. [84] Copyright 2016, WILEY-VCH. (F-I) Reproduced with permission. [76] Copyright 2018, National Academy of Sciences.
2.3. pH responsiveness
pH is a parameter in chemical and biomedical media that affects the host matrix assembly morphology, alignment conformation, and optical properties of plasmonic NPs. In general, the stability of plasmonic NPs in suspensions largely relies on the surface charge that usually originates from the terminal groups of surface modifiers, plasmonic nanoassemblies with high negative or positive surface charges are extremely unstable in solvents because of electrostatic repulsion; however, the surface charges may be partially converted to the opposite charge under acid or alkaline conditions via protonation or deprotonation, thus reducing the surface charge absolute value to trigger and control NP self-assembly.[86–94] Responsive PAA,[95] polymer gel,[96] copolymers,[95] GSH,[97] and polymethacrylate (PMAA)[98] have been used as surface modifiers for the pH-responsive assembly of NPs. Yin et al.[99] reported a limited ligand-protection strategy to prepare Ag nanoassemblies with adjustable plasmonic characteristics between UV and NIR regions. The existence of limited PAA ligands played an important role in generating Ag nanoassemblies in the solution: it decreased the surface charge of plasmonic NPs, weakened interparticle electrostatic repulsion, and promoted spontaneous aggregation. In addition, PAA on the surface of AgNP enabled a dynamic color variation and highly reversible assembly in the system because the surface charge could be easily manipulated by regulating the protonation of carboxyl groups by pH changes (Fig. 3A-D). To further control the assembly behavior, a pH-responsive co-assembly system with functionalized plasmonic NPs and additional polymers as external mediators was developed. Ijiro et al.[100] confirmed that AuNP modified with oligoethylene glycol (OEG) held pH-responsive co-assembly with PAA. In this system, the protonation of PAA on pH change induced the generation of a hydrogen bond between polymers and OEG-AuNP, leading to sensitive pH responsiveness (Fig. 3E). Hydrogen bond-mediated co-assemblies of external anionic polymers with OEG-AuNP were monitored by measuring the extinction spectra (Fig. 3F). The size and plasmonic properties of nanoassemblies were highly dependent on both the molecular weight of PAA and the ratio of PAA to AuNP (Fig. 3G). Currently, pH-responsive reversible assembly remains the focus of research. Yuan et al.[101] employed two different types of amines and polyethylene glycol (PEG)-ligands co-functionalized on the surface of AuNP to reversibly shield/deshield different ligands by pH-responsive self-assembly. Amines were deprotonated with pH values ≥7.2; the steric repulsion was lower than the hydrophobic PEG interaction, and the hydrophobic interaction was dominant, leading to the aggregation of AuNP. In contrast, amines were protonated at pH values ≤6.8, and the Au nanoassemblies disassembled (Fig. 3H, I). In summary, responsive plasmonic nanoassemblies were mainly dependent on the polymer properties such as protonation of ionizable groups or acid degradation of chemical bonds.
Fig. 3.

(A) Schematic illustration of assemblies of AgNP. (B) Digital photographs of pH-induced aggregation of AgNP obtained with various reaction times. (C) Extinction spectra exhibiting the tuning of the Ag plasmon bands. (D) Digital photographs of pH-responsive Ag nanoassemblies. (E) Schematic illustration of pH-responsive assembly/disassembly of the AuNP via hydrogen bonding. (F) Extinction spectra of 11-mercaptoundecyl hexaethylene glycol ether (EG6)-AuNP under different pH conditions in the absence or presence of PAA. (G) Proposed mechanism of the [PAA]/[AuNP] ratio-dependent aggregation behavior. (H) Schematic illustrations of pH triggered assembled and disassembly of AuNP. (I) TEM images at different pH values of functionalized AuNP. (A-D) Reproduced with permission.[99] Copyright 2018, American Chemical Society. (E-G) Reproduced with permission.[100] Copyright 2017, American Chemical Society. (H, I) Reproduced with permission.[101] Copyright 2017, American Chemical Society.
2.4. Temperature responsiveness
Temperature is as an exogenous trigger that stimulates functionalized plasmonic NPs by adjusting the structural transformation between expansion and contraction forms of modified molecules on AuNP surface with changing temperatures. By adjusting the lower critical solution temperature (LCST) of thermosensitive polymers, the functionalized plasmonic NPs of thermosensitive polymers can selectively shrink and expand.[102–107] For example, poly(Nisopropylacrylamide) (PNIPAM) is a temperature-responsive polymer with an LCST of 32 °C. PNIPAM shows a volume-phase transition at LCST.[108–112] Cho et al.[113] designed a facile method to broadly and reversibly tune the optical bandwidth of PNIPAM gel supporting AuNP to show a series of color variations with changing temperatures (Fig. 4A). The extinction spectra and assembly shapes of hybrid colloids could also reversibly change with temperature. The amount of switchable colors and temperature reversible plasmonic shifts rely highly on the size of AuNP (Fig. 4B). Additional free PNIPAM added to the functionalized plasmonic system can facilitate NP assembly. Gooding et al.[114] developed an approach for preparing and adjusting the optical properties of temperature-responsive plasmonic nanogels. The magnitude of optical bandwidth changes could easily be regulated by adding free PNIPAM to the solution, with larger redshifts by adding more PNIPAM (Fig. 4C). Meanwhile, reversible assembly or disassembly could be observed with temperatures above or below LSCT, respectively (Fig. 4D). Moreover, the properties of these assembled nanostructures could be adjusted by the temperature. Baumberg et al.[115] developed a strategy to dynamically control the distance between PNIPAM-functionalized AuNP, enabling the triggering of strong color variations from plasmon shifts. They also revealed how the local charge attributed to AuNP by thermoresponsive ligands and the environment determined the aggregation of NPs, and thus the tunability of the plasmon. Besides traditional means such as direct heating, the heating source can also be obtained through light-heat conversion. Scherman et al.[116] combined light-induced temperature-responsive polymers with charged AuNP to store elastic energy that could be rapidly released under light regulation in repeatable isotropic nanotransducers. Temperature-responsive PNIPAM-functionalized 60 nm diameter citrate-stabilized AuNP were irradiated with a 532 nm laser. The NP temperature increased to over 35 °C, thus resulting in the aggregation of NPs and causing a huge redshift in the absorption peak to 645 nm. The extinction peak shifted steadily to 670 nm after laser irradiation for 60 s because of the hydrophobic collapse of PNIPAM above LSCT; and a rapid blue-shift to 539 nm when irradiation ceased because of the rapid swelling of PNIPAM below LSCT. In addition to PNIPAM, other thermally responsive polymers could also be used for the assembly of plasmonic NPs.[107, 117] Furthermore, plasmonic nanoassemblies with different morphologies can be implemented via step-by-step assembly. For instance, Ijiro et al.[118] reported the temperature-responsive two-step aggregation of AuNRs functionalized with alkyl-headed hexa(ethylene glycol) derivatives, which was triggered by stepwise dehydration. When AuNRs were aggregated by heating, they primarily produced side-by-side assemblies. Subsequently, the aggregated assemblies were created in a time-dependent pattern via two-step self-assembly.
Fig. 4.

(A) Schematic outlining of the thermoreversible optical bandwidth changes and a series of switchable colors were highly dependent on the diameters of the AuNP. (B) SEM images of the hybrid colloids prepared at room temperature and 50°C and temperaturedependent variations in the UV–vis spectra and the corresponding colors. (C) Schematic illustrating the temperature mediated aggregation of AuNP. (D) TEM images for the nanogel at 25°C (left) and at 40°C (right). (E) (a) Chemical structures of the hexa(ethylene glycol) derivatives; (b) Schematic illustration of the ligands functionalized on the AuNRs surface; (c) Proposed mechanism of temperature-resopnsive assembly of AuNRs. (F) STEM images of AuNRs dried at 30 and 60°C. (A, B) Reproduced with permission.[113] Copyright 2014, American Chemical Society. (C, D) Reproduced with permission.[114] Copyright 2016, American Chemical Society. (E, F) Reproduced with permission.[118] Copyright 2018, WILEY-VCH.
The aggregated assemblies were disassembled into the side-by-side aggregations on cooling, and then dispersed into mono-AuNRs. The two-step self-assembly process was repeatable and reversible (Fig. 4E, F).
2.5. Metal ion responsiveness
Metal ion-triggered assembly is mainly associated with the properties of molecules modified on the surface of plasmonic NPs. The modified molecules are classified into click chemistry, coordination-based recognition, and ligand exchange reactions according to the responsive mechanism.[119] Click chemistry is a highly reliable and selective approach in synthesis chemistry, and can also play an important role in the field of material chemistry.[120–123] For example, Jiang et al.[124] developed an approach for the aggregation of azide- and terminal alkyne-functionalized AuNP in aqueous solutions via Cu(I) ion catalyzed click reaction (Fig. 5A). Briefly, when sodium ascorbate (reductant) and Cu(II) were both introduced into the mixture of azide- and terminal alkyne-functionalized AuNP at room temperature, the aggregation of AuNP occurred and the color of solution became clear, causing precipitate formation and absorption red-shift (Fig. 5B). Click reactions can be monitored in real time using spectrograms and imaging techniques. Long et al.[125] reported a facile yet effective approach for real-time monitoring of a Cu(I)-catalyzed click reaction at the mono-particle level via plasmon resonance Rayleigh scattering (PRRS) spectra and dark-field microscopy (DFM). The click reaction on the surface of terminal azide- and alkyne-functionalized AuNP lead to interparticle coupling, causing color variations on DFM and absorption red-shift in PRRS spectroscopy. Coordination-based recognition uses the chelation ability of coordination groups modified on the plasmonic NPs and metal ions to induce plasmonic NP assembly.[126–127] Qi et al.[128] prepared the PNIPAM@AuNP with excellent dispersion and stability via a microfluidic droplet synthesis system. The synthesized PNIPAM@AuNP were sensitive to Cu(II) ions. When Cu(II) ions were injected into the aqueous solution containing PNIPAM@AuNP, the lone pair electrons of amino groups in the PNIPAM-functionalized AuNP chelated with Cu(II) because of the empty orbital of the Cu(II) ions, resulting in an absorption redshift and aggregation of NPs via Cu-N bonds (Fig. 5C). Arginine-functionalized AuNP (Arg-AuNP) can also be used for GSH sensing via coordination-based recognition. Jiang et al.[129] reported a dispersion-dominated chromogenic approach for the detection of GSH. In their study, arginine on the Arg-AuNP competed with GSH for Hg(II) binding. Hg(II) triggered the aggregation of Arg-AuNP in the absence of GSH because of the molecular interactions between Hg(II) and amino acids. Aggregation of Arg-AuNP was prevented in the presence of GSH because of specific interactions between Hg(II) and GSH, thus resulting in a visible color variation for GSH sensing (Fig. 5D). The meso-2,3-dimercaptosuccinic acid (DMSA)-modified AuNP (DMSA-AuNP) can be used for the detection of Cr3+ and Cr2O72− via coordination-based recognition.[130] DMSA consists of two −SH groups and two −COOH groups, and the −COOH groups in DMSA are chelated with Cr3+ or Cr2O72− through strong OH···O hydrogen bonds, resulting in the aggregation of DMSA-AuNP; therefore, these groups were used for Cr(III) or Cr(VI) sensing (Fig. 5E). Further, a ligand exchange reaction, as the name implies, indicates that the ligand in the coordination compound can be replaced by other ligands, thus mediating the assembly of ligand-modified plasmonic NPs. For example, Huang et al.[131] developed (11-mercaptoundecyl)-trimethylammonium (MTA)-modified AuNRs with sulfhydryl groups for aggregation-induced PA sensing of Hg(II). MTA was rapidly displaced from the surface of AuNRs because of the high affinity of Hg(II) toward the sulfhydryl group in the presence of Hg(II). Subsequently, AuNRs were aggregated because of the loss of ligand protection on their surface, leading to enhanced PA signals (Fig. 5F). Similarly, other metal ions such as Pd(II)[132–133] and Al(III)[134] can also induce the aggregation of plasmonic NPs.
Fig. 5.

(A) The Cu(II) ions mediated aggregation between two kinds of AuNP using click chemistry. (B) Photographs of the solution containing the mixture of functionalized AuNP before (left) and after the addition of Cu(II) (right), and corresponding absorbance spectra. (C) Proposed mechanism of the Cu(II) ions-triggered aggregation of PNIPAm@AuNP. (D) Arginine-functionalized AuNP for detection of GSH. (E) DMSA-modified AuNP for both Cr(III) and Cr(VI) sensing. (F) Schematic illustrating photoacoustic sensing of Hg(II) based on ligand exchange reaction. (A, B) Reproduced with permission.[124] Copyright 2008, WILEY-VCH. (C) Reproduced with permission.[128] Copyright 2017, American Chemical Society. (D) Reproduced with permission.[129] Copyright 2015, WILEY-VCH. (E) Reproduced with permission.[130] Copyright 2015, The Royal Society of Chemistry. (F) Reproduced with permission.[131] Copyright 2018, ELSEVIER.
2.6. External field responsiveness
Several in vitro responsive assembly methods mentioned above are based on surface molecule-modified plasmonic NPs and lack the active induction function. Their responsive behavior highly depends on the chemical composition of surface molecules (e.g., polymer, small molecules, polypeptide, and DNA)-functionalized plasmonic NPs. Many external fields such as magnetic and electric fields and US are used to induce a more precise preparation of nanoassemblies. Modulation of their properties by controlling nanostructured units (e.g., morphology, size, and crystal structure) is a characteristic and promising method in the field of modern material synthesis.[16, 135] Magnetic-plasmon NPs with both plasmonic and magnetic properties enable the induction of plasmonic NP assemblies under an applied magnetic field.[136–139] For example, Liz-Marzan et al.[140] prepared magnetic-plasmonic NPs through co-encapsulation of iron oxide nanocrystals and Au nanostars within a protective layer of polystyrene (PS)-block-PAA (Fig. 6A). The magnetic-plasmonic NPs possessed specific magnetic responsiveness. Aggregation of NPs was observed on applying a magnetic field. In contrast, NP dispersion was observed without an applied magnetic field (Fig. 6B). Furthermore, the surface enhanced Raman scattering signal intensity under an applied magnetic field was stronger than that without a magnetic field (Fig. 6C). Studies concerning the effect of an electric field on assemblies have mainly explored the effects of different field strengths, directions, and induction time on the orientation and structural transformations of assemblies.[141–143] Zhu et al.[144] developed a simple strategy to regulate the confined assembly of PS-tethered AuNRs (AuNRs@PS) under anodic aluminum oxide (AAO) channels with the help of an electric field (Fig. 6D). Various AuNRs@PS nanoassemblies with well-ordered structures and excellent surface plasmonic properties could be achieved by adjusting the orientation of the electric field, pore size of the AAO channel, and molecular weight of PS brushes (Fig. 6E). Research on US-induced assembly has also become a new research hotspot in the preparation of assemblies. Son et al.[145] reported US-mediated self-assembled immobilization nanogap arrays of monomeric Au and AgNP arrays inside AAO nanopores. The ultrasonic interaction with the solution produced a large amount of energy, which offered the necessary power for the movement and regulation of Au or AgNP into AAO nanopores. This implosion in all orientations allowed plasmonic NP internalization on both sides of AAO nanopores, thus achieving monomeric ordered alignment (Fig. 6F). Furthermore, a 4-mercaptophenylboronic acid-functionalized AgNP array was used to capture bacteria through reversible binding between the bacterial peptidoglycan and boronic acid group. Upon a 514 nm light irradiation, the generated Ag+ ions could destruct various bacteria by photocatalytic bacterial (Fig. 6G).
Fig. 6.
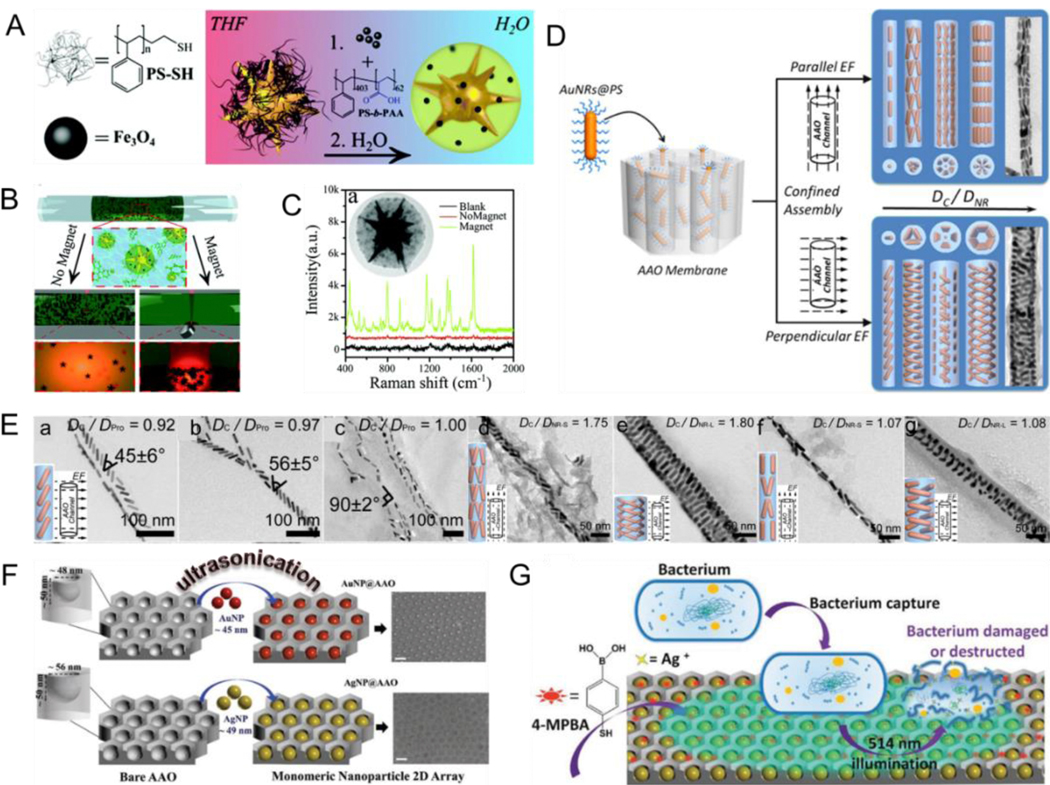
(A) Schematic illustrating the fabrication for Au nanostars decorated with polymer shells consisting of Fe3O4 NPs. (B) The assembly of the NPs near a magnet; and (C) Raman spectra of probe molecules. (D) Schematic illustrating the strategy for assembly of Au nanorods in cylindrical channel with the assistance of electric field. (E) TEM images of AuNRs@PS assemblies with the identical packing mode under analogic confinement strength. (F) Schematic illustrating US-induced self-assembly of plasmonic NPs. (G) Bacteria annihilation process. (A-C) Reproduced with permission.[140] Copyright 2015, The Royal Society of Chemistry. (D, E) Reproduced with permission.[144] Copyright 2016, American Chemical Society. (F, G) Reproduced with permission.[145] Copyright 2019, WILEY-VCH.
