

ABSTRACT
Animal germ cells communicate directly with each other during gametogenesis through intercellular bridges, often called ring canals (RCs), that form as a consequence of incomplete cytokinesis during cell division. Developing germ cells in Drosophila have an additional specialized organelle connecting the cells called the fusome. Ring canals and the fusome are required for fertility in Drosophila females, but little is known about their roles during spermatogenesis. With live imaging, we directly observe the intercellular movement of GFP and a subset of endogenous proteins through RCs during spermatogenesis, from two-cell diploid spermatogonia to clusters of 64 post-meiotic haploid spermatids, demonstrating that RCs are stable and open to intercellular traffic throughout spermatogenesis. Disruption of the fusome, a large cytoplasmic structure that extends through RCs and is important during oogenesis, had no effect on spermatogenesis or male fertility under normal conditions. Our results reveal that male germline RCs allow the sharing of cytoplasmic information that might play a role in quality control surveillance during sperm development.
KEY WORDS: Drosophila spermatogenesis, Ring canal, Fusome, Intercellular movement
Highlighted Article: An investigation of the function of Drosophila male germline ring canals and fusomes during spermatogenesis, documenting protein movement through ring canals and determining that fusomes are largely dispensable for male fertility.
INTRODUCTION
Germ cells throughout the animal kingdom maintain direct cytoplasmic intercellular connections during gametogenesis. Germ cell intercellular bridges (ICBs) were first observed in cat testes by electron microscopy that revealed channels connecting spermatids of ∼1 μm in diameter lined with an electron-dense plasma membrane compartment (Burgos and Fawcett, 1955). Subsequent reports described similar ICBs in other species, ranging from Hydra to mammals (Dym and Fawcett, 1971; Fawcett et al., 1959).
In Drosophila, intercellular bridges are called ring canals (RCs) and were first discovered in ovarian germline cells (Brown and Smith, 1964; Koch and King, 1966, 1969), where they are essential for oocyte growth during oogenesis (Greenbaum et al., 2011; Robinson and Cooley, 1996). Moreover, previous work from our lab has shown that RCs in Drosophila somatic ovarian follicle cells allow movement of cytoplasmic contents between connected cells (Airoldi et al., 2011) that contributes to protein level equilibration (McLean and Cooley, 2013). In contrast, the functional significance of male RCs during spermatogenesis remains less well characterized.
During Drosophila spermatogenesis, germline stem cells (GSCs) located at the hub of the testis divide asymmetrically to produce another GSC and a spermatogonial cell that divides mitotically four times to form a cluster of 16 primary spermatocytes connected by RCs (Fig. 1A′,B′). Mature spermatocytes enter meiosis synchronously to produce 64 spermatids that remain connected by RCs (Fig. 1A″) (Fuller, 1993; Hime et al., 1996). RCs form as the result of incomplete cytokinesis during mitotic and meiotic cell divisions, during which cleavage furrows ingress but do not complete the final cytokinetic step of abscission leaving bridges of 1-2 μm in diameter (Fig. 1B,B′). Spermatids remain connected via RCs during the subsequent processes of spermatid tail elongation and individualization that are required for the production of mature motile spermatozoa (Fig. 1B,B′). Proteins identified at RCs include several that persist from cleavage furrows during cytokinesis, as well as proteins recruited after furrow ingression. Unlike the actin-rich intercellular bridges in male mice or Drosophila females, Drosophila male germline RCs have a septin-rich cytoskeleton, which includes Pnut, Sep1 and Sep2, in addition to Pavarotti (Pav, a kinesin-like protein) and its obligate binding partner Tumbleweed (a RacGAP), the cytoskeletal scaffolding proteins Cindr and Anillin, Nessun dorma, and Orbit/CLASP (for reviews, see Haglund et al., 2011; Lu et al., 2017; Yamashita, 2018).
Fig. 1.

Overview of spermatogenesis in the Drosophila testis. (A) Cartoon depicting spermatid development in Drosophila. Germline stem cells (red) are located at the hub (green) of the testis. (A′) Spermatogonia divide mitotically four times to form a 16-cell cyst. (A″) These cysts undergo a growth phase of ∼3 days before undergoing two rounds of meiosis to form 64-cell cysts. Following this, each cell elongates a tail to form bundles of mature spermatids. (B-B″) Immunofluorescence shows that ring canals marked by Pav::GFP (B′) and the fusome stained with Adducin antibody (B″) are present throughout spermatogenesis. Scale bar: 100 μm; inset 10 μm.