2.7. Multi-stimuli responsiveness
In the abovementioned sections, we have systematically discussed plasmonic assembly/disassembly in response to a single stimulus such as enzymes, light, pH, environmental temperatures, metal ions, and external fields. However, this single stimulus responsiveness has a single function and often does not meet the needs of complex assemblies. Alternatively, the current trend is shifting to stimuli-responsiveness systems that respond to more than one stimulus, thus forming a multi-stimuli-responsive system. In this way, the advantages of various single stimulus responsiveness units complement each other to achieve precise control over the assembly process, resulting in better properties and more functional assemblies.[146–151] For instance, Chen et al.[152] prepared a monolayer AuNP film with a dual-responsive plasmonic property for pH and temperature by modifying an OEG-based dendronized copolymer through a facile yet effective self-assembly method at the oil–water interface (Fig. 7A). The large-area free-standing film showed a reversible plasmonic displacement of approximately 77 nm and color variations with pH and temperature changes (Fig. 7B, C). Recently, single-NP-thick, free-standing, stimuli-responsive plasmonic superlattice films have been developed for various applications in the fields of sensor design, microanalysis, and biological diagnostics. Chen et al.[153] developed a facile yet effective approach by employing AuNP@polyaniline (PANI) core-shell structures for constructing a free-standing two-dimensional superlattice monolayer film at the air–liquid interface. PANI played a crucial role in the film, as it not only served as a physical barrier to prevent the van der Waals attraction between AuNP but also provided the film with dual-stimuli (pH and potential variation), resulting in reversible plasmonic and optical responsiveness for regulating color changes (Fig. 7D, E). The two-dimensional superlattice film with dual-responsive plasmonic switches could be used in plasmonic and colorimetric sensors. Nanocomposite tectons (NCTs), with programmed self-assembly capability, are the building blocks for polymer-NP composites developed in recent years and are an ideal choice for the synthesis of these complex nanoassemblies. Macfarlane et al.[154] designed two kinds of NCT samples: 28-nm AuNP functionalized with Hamilton wedge (HW) and terpyridine (Tpy) groups (HW-Tpy-NCTs) and 16-nm AuNP functionalized with cyanuric acid (CA) groups (CA-NCTs) (Fig. 7F, G). As per their design, the mixture of HW-Tpy-NCTs and CA-NCTs forms a multi-stimuli-responsive system to induce assembly and disassembly. HW/CA pairs can form hydrogen bonds at room temperature but heating can break the hydrogen bond. Further, metal coordination bonds form when Zn2+ ions encounter Tpy, but adding an extra Tpy to the system solution breaks the coordination bond. Thus, temperature, Zn2+ and extra Tpy can act as stimulus to control the morphology of responsive NCT assemblies. As shown in Fig. 7H-J, NCT can pathway-dependent assembly/disassembly in which the resulting assemblies (e.g. Tpy complexes, HW/CA complexes, both, or none) relied on the order in which various stimuli were introduced.
Fig. 7.
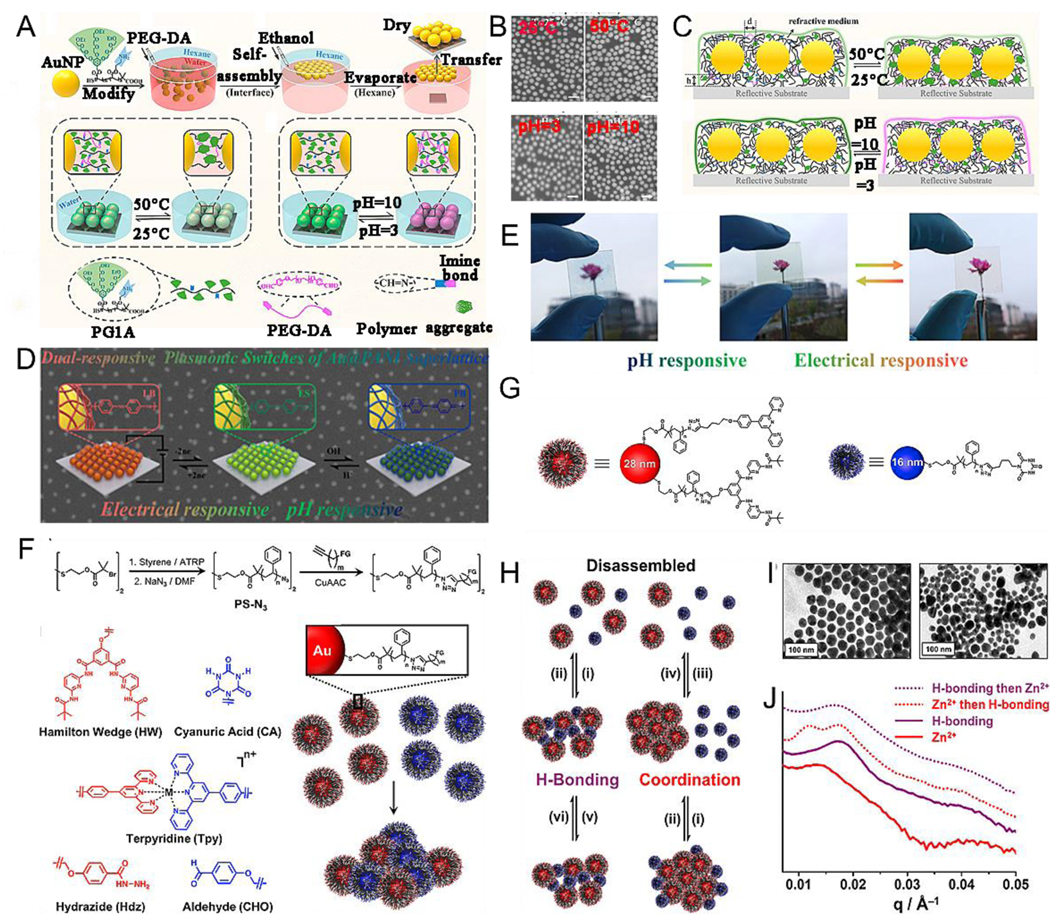
Proposed mechanism of the preparation process of freestanding AuNP self-assembled monolayer films (SAMFs) at the air–water interface. (B) SEM images of SAMFs on a silicon wafer under the aqueous solution with different temperature and pH. (C) Schematic illustration of SAMFs transferred on silicon wafer under various conditions. (D) Schematic illustration of electrical and pH responsive plasmonic switches. (E) pH- and electrical-responsive color change. (F) General synthetic route, chemical structures of the different functional groups and proposed mechanism of the assembly. (G) Schematic illustration of the nanocomposite tectons self-assembled systems for examining multi-stimuli responsiveness. (H) A proposed mechanism of the different responsiveness (i) cool; (ii) heat; (iii) heat, addition of Zn2+ ion; (iv) heat, addition of Tpy; (v) addition of Zn2+ ion; (vi) addition of Tpy. (I) TEM images of the assembly triggered by hydrogen bonding interactions only (right) and coordination interactions only (left). (J) SAXS traces for the hamilton wedge-Tpy and cyanuric acid-based multistimuli responsive nanocomposite tectons assemblies under various conditions. (A-C) Reproduced with permission.[152] Copyright 2018, American Chemical Society. (D, E) Reproduced with permission.[153] Copyright 2020, American Chemical Society. (F-J) Reproduced with permission.[154] Copyright 2019, American Chemical Society.
3. Responsive in vivo in situ assembly/disassembly of plasmonic NPs
With the development of nanotechnology and nanomedicine, increasing attention has been paid to the concept of building plasmonic nanoassemblies in situ in the body. Achieving controllable assembly/disassembly regulation of plasmonic NPs is of great significance for improving delivery efficiency, enhancing imaging signals at lesion sites, and promoting healing ability. Cancers with rapid cell proliferation and metabolism give rise to various abnormal cellular or extracellular microenvironments, such as tumor acidic microenvironment, overexpressed enzymes, abnormal metal ion enrichment, and high levels of reduced GSH and ROS; these can be employed as biological endogenous stimuli for in situ assembly/disassembly in vivo. In addition, light, temperature, and US can also be used as external stimuli to induce in vivo assembly/disassembly of NPs.
3.1. External stimuli-induced in situ assembly
Of the external stimuli, light is deemed to be the most attractive one due to its noninvasive excitation, instant input and removal, wavelength variation, remote control, and site specificity. Because of these intrinsic characteristics, several studies have focused on light-responsive systems and materials. For example, Gao et al.[155] developed photolabile diazirine-decorated AuNP (dAuNP) that were covalently cross-linked upon laser excitation at 405 nm by a diazirine terminal group of PEG on the surface of AuNP. The diazirine group on the Au surface was initially transformed into a carbene on laser irradiation at 405 nm. Following this, the carbene moieties formed bonds with diazirine-functionalized AuNP through C-H, C-C, O-H, and heteroatom-H insertions, resulting in the generation of covalently crosslinked AuNP assemblies. However, no aggregation was observed at the sites that were not covered by the laser: the laser beams could precisely and efficiently focus on regions of very tiny tumors, thus mediating in situ aggregation of NPs and precise PA imaging and photothermal therapy (PTT) (Fig. 8A). The AuNP were aggregated and their hydrodynamic size became larger when a 405-nm laser was applied (Fig. 8B, C). Meanwhile, the surface plasmon resonance peak gradually redshifted to a longer wavelength because of plasmonic coupling between the closely packed AuNP (Fig. 8D). The diazirine-decorated AuNPs were enriched in the tumor region after intravenous injection, followed by 405 nm light irradiation to trigger aggregation of AuNP. Once exposure to 808 nm laser irradiation for 10 min, the tumor temperature was raised by 26.7 °C. Plasmonic NPs modified with light-responsive molecules can self-assemble upon light irradiation; further, unmodified plasmonic NPs can also aggregate upon light irradiation. Zhou et al.[156] reported on laser-induced in situ aggregation of cubic α-Fe2O3@Au nanocomposites for imaging-guided photothermal and enhanced radiation therapy. Interestingly, the α-Fe2O3 substrate of α-Fe2O3@Au nanocomposites exhibited partial ablation with the assistance of 808-nm laser irradiation, and then the ultrasmall AuNP on the surface of α-Fe2O3@Au nanocomposites in situ aggregated into a larger size (Fig. 8E). Further, 96% of ultrasmall AuNPs showed in situ aggregation to approximately 13 nm in size and simultaneously α-Fe2O3@Au nanocomposites were disintegrated after 808 nm laser irradiation (Fig. 8F-I). In addition to light, other external stimuli such as temperature can also induce in vivo in situ aggregation of plasmonic NPs. Gao et al.[157] designed thermally sensitive, elastin-like, polypeptide-functionalized AuNP that could aggregate in situ into Au nanoassemblies in the tumor region at elevated temperatures for tumor multimodal imaging and PTT.
Fig. 8.
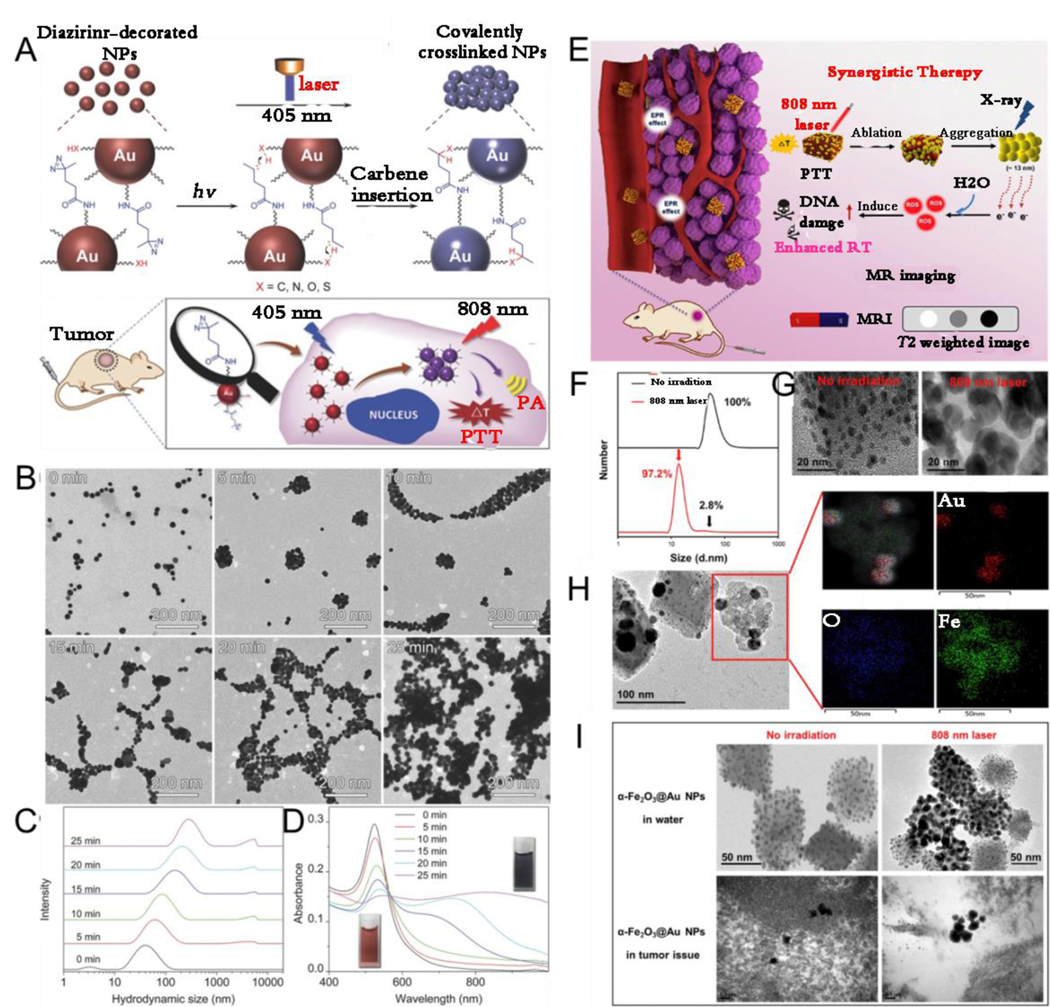
(A) Schematic illustration of light-induced in vivo in situ self-assembly of AuNP. (B) TEM images, (C) hydrodynamic size profiles and (D) UV-Vis spectra of AuNP before and after laser irradiation. (E) Schematic illustration of NIR laser responsive imaging-guided synergistic therapy. (F) Hydrodynamic size profiles, (G) TEM images of α-Fe2O3@Au. (H) Element mapping on the surface of laser-irradiated α-Fe2O3@Au. (I) TEM images of α-Fe2O3@Au with and without laser irradiation. (A-D) Reproduced with permission.[155] Copyright 2017, WILEY-VCH. (E-I) Reproduced with permission.[156] Copyright 2019, ELSEVIER.
3.2. Tumor microenvironment-induced in vivo assembly of plasmonic NPs
3.2.1. Enzyme responsiveness
It is a well-known fact that enzymes play a critical role in physiological metabolism. The concentration and activity of enzymes under some pathological conditions, such as inflammation, infection, and cancer, are abnormally higher than those under normal conditions because of the up-regulation and overexpression of enzymes under abnormal conditions.[158–161] Such abnormalities can be used as a trigger for designing enzyme-responsive assembly/disassembly of plasmonic NPs functionalized with response-specific esters or peptide molecules with high sensitivity and selectivity for biomedical applications. Tumor-derived ALP, which can regulate tumor growth, epithelial plasticity, and disease-free survival, plays an important role in metastatic prostate cancer.[162] Wang et al.[64] designed an ALP enzyme-responsive AuNP platform modified by CREKAYPFFK(Nph) peptide (AuNP@Peptide) that can cause in vivo in situ AuNP aggregation into large-sized AuNP for tumor-specific targeting, enhanced retention, and PTT (Fig. 9A). In the work, the authors first reported aggregation triggered by ALP in vitro, and then confirmed that AuNP@Peptide showed a sensitive response to ALP (Fig. 9B-E). Further, the in vivo tumor experiments demonstrated that ALP-responsive in situ self-assembly of AuNP@Peptide could enhance the PTT effect.
Fig. 9.
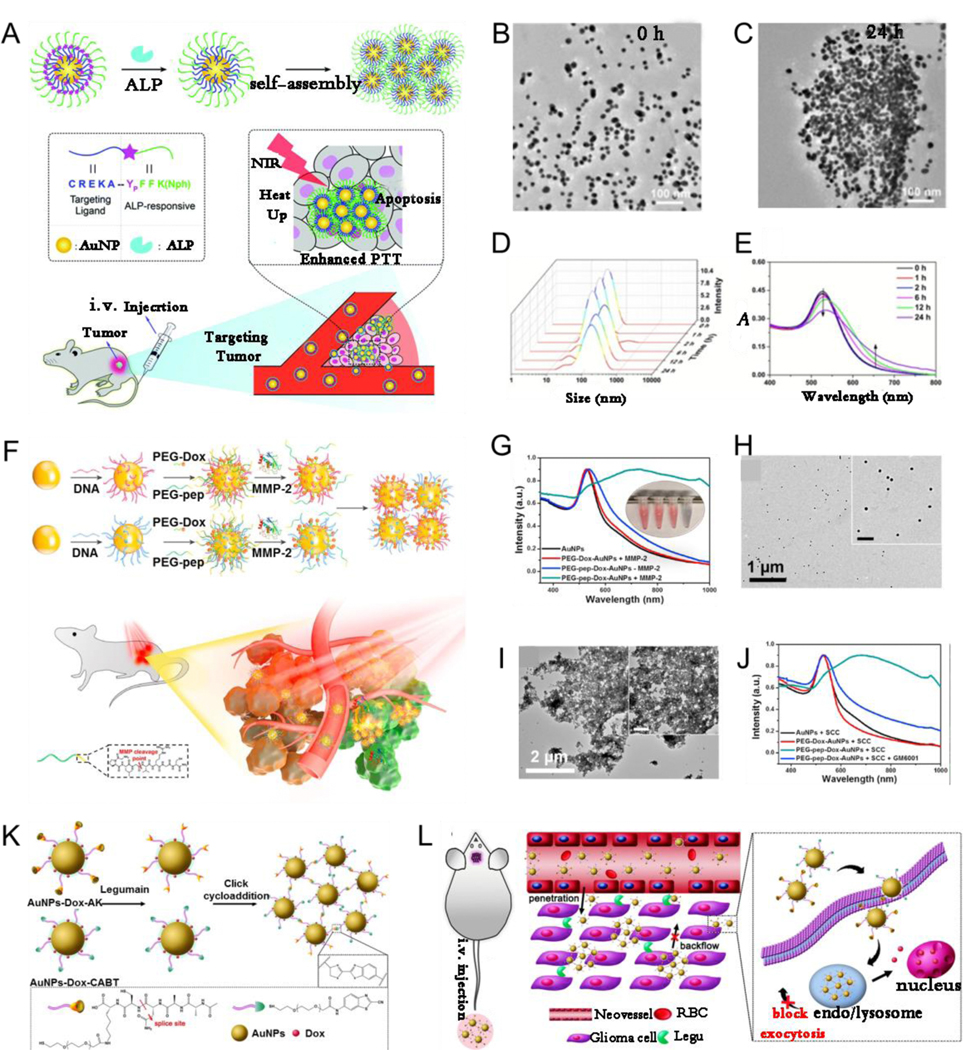
(A) Schematic illustration of enzyme-responsive intracellular self-assembly. TEM images before (B) and after (C) aggregation. (D) Hydrodynamic diameters, and (E) Absorption spectra of AuNP@Peptide before and after addition of ALP. (F) Schematic illustration of MMP-triggered aggregation of AuNP in vivo for PA imaging and PTT. (G) UV–Vis absorption of PEG-Dox-AuNP with MMP-2, AuNP, PEG-pep-Dox-AuNP with and without MMP-2. TEM images of PEG-pep-Dox-AuNP after incubating for 2 h (H) without and (I) with MMP-2. (J) UV–Vis spectra of AuNP, PEG-Dox-AuNP with cell culture media, PEG-pep-Dox-AuNP with cell culture media but with or without MMP-2 inhibitor. (K) Schematic illustrating the legumain-induced aggregation of AuNP-Dox-A&C. (L) Diagram depicting in vivo behavior of AuNP-Dox-A&C after injection for enhanced accumulation. (A-E) Reproduced with permission.[64] Copyright 2018, The Royal Society of Chemistry. (F-J) Reproduced with permission.[163] Copyright 2019, ELSEVIER. (K, L) Reproduced with permission.[164] Copyright 2016, American Chemical Society.
Enzyme-responsive in vivo aggregation can be used for drug delivery in synergistic chemo-photothermal therapy. Nie et al.[163] designed matrix metalloproteinase (MMP)responsive nanoprobe (PEG-pep-Dox-AuNP) by grafting complementary DNA strands tethered with PEG linked doxorubicin (Dox) on the AuNP surface. The nanoprobes quickly aggregated in situ within MMP-abundant tumor region, but maintained well isolated form in normal tissues, and could be used for PA imaging-guided PTT and drug delivery (Fig. 9F). As shown in Fig. 9G-J, the absorption peak red-shifted from 528 nm to 724 nm and clear aggregation of NPs was observed after PEG-pep-Dox-AuNP were incubated with MMP-2 enzyme, indicating that MMP-2 can act as trigger to induce AuNP assembly. In addition, PEG-pep-Dox-AuNP exhibited an obvious red-shift in absorption to approximately 705 nm in SCC-7 epithelioid cells with a high-MMP-2 expression culture medium, but no aggregation was observed when an MMP inhibitor (GM6001) was added to the SCC-7 culture medium, indicating MMP specificity of PEG-PEP-Dox-AuNP. Furthermore, enhanced PA intensity and PTT efficiency in vivo using the nanoprobe compared with those using the MMP-inert nanoprobe indicated that using an enzyme-responsive nanoprobe is a promising strategy for cancer theranostics.