In addition to RCs, cells within a cyst are connected by the fusome: a large branched organelle that extends through each RC in the cyst (Fig. 1B,B″). Fusomes contain several cytoskeletal proteins, including α- and β-Spectrin, as well as Adducin [encoded by the hu-li tai shao (hts) gene] (De Cuevas et al., 1996; Lin et al., 1994; Yue and Spradling, 1992), all three of which are otherwise cortically located in somatic cells. In electron micrographs of Drosophila ovaries, fusomes exclude most ribosomes and mitochondria, and also contain abundant endoplasmic reticulum (ER) cisternae (Lin et al., 1994). Several ER proteins are concentrated in female fusomes, including PDI, Sec61ɑ, Rtnl1 and TER94, as well as the KDEL ER reporter (León and McKearin, 1999; Lighthouse et al., 2008; Röper, 2007; Snapp et al., 2004). In females, the fusome disappears soon after cells exit the mitotic cell cycle during oogenesis. Genetic analysis has shown that the fusome is necessary to orient mitotic spindles during cell division and to mediate the transfer of mRNAs and proteins into the pro-oocyte from nurse cells during oocyte specification (De Cuevas et al., 1996; Lin et al., 1994; Lin and Spradling, 1995; McKearin and Ohlstein, 1995). Mutations in the hts gene cause egg chamber arrest and female sterility.
The fusomes in males differ from female fusomes both in their composition and how long they persist during gametogenesis. The same ER proteins found in female fusomes are not concentrated in the male fusome (Lighthouse et al., 2008). The male fusome does not disappear after the completion of spermatogonial mitotic divisions; instead, it grows with each meiotic division and persists throughout spermatogenesis, including during spermatid elongation (Fig. 1B″, bottom inset), where it has been proposed to gather RCs to the distal tip of elongating spermatids (Hime et al., 1996). The male fusome has been implicated in the coordinated cell death response of germ cell cysts to DNA damage (Lu and Yamashita, 2017). However, the extent to which the fusome is required for sperm development under normal conditions remains unclear.
Here, using a combination of extensive live cell imaging, genetics and electron microscopy, we have explored the function of Drosophila male germline RCs and fusomes during spermatogenesis. We directly observed movement of GFP and endogenous proteins through RCs. Movement between neighboring cells within a germline cyst occurred throughout all stages of spermatogenesis, including post-meiotically, and independently of protein size. Furthermore, we found that fusome disruption specifically in male germline cells did not have a major effect on fertility, RC formation or intercellular protein movement.
RESULTS
RCs allow intercellular movement of proteins in mitotic spermatogonial cells
To investigate intercellular protein movement in the Drosophila testis, we expressed photoactivatable GFP (PA-GFP) (Pfeiffer et al., 2012) using nos- or bam-Gal4 drivers in germline cells also expressing GFP-tagged Pav (Pav::GFP) to mark RCs. Following activation in a single cell within a spermatogonial cyst, we captured time-lapse movies of PA-GFP localization (Fig. 2, Movies 1-4). Within 30 s of photoactivation, we observed movement of PA-GFP from the activated cell through RCs to other cells within mitotically active cysts (Fig. 2A-L). Within 10 min of photoactivation, we observed GFP signal throughout most, if not all, cells in all spermatogonial cysts from 2-cell through 16-cell cysts (Fig. 2M-P, n=94). These data demonstrate that RCs are persistent open channels that allow diffusion of GFP between spermatogonial cells.
Fig. 2.

RCs allow movement of GFP between germline cells in a cyst. (A-L) Live imaging of activated PA-GFP at various stages of spermatogenesis reveals sharing of GFP between cells in a cyst (red outline) through the ring canals (marked with Pav::GFP, white arrow). After activation of PA-GFP in a single cell or small region of cells (yellow outline), GFP was found in most of the cells in that cyst after 10 min (white outline). Scale bars: 10 μm. (M-P) Quantification of PA-GFP movement following photoactivation from a single donor cell (solid line) to other cells within the cyst (dashed line represents the fluorescence intensity from an average of all other non-activated cells within the same cyst). Normalized fluorescence intensity (AU) was plotted with respect to time. Error bars represent s.e.m.
To examine the movement of endogenous Drosophila proteins through RCs, we performed fluorescence loss in photobleaching (FLIP) experiments in 16-cell post-mitotic spermatocyte cysts expressing either GFP or GFP-fusion proteins. We selected FlyTrap lines with GFP-tagged proteins (Buszczak et al., 2007; Lowe et al., 2014; Nagarkar-Jaiswal et al., 2015; Quiñones-Coello et al., 2007) that had high GFP expression in the male germline (Figs S1 and S2) or had been previously assayed for movement in female somatic follicle cells (Airoldi et al., 2011). A region of interest was repeatedly photobleached while we simultaneously captured images of the GFP fluorescence in the entire cyst. Movement of proteins into the bleaching zone was determined by a loss of GFP fluorescence in neighboring cells. We first demonstrated by FLIP that, consistent with PA-GFP results, cytoplasmically expressed GFP moved through the RCs (Fig. 3A-C,M). Similarly, we observed movement of several GFP-fusion proteins, including Kra, Oda, Men-B and β-Tub56D (Fig. 3D-I, Table 1). However, we did not detect movement of most of the selected GFP-fusion proteins within the 60 min time-frame (n=2-4 cysts per genotype; Table 1). The sizes of the different proteins did not appear to correlate with the ability to move through the RCs between the cells. Remarkably, GFP::CaM, which we have previously shown moves through RCs in female follicle cells (Airoldi et al., 2011), did not move between male germline cells (Fig. 3J-L,P). These, albeit limited, data indicate that only a subset of proteins freely diffuses between cells, despite the open RCs, and suggest that there are some, as yet unknown, criteria for this ability to move.