Enzyme-responsive aggregation occurs not only in subcutaneous solid tumors but also in brain tumors. For example, Gao et al.[164] designed an enzyme-responsive nanoplatform (AuNP-A&C) consisting of 2-cyano-6-aminobenzothiazole-functionalized AuNP (AuNPCABT) and peptide (Ala-Ala-AsnCys-Lys)-functionalized AuNP (AuNP-AK). Legumain enzyme induced the hydrolysis of AuNP-AK to expose 1,2-thiolamino groups, which then underwent click cycloaddition with the cyano group on AuNP-CABT, resulting in the self-assembly of AuNP (Fig. 9K, L). Aggregation-based methods could increase the chemotherapeutic enrichment in the glioma and showed an improved therapeutic outcome.
3.2.2. Tumor acidic environment responsiveness
Acid–base homeostasis in living organisms is reflected by the pH gradients across multifarious biological systems. For example, the pH values differ between healthy tissues and certain pathological tissues and between extracellular environment and some intracellular microenvironments: the pH of extracellular environments is 7.4, but that of intracellular compartments is lower (e.g., endosomes, 5.5–6.0; lysosomes, 4.5–5.5; and Golgi apparatus, 6.4), except for cytosol, which is 7.4.[165–166] In addition, the extracellular pH in inflammatory or tumor tissues is approximately 6.5–6.9 because of rapid cell proliferation and metabolism, which is lower than that in healthy tissues (pH 7.4).[167–169] Based on these pH gradients, it is clear that pH often acts as an endogenous trigger to mediate in vivo assembly of NPs.[170–173] Zhang et al.[174] reported that an AuNP system consisting of AuNP-A modified with a Lys-Gly-Gly-Lys-Gly-Gly-Lys-Cys (peptide1) covalently crosslinked with 2, 3-dimethylmaleic anhydride (DMMA) (Fig. 10A) and that AuNP-B was modified with an Asp-Asp-Asp-Asp-Asp-Cys (peptide2) (Fig. 10B). In tumor acidic environment, the surface of the negatively charged AuNP-A was converted to a positively charged state, which then electrostatically interacted with the negatively charged AuNP-B to cause AuNP accumulation in situ (Fig. 10C). To explore pH-responsive assemblies for pH-activated drug release, Kim et al.[175] developed a “smart” AuNP Dox conjugate (SANDC) consisting of a smart AuNP and covalently conjugated Dox, which could release the Dox via a pH-induced linker cleavage in a tumor acidic microenvironment and simultaneously transform the surface charge from negative to a mixture of positive and negative charges, thus triggering in situ accumulation among the NPs through electrostatic interactions (Fig. 10D). The SANDC was sensitive to pH. As shown in Fig. 10E and F, a time-dependent redshift in absorption was observed at pH 5.5, but no change was observed at pH 7.4. The pH responsiveness strategy could be used to achieve irreversible aggregation and pH-specific drug release in a tumor microenvironment. Yuan et al.[176] designed acid-responsive assembled and drug-loading AuNP (Au@PAH-Pt/DMMA) consisting of a polyallylamine drug carrying arm, covalently coupled with Pt(IV) prodrugs and AuNP carriers. The DMMA exfoliated hydrophilic layer began to fall off in acidic tumor microenvironment, exposing the protonated amino groups and accelerating in vivo aggregation of NPs via electrostatic interactions, followed by the release of Pt(IV) prodrugs (Fig. 10G). A tumor acidic microenvironment is an endogenous stimulus that not only triggers assembly but also induces disassembly of plasmonic NPs, thus meeting various needs. For example, Song et al.[177] designed plasmonic vesicles assembled from SERS-encoded amphiphilic AuNP for drug delivery. The plasmonic vesicle could be disassembled in acidic intracellular compartment to trigger drug release, which could be monitored by Raman spectroscopy and plasmonic imaging. Self-assembled responsive magnetic-plasmonic vesicles were pH responsive and disassembled in the tumor acidic environment, realizing multimodal imaging-guided combined cancer therapy. Recently, Song et al.[178] reported on asymmetric iron oxide-gold Janus NPs (Fe3O4-Au JNPs), wherein the hydrophilic PEG and ROS-generating poly(lipid hydroperoxide) (PLHP) were sequentially modified on the surface of Fe3O4-Au JNPs, forming a magnetic-plasmonic bilayer vesicle similar to the liposome membrane. Its position in the vesicle shell could be arbitrarily regulated, and the vesicle cavity was used to load anticancer drugs. The vesicle achieved high enrichment at the tumor site after intravenous administration and disassembled to release drug molecules in the acidic tumor environment. Simultaneously, a low pH induced the release of Fe2+ ions, which could produce singlet oxygen by reacting with PLHP, thus realizing multimodality imaging-guided combined ROS and chemotherapy for cancer (Fig. 10H-L).
Fig. 10.
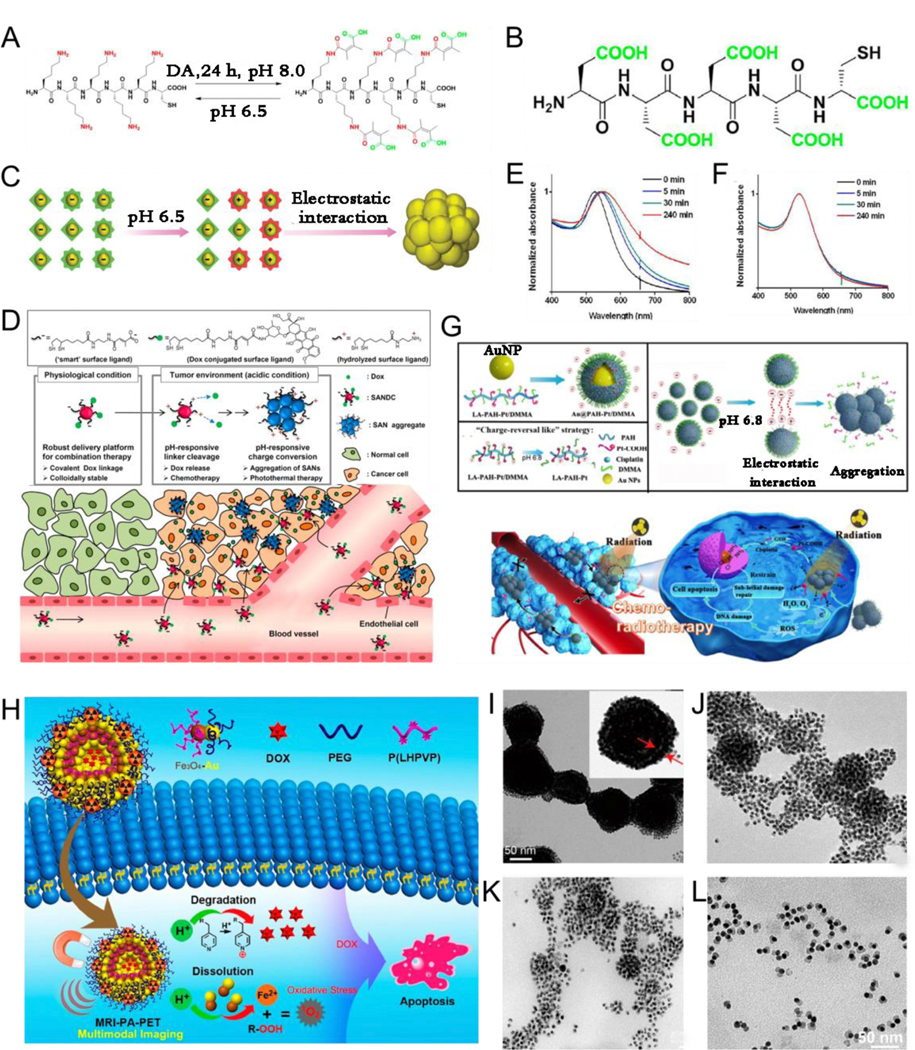
Chemical structural formula of acid responsive peptides1 (A) and non- responsive peptides2 (B). (C) Schematic illustration of the pH-induced aggregation of AuNP. (D) Diagram depicting the pH-triggered aggregation of AuNP Dox conjugate (SANDC) and simultaneous drug release. Time evolution of UV-Vis spectra for SANDC in pH=5.5 (E), and pH=7.4 (F) solutions. (G) Schematic illustration of pH-responsive aggregated cisplatin-loaded AuNP (Au@PAH-Pt/DMMA) and their in vivo working behavior. (H) Proposed mechanism of the anticancer effect of Dox-loaded double-layered magnetic-plasmonic vesicles and acid-triggered disassembly. TEM images of the magnetic-plasmonic vesicles (I) and after incubation in an acidic environment (pH 5.4) for 30 (J), 60 (K), and 90 min (L). (A-C) Reproduced with permission.[174] Copyright 2019, WILEY-VCH. (D-F) Reproduced with permission.[175] Copyright 2013, American Chemical Society. (G) Reproduced with permission.[176] Copyright 2019, Springer. (H-L) Reproduced with permission.[178] Copyright 2019, American Chemical Society.
3.2.3. Dual-stimuli responsiveness
Because of the intricate microenvironment of the living organisms, single stimulus responsiveness may not be able to meet the multiple demands of ratiometric imaging, drug delivery or release, and combined therapy guided by multimodal imaging, among others. Nonetheless, multi-stimuli-responsive systems comprising two or more diverse stimulations have been exploited to accommodate pathological conditions.[179–181] Herein, the dual-stimuli-responsive system with different stimuli of plasmonic nanoassemblies has been highlighted. Song et al.[182] designed a tumor pH and GSH responsive AuNR nanoplatform that was assembled by amphiphilic pH responsive polymer, PA activating croconium dye, hydrophilic PEG, and GSH responsive prodrug Dox monomer (AuNR@PDox), for increasing tumor accumulation efficiency and penetration depth. The nanoplatform could be disassembled into a single AuNR and polymers in a tumor acidic microenvironment and reducing physiological environment, respectively. First, the NPs were approximately 70 nm in size with relatively long blood circulation time under physiological acidity. Second, the enhanced permeability and retention effect enabled the NPs to reach the tumor site, the PEG-PCRVP shell was removed and AuNR@PDox clusters were broken down into single AuNR@PDox NPs because of the extracellular acidic environment of the tumor. Finally, the AuNR@PDox NPs less than 12 nm in size reached the solid tumor site and released the drug as a result of a GSH-reducing environment, leading to enhanced chemotherapy effect (Fig. 11A-E). The hybrid assemblies of plasmonic NPs and other inorganic materials not only inherit individual properties of plasmonic NPs and the other inorganic materials but also show specific responsiveness compared with those of a single plasmonic building block; thus, these hybrid assemblies can be used in stimuli responsive bioimaging, diagnosis and treatment of diseases. Yang et al.[183] reported on the release and aggregation of Au nanospheres based on pH-GSH-reducing environment responsiveness in tumor theranostics. Au nanospheres were encapsulated in the Zeolitic imidazolate framework-8 (ZIF-8) to form a theranostic agent (Au@ZIF-8) with weak coupling plasmonic resonance absorption because of large gaps between the dispersed Au nanospheres. When hybrid Au@ZIF-8 entered the tumor space, Au nanospheres were released from ZIF-8 because of the instability of ZIF-8 at low pH or overexpressed GSH; the released Au aggregated at the tumor region following Zwitter ion electrostatic interactions, resulting in the formation of interparticle hydrogen bonds (Fig. 11F). In addition, Yang et al.[184] further designed a hybrid gold@manganese dioxide core-shell nanostructure (Au@MnO2) probe for photothermally enhanced chemodynamic therapy (CDT). Au@MnO2 probe accumulated in tumor region after intravenous administration, AuNP core was released after the MnO2 shell was degraded, and the released AuNP assembled in situ into large-size aggregation in the tumor in the presence of the overexpressed GSH and low pH. Sonodynamic therapy (SDT) with high precision, deeper tissue penetration, excellent patient compliance, and lower side effects is a new therapeutic method, which can eliminate malignant tumors by using both sonosensitizers and low frequency US.[185] In addition, US can be used as an external stimulus to mediate in vivo in situ disassembly of NPs. Song et al.[186] developed Janus Au-MnO NPs modified with PEG and ROS-sensitive polymers. First, US triggered these vesicles to disassemble into small dispersed Janus Au-MnO NPs, which then further degraded into smaller individual AuNP and released Mn2+ ions in response to GSH overexpression in the tumor region. These were then used for combined therapy with SDT and CDT.
Fig. 11.
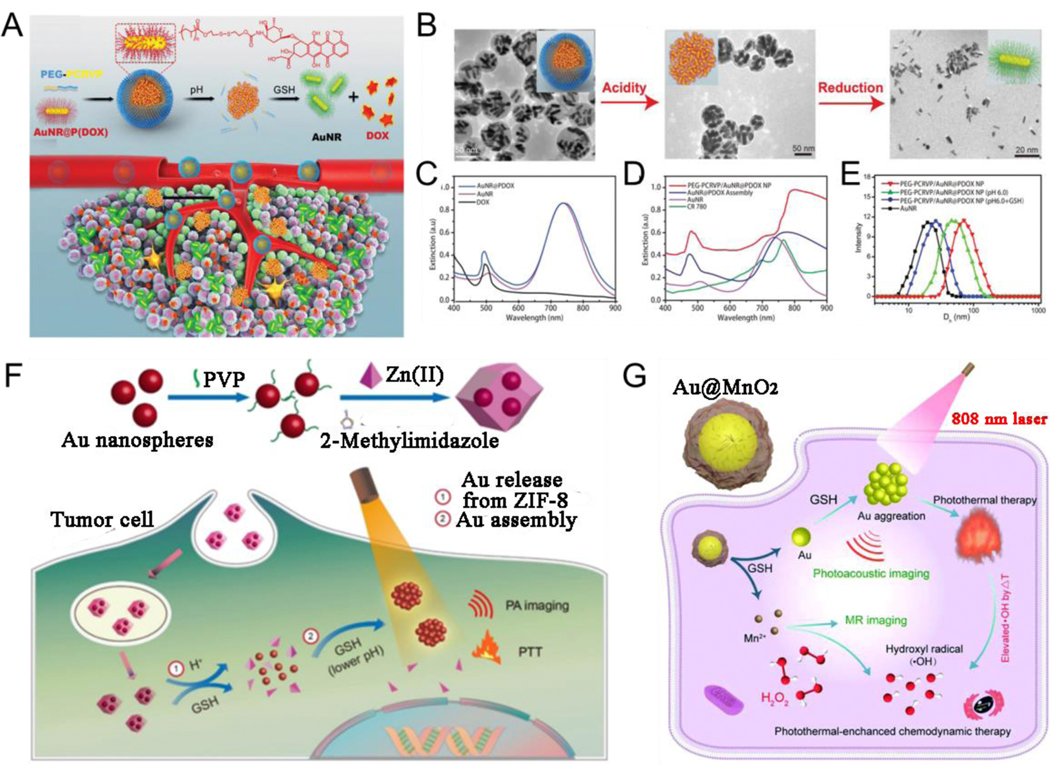
(A) Fabrication of the hybrid photoacoustic activating polymer and Au nanorod cluster assembly, and acidic and reduction environment triggered disassembly. (B) TEM images of the PEG-PCRVP/AuNR@PDox plasmonic vesicles, the nanoassemblies of AuNR@PDox after removing the polymer shell, and single AuNR trigged by pH and GSH. Absorption of (C) Dox, AuNR, and AuNR@PDox, and (D) hybrid NPs and its components. (E) DLS data of the hybrid NPs before (red line) and after treated with pH 6.0 (green line), pH 6.0 and GSH (blue line), and AuNR (black line). (F) Schematic Illustration of the preparaation and release and aggregatiom Au nanospheres from Au@ZIF-8. (G) GSH-mediated Au@MnO2 theranostic agent for PA and magnetic resonance imaging-guided therapy. (A-E) Reproduced with permission.[182] Copyright 2019, WILEY-VCH. (F) Reproduced with permission.[183] Copyright 2020, American Chemical Society. (G) Reproduced with permission.[184] Copyright 2020, The Royal Society of Chemistry.
4. Applications of responsive plasmonic nanoassemblies
In the abovementioned sections, we systematically reviewed stimuli-responsive in vitro and in vivo assembly and disassembly of plasmonic NPs; however, irrespective of the form of assembly/disassembly used, the ultimate goals are to achieve a means for practical application. Therefore, in the following sections, we will focus on various biomedical applications of these stimuli-responsive plasmonic assemblies.
4.1. In vitro/in vivo biosensors
Plasmonic assemblies can serve as detection platforms with high sensitivity and selectivity and rapid responsiveness, making them a promising choice for biosensors.[35, 41, 187–188] At present, in vitro biosensors are mainly used in food safety, environmental monitoring, and biomedical fields. For example, Abbas et al.[189] reported a liposome-amplified plasmonic immunoassay wherein bacteria or antigens could rapidly mediate a chemical cascade reaction, resulting in chromogenic AuNP aggregation for visually sensing pathogens of genus Listeria and Salmonella and species E. coli O157 in food or water samples (Fig. 12A). Briefly, a chemical cascade reaction was triggered by a single pathogen in the sample causing a visual colorimetric variation from red to dark-blue. The rapid and enhanced response was initiated by the triggered decomposition of cysteine-loaded liposomes and the accumulation of AuNP. Given the fact that sensitive and precise detection of viral infection is essential for public health and safety, recently, Xiong et al.[190] developed a colorimetric sensing platform with explosive catalysis to accelerate the disassembly of the Au nanoassemblies from an enzyme muster, containing multiple enzymes encapsulated into a single active unit by liposomes, for the specific and ultrasensitive detection of enterovirus 71 virus (Fig. 12B). Metal ions affect various aspects of cellular life; for example, high levels of copper ion are accumulated in the tissues of patients with Wilson disease, harming their liver and nervous system.[191–193] Therefore, it is important to accurately sense metal ion signals. Li et al.[194] prepared cysteine-coated Au nanostars (Cys-AuNSs) for SERS-based Cu2+ detection (Fig. 12C). Au nanostar aggregation caused by the strong coordination capacity of Cu2+ with cysteine resulted in distinct SERS signals, thus enabling the sensing of Cu2+ with high selectivity and sensitivity. pH is associated with pathological behavior; therefore, the use of pH sensors is practically interesting for monitoring the acidity of the tumor microenvironment for better disease theranostics and prognosis. Zheng et al.[195] designed a ratiometric pH nanosensor in which the conjugation of a pH-insensitive dye, tetramethyl-rhodamine (TAMRA), with pH-insensitive luminescent GSH-coated AuNP (GS-AuNP) fluorescently and quantitatively reported pH variations in vivo. Such pH-responsive biosensors could be attributed to the dimerization of TAMRA dyes on AuNP surface, the geometries of which were very sensitive to surface charges and could be reversely modulated via the protonation of GSH ligands on AuNP surface (Fig. 12D). Hydrogen sulfide (H2S) as a gas signaling molecule possesses multipurpose roles in the biosome. However, efficient detection of H2S remains a challenge. Li et al.[196] developed a 4-acetamidobenzenesulfonyl azide (4-AA)-modified AuNP (AuNP/4-AA) nanosensor for high-selectivity sensing of endogenous H2S at cellular level. The sensing was accomplished with a SERS spectrum resulting from a variety of AuNP/4-AA nanosensors originating from the inter-reaction between H2S and 4-AA (Fig. 12E). In addition, the microRNA (miRNA) nanosensors and ROS nanosensors have been developed. For example, Kanaras et al.[197] designed DNA-coated AuNPs for the miRNA sensing. In this study, Cy3 dye-modified oligonucleotide strand was hybridized to carboxyfluorescein dye-modified oligonucleotide strand, then functionalized to AuNPs’ surface. The DNA-coated AuNPs with close proximity of dyes showed a FL off state as the signal was quenched by the Au core, miRNA can be complementary to the carboxyfluorescein dye-modified oligonucleotide strand, thereby releasing Cy3 dye-modified oligonucleotide strand, leading to FL on state. Chen et al.[198] developed FL Ag nanoclusters (AgNCs), based on the fact that emission of AgNCs is sensitive and selective to the oxidation state, for the detection of various species of ROS.
Fig. 12.
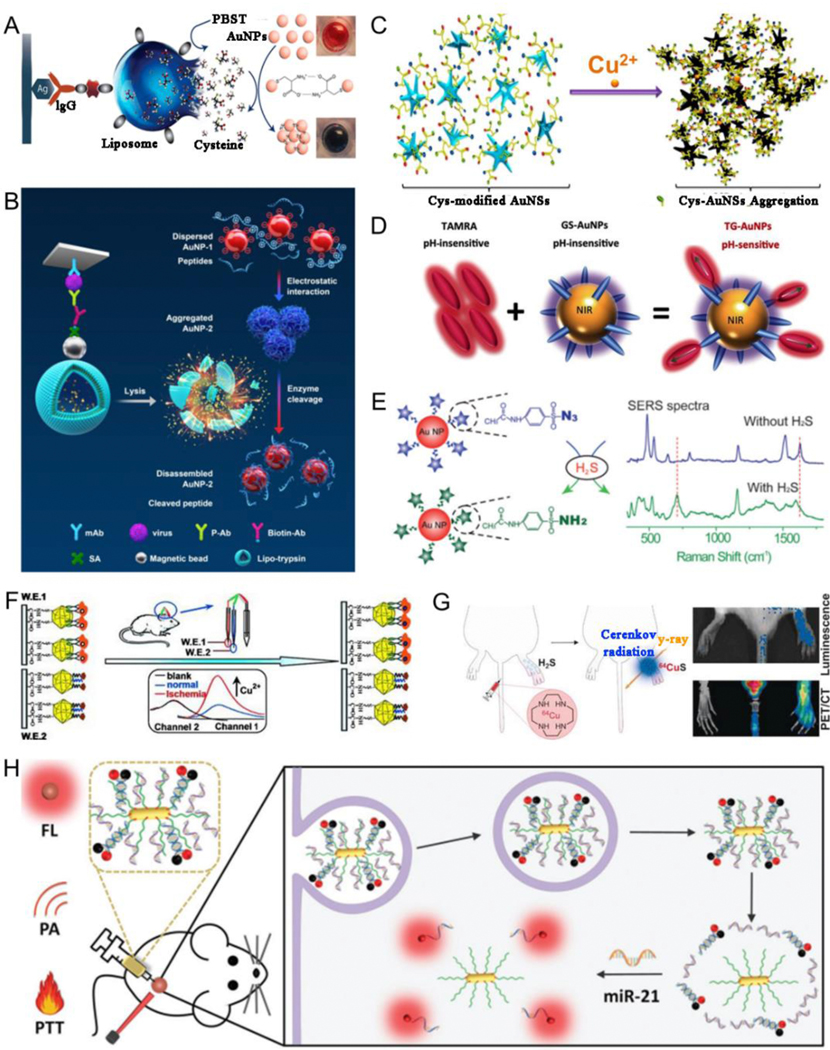
(A) Pathogen sensing using liposome-amplified plasmonic immunoassay. (B) Proposed mechanism of colorimetric immunoassay of virus using enzyme to catalyze the Au-nanoaggregate disassembly. (C) Schematic illustration of Cu2+ sensing using Cys-AuNSs. (D) Schematic illustration of a ratiometric pH sensor. (E) Sensing mechanism of Au-based nanosensor for H2S gas. (F) Schematic illustration of a biosensor for in vivo sensing of cerebral Cu2+ in rat brain. (G) Schematic illustration of radioactive Cu complexes and their application in H2S sensing. (H) Schematic illustration of a biosensor for miRNA detection. (A) Reproduced with permission.[189] Copyright 2015, American Chemical Society. (B) Reproduced with permission.[190] Copyright 2020, American Chemical Society. (C) Reproduced with permission.[194] Copyright 2014, American Chemical Society. (D) Reproduced with permission.[195] Copyright 2016, WILEY-VCH. (E) Reproduced with permission.[196] Copyright 2015, WILEY-VCH. (F) Reproduced with permission.[199] Copyright 2013, WILEY-VCH. (G) Reproduced with permission.[200] Copyright 2016, WILEY-VCH. (H) Reproduced with permission.[201] Copyright 2018, WILEY-VCH
Biosensors are currently used mainly for in vitro detection. However, in vitro detection is only the most basic application of the sensing technology in the biomedical field. The development of a highly active, selective, and stable in vivo bioresponsive nanosensor is highly significant for the accurate detection of diseases and maintenance of human health. Tian et al.[199] reported a ratiometric electrochemical sensor for in vivo detection of Cu2+ ions in rat brain using nanograin-rich Au truncated octahedral microcages, which remarkably improved the detection sensitivity given the large surface area and excellent electrocatalytic activity of Au microcages (Fig. 12F). In this study, first, the bovine erythrocyte copper-zinc superoxide dismutase (E2Zn2SOD) and Cu2+ interacted with good specificity to reconstitute superoxide dismutase. Second, a 6-(ferrocenyl) hexanethiol-functionalized electrode was used as a correction electrode for guarding against environmental interference, and it further associated with a E2Zn2SOD-modified electrode to generate a dual channel ratiometric electrochemical biosensor, hence improving the precision for sensing cerebral Cu2+ ions. Gas-responsive nanosensors have attracted increasing attention in recent years because of their high sensitivity and biosafety. For example, Yoo et al.[200] designed macrocyclic 64Cu–cyclen complexes that reacted immediately with H2S to engender insoluble 64CuS for in vivo sensing, quantification, and imaging of H2S. When 64Cu–cyclen complexes were injected into the mice, positron emission tomography (PET) and Cerenkov luminescence imaging clearly visualized and quantified overexpressed H2S content in inflamed claws of mice (Fig. 12G). The miRNAs, noncoding RNA molecules, can act as diagnostic biomarkers for cancer sensing because their aberrant overexpression is associated with tumor growth and development. Liang et al.[201] designed a polyethylenimine (PEI)-functionalized AuNR fuel and developed a miRNA explorer (FIRE) (AuNR-PEI/FIRE) probe with strong signal for the in vivo detection of miRNA-21 and efficient discrimination of cancer cells and normal cells. The AuNR-PEI/FIRE probe improved cancer sensing capability in vivo and simultaneously showed outstanding tumor elimination efficacy under fluorescence (FL) imaging and PA imaging-guided PTT (Fig. 12H).
4.2. Surface-enhanced Raman scattering
The significant enhancement in SERS is originated from the result of the electromagnetic enhancement from the LSPR mode or chemical enhancement wherein interactions between the molecule and NPs lead to a molecular electronic state change.[202–206] Effective control of SERS signals can be achieved by regulating the molecular electronic state or LSPR mode.[24, 98, 207–211] For example, Lohmuller et al.[212] reported that photothermally induced atrophy of the DNA origami template between Au dimers can be used to decrease the gap size to <2 nm, leading to the augmentation of the field enhancement in NP gap, and therefore can be used to quantitatively sense single molecules via SERS. The SERS peak of a single molecule (Cy3.5) located in the dimer hot spot was not observed before laser irradiation, whereas the redshift of the SERS peak with decreased interparticle gap was achieved after laser irradiation, and the Raman spectra showed the signature peaks of Cy3.5 dyes (Fig. 13A). Stimuli-responsive reversible assembly has always been the focus of research because of its unique properties and excellent application prospects. A reversible SERS signal variation was achieved via reversible assembly to control aggregation and disaggregation of NPs. Mangeney et al.[213] reported a temperature-responsive plasmonic device consisting of Au colloidal NPs and an Au flat film with a thermosensitive polymer brush layer that could control the distance between NPs and the substrate. PNIPAM with an inverse and reversible phase transition at an LCST of approximately 32 °C was used as a linker because of its exceptional phase transition capability. PNIPAM was hydrated; the polymer chains adopted extended structures below the LCST and showed a state of hydrophobic collapse above the LCST. The different conformations of PNIPAM connector between the Au surface and the Au nanoassemblies were supposed to cause drastic variations in the optical performance of the substrate. The colloidal NPs on the Au film were located far from each other (120 nm) below the LCST, and no coupling was observed because of the weak SERS spectra stemming from non-coupled NPs. In contrast, the closely proximity of the Au surface and the AuNP (20 nm) above the LCST resulted in strong interactions with a higher SERS spectrum intensity (Fig. 13B-E).
Fig. 13.

(A) Schematic diagram, Rayleigh and Raman scattering spectra of an AuNP dimer functionalized with a Cy3.5 dye molecule located at the center of hot spot before and after laser illumination (B) Schematic illustration of temperature-induced electromagnetic coupling in Au/polymer hybrid plasmonic assembly. (C) SERS spectra at different temperatures. (D) Integrated SERS signal intensity of methylene blue molecules versus temperature. (E) SERS spectra of methylene blue adsorbed on Au-PNIPAM-NPs substrates and recorded at different temperatures. (A) Reproduced with permission.[212] Copyright 2016, American Chemical Society. (B-E) Reproduced with permission.[213] Copyright 2010, American Chemical Society.
4.3. Photoacoustic imaging
PA imaging can be used to study metabolic functions and physiological and pathological characteristics of organisms, and is especially beneficial for early sensing of cancer and therapeutics.[214–220] Stimuli-responsive PA contrast agents can cause in vivo in situ aggregation or switch, suppressing or eliminating inherent background noise, and can therefore enhance PA signals or modulate imaging information more accurately. For example, Emelianov et al.[221] developed PNIPAM nanogel-functionalized plasmonic NPs to construct two temperature-responsive PA contrast agents, PNIPAM-CuS and PNIPAM-AuNR (Fig. 14A). It is well known that PNIPAM is a thermosensitive polymer with a reversible phase transition hydrophobic chain, the diameter of which can decrease or increase rapidly by changing the temperature above or below its critical temperature (32 °C, Fig. 14B). Thus, the size of PNIPAM-AuNRs decreased obviously and the absorption peak redshifted when the temperature was above the critical temperature (Fig. 14C, D). An enhanced PA signal was observed with an increase in the loading concentration of AuNRs because of an enhanced LSPR effect (Fig. 14E). For the in vivo assay (Fig. 14F), PA signal significantly increased at 24 h after PNIPAM-AuNR injection. The continuous-wave laser stimulus-induced AuNP heating of the PNIPAM cage led to in situ aggregation of PNIPAM-AuNR contrast agent and resulted in a 5.3-fold PA signal enhancement in the tumor region. Further, the background signal around the tumor was eliminated by dynamic contrast-enhanced PA imaging.
Fig. 14.
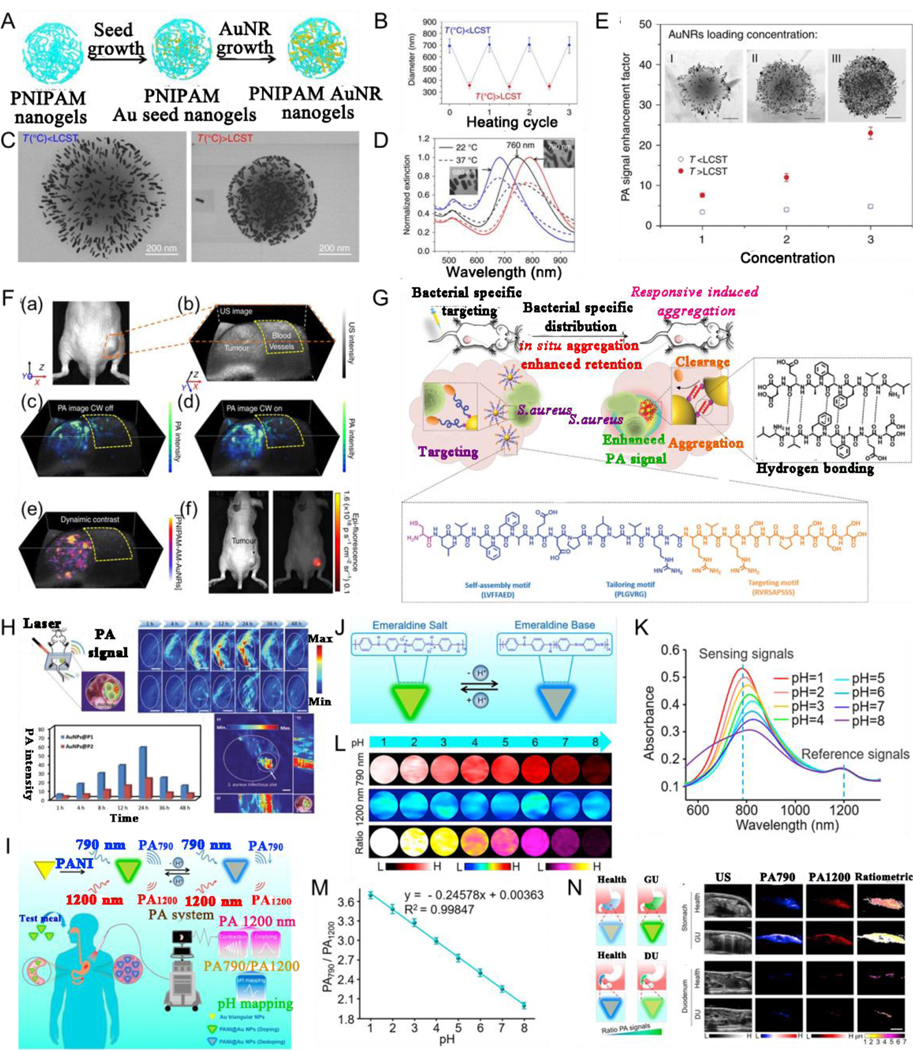
(A) Schematic illustration of the PNIPAM-AuNR preparation. (B) Averaged diameter of PNIPAM below and above lower critical solution temperature (LCST). (C) TEM images of PNIPAM-AuNR nanostructures fabricated at temperature below and above LCST. (D) Absorption of PNIPAM-AuNR with different resonance peaks by altering the nanorod aspect ratios. (E) Photoacoustic signal enhancement factor as a function of AuNR loading concentration below and above LCST. (F) In vivo photoacoustic imaging of a tumor-bearing mouse. (a) A photograph of a tumor-bearing mouse. (b) Ultrasound image of the tumor. (c) Photoacoustic image 24 h post-injection exhibits the tumor, and blood vessels near the tumor. (d) Photoacoustic intensity from tumor is enhanced when the continuous-wave laser is on. (e) Dynamic contrast-enhanced PAimaging. (f) In vivo FL imaging 24 h after injection. (G) Schematic illustration of bacterial infection sensing in vivo by AuNP@P1. (H) Photoacoustic imaging of bacterial infection in vivo. (I) Schematic illustration of diagnosis strategy of gastric and intestinal diseases via quantitative PA imaging. (J) Schematic illustrating a pH responsiveness of nanosensor. (K) Different pH’s absorption, and (L) PA imaging of nanosensor, and (M) PA790/PA1200 ratios of nanosensor. (N) Schematic diagram, US imaging, PA imaging at 790 nm, at 1200 nm, and ratiometric PA imaging of stomach or duodenum. (A-F) Reproduced with permission.[221] Copyright 2017, Springer Nature Limited. (G, H) Reproduced with permission.[222] Copyright 2020, WILEY-VCH. (I-N) Reproduced with permission.[236] Copyright 2019, American Chemical Society.
The intermediate propagation of drug-resistant bacteria is changing the focus of global public health. Early-stage pathogen bioimaging can provide a unique perspective for obtaining accurate infection information in patients. Lu et al.[222] designed a bacteria-instructed enzyme-responsive PA contrast agent (AuNP@P1) consisting of AuNP surface coated with peptide P1 sequence with CLVFFAED-PLGVRG-RVRSAPSSS, with CLVFFAED as a self-assembly fragment, PLGVRG as an enzyme-responsive linker, and RVRSAPSSS as a targeting ligand. The overexpressed bacterial enzymes specifically tailor peptides on the AuNP surface, triggering the in situ aggregation of NPs and leading to a dramatic increase in PA signal for in vivo bioimaging of bacterial infection with high specificity and sensitivity (Fig. 14G, H).
Ratiometric PA imaging with low impact from environmental factors of interfering PA signals offers an effective approach for the relative quantification of physiological and pathological information of organisms both in vitro and in vivo.[223–233] Gastrointestinal diseases affect most of the world’s population, seriously impair the quality of life of individuals, and increase the burden on the healthcare system.[234–235] Functional parameters such as gastrointestinal motility and pH can effectively reflect the variations in gastrointestinal activity under physiological and pathological conditions. Therefore, it is necessary to use a real-time, non-invasive method to quantitatively measure gastrointestinal functional parameters. Nie et al.[236] designed a broad-band pH-responsive ratiometric sensor based on PANI-functionalized Au triangular nanosheets using PA imaging as an advanced technique to quantitatively measure various gastrointestinal functional parameters, such as peristaltic frequency and amplitude, and pH values (Fig. 14I). The pH-responsive mechanism of PANI was that when the pH transformed from alkaline into acidic, PANI was reversibly protonated from an emeraldine base to an emeraldine salt with a decreased absorption peak at approximately 800 nm and an unchanged absorption peak at 1200 nm (Figure 14J, K). PA imaging at various pHs and PA790/PA1200 signal ratios (Figure 14L, M) demonstrated that the pH-responsive sensor could act as a credible probe for monitoring gastrointestinal pH. The pH-responsive ratiometric PA imaging technique could be used to quantitatively measure gastric and duodenal pH in vivo, showing higher PA790/PA1200 ratiometric PA signals in both gastric and duodenal ulcers than those in the normal condition (Figure 14N).
4.4. Stimuli-responsive multimodal imaging
Molecular imaging is the use of imaging methods to display specific molecules at a tissue level, cellular level, and/or subcellular level, reflecting changes of molecular signatures in the living state. Molecular imaging is a rapidly growing interdisciplinary field, which combines molecular biology, physics, chemistry, radiology, nuclear medicine, computer medicine, and other disciplines. The main imaging methods included in molecular imaging can be classified into five categories: optical imaging including PA imaging and FL imaging, radionuclide imaging including PET and single-photon emission computed tomography (SPECT), magnetic resonance imaging (MRI), computed tomography (CT) imaging, and US imaging.[237–240] A single imaging method often has limitations, and it is difficult to simultaneously meet the requirements of sensitivity, specificity, targeting etc. with a single method.[241–244] Stimuli-responsive molecular contrast agents in multimodal imaging can be used for multiple imaging methods; this in turn helps overcome the deficiencies of a single imaging mode, realizes complementary advantages, and broadens the application range of molecular imaging technology.[245–246] For example, Chen et al.[247] developed tumor microenvironment-responsive magnetic Au nanowreaths (AuNWs) to achieve MRI and PA imaging-guided PTT (Fig. 15A). The magnetic AuNWs were prepared by a layer-by-layer self-assembly method wherein positively charged polymeric materials containing disulfide bonds were assembled with negatively charged exceedingly small magnetic iron oxide nanoparticles (ES-MIONs) on silica-coated AuNWs, and then modified with PEG. The T1 contrast ability of ES-MION was inhibited in the magnetic AuNWs assembly because of the increased T2 decaying effect. After intravenous injection, overexpressed GSH in the tumor microenvironment cut off the disulfide bond of the polymer, released ES-MIONs from the assembly, and enabled T1 contrast (Fig. 15B). PA signals in the tumors at 24 h post-injection was 5-fold higher than the background signals (Fig. 15C). To explore applications of multimodal imaging in orthotopic cancer, Song et al.[186] reported an US- and GSH-responsive Janus Au-MnO vesicle (JNP Ve) for improving tumor penetration depth and sono-chemodynamic synergistic therapy in orthotopic liver carcinoma (Fig. 15D). In vitro experiments showed that under US and GSH dual stimulations, JNP Ve was first decomposed into small Au-MnO NPs to enhance tumor penetration and then further decomposed into smaller AuNP and Mn2+ ions to improve PA and MR imaging effect, thus proving to be a promising dual-responsive contrast agent for in vivo dual-modal MRI and PA imaging (Fig. 15E-G). The in vivo imaging effect under the dual stimuli of US and GSH was assessed in an orthotopic liver tumor-bearing mouse model after intravenous injection of JNP Ve, and enhanced in vivo MRI and PA imaging effects were observed (Fig. 15H-J).