Fig. 3.

RCs allow for sharing of some, but not all, proteins. (A-L) Fluorescence loss in photobleaching (FLIP) demonstrated that not all GFP-tagged proteins move between the cells in a 16-cell cyst. Several cells within a 16-cell cyst (red outline) expressing GFP or a GFP-tagged protein were continuously bleached (yellow outline) over the course of 1 h. Protein movement was determined by a loss in GFP fluorescence from neighboring cells within that cyst (white outline), indicating that GFP from non-bleached cells moved into the bleached region. (M-P) Quantification of GFP from the representative images (A-L) in the bleached (solid line), non-bleached (dotted line) and neighboring (dashed line) regions in a spermatocyte cyst. FLIP was detected for GFP, GFP::Oda, GFP::Men-B but not GFP::CaM. Mean fluorescence intensity (AU) is plotted with respect to time. Intermittent peaks on the graphs represent quick recovery of GFP in the sample while the microscope switches between capture and bleach modes. Scale bar: 10 μm.
Table 1.
Protein movement summary
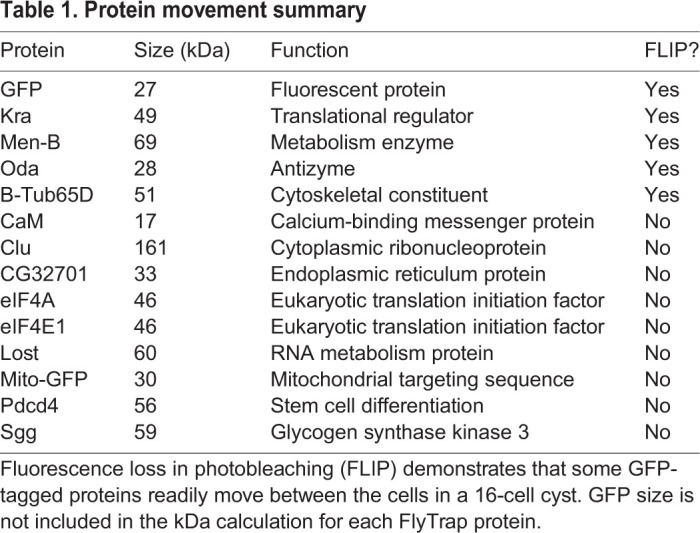
Proteins can move between cells in meiotic and post-meiotic cysts
One hypothesis for the function of RCs during spermatogenesis is to allow sharing of X-linked gene products to Y-bearing cells after meiosis (Braun et al., 1989; Morales et al., 2002). To assess post-meiotic sharing, we performed photoactivation studies in meiotic and post-meiotic cysts, as well as in haploid spermatids in the same manner as before. One notable difference, however, is that, in the later stages, multiple cells, rather than a single cell, were irradiated to increase the amount of visible PA-GFP in the smaller cells. In 32- and 64-cell cysts, where we illuminated cells in two separate locations, we could easily detect the spread of photoactivated PA-GFP between cells within 10 min of photoactivation (Fig. 4A-F, Movies 5, 6). Interestingly, not all of the cells of the post-meiotic cysts were fluorescent after 10 min of imaging (Fig. 4C,F). This is most likely because the ratio of photoactivated GFP to overall cyst size is smaller in the post-meiotic cells than in the earlier cysts, and diffused GFP becomes undetectable further away from the original region of activation. Additionally, we performed FLIP on endogenous GFP-tagged proteins in 64-cell cysts, and found their ability to move between cells just as we observed in primary spermatocytes (GFP::Men-B shown in Fig. 4J-L).
Fig. 4.