Fig. 15.
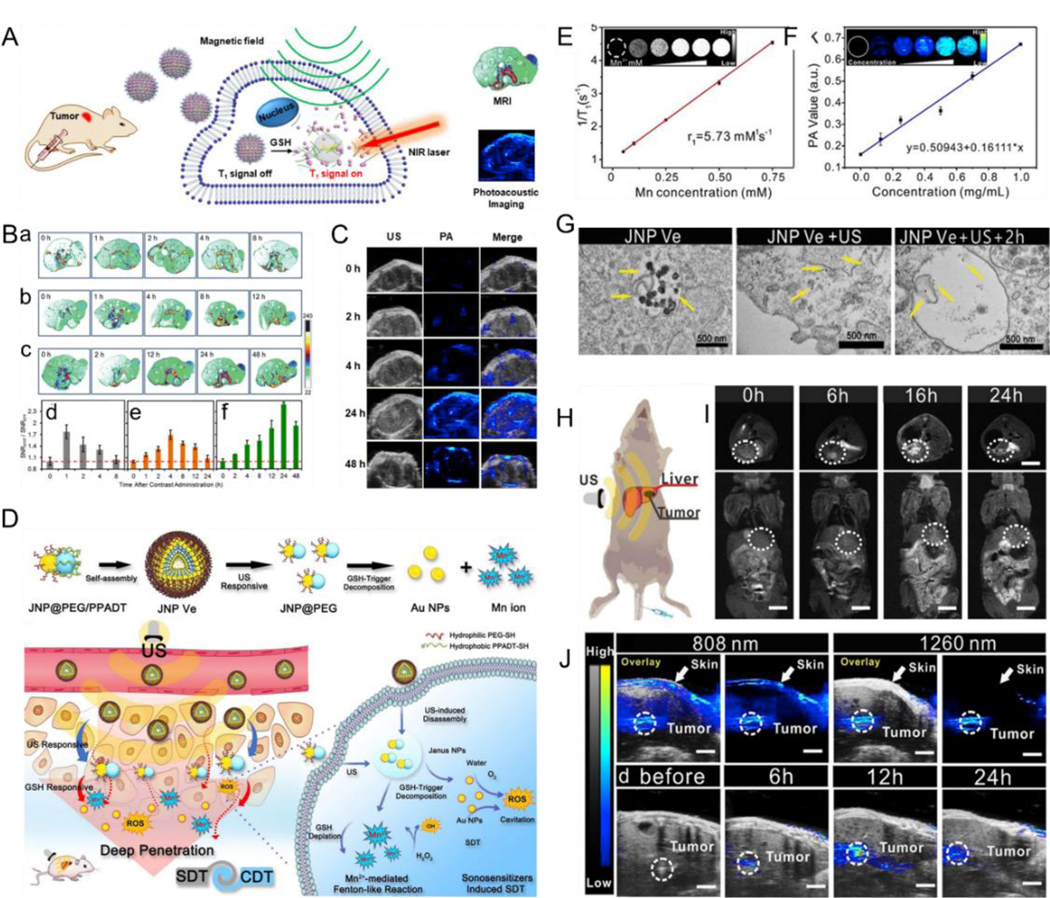
(A) Proposed mechanism of magnetic Au nanowreath and their applications as GSH-responsive imaging contrast agents. (B) In vivo MRI of tumors. (a–c) MR images of tumor-bearing mice and (d–f) corresponding quantificational signals analysis after injection of (a,d) magnevist, (b,e) ES-MIONs, and (c,f) magnetic AuNWs. (C) US, PAimaging, and merged tumor images at various time after injection of magnetic AuNWs upon illumination by an 808 nm laser. (D) Schematic illustration of self-assembly and ultrasound-GSH triggered disassembly of amphiphilic Janus Au-MnO vesicles. (E) Longitudinal relaxation rates and MR images (inset) of JNP Ve after GSH treatment. (F) NIR-II PAimaging signals and images (inset) of the JNP Ve. (G) Images of sections of cells with different treatment. (H) Diagram showing the mice model of orthotopic liver tumor. (I) In vivo MR images of mice obtained at 24 h post-injection of JNP Ve. (J) PAimaging of orthotopic liver tumor. (A-C) Reproduced with permission.[247] Copyright 2018, American Chemical Society. (D-J) Reproduced with permission.[186] Copyright 2020, WILEY-VCH.
4.5. Stimuli-responsive photo-activated therapy
In recent years, photo-activated therapy has attracted the attention of researchers because of its excellent efficacy and low toxic side effects.[248–250] Currently, PTT and photodynamic therapy (PDT) are the two most important photo-activated therapy methods. PTT and PDT use a series of photosensitizers to convert light energy into local hyperpyrexia and ROS under specific wavelength laser irradiations, respectively, for destroying tumor cells as well as tumor vasculature to remove the tumor tissue.[251] Such photosensitizers are extremely low in toxicity and thus do not lead to systemic toxicity by non-specific distribution to other normal tissues without laser irradiation, ensuring accurate and effective tumor-specific killing effects.[30, 252] Herein, the stimuli-responsive photo-activated therapies such as PTT, PDT, and related synergistic therapy are highlighted. Endogenous stimuli such as a tumor acidic microenvironment can induce the in situ aggregation of NPs and then cure the tumor via PTT. Kim et al.[253] reported that mesenchymal stem cells (MSCs) could assemble pH-responsive AuNP (PSAuNP) in endosomes, target tumors, and enhance PTT. These nanoassemblies possessed a higher cellular retention capability compared with pH-insensitive AuNP (cAuNP). The surface charge of PSAuNP could be transformed from negative charge to a mixture of positive and negative charges, resulting in the aggregation of AuNP through electrostatic interactions (Fig. 16A). After intravenous injection, the tumor-targeting efficiency of PSAuNP-laden MSCs (MSC-PSAuNP) was reported to improve by 37-fold and the temperature increase by 8.3 °C in comparison to injections of cAuNP after irradiation, leading to a significantly enhanced PTT effect. Further, apart from PTT, PDT is a vital method to ablate tumors. Cai et al.[181] designed core-shell Au nanocage@manganese dioxide (AuNC@MnO2) NPs as tumor pH/H2O2 microenvironment-responsive oxygen generators and NIR-mediated ROS producers for oxygen-enhanced immunogenic PDT against metastatic triple-negative breast cancer (Fig. 16B). Herein, MnO2 on the surface of AuNC@MnO2 degraded in the tumor pH/H2O2 microenvironment and produced substantial amount of oxygen to enhance PDT effect under laser irradiation in situ. The in vivo experiment showed that AuNC@MnO2 with laser illumination not only eliminated the primary tumor but also inhibited tumor recurrence and metastasis (Fig. 16 C-F).
Fig. 16.
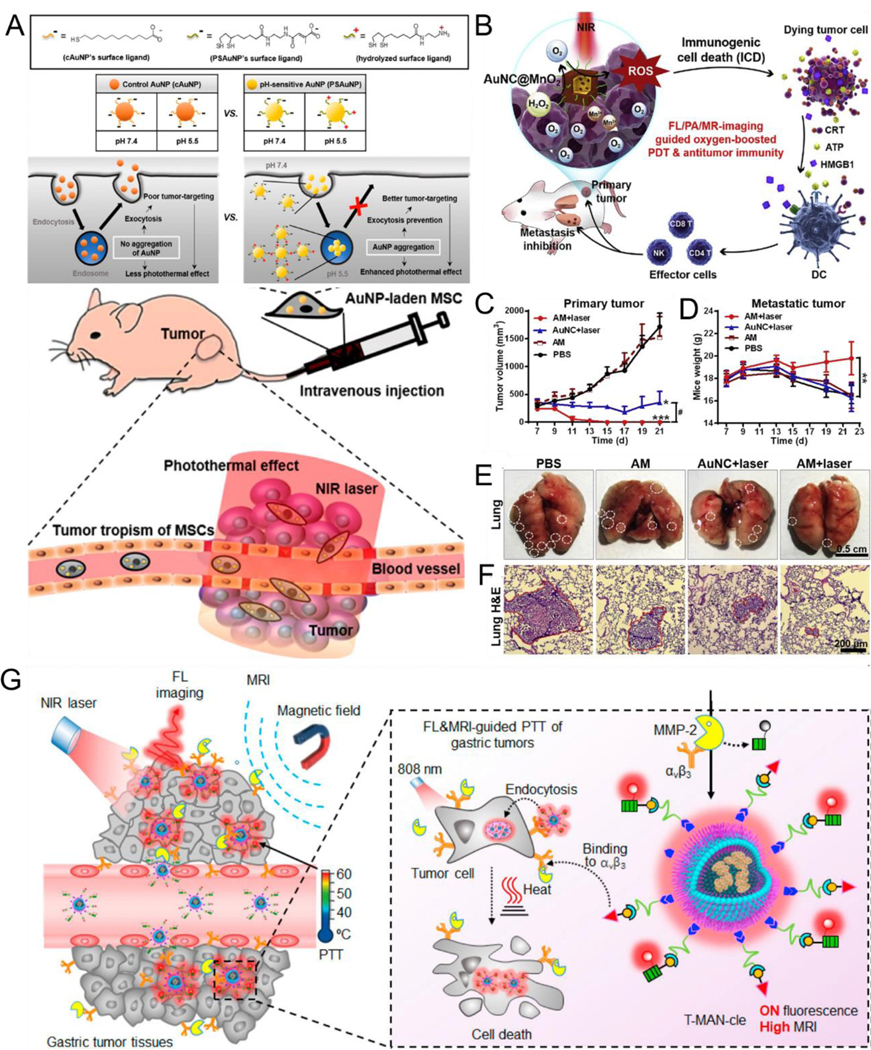
(A) Proposed mechanisms of the enhanced targeting and photothermal effect of the AuNP using pH-sensitive AuNP-laden mesenchymal stem cells (MSC-PSAuNP). (B) The therapeutic mechanism of AuNC@MnO2 for imaging-guided pH/H2O2-responsive oxygen-boosted PDT-immunotherapy. (C) Tumor growth curves and (D) body weight of various treated groups. (E) The images and (F) corresponding H&E staining of metastatic lesions. (G) Schematic illustration of matrix metalloprotease-2 (MMP-2)-activatable nanoprobe (T-MAN) for imaging-guided PTT of gastric tumor. (A) Reproduced with permission.[253] Copyright 2015, American Chemical Society. (B-F) Reproduced with permission.[181] Copyright 2018, ELSEVIER. (J) Reproduced with permission.[254] Copyright 2019, American Chemical Society.
In addition to Au and Ag, copper sulfide (CuS) is also a representative plasmonic assembly material. Ye et al.[254] designed a tumor-targeted and MMP-2-responsive nanoprobe (T-MAN) that was prepared by using Gd-doping CuS micellar NPs modified with Cyclo(Arg-Gly-Asp) (cRGD) peptide and an MMP-2-cleavable fluorescent substrate (Cy5.5 fluorophores). The nanoplatform exhibited excellent photothermal conversion efficiency ( 70.1%) under 808 nm laser irradiation. Upon intravenous administration, T-MAN could preferentially accumulate in the gastric tumors ( 23.4% ID%/g) through αvβ3 integrin-guided active targeting. Moreover, in response to overexpressed MMP-2 in the tumor matrix, T-MAN could significantly increase NIR fluorescence (Cy5.5, 185-fold) and show a high r1 relaxivity ( 60.0 mM−1 s−1 per Gd3+ at 1 T) to precisely outline gastric tumors. Further, laser-triggered ablation of orthotopic gastric cancer was achieved through bimodal FL/MR imaging guided PTT (Fig. 16G).
4.6. Enhanced X-ray therapy
Radiotherapy such as X-ray therapy is a common modality in cancer therapy, with the main purpose of killing cancer cells and shrinking tumors.[255–258] The use of radiosensitizers can improve local treatment efficacy by using relatively safe and low radiation doses, and thus makes tumor cells more sensitive to radiation.[259] An ideal radiosensitizer should meet the requirement of long-term tumor retention and rapid clearance in vivo, while increasing the specificity of radiotherapy to maximize radiosensitivity.[260] Assemblies sized 100 nm are too large to enter the dense region of tumors, whereas assemblies sized less than 30 nm are small enough to enter into tumors but still flow back into the blood circulation or the surrounding tissues and show weak long-term tumor retention capability.[261–264] If plasmonic NPs sized less than 30 nm could accumulate or assemble into larger-sized aggregates after entering the tumor in a stimuli-responsive manner, the plasmonic NP aggregates would be trapped into the tumor region and the diffusion or backflow would be prevented, thus improving the tumor retention and X-ray efficiency of sensibilization. Based on this, Liang et al.[174] designed an AuNP system wherein pH-triggered aggregation of approximately 30-nm AuNP showed efficient radiosensitization for enhanced X-ray radiotherapy (Fig. 10F, Fig. 17A). The pH-responsive in vivo in situ aggregation strategy reduced the non-specific diffusion of individual dispersed AuNP into the peripheral tissues of the tumor and the backflow into the blood circulation, increasing the sensitizer enhancement ratio value of AuNP from 1.16 to 1.73; this helped achieve good radiotherapy efficiency (Fig. 17B-D). Meanwhile, the damage caused by radiation to normal tissues was greatly reduced because of the decrease in the radiation dose and the rapid removal of monodispersed small-size AuNP from normal tissues. In addition, X-ray could be used as an external stimulus to induce ROS generation, thus promoting PDT. For example, Chen et al.[265] developed photosensitizer-associated aggregation-induced emission (AIE) of heterogeneous Au clustoluminogens (AIE-Au) using GSH-protected Au clusters to achieve efficient low-dose X-ray-induced PDT with low side effects (Fig. 17E). AIE-Au was assembled via cationic polymers on its surface, resulting in AIE that enhanced X-ray-excited luminescence by 5.2 times. AIE-Au showed strong absorption ability to X-ray and thus could effectively produce hydroxyl radicals under irradiation with low-dose X-ray, and enhanced the effect of radiotherapy. In addition, AIE-Au could convert X-rays to optical luminescence to stimulate conjugated photosensitizers for PDT. Low-dose X-ray-mediated ROS generation by AIE-Au resulted in high-performance therapy for radioresistant tumors (Fig. 17F-I).
Fig. 17.

(A) Proposed mechanisms of in vivo behavior of AuNP system after intravenous injection for enhanced tumor retention and increased RT. (B) Tumor volumes, (C) bioluminescent imaging and (D) Tumor weight in the mice after 20 days treatment. (E) Proposed mechanisms of R-AIE-Au for CT and FL imaging-guided X-ray-triggered enhanced RT and PDT. (F) Schematic illustration of in vivo anticancer treatment (G) Tumor volumes of various groups of tumor-bearing mice after treatments. (H) Photographs of tumors obtained mice in various treatment groups. (I) Body weight of tumor-bearing mice in various treatment groups. (A-D) Reproduced with permission.[174] Copyright 2019, WILEY-VCH. (E-I) Reproduced with permission.[265] Copyright 2019, WILEY-VCH.
4.7. Stimuli-responsive drug release
Drug resistance is a main barrier for effective cancer treatment. Various drug delivery systems offer promising methods to reverse multidrug resistance, wherein inefficient cellular uptake and deficient intracellular drug release are still the rate-limiting steps for achieving the desired drug concentration level within the treatment window.[266–267] For improving drug targeting and reducing drug release into normal tissues, stimuli-responsive drug delivery systems, stimulated by changes in conditions such as temperature, pH, and enzymes, can be used for releasing drugs into specific tumor tissues, increasing the accumulation of drugs in tumor cells, and reducing the toxic side effects of drugs while increasing their efficacy.[175, 177, 268–275] Early studies of stimuli-responsive drug release was applied at the cellular level; for example, Del Pino et al.[276] developed a plasmonic core-shell Au nanostar/ZIF-8 nanocomposite loaded with functional cargo bisbenzimide compound Hoechst H 33258 (HOE) molecules trapped in an amphiphilic polymer for preventing the leakage of bisbenzimide inside living cells. Upon NIR light irradiation, the active cargo was released owing to the plasmonic absorption of Au nanostars, which resulted in a local increase in temperature and thereby HOE thermodiffusion. More recently, stimuli-responsive drug release has been tested in the living body. For example, a type of tumor cell membrane-encapsulated Au nanocage loaded with Dox (CDAuN) was synthesized by Wang et al.[277], which adopted the homotypic targeted natural tumor cell membranes and was hyperthermia responsive, rendering the Dox-incorporated Au nanocages with the photothermal property. This nanocage could achieve selective and specific targeting of homotypic cancer cells, hyperthermia-induced drug release under NIR irradiation, and the synergism of PTT/chemotherapy (Fig. 18A). The tumor microenvironment, such as pH and enzyme, is also an efficient stimulus for triggering drug release in vivo. Qiu et al.[278] developed in situ-formed AuNP hybrid polymersomes consisting of amphiphilic polyphosphazenes with a hydrophobic 2-diethylaminoethyl 4-aminobenzoate (DEAB) and hydrophilic amino-terminal PEG and Dox for pH-responsive intracellular release into a tumor cell and inhibited drug leakage into blood circulation. Drug leakage of the obtained nanoplatform was effectively relieved at a pH of 7.4, and the systemic blood circulation time of Dox in mice was significantly prolonged. When the nanoplatform arrived at the tumor site, the drug was released owing to the protonation of the tertiary amine in DEAB of the nanoplatform in the acidic tumor microenvironment (Fig. 18B).
Fig. 18.

(A) Proposed mechanisms of the mouse breast cancer cell membrane-functionalized gold nanocage for the hyperthermia-induced release of DOX and homotypic targeted therapy of 4T1 cancer cell growth and metastasis. (B) Schematic illustration of strategy to introduce AuNP into polyphosphazenes vesicles and acid-sensitive intracellular release. (C) Schematic illustration of an enzyme-responsive DOX release from ICG/DOX@Gel-CuS NMs. (D) Schematic illustration of pH/NIR light responsive sequential drug release from amphiphilic Janus NPs. (A) Reproduced with permission.[277] Copyright 2017, WILEY-VCH. (B) Reproduced with permission.[278] Copyright 2017, WILEY-VCH. (C) Reproduced with permission.[279] Copyright 2019, American Chemical Society. (D) Reproduced with permission.[280] Copyright 2018, ELSEVIER.
Realizing in vivo spatial and temporal monitoring of nanopharmaceutical drug release remains a tremendous challenge, but is critical in disease treatment; however, enzyme-responsive drug release provides a feasible solution to this problem. Liang et al.[279] designed core-satellite indocyanine green (ICG)/Dox@Gel-CuS nanopharmaceutical consisting of gelatin (Gel) NPs with payloads of NIR fluorochrome ICG and Dox and satellite CuS NPs. The fluorescence of ICG was concealed within the holonomic ICG/DOX@Gel-CuS system. Upon in vivo administration, the FL signal gradually recovered with the release of the chemodrug Dox from ICG/Dox@Gel-CuS via enzyme-responsive degradation. Thus, FL imaging could be used to quantitatively monitor Dox release in vivo. In addition, the PA signal originating from CuS NPs was independent of the assembly/disassembly status of ICG/DOX@Gel-CuS system, which was used for real-time nanopharmaceutical tracking (Fig. 18C). Co-delivery of dual drugs with different physicochemical characteristics and in a specific drug release order for cancer treatment is crucial for overcoming drug resistance and reducing side effects; therefore, the development of dual drug delivery and sequential release system for cancer theranostics is of considerable clinical significance. To this end, Wang et al.[280] synthetized amphiphilic poly(3-caprolactone) (PCL)-AuNC/Fe(OH)3-PAA Janus NPs to simultaneously preserve the hydrophobic (docetaxel) and hydrophilic (Dox) drugs in their diverse domains. The PCL-AuNC/Fe(OH)3-PAA nanoplatform, with independent pH and NIR light-responsive properties, realized sequential and controlled drug release, and in vivo synergistic chemo-photothermal therapy (Fig. 18D).
4.8. Stimuli-responsive aggregation induced cancer therapy
We systematically summarized in vitro and in vivo aggregation of pH-responsive plasmonic NPs; however, these aggregates can only eliminate tumors with the combined use of a corresponding treatment (e.g., PTT, PDT, or X-ray therapy). Stimuli-responsive aggregation induced cancer therapy has rarely been reported. Recently, Grzybowski et al.[281] prepared a kind of mixed-charge AuNP (defined as [+/−] NP) with pH-responsive aggregation capability by a 4:1 ratio modification of positively charged N, N, N-trimethyl(11-mercaptoundecyl) ammonium chloride and negatively charged 11-mercaptoundecanoic acid ligands on the surface of 5 nm AuNP. The [+/−] NPs rapidly aggregated into 50–100 nm clusters on cell surface via endocytosis and gradual accumulation in endosomes, and the formed clusters were then transported to more acidic lysosomes, followed by aggregation into approximately 2 μm supracrystals; this induced osmotic flow and lysosomal swelling, and the lysosomal membrane integrity was gradually lost, eventually leading to cell death. Conversely, [+/−] NP accumulation was limited inside normal cells owing to the higher pH and the NPs could be excluded from normal cells through exocytosis, resulting in almost no harm to these cells (Fig. 19A-D). In addition, this strategy was applied to cells in various types of cancers such as sarcoma, breast cancer, prostate and lung cancer, and melanoma. This work utilized the pH-dependent aggregation of NPs to selectively destroy cancer cells, which may be a good idea for advancing cancer therapy. However, before considering the clinical application of this strategy, the following problems need to be addressed: First, in vivo efficacy needs to be certified; second, biodegradation and clearance capabilities of Au supracrystals need to be considered. Finally, pH-responsive aggregation mechanism of mixed charge NPs remains to be further investigated.[282]
Fig. 19.
(A) Schematic structure and pH-responsive cascaded aggregation of mixed-charge NPs ([+/−] NP). (B) Summary of how crystallization of [+/−] NP in lysosomes results in selective destroying of cancer cells. (C) Proposed aggregation mechanism of cancer (left) versus normal (right) cells. (D) TEM images of HT1080 (left) or MEF (right) cells at 24 h post-treated with 4:1 NPs. (A-D) Reproduced with permission.[281–282] Copyright 2020, Springer Nature Limited.
5. Conclusions and perspectives
Stimuli-responsive assemblies are molecules with a “responsive” behavior that can produce response signals such as color variation, electronic performance, mechanical properties, and other physicochemical property changes, to external environmental stimuli such as temperature, pressure, light, humidity, and tumor microenvironment effect.[283–288] Therefore, they have a wide range of applications in information technology, catalysis, biosensors, drug delivery, bioimaging, therapy, and other fields.[289–292] Plasmonic assemblies functionalized with “responsiveness” molecule are also sensitive to stimuli. The “responsive” molecule structure changes after receiving the stimulus signal, which further induces the reversible or irreversible aggregation of the plasmonic nanosystem, thus broaden its application range. In this study, we have summarized and reviewed stimuli-responsive plasmonic assemblies, including both in vitro and in vivo responsiveness, as well as their applications in biomedicine. Various stimulus factors including external stimuli such as light, US, metal ions, magnetic and electric fields, and tumor microenvironment including overexpressed enzymes, pH, and high levels of ROS and GSH, as well as their biomedical applications in biosensors, SERS, PA imaging, multimodal imaging, photo-activated therapy, enhanced X-ray therapy, drug release, and stimuli-responsive aggregation-induced cancer therapy have been elaborated.
Currently, although stimuli-responsive plasmonic assemblies have shown promising outcomes in both preparation and application, there are endless possibilities for the development of multifunctional new materials that meet the application needs of different individuals. Accordingly, three research directions deserve our special attention: (1) traditional stimuli conditions such as pH, light, temperature, enzyme, and single stimulus models no longer meet the needs of rapidly developing plasmonic assemblies. Therefore, the development of novel stimuli conditions and multi-stimuli methods with functional responsive characteristics has become a trending topic in the construction of stimulire-sponsive plasmonic nanosystems. This will open a new avenue for the rational use of plasmonic assemblies and the development of new materials; (2) the combination of in vitro assembly and in vivo assembly/disassembly is another interesting nanotechnology research direction because this combination can meet more application needs. These are expected to provide new methods and strategies for future clinical oncology nanomedicine, diagnosis, and treatment technology; (3) improving the stability, biocompatibility, and biodegradability; ensuring non-toxic, timed turns on and off, and precise control of response sites of stimuli-responsive systems is another direction of effort. This contributes to precision therapy and clinical translation.
In brief, with the increasing progress and the continuous innovation of science and technology, we have reason to believe that stimuli-responsive plasmonic assemblies will surely lead to effective strategies, and bring substantial benefits to mankind in many fields including biomedicine in near future.
Highlights.
In this review, we summarized the recent progress of stimuli-responsive plasmonic nanoassemblies and their properties and biological functions of with respect to their categories, responsive mechanisms, as well as their biomedical applications. Particular attention was payed to the in vitro and in vivo assembly/disassembly at different stimulus and their biomedicine applications at individual and synergistic stimuli.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 21874024 and 21635002), the joint research projects of Health and Education Commission of Fujian Province (2019-WJ-20), and the Intramural Research Program (IRP), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH).
Biography

Xiaoyuan (Shawn) Chen received his PhD in chemistry from the University of Idaho in 1999. He joined the University of Southern California as an Assistant Professor of Radiology in 2002. He then moved to Stanford University in 2004 and was promoted to Associate Professor in 2008. In 2009, he joined the Intramural Research Program of the NIBIB/NIH as a Senior Investigator and Chief of the Laboratory of Molecular Imaging and Nanomedicine (LOMIN). He has authored/co-authored over 800 peer-reviewed papers and the total citations exceed 78,000 (H-Index: 146). He is also the founding editor of journal Theranostics.

Jibin Song obtained his PhD degree in Chemical and Biomedical Engineering from Nanyang Technological University, Singapore, in 2014. Then he worked with Prof. Xiaoyuan (Shawn) Chen as a postdoctoral fellow at the National Institutes of Health (NIH). He joined the Fuzhou University as a “Min Jiang Scholar” Professor of analytical chemistry in 2018. Prof. Song has published over 70 papers in high impact journals. His research mainly focuses on developing molecular imaging nanoprobes for bioimaging, biosensing and drug/gene delivery

Prof. Huanghao Yang is a fellow of the Royal Society of Chemistry, who received his PhD from Xiamen University in 2002 and engaged postdoctoral research in Hong Kong University of Science and Technology (2002–2004). He joined Fuzhou University in 2008. He has been supported by the National Science Foundation for Distinguished Young Scholars of China in 2011, the National Award for the Chang Jiang Scholar Program in 2012. His research interests mostly focus on bioanalysis, chemical biology, and nanomedicine.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Qinrui Fu, MOE key laboratory for analytical science of food safety and biology, College of Chemistry, Fuzhou University, Fuzhou 350108, China.
Zhi Li, MOE key laboratory for analytical science of food safety and biology, College of Chemistry, Fuzhou University, Fuzhou 350108, China.
Fengfu Fu, MOE key laboratory for analytical science of food safety and biology, College of Chemistry, Fuzhou University, Fuzhou 350108, China
Xiaoyuan Chen, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), Bethesda, Maryland 20892, United States
Jibin Song, MOE key laboratory for analytical science of food safety and biology, College of Chemistry, Fuzhou University, Fuzhou 350108, China
Huanghao Yang, MOE key laboratory for analytical science of food safety and biology, College of Chemistry, Fuzhou University, Fuzhou 350108, China
References
- [1].Kolwas K, Plasmonics 14 (2019) 1629–1637. [Google Scholar]
- [2].Rycenga M, Cobley CM, Zeng J, Li W, Moran CH, Zhang Q, Qin D, Xia Y, Chem. Rev 111 (2011) 3669–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hsu SW, Rodarte AL, Som M, Arya G, Tao AR, Chem. Rev 118 (2018) 3100–3120. [DOI] [PubMed] [Google Scholar]
- [4].Chao J, Lin Y, Liu H, Wang L, Fan C, Mater. Today 18 (2015) 326–335. [Google Scholar]
- [5].Jiang L, Sun Y, Huo F, Zhang H, Qin L, Li S, Chen X, Nanoscale 4 (2012) 66–75. [DOI] [PubMed] [Google Scholar]
- [6].Klinkova A, Choueiri RM, Kumacheva E, Chem. Soc. Rev 43 (2014) 3976–3991. [DOI] [PubMed] [Google Scholar]
- [7].Yin T, Jiang L, Shen Z, Chin. Phys. B 27 (2018) 097803. [Google Scholar]
- [8].Zhang T, Su D, Li R-Z, Wang S-J, Shan F, Xu J-J, Zhang X-Y, Photon J. Energy 6 (2016) 042504. [Google Scholar]
- [9].Kumar A, Kim S, Nam JM, J. Am. Chem. Soc 138 (2016) 14509–14525. [DOI] [PubMed] [Google Scholar]
- [10].Oh SH, Altug H, Nat. Commun 9 (2018) 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gong J, Li G, Tang Z, Nano Today 7 (2012) 564–585. [Google Scholar]
- [12].Song J, Lin L, Yang Z, Zhu R, Zhou Z, Li Z-W, Wang F, Chen J, Yang H, Chen X, J. Am. Chem. Soc 141 (2019) 8158–8170. [DOI] [PubMed] [Google Scholar]
- [13].Zhang D, Ronson TK, Nitschke JR, Acc. Chem. Res 51 (2018) 2423–2436. [DOI] [PubMed] [Google Scholar]
- [14].Lan X, Wang Q, Adv. Mater. 28 (2016) 10499–10507. [DOI] [PubMed] [Google Scholar]
- [15].Thorkelsson K, Bai P, Xu T, Nano Today 10 (2015) 48–66. [Google Scholar]
- [16].Walther A, Muller AH, Chem. Rev 113 (2013) 5194–5261. [DOI] [PubMed] [Google Scholar]
- [17].Boles MA, Engel M, Talapin DV, Chem. Rev 116 (2016) 11220–11289. [DOI] [PubMed] [Google Scholar]
- [18].Ariga K, Nishikawa M, Mori T, Takeya J, Shrestha LK, Hill JP, Sci. Techno. Adv. Mater. 20 (2019) 51–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chakraborty P, Nag A, Chakraborty A, Pradeep T, Acc. Chem. Res 52 (2019) 2–11. [DOI] [PubMed] [Google Scholar]
- [20].Khor SY, Quinn JF, Whittaker MR, Truong NP, Davis TP, Macromol. Rapid Commun 40 (2019) 1800438. [DOI] [PubMed] [Google Scholar]
- [21].Mayer M, Schnepf MJ, Koenig TAF, Fery A, Adv. Opt. Mater. 7 (2019) 1800564. [Google Scholar]
- [22].Zhang W-J, Hong C-Y, Pan C-Y, Macromol. Rapid Commun 40 (2019) 1800279. [DOI] [PubMed] [Google Scholar]
- [23].Kakuta T, Yamagishi T-A, Ogoshi T, Acc. Chem. Res 51 (2018) 1656–1666. [DOI] [PubMed] [Google Scholar]
- [24].Liu N, Liedl T, Chem. Rev 118 (2018) 3032–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ha M, Kim JH, You M, Li Q, Fan C, Nam JM, Chem. Rev 119 (2019) 12208–12278. [DOI] [PubMed] [Google Scholar]
- [26].Liu G, Zhu J, Guo H, Sun A, Chen P, Xi L, Huang W, Song X, Dong X, Angew. Chem. Int. Ed 58 (2019) 18641–18646. [DOI] [PubMed] [Google Scholar]
- [27].Liu Y, Zhen W, Wang Y, Liu J, Jin L, Zhang T, Zhang S, Zhao Y, Song S, Li C, Zhu J, Yang Y, Zhang H, Angew. Chem. Int. Ed 58 (2019) 2407–2412. [DOI] [PubMed] [Google Scholar]
- [28].Liu C, Zhang S, Li J, Wei J, Mullen K, Yin M, Angew. Chem. Int. Ed 58 (2019) 1638–1642. [DOI] [PubMed] [Google Scholar]
- [29].Chen YS, Zhao Y, Yoon SJ, Gambhir SS, Emelianov S, Nat. Nanotechnol 14 (2019) 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kim M, Lee JH, Nam JM, Adv. Sci 6 (2019) 1900471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang S, Geryak R, Geldmeier J, Kim S, Tsukruk VV, Chem. Rev 117 (2017) 12942–13038. [DOI] [PubMed] [Google Scholar]
- [32].Song Z, Han Z, Lv S, Chen C, Chen L, Yin L, Cheng J, Chem. Soc. Rev 46 (2017) 6570–6599. [DOI] [PubMed] [Google Scholar]
- [33].Darabi A, Jessop PG, Cunningham MF, Chem. Soc. Rev 45 (2016) 4391–4436. [DOI] [PubMed] [Google Scholar]
- [34].Grzelczak M, Vermant J, Furst EM, Liz-Marzan LM, ACS Nano, 4 (2010) 3591–3605 [DOI] [PubMed] [Google Scholar]
- [35].Wu X, Hao C, Kumar J, Kuang H, Kotov NA, Liz-Marzan LM, Xu C, Chem. Soc. Rev 47 (20^) 4677–4696. [DOI] [PubMed] [Google Scholar]
- [36].Grzelczak M, Liz-Marzan LM, Klajn R, Chem. Soc. Rev 48 (2019) 1342–1361. [DOI] [PubMed] [Google Scholar]
- [37].Gon ‘alez-Rubio G, Guerrero-Martinez A, Liz-Marzan LM, Acc. Chem. Res 49 (2016) 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Watanabe K, Kuroda K, Nagao D, Materials 11 (2018) 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhou W, Gao X, Liu D, Chen X, Chem. Rev 115 (2015) 10575–10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu J, He H, Xiao D, Yin S, Ji W, Jiang S, Luo D, Wang B, Liu Y, Materials 11 (2018) 1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mejia-Salazar JR, Oliveira ON Jr., Chem. Rev 118 (2018) 10617–10625. [DOI] [PubMed] [Google Scholar]
- [42].Wu D, Duan X, Guan Q, Liu J, Yang X, Zhang F, Huang P, Shen J, Shuai X, Cao Z, Adv. Funct. Mater. 29 (2019) 1900095. [Google Scholar]
- [43].Kelley EG, Albert JNL, Sullivan MO, Epps TH III, Chem. Soc. Rev 42 (2013) 7057–7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lee GY, Qian WP, Wang L, Wang YA, Staley CA, Satpathy M, Nie S, Mao H, Yang L, ACS Nano 7 (2013) 2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ma T, Hou Y, Zeng J, Liu C, Zhang P, Jing L, Shangguan D, Gao M, J. Am. Chem. Soc 140 (2018) 211–218. [DOI] [PubMed] [Google Scholar]
- [46].Zhang P, Wang J, Chen H, Zhao L, Chen B, Chu C, Liu H, Qin Z, Liu J, Tan Y, Chen X, Liu G, J. Am. Chem. Soc 140 (2018) 14980–14989. [DOI] [PubMed] [Google Scholar]
- [47].Li Z, Yin S, Cheng L, Yang K, Li Y, Liu Z, Adv. Funct. Mater. 24 (2014) 2312–2321. [Google Scholar]
- [48].Mou X, Ali Z, Li S, He N, J. Nanosci. Nanotechno 15 (2015) 54–62. [DOI] [PubMed] [Google Scholar]
- [49].Lei Z, Ding L, Yao C, Mo F, Li C, Huang Y, Yin X, Li M, Liu J, Zhang Y, Ling C, Wang Y, Adv. Mater. 31 (2019) e1807456. [DOI] [PubMed] [Google Scholar]
- [50].Teng L, Song G, Liu Y, Han X, Li Z, Wang Y, Huan S, Zhang XB, Tan W, J. Am. Chem. Soc 141 (2019) 13572–13581. [DOI] [PubMed] [Google Scholar]
- [51].Liu F, Lin L, Zhang Y, Wang Y, Sheng S, Xu C, Tian H, Chen X, Adv. Mater. 31 (2019) 1902885. [DOI] [PubMed] [Google Scholar]
- [52].Feng W, Han X, Wang R, Gao X, Hu P, Yue W, Chen Y, Shi J, Adv. Mater. 31 (2019) 1805919. [DOI] [PubMed] [Google Scholar]
- [53].Zhao M, Li B, Fan Y, Zhang F, Adv. Healthc. Mater. 8 (2019) e1801650. [DOI] [PubMed] [Google Scholar]
- [54].Mikolajczak DJ, Berger AA, Koksch B, Angew. Chem. Int. Ed (2020) DOI: 10.1002/anie.201908625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wolfenden R, Snider MJ, Acc. Chem. Res 34 (2001) 938–945. [DOI] [PubMed] [Google Scholar]
- [56].Nanda V, Koder RL, Nat. Chem 2 (2010) 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ding Z, Sun H, Ge S, Cai Y, Yuan Y, Hai Z, Tao T, Hu J, Hu B, Wang J, Liang G, Adv. Funct. Mater. 29 (2019) 1903860. [Google Scholar]
- [58].Yuan Y, Ding Z, Qian J, Zhang J, Xu J, Dong X, Han T, Ge S, Luo Y, Wang Y, Zhong K, Liang G, Nano Lett. 16 (2016) 2686–2691. [DOI] [PubMed] [Google Scholar]
- [59].Gallo J, Kamaly N, Lavdas I, Stevens E, Nguyen QD, Wylezinska-Arridge M, Aboagye EO, Long NJ, Angew. Chem. Int. Ed 53 (2014) 9550–9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gao Z, Hou Y, Zeng J, Chen L, Liu C, Yang W, Gao M, Adv. Mater. 29 (2017) 1701095. [DOI] [PubMed] [Google Scholar]
- [61].Liu R, Aw J, Teo W, Padmanabhan P, Xing B, New J. Chem. 34 (2010) 594–598. [Google Scholar]
- [62].Gupta S, Andresen H, Ghadiali JE, Stevens MM, Small 6 (2010) 1509–1513. [DOI] [PubMed] [Google Scholar]
- [63].Li H, Wang P, Deng Y, Zeng M, Tang Y, Zhu WH, Cheng Y, Biomaterials 139 (2017) 30–38. [DOI] [PubMed] [Google Scholar]
- [64].Yang S, Yao D, Wang Y, Yang W, Zhang B, Wang D, Chem. Commun 54 (2018) 9841–9844. [DOI] [PubMed] [Google Scholar]
- [65].Zuo YM, Yan X, Xue J, Guo LY, Fang WW, Sun TC, Li M, Zha Z, Yu Q, Wang Y, Zhang M, Lu Y, Cao B, He T, ACS Appl. Mater. Interfaces 12 (2020) 4333–4342. [DOI] [PubMed] [Google Scholar]
- [66].Liu Y, Pan M, Wang W, Jiang Q, Wang F, Pang DW, Liu X, Anal. Chem 91 (2019) 2086–2092. [DOI] [PubMed] [Google Scholar]
- [67].Hu Q, Katti PS, Gu Z, Nanoscale 6 (2014) 12273–12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jiang D, Ni D, Rosenkrans ZT, Huang P, Yan X, Cai W, Chem. Soc. Rev 48 (2019) 3683–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chen Y, Xianyu Y, Dong M, Zhang J, Zheng W, Qian Z, Jiang X, Anal. Chem 90 (2018) 6906–6912. [DOI] [PubMed] [Google Scholar]
- [70].Xianyu Y, Wang Z, Jiang X, ACS Nano, 8 (2014) 12741–12747. [DOI] [PubMed] [Google Scholar]
- [71].Zeng Z, Mizukami S, Fujita K, Kikuchi K, Chem. Sci 6 (2015) 4934–4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Li J, Li X, Shi X, He X, Wei W, Ma N, Chen H, ACS Appl. Mater. Interfaces. 5 (2013) 9798–9802. [DOI] [PubMed] [Google Scholar]
- [73].Bian T, Chu Z, Klajn R, Adv. Mater. 31 (2019) 1905866. [DOI] [PubMed] [Google Scholar]
- [74].Xiao Z, Ji C, Shi J, Pridgen EM, Frieder J, Wu J, Farokhzad OC, Angew. Chem. Int. Ed, 51 (2012) 11853–11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kim JY, Yeom J, Zhao G, Calcaterra H, Munn J, Zhang P, Kotov N, J. Am. Chem. Soc 141 (2019) 11739–11744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Chen Y, Wang Z, He Y, Yoon YJ, Jung J, Zhang G, Lin Z, Proc. Natl. Acad. Sci. U. S. A 115 (2018) E1391–E1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Guarrotxena N, Quijada-Garrido I, Chem. Mater. 28 (2016) 1402–1412. [Google Scholar]
- [78].Jaquay E, Martínez LJ, Huang N, Mejia CA, Sarkar D, Povinelli ML, Nano Lett. 14 (2014) 5184–5188. [DOI] [PubMed] [Google Scholar]
- [79].Lin L, Peng X, Wang M, Scarabelli L, Mao Z, Liz-Marzan LM, Becker MF, Zheng Y, ACS Nano,10 (2016) 9659–9668. [DOI] [PubMed] [Google Scholar]
- [80].Qian Z, Ginger DS, J. Am. Chem. Soc 139 (2017) 5266–5276. [DOI] [PubMed] [Google Scholar]
- [81].Wang Q, Li D, Xiao J, Guo F, Qi L, Nano Res. 12 (2019) 1563–1569. [Google Scholar]
- [82].Xia H, Gao Y, Yin L, Cheng X, Wang A, Zhao M, Ding J, Shi H, ChemBioChem 20 (2019) 667–671. [DOI] [PubMed] [Google Scholar]
- [83].Trenor SR, Shultz AR, Love BJ, Long TE, Chem. Rev 104 (2004) 3059–3077. [DOI] [PubMed] [Google Scholar]
- [84].He H, Feng M, Chen Q, Zhang X, Zhan H, Angew. Chem. Int. Ed 55 (2016) 936–940. [DOI] [PubMed] [Google Scholar]
- [85].Zhang Q, Qu DH, Tian H, Adv. Opt. Mater. 7 (2019) 1900033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Liu H, Jiang Q, Pang J, Jiang Z, Cao J, Ji L, Xia X, Wang K, Adv. Funct. Mater. 28 (2018) 1703847. [Google Scholar]
- [87].Park S, Lee WJ, Park S, Choi D, Kim S, Park N, Sci. Rep 9 (2019) 20180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Swami A, Mittal S, Chopra A, Sharma RK, Wangoo N, Appl. Nanosci 8 (2018) 467–473. [Google Scholar]
- [89].Lopatina LI, Tsarkova LA, Colloid Polym. Sci 297 (2019) 339–350. [Google Scholar]
- [90].Ma J, Hu Z, Wang W, Wang X, Wu Q, Yuan Z, ACS Appl. Mater. Interfaces 9 (2017) 16768–16778. [DOI] [PubMed] [Google Scholar]
- [91].Fan C, Bian T, Shang L, Shi R, Wu L-Z, Tung C-H, Zhang T, Nanoscale 8 (2016) 3923–3925. [DOI] [PubMed] [Google Scholar]
- [92].Ju E, Li T, Silva SR, Gao S-J, ACS Appl. Mater. Interfaces 11 (2019) 34717–34724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Xu Y, Wang X, Ma X, Dyes Pigm. 145 (2017) 385–390. [Google Scholar]
- [94].Dong B, Du S, Wang C, Fu H, Li Q, Xiao N, Yang J, Xue X, Cai W, Liu D, ACS Nano 13 (2019) 1421–1432. [DOI] [PubMed] [Google Scholar]
- [95].Li W, Kanyo I, Kuo CH, Thanneeru S, He J, Nanoscale 7 (2015) 956–964. [DOI] [PubMed] [Google Scholar]
- [96].Tokarev I, Tokareva I, Minko S, Adv. Mater. 20 (2008) 2730–2734. [DOI] [PubMed] [Google Scholar]
- [97].Sun Z, Ni W, Yang Z, Kou X, Li L, Wang J, Small 4 (2008) 1287–1292. [DOI] [PubMed] [Google Scholar]
- [98].Qian X, Li J, Nie S, J. Am. Chem. Soc 131 (2009) 7540–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Liu L, Gao Z, Jiang B, Bai Y, Wang W, Yin Y, Nano Lett. 18 (2018) 5312–5318. [DOI] [PubMed] [Google Scholar]
- [100].Torii Y, Sugimura N, Mitomo H, Niikura K, Ijiro K, Langmuir, 33 (2017) 5537–5544 [DOI] [PubMed] [Google Scholar]
- [101].Ma J, Hu Z, Wang W, Wang X, Wu Q, Yuan Z, ACS Appl. Mater. Interfaces 9 (2017) 16767–16777. [DOI] [PubMed] [Google Scholar]
- [102].Bajpai AK, Shukla SK, Bhanu S, Kankane S, Prog. Polym. Sci 33 (2008) 1088–1118 [Google Scholar]
- [103].Wang X, Wang X, Jin S, Muhammad N, Guo Z, Chem. Rev 119 (2019) 1138–1192. [DOI] [PubMed] [Google Scholar]
- [104].Lewandowski W, Fruhnert M, Mieczkowski J, Rockstuhl C, Górecka E, Nat. Commun 6 (2015) 1–9. [DOI] [PubMed] [Google Scholar]
- [105].Yao LY, Lee TK, Yam VW, J. Am. Chem. Soc 138 (2016) 7260–7263. [DOI] [PubMed] [Google Scholar]
- [106].Boyer C, Whittaker MR, Luzon M, Davis TP, Macromolecules 42 (2009) 6917–6926. [Google Scholar]
- [107].Durand-Gasselin C, Sanson N, Lequeux N, Langmuir 27 (2011) 12329–12335. [DOI] [PubMed] [Google Scholar]
- [108].Yeshchenko OA, Naumenko AP, Kutsevol NV, Maskova DO, Harahuts II, Chumachenko VA, Marinin AI, J. Phys. Chem. C 122 (2018) 8003–8010. [Google Scholar]
- [109].Schweizerhof S, Demco DE, Mourran A, Fechete R, Moller M, Langmuir 34 (2018) 8031–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Zhang CL, Cao FH, Wang JL, Yu ZL, Ge J, Lu Y, Wang ZH, Yu SH, ACS Appl. Mater. Interfaces 9 (2017) 24857–24863. [DOI] [PubMed] [Google Scholar]
- [111].Ding T, Rüttiger C, Zheng X, Benz F, Ohadi H, Vandenbosch GAE, Moshchalkov VV, Gallei M, Baumberg JJ, Adv. Opt. Mater. 4 (2016) 877–882. [Google Scholar]
- [112].Cormier S, Ding T, Turek V, Baumberg JJ, Adv. Opt. Mater. 6 (2018) 1800208. [Google Scholar]
- [113].Lim S, Song JE, La JA, Cho EC, Chem. Mater. 26 (2014) 3272–3279. [Google Scholar]
- [114].Han F, Soeriyadi AH, Vivekchand SRC, Gooding JJ, ACS Macro Lett. 5 (2016) 626–630. [DOI] [PubMed] [Google Scholar]
- [115].Turek VA, Cormier S, Sierra-Martin B, Keyser UF, Ding T, Baumberg JJ, Adv. Opt. Mater. 6 (2018) 1701270. [Google Scholar]
- [116].Ding T, Valev VK, Salmon AR, Forman CJ, Smoukov SK, Scherman OA, Frenkel D, Baumberg JJ, Proc. Natl. Acad. Sci. U. S. A 113 (2016) 5503–5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Zhu M-Q, Wang L-Q, Exarhos GJ, Li ADQ, J. Am. Chem. Soc 126 (2004) 2656–2657. [DOI] [PubMed] [Google Scholar]
- [118].Iida R, Mitomo H, Niikura K, Matsuo Y, Ijiro K, Small 14 (2018) e1704230. [DOI] [PubMed] [Google Scholar]
- [119].Chen Y, Xianyu Y, Jiang X, Acc. Chem. Res 50 (2017) 310–319. [DOI] [PubMed] [Google Scholar]
- [120].Yi G, Son J, Yoo J, Park C, Koo H, Biomater. Res 22 (2018) 13–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Hapuarachchige S, Artemov D, Top Magn. Reson. Imaging 25 (2016) 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Kim E, Koo H, Chem. Sci 10 (2019) 7835–7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Zhang T, Zheng Z, Cheng X, Ding X, Peng Y, Prog. Chem 20 (2008) 1090–1101. [Google Scholar]
- [124].Zhou Y, Wang S, Zhang K, Jiang X, Angew. Chem. Int. Ed 47 (2008) 7454–7456. [DOI] [PubMed] [Google Scholar]
- [125].Shi L, Jing C, Ma W, Li DW, Halls JE, Marken F, Long YT, Angew. Chem. Int. Ed 52 (2013) 6011–6014. [DOI] [PubMed] [Google Scholar]
- [126].Si S, Raula M, Paira TK, Mandal TK, ChemPhysChem 9 (2008) 1578–1584. [DOI] [PubMed] [Google Scholar]
- [127].Xin J-Y, Li C-Y, Zhang S, Wang Y, Zhang W, Xia C-G, IET Nanobiotechnol. 12 (2018) 915–921.30247130 [Google Scholar]
- [128].Qiao J, Ding H, Liu Q, Zhang R, Qi L, Anal. Chem 89 (2017) 2080–2085. [DOI] [PubMed] [Google Scholar]
- [129].Xianyu Y, Xie Y, Wang N, Wang Z, Jiang X, Small 11 (2015) 5510–5514. [DOI] [PubMed] [Google Scholar]
- [130].Chen W, Cao F, Zheng W, Tian Y, Xianyu Y, Xu P, Zhang W, Wang Z, Deng K, Jiang X, Nanoscale 7 (2015) 2042. [DOI] [PubMed] [Google Scholar]
- [131].Huang Y, Li F, Ma G, Yang W, Zhang X, Lin J, Luo Y, Huang P, Talanta 187 (2018) 65–72. [DOI] [PubMed] [Google Scholar]
- [132].Gunupuru R, Maity D, Bhadu GR, Chakraborty A, Srivastava DN, Paul P, J. Chem. Sci 126 (2014) 627–635. [Google Scholar]
- [133].Chai F, Wang C, Wang T, Li L, Su Z, ACS Appl. Mater. Interfaces 2 (2010) 1466–1470. [DOI] [PubMed] [Google Scholar]
- [134].Luo X, Xie X, Meng Y, Sun T, Ding J, Zhou W, Anal. Chim. Acta. 1087 (2019) 76–85. [DOI] [PubMed] [Google Scholar]
- [135].Hermanson KD, Lumsdon SO, Williams JP, Kaler EW, Velev OD, Science 294 (2001) 1082–1086. [DOI] [PubMed] [Google Scholar]
- [136].Guo QH, Zhang CJ, Wei C, Xu MM, Yuan YX, Gu RA, Yao JL, Spectrochim. Acta A: Mol. Biomol. Spectrosc. 152 (2016) 336–342. [DOI] [PubMed] [Google Scholar]
- [137].Scaramuzza S, Polizzi S, Amendola V, Nanoscale Adv. 1 (2019) 2681–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Wang C, Irudayaraj J, Small 6 (2010) 283–289. [DOI] [PubMed] [Google Scholar]
- [139].Xue X, Liu K, Furlani EP, J. Phys. Chem. C 121 (2017) 9489–9496. [Google Scholar]
- [140].La Porta A, Sanchez-Iglesias A, Altantzis T, Bals S, Grzelczak M, Liz-Marzan LM, Nanoscale 7 (2015) 10377–10381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Cha SH, Kang SH, Lee YJ, Kim JH, Ahn EY, Park Y, Cho S, Sci. Rep 9 (2019) 3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Liu L, Chen K, Xiang N, Ni Z, Electrophoresis 40 (2019) 873–889. [DOI] [PubMed] [Google Scholar]
- [143].Fu K, Chen S, Zhao J, Willis BG, ACS Sensors 1 (2016) 444–450. [Google Scholar]
- [144].Wang K, Jin SM, Xu J, Liang R, Shezad K, Xue Z, Xie X, Lee E, Zhu J, ACS Nano 10 (2016) 4954–4960. [DOI] [PubMed] [Google Scholar]
- [145].Nam NN, Bui TL, Son SJ, Joo SW, Adv. Funct. Mater. 29 (2019) 1809146. [Google Scholar]
- [146].Strozyk MS, Chanana M, Pastoriza-Santos I, Pérez-Juste J, Liz-Marzán LM, Adv. Funct. Mater. 22 (2012) 1436–1444. [Google Scholar]
- [147].Cao Z-Q, Wang G-J, Chem. Rec 16 (2016) 1398–1435. [DOI] [PubMed] [Google Scholar]
- [148].Guragain S, Bastakoti BP, Malgras V, Nakashima K, Yamauchi Y, Chem-Eur. J 21 (2015) 13164–13174. [DOI] [PubMed] [Google Scholar]
- [149].Zhuang J, Gordon MR, Ventura J, Li L, Thayumanavan S, Chem. Soc. Rev 42 (2013) 7421–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Cheng R, Meng F, Deng C, Klok H-A, Zhong Z, Biomaterials 34 (2013) 3647–3657. [DOI] [PubMed] [Google Scholar]
- [151].Schattling P, Jochum FD, Theato P, Polym. Chem 5 (2014) 25–36. [Google Scholar]
- [152].Liu B, Lu X, Qiao Z, Song L, Cheng Q, Zhang J, Zhang A, Huang Y, Chen T, Langmuir 34 (2018) 13047–13056. [DOI] [PubMed] [Google Scholar]
- [153].Lin H, Song L, Huang Y, Cheng Q, Yang Y, Guo Z, Su F, Chen T, ACS Appl. Mater. Interfaces 12 (2020) 11296–11304. [DOI] [PubMed] [Google Scholar]
- [154].Wang Y, Santos PJ, Kubiak JM, Guo X, Lee MS, Macfarlane RJ, J. Am. Chem. Soc 141 (2019) 13234–13243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Cheng X, Sun R, Yin L, Chai Z, Shi H, Gao M, Adv. Mater. 29 (2017) 1604894. [DOI] [PubMed] [Google Scholar]
- [156].Zhong D, Zhao J, Li Y, Qiao Y, Wei Q, He J, Xie T, Li W, Zhou M, Biomaterials 219 (2019) 119369. [DOI] [PubMed] [Google Scholar]
- [157].Sun M, Peng D, Hao H, Hu J, Wang D, Wang K, Liu J, Guo X, Wei Y, Gao W, ACS Appl. Mater. Interfaces 9 (2017) 10453–10460. [DOI] [PubMed] [Google Scholar]
- [158].Yang K, Zhu L, Nie L, Sun X, Cheng L, Wu C, Niu G, Chen X, Liu Z, [Google Scholar]
- [159].Baruch A, Jeffery DA, Bogyo M, Trends Cell Biol. 14 (2004) 29–35. [DOI] [PubMed] [Google Scholar]
- [160].Yan R, Ye D, Sci. Bull 61 (2016) 1672–1679. [Google Scholar]
- [161].Tung CH, Mahmood U, Bredow S, Weissleder R, Cancer Res. 60 (2000) 4953–4958. [PubMed] [Google Scholar]
- [162].Rao SR, Snaith AE, Marino D, Cheng X, Lwin ST, Orriss IR, Hamdy FC, Edwards CM, Br. J. Cancer 116 (2017) 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Yang K, Liu Y, Wang Y, Ren Q, Guo H, Matson JB, Chen X, Nie Z, Biomaterials 223 (2019) 119460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Ruan S, Hu C, Tang X, Cun X, Xiao W, Shi K, He Q, Gao H, ACS Nano 10 (2016) 10086–10098. [DOI] [PubMed] [Google Scholar]
- [165].Crucho CI, ChemMedChem 10 (2015) 24–38. [DOI] [PubMed] [Google Scholar]
- [166].Felber AE, Dufresne MH, Leroux JC, Adv. Drug Deliv. Rev 64 (2012) 979–992. [DOI] [PubMed] [Google Scholar]
- [167].Gatenby RA, Gillies RJ, Nat. Rev. Cancer 4 (2004) 891–899. [DOI] [PubMed] [Google Scholar]
- [168].Grinstein S, Swallow CJ, Rotstein OD, Clin. Biochem 24 (1991) 241–247. [DOI] [PubMed] [Google Scholar]
- [169].Jo J, Lee CH, Kopelman R, Wang X, Nat. Commun 8 (2017) 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Yu Z, Wang M, Pan W, Wang H, Li N, Tang B, Chem. Sci 8 (2017) 4896–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Cheng M, Zhang Y, Zhang X, Wang W, Yuan Z, Biomater. Sci 7 (2019) 2009–2022. [DOI] [PubMed] [Google Scholar]
- [172].Zhang Y, Chang J, Huang F, Yang L, Ren C, Ma L, Zhang W, Dong H, Liu J, Liu J, ACS Biomater. Sci. Eng 5 (2019) 1589–1601. [DOI] [PubMed] [Google Scholar]
- [173].Ma J, Li X, Hu Z, Wang X, Zhang Y, Wang W, Wu Q, Yuan Z, Sci. China Chem 62 (2019) 105–117. [Google Scholar]
- [174].Zhang Y, Huang F, Ren C, Liu J, Yang L, Chen S, Chang J, Yang C, Wang W, Zhang C, Liu Q, Liang X-J, Liu J, Adv. Sci 6 (2019) 1801806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Nam J, La W-G, Hwang S, Ha Y. Su, Park N, Won N, Jung S, Ho Bhang S, Ma Y-J, Cho Y-M, Jin M, Han J, Shin J-Y, Wang EK, Kim SG, Cho S-H, Yoo J, Kim BS, Kim S, ACS Nano 7 (2013) 3388–3402. [DOI] [PubMed] [Google Scholar]
- [176].Zhang X, Zhang C, Cheng M, Zhang Y, Wang W, Yuan Z, Nano Res. 12 (2019) 2815–2826. [Google Scholar]
- [177].Song J, Zhou J, Duan H, J. Am. Chem. Soc 134 (2012) 13458–13469. [DOI] [PubMed] [Google Scholar]
- [178].Song J, Lin L, Yang Z, Zhu R, Zhou Z, Li ZW, Wang F, Chen J, Yang H, Chen X, J. Am. Chem. Soc 141 (2019) 8158–8170. [DOI] [PubMed] [Google Scholar]
- [179].Song J, Pu L, Zhou J, Duan B, Duan H, ACS Nano 7 (2013) 9947–9960. [DOI] [PubMed] [Google Scholar]
- [180].Ma B, Wang S, Liu F, Zhang S, Duan J, Li Z, Kong Y, Sang Y, Liu H, Bu W, Li L, J. Am. Chem. Soc 141 (2019) 849–857. [DOI] [PubMed] [Google Scholar]
- [181].Liang R, Liu L, He H, Chen Z, Han Z, Luo Z, Wu Z, Zheng M, Ma Y, Cai L, Biomaterials 177 (2018) 149–160. [DOI] [PubMed] [Google Scholar]
- [182].Liu T, Tong L, Lv N, Ge X, Fu Q, Gao S, Ma Q, Song J, Adv. Funct. Mater. 29 (2019) 1806429. [Google Scholar]
- [183].An L, Cao M, Zhang X, Lin J, Tian Q, Yang S, ACS Appl. Mater. Interfaces 12 (2020) 8050–8061. [DOI] [PubMed] [Google Scholar]
- [184].Wang H, An L, Tao C, Ling Z, Lin J, Tian Q, Yang S, Nanoscale 12 (2020) 5139–5150. [DOI] [PubMed] [Google Scholar]
- [185].Liang S, Deng X, Chang Y, Sun C, Shao S, Xie Z, Xiao X, Ma P, Zhang H, Cheng Z, Lin J, Nano Lett. 19 (2019) 4134–4145. [DOI] [PubMed] [Google Scholar]
- [186].Lin X, Liu S, Zhang X, Zhu R, Chen S, Chen X, Song J, Yang H, Angew. Chem. Int. Ed 59 (2020) 1682–1688. [DOI] [PubMed] [Google Scholar]
- [187].Park SG, Xiao X, Min J, Mun C, Jung HS, Giannini V, Weissleder R, Maier SA, Im H, Kim DH, Adv. Funct. Mater. 29 (2019) 1904257. [Google Scholar]
- [188].Saha K, Agasti SS, Kim C, Li X, Rotello VM, Chem Rev, 112 (2012) 2739–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Bui MP, Ahmed S, Abbas A, Nano Lett. 15 (2015) 6239–6246. [DOI] [PubMed] [Google Scholar]
- [190].Xiong LH, Huang S, Huang Y, Yin F, Yang F, Zhang Q, Cheng J, Zhang R, He X, ACS Appl. Mater. Interfaces 12 (2020) 12525–12532. [DOI] [PubMed] [Google Scholar]
- [191].Hirayama T, Van de Bittner GC, Gray LW, Lutsenko S, Chang CJ, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 2228–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Wang S, Yu G, Ma Y, Yang Z, Liu Y, Wang J, Chen X, ACS Appl. Mater. Interfaces, 11 (2019) 1917–1923. [DOI] [PubMed] [Google Scholar]
- [193].Starling S, Marshall L, Nat. Rev. Dis. Primers 4 (2018) 22.30190465 [Google Scholar]
- [194].Ndokoye P, Ke J, Liu J, Zhao Q, Li X, Langmuir 30 (2014) 13491–13497. [DOI] [PubMed] [Google Scholar]
- [195].Sun S, Ning X, Zhang G, Wang YC, Peng C, Zheng J, Angew. Chem. Int. Ed 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196]. Li DW, Qu LL, Hu K, Long YT, Tian H, Angew. Chem. Int. Ed 54 (2015) 12758–12761. [DOI] [PubMed] [Google Scholar]
- [197].Moros M, Kyriazi ME, El-Sagheer AH, Brown T, Tortiglione C, Kanaras AG, ACS Appl. Mater. Interfaces 11 (2019) 13905–13911. [DOI] [PubMed] [Google Scholar]
- [198].Chen X, Wang T, Le W, Huang X, Gao M, Chen Q, Xu S, Yin D, Fu Q, Shao C, Chen B, Shi D, Theranostics 10 (2020) 3430–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Chai X, Zhou X, Zhu A, Zhang L, Qin Y, Shi G, Tian Y, Angew. Chem. Int. Ed 52 (2013) 8129–8133. [DOI] [PubMed] [Google Scholar]
- [200].Sarkar S, Ha YS, Soni N, An GI, Lee W, Kim MH, Huynh PT, Ahn H, Bhatt N, Lee YJ, Kim JY, Park KM, Ishii I, Kang SG, Yoo J, Angew. Chem. Int. Ed 55 (2016) 9365–9370. [DOI] [PubMed] [Google Scholar]
- [201].Yan N, Wang X, Lin L, Song T, Sun P, Tian H, Liang H, Chen X, Adv. Funct. Mater. 28 (2018) 1800490. [Google Scholar]
- [202].Bantz KC, Meyer AF, Wittenberg NJ, Im H, Kurtulus O, Lee SH, Lindquist NC, Oh S-H, Haynes CL, Phys. Chem. Chem. Phys 13 (2011) 11551–11567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Henry A-I, Sharma B, Cardinal MF, Kurouski D, Van Duyne RP, Anal. Chem 88 (2016) 6638–6647. [DOI] [PubMed] [Google Scholar]
- [204].Laing S, Jamieson LE, Faulds K, Graham D, Nat. Rev. Chem 1 (2017) 0060. [Google Scholar]
- [205].Li Y, Wei Q, Ma F, Li X, Liu F, Zhou M, Acta Pharm. Sin. B 8 (2018) 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Moore TJ, Moody AS, Payne TD, Sarabia GM, Daniel AR, Sharma B, Biosensors. 8 (2018) 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Ma Y, Sikdar D, Fedosyuk A, Velleman L, Klemme DJ, Oh SH, Kucernak ARJ, Kornyshev AA, Edel JB, ACS Nano 14 (2020) 328–336. [DOI] [PubMed] [Google Scholar]
- [208].Hu Y, Cheng H, Zhao X, Wu J, Muhammad F, Lin S, He J, Zhou L, Zhang C, Deng Y, Wang P, Zhou Z, Nie S, Wei H, ACS Nano 11 (2017) 5558–5566. [DOI] [PubMed] [Google Scholar]
- [209].Gupta P, Luan J, Wang Z, Cao S, Bae SH, Naik RR, Singamaneni S, ACS Appl. Mater. Interfaces 11 (2019) 37939–37946. [DOI] [PubMed] [Google Scholar]
- [210].Fernandez-Lopez C, Polavarapu L, Solis DM, Taboada JM, Obelleiro F, Contreras-Caceres R, Pastoriza-Santos I, Perez-Juste J, ACS Appl. Mater. Interfaces 7 (2015) 12530–12538. [DOI] [PubMed] [Google Scholar]
- [211].Curtis T, Taylor AK, Alden SE, Swanson C, Lo J, Knight L, Silva A, Gates BD, Emory SR, Rider DA, ACS Omega 3 (2018) 10572–10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Simoncelli S, Roller EM, Urban P, Schreiber R, Turberfield AJ, Liedl T, Lohmuller T, ACS Nano, 10 (2016) 9809–9815. [DOI] [PubMed] [Google Scholar]
- [213].Gehan H, Fillaud L, Chehimi MM, Aubard J, Hohenau A, Felidj N, Mangeney C, ACS Nano, 4 (2010) 6491–6500. [DOI] [PubMed] [Google Scholar]
- [214].Fu Q, Zhu R, Song J, Yang H, Chen X, Adv. Mater. 31 (2019) e1805875. [DOI] [PubMed] [Google Scholar]
- [215].Li W, Chen X, Nanomedicine 10 (2015) 299–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Taruttis A, Ntziachristos V, Nat. Photonics 9 (2015) 219–227. [Google Scholar]
- [217].Wang S, Lin J, Wang T, Chen X, Huang P, Theranostics 6 (2016) 2394–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Weber J, Beard PC, Bohndiek SE, Nat. Methods, 13 (2016) 639–650. [DOI] [PubMed] [Google Scholar]
- [219].Wang LV, Hu S, Science 335 (2012) 1458–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Fu Q, Li Z, Ye J, Li Z, Fu F, Lin S-L, Chang CA, Yang H, Song J, Theranostics 10 (2020) 4997–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Chen YS, Yoon SJ, Frey W, Dockery M, Emelianov S, Nat. Commun 8 (2017) 15782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Lu SZ, Guo XY, Zou MS, Zheng ZQ, Li YC, Li XD, Li LL, Wang H, Adv. Healthc. Mater. 9 (2020) e1901229. [DOI] [PubMed] [Google Scholar]
- [223].Chen Z, Mu X, Han Z, Yang S, Zhang C, Guo Z, Bai Y, He W, J. Am. Chem. Soc 141 (2019) 17973–17977. [DOI] [PubMed] [Google Scholar]
- [224].Zheng J, Zeng Q, Zhang R, Xing D, Zhang T, J. Am. Chem. Soc 141 (2019) 19226–19230. [DOI] [PubMed] [Google Scholar]
- [225].Zhang K, Meng X, Yang Z, Cao Y, Cheng Y, Wang D, Lu H, Shi Z, Dong H, Zhang X, Adv. Mater. 31 (2019) 1807888 [DOI] [PubMed] [Google Scholar]
- [226].Yin L, Sun H, Zhang H, He L, Qiu L, Lin J, Xia H, Zhang Y, Ji S, Shi H, Gao M, J. Am. Chem. Soc 141 (2019) 3265–3273. [DOI] [PubMed] [Google Scholar]
- [227].Huang X, Song J, Yung BC, Huang X, Xiong Y, Chen X, Chem. Soc. Rev 47 (2018) 2873–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Yang Z, Dai Y, Yin C, Fan Q, Zhang W, Song J, Yu G, Tang W, Fan W, Yung BC, Li J, Li X, Li X, Tang Y, Huang W, Song J, Chen X, Adv. Mater. 30 (2018) 1707509. [DOI] [PubMed] [Google Scholar]
- [229].Liu Y, Wang S, Ma Y, Lin J, Wang H-Y, Gu Y, Chen X, Huang P, Adv. Mater. 29 (2017) 1606129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Yin C, Zhen X, Fan Q, Huang W, Pu K, ACS Nano 11 (2017) 4174–4182. [DOI] [PubMed] [Google Scholar]
- [231].Zhang J, Zhen X, Upputuri PK, Pramanik M, Chen P, Pu K, Adv. Mater. 29 (2017) 1604764. [DOI] [PubMed] [Google Scholar]
- [232].Chen Q, Liu X, Chen J, Zeng J, Cheng Z, Liu Z, Adv. Mater. 27 (2015) 6820. [DOI] [PubMed] [Google Scholar]
- [233].Li H, Zhang P, Smaga LP, Hoffman RA, Chan J, J. Am. Chem. Soc 137 (2015) 15628–15631. [DOI] [PubMed] [Google Scholar]
- [234].Joensuu H, Hohenberger P, Corless CL, Lancet 382 (2013) 973–983. [DOI] [PubMed] [Google Scholar]
- [235].Dedhia PH, Bertaux-Skeirik N, Zavros Y, Spence JR, Gastroenterology 150 (2016) 1098–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Huang W, Chen R, Peng Y, Duan F, Huang Y, Guo W, Chen X, Nie L, ACS Nano 13 (2019) 9561–9570. [DOI] [PubMed] [Google Scholar]
- [237].Shin T-H, Choi Y, Kim S, Cheon J, Chem. Soc. Rev 44 (2015) 4501–4516. [DOI] [PubMed] [Google Scholar]
- [238].Wu Y, Ali MRK, Chen K, Fang N, El-Sayed MA, Nano Today 24 (2019) 120–140. [Google Scholar]
- [239].Smith BR, Gambhir SS, Chem. Rev 117 (2017) 901–986. [DOI] [PubMed] [Google Scholar]
- [240].Mahan MM, Doiron AL, J. Nanomater. (2018) 5837276 [Google Scholar]
- [241].Key J, Leary JF, Int. J. Nanomedicine 9 (2014) 711–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Kim J, Piao Y, Hyeon T, Chem. Soc. Rev 38 (2009) 372–390. [DOI] [PubMed] [Google Scholar]
- [243].Lee D-E, Koo H, Sun I-C, Ryu JH, Kim K, Kwon IC, Chem. Soc. Rev 41 (2012) 2656–2672. [DOI] [PubMed] [Google Scholar]
- [244].Wallyn J, Anton N, Akram S, Vandamme TF, Pharm. Res-Dordr 36 (2019) 78. [DOI] [PubMed] [Google Scholar]
- [245].He X, Zhao Z, Xiong LH, Gao PF, Peng C, Li RS, Xiong Y, Li Z, Sung HH, Williams ID, Kwok RTK, Lam JWY, Huang CZ, Ma N, Tang BZ, J. Am. Chem. Soc 140 (2018) 6904–6911. [DOI] [PubMed] [Google Scholar]
- [246].He X, Peng C, Qiang S, Xiong LH, Zhao Z, Wang Z, Kwok RTK, Lam JWY, Ma N, Tang BZ, Biomaterials 238 (2020) 119834. [DOI] [PubMed] [Google Scholar]
- [247].Liu Y, Yang Z, Huang X, Yu G, Wang S, Zhou Z, Shen Z, Fan W, Liu Y, Davisson M, Kalish H, Niu G, Nie Z, Chen X, ACS Nano 12 (2018) 8129–8137. [DOI] [PubMed] [Google Scholar]
- [248].Liu Y, Bhattarai P, Dai Z, Chen X, Chem. Soc. Rev 48 (2019) 2053–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Vines JB, Yvon J-H, Ryu N-E, Lim D-J, Park H, Front. Chem 7 (2019) 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Melancon MP, Zhou M, Li C, Acc. Chem. Res 44 (2011) 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Son J, Yi G, Yoo J, Park C, Koo H, Choi HS, Adv. Drug Deliv. Rev 138 (2019) 133–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Chen D, Tang Y, Zhu J, Zhang J, Song X, Wang W, Shao J, Huang W, Chen P, Dong X, Biomaterials 221 (2019) 119422. [DOI] [PubMed] [Google Scholar]
- [253].Kang S, Bhang SH, Hwang S, Yoon J-K, Song J, Jang H-K, Kim S, Kim B-S, ACS Nano 9 (2015) 9678–9690. [DOI] [PubMed] [Google Scholar]
- [254].Shi H, Sun Y, Yan R, Liu S, Zhu L, Liu S, Feng Y, Wang P, He J, Zhou Z, Ye D, Nano Lett. 19 (2019) 937–947. [DOI] [PubMed] [Google Scholar]
- [255].Chen X, Song J, Chen X, Yang H, Chem. Soc. Rev 48 (2019) 3073–3101. [DOI] [PubMed] [Google Scholar]
- [256].Fan W, Tang W, Lau J, Shen Z, Xie J, Shi J, Chen X, Adv. Mater. 31 (2019) 1806381 [DOI] [PubMed] [Google Scholar]
- [257].Pogue BW, Wilson BC, J. Biomed. Opt 23 (2018) 121610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Zhou Z, Chan A, Wang Z, Huang X, Yu G, Jacobson O, Wang S, Liu Y, Shan L, Dai Y, Shen Z, Lin L, Chen W, Chen X, Angew. Chem. Int. Ed 57 (2018) 8463–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Ren X-D, Hao X-Y, Li H-C, Ke M-R, Zheng B-Y, Huang J-D, Drug Discov. Today 23 (2018) 1791–1800. [DOI] [PubMed] [Google Scholar]
- [260].Goswami N, Luo Z, Yuan X, Leong DT, Xie J, Mater. Horizons 4 (2017) 817–831. [Google Scholar]
- [261].Sun Q, Sun X, Ma X, Zhou Z, Jin E, Zhang B, Shen Y, Van Kirk EA, Murdoch WJ, Lott JR, Lodge TP, Radosz M, Zhao Y, Adv. Mater. 26 (2014) 7615–7621. [DOI] [PubMed] [Google Scholar]
- [262].Liu X, Chen Y, Li H, Huang N, Jin Q, Ren K, Ji J, ACS Nano, 7 (2013) 6244–6257. [DOI] [PubMed] [Google Scholar]
- [263].Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WCW, Nano Lett. 9 (2009) 1909–1915. [DOI] [PubMed] [Google Scholar]
- [264].Ding Y, Sun Z, Tong Z, Zhang S, Min J, Xu Q, Zhou L, Mao Z, Xia H, Wang W, Theranostics 10 (2020) 5195–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Sun W, Luo L, Feng Y, Cai Y, Zhuang Y, Xie RJ, Chen X, Chen H, Angew. Chem. Int. Ed 58 (2019) 1–8. [Google Scholar]
- [266].Zhou L, Wang H, Li Y, Theranostics 8 (2018) 1059–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Kunjachan S, Rychlik B, Storm G, Kiessling F, Lammers T, Adv. Drug Deliv. Rev 65 (2013) 1852–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Song J, Yang X, Jacobson O, Lin L, Huang P, Niu G, Ma Q, Chen X, ACS Nano, 9 (2015) 9199–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Mura S, Nicolas J, Couvreur P, Nat. Mater. 12 (2013) 991–1003. [DOI] [PubMed] [Google Scholar]
- [270].Yin Q, Shen J, Zhang Z, Yu H, Li Y, Adv. Drug Deliv. Rev 65 (2013) 1699–1715. [DOI] [PubMed] [Google Scholar]
- [271].Wang P, Sun J, Lou Z, Fan F, Hu K, Sun Y, Gu N, Adv. Mater. 28 (2016) 10801–10808. [DOI] [PubMed] [Google Scholar]
- [272].Xu W, Qian J, Hou G, Suo A, Wang Y, Wang J, Sun T, Yang M, Wan X, Yao Y, ACS Appl. Mater. Interfaces 9 (2017) 36533–36547. [DOI] [PubMed] [Google Scholar]
- [273].Gao S, Tang G, Hua D, Xiong R, Han J, Jiang S, Zhang Q, Huang C, J. Mater. Chem. B 7 (2019) 709–729. [DOI] [PubMed] [Google Scholar]
- [274].Hossen S, Hossain MK, Basher MK, Mia MNH, Rahman MT, Uddin MJ, J. Adv. Res 15 (2019) 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Mi P, Theranostics 10 (2020) 4557–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Carrillo-Carrion C, Martinez R, Navarro Poupard MF, Pelaz B, Polo E, Arenas-Vivo A, Olgiati A, Taboada P, Soliman MG, Catalan U, Fernandez-Castillejo S, Sola R, Parak WJ, Horcajada P, Alvarez-Puebla RA, Del Pino P, Angew. Chem. Int. Ed 58 (2019) 7078–7082. [DOI] [PubMed] [Google Scholar]
- [277].Sun H, Su J, Meng Q, Yin Q, Chen L, Gu W, Zhang Z, Yu H, Zhang P, Wang S, Li Y, Adv. Funct. Mater. 27 (2017) 1604300. [Google Scholar]
- [278].Fu J, Liang L, Qiu L, Adv. Funct. Mater. 27 (2017) 1604981. [Google Scholar]
- [279].Li X, Bottini M, Zhang L, Zhang S, Chen J, Zhang T, Liu L, Rosato N, Ma X, Shi X, Wu Y, Guo W, Liang XJ, ACS Nano, 13 (2019) 176–186. [DOI] [PubMed] [Google Scholar]
- [280].Zhang L, Zhang M, Zhou L, Han Q, Chen X, Li S, Li L, Su Z, Wang C, Biomaterials 181 (2018) 113–125. [DOI] [PubMed] [Google Scholar]
- [281].Borkowska M, Siek M, Kolygina DV, Sobolev YI, Lach S, Kumar S, Cho YK, Kandere-Grzybowska K, Grzybowski BA, Nat. Nanotechnol (2020) DOI: 10.1038/s41565-41020-40643-41563. [DOI] [PubMed] [Google Scholar]
- [282].Qiu J, Xia Y, Nat. Nanotechnol (2020) DOI: 10.1038/s41565-41020-40639-z. [DOI] [PubMed] [Google Scholar]
- [283].Liu L, Wang X, Wang N, Peng T, Wang S, Angew. Chem. Int. Ed 56 (2017) 9160–9164. [DOI] [PubMed] [Google Scholar]
- [284].Shen J, Luan B, Pei H, Yang Z, Zuo X, Liu G, Shi J, Wang L, Zhou R, Cheng W, Fan C, Adv. Mater. 29 (2017) 1606796. [DOI] [PubMed] [Google Scholar]
- [285].Ali I, Chen L, Huang Y, Song L, Lu X, Liu B, Zhang L, Zhang J, Hou L, Chen T, Langmuir 34 (2018) 4908–4913. [DOI] [PubMed] [Google Scholar]
- [286].Su CH, Chiu HL, Chen YC, Yesilmen M, Schulz F, Ketelsen B, Vossmeyer T, Liao YC, Langmuir 35 (2019) 3256–3264. [DOI] [PubMed] [Google Scholar]
- [287].Jiang Q, Liu Q, Shi Y, Wang ZG, Zhan P, Liu J, Liu C, Wang H, Shi X, Zhang L, Sun J, Ding B, Liu M, Nano Lett. 17 (2017) 7125–7130. [DOI] [PubMed] [Google Scholar]
- [288].Liu L, Aleisa R, Zhang Y, Feng J, Zheng Y, Yin Y, Wang W, Angew. Chem. Int. Ed 58 (2019) 16307–16313. [DOI] [PubMed] [Google Scholar]
- [289].Vassalini I, Alessandri I, Catalysts 8 (2018) 569. [Google Scholar]
- [290].Zhang J, Zhang M, Tang K, Verpoort F, Sun T, Small 10 (2014) 32–46. [DOI] [PubMed] [Google Scholar]
- [291].Hoffman AS, Adv. Drug Deliv. Rev 65 (2013) 10–16. [DOI] [PubMed] [Google Scholar]
- [292].Liu Z, Wang W, Xie R, Ju X-J, Chu L-Y, Chem. Soc. Rev 45 (2016) 460–474. [DOI] [PubMed] [Google Scholar]