Movement of proteins in meiotic cysts and haploid spermatids. (A-I) After meiosis I and II, and during elongation of spermatid tails, PA-GFP moved between cells of a cyst (red outline). PA-GFP activated within a small region of the cyst (yellow outline) appeared in neighboring cells within that cyst (white outline). Cells activated previously in D are marked with blue asterisks. White arrow indicates RC end of spermatid bundle. (J-L) Movement of endogenous GFP::Men-B occurred in post-meiotic 64-cell cysts. Bleaching zone in this FLIP experiment is outlined in yellow. Cells with loss of fluorescence are outlined in white. Scale bars: 25 μm. (M) Cartoon of spermatid bundle depicting possible pathways of PA-GFP spread after activation (marked by star at position 1). (N-S) PA-GFP activation in a spermatid bundle over the course of 10 min with activation occurring in region 1 (shown in O). Scale bar: 25 μm. (T) Cartoon of actual PA-GFP spread showing that movement is predominantly through RCs rather than through lateral perforations. (U) Normalized fluorescence intensity of PA-GFP over time measured at the regions indicated in O. GFP fluorescence increased in region 4 (yellow line) before region 5 (dark-blue line). The RC end of spermatid bundles is marked by an arrow.
Finally, we tracked movement of PA-GFP in elongated spermatids after activation in a central zone of a spermatid bundle. We observed the spreading of GFP along the length of the bundle and to outer spermatids (Fig. 4G-I, Movie 7). As RCs were located at one end of the spermatid bundle (Fig. 4I, arrow), GFP would need to reach RCs before spreading to lateral cells. However, in addition to the RCs, elongated spermatids contain lateral membrane perforations throughout the length of the tails (Fabrizio et al., 1998; Tokuyasu et al., 1972), making it possible that GFP moved laterally through the perforations independent of RCs (Fig. 4M). To investigate protein movement further, we carefully tracked the spread of PA-GFP over a period of 10 min following activation (Fig. 4N-S). We reasoned that if movement to lateral cells depends on RCs, GFP fluorescence would increase near RCs (Fig. 4O, region 4) before it accumulated at a location directly lateral to the activation zone (Fig. 4O, region 5). In all samples examined (n=3), PA-GFP was first observed nearest the RCs, in region 4 (Fig. 4P,U; Fig. S3). Although we cannot rule out the possibility of some lateral movement through perforations, these data suggest that intercellular movement is predominantly through RCs (Fig. 4T). Our observation of intercellular protein movement during and after meiosis and during spermatid tail elongation provides evidence that developing sperm are in direct communication throughout spermatogenesis.
RNAi inhibition of α-Spectrin or Adducin severely disrupts fusome structure
During Drosophila oogenesis, the fusome is necessary for the production of viable gametes but its role in spermatogenesis remains largely unknown (De Cuevas and Spradling, 1998; Lin et al., 1994). To investigate the role of the fusome in males, we carried out germline-specific RNAi of genes encoding two structural proteins in the fusome: α-Spectrin and Adducin (encoded by the hts gene). Using a Pav::GFP background to mark RCs, we used two different Gal4 drivers to express these RNAi lines throughout development and in adults: nos-Gal4, which expresses in the early mitotic stages; or bam-Gal4, which begins expression in eight-cell cysts (Lu and Yamashita, 2017). We stained control, α-Spectrin knockdown and hts knockdown testes using antibodies to either α-Spectrin (3A9) or Adducin (Hts1B1) to monitor the fusome by immunofluorescence.
In wild-type testes, the fusome was present throughout all stages of spermatogenesis and extended through RCs connecting all cells within a cyst, and was present at the growing end of spermatid tails (Fig. 5A-E). In both nos> and bam>ɑSpec RNAi testes, we did not detect either Adducin (Fig. 5F-J, Fig. S4) or α-Spectrin (Figs S5-S6) fusome labeling in the mitotic region where nos and bam are expressed (Fig. 5F,G, zone 1), indicating the fusome was disrupted. We did, however, observe Adducin cortically at membranes in nos> and bam>ɑSpec RNAi testes and in elongating spermatid tails (Fig. 5G,I, Fig. S4F,H). In both genotypes, α-Spectrin protein remained absent from a fusome structure in zones 2 and 3 (Figs S5-S6), demonstrating the long-term effectiveness of RNAi driven by nos-Gal4 and bam-Gal4. Interestingly, in both nos> and bam>aSpec RNAi testes, we did observe Adducin-containing fusome-like fragments in primary spermatocytes (Fig. 5H, zone 2) and in meiotically dividing spermatocytes (Fig. 5J, zone 3), despite the absence of α-Spectrin (see also Fig. S4). Only a small subset of the fusome-like fragments was associated with RCs (Fig. 5H″, inset). These results suggested that cells began accumulating Adducin in fusome-like fragments independent of α-Spectrin in post-mitotic cysts.
Fig. 5.

nos Gal4-driven knockdown of α-Spectrin is sufficient to compromise the fusome throughout spermatogenesis. (A-A″) Wild-type testis with RCs marked by Pav::GFP and fusomes labeled with Adducin antibody (1B1). (B-C″,E-E″) Detailed images of the regions marked by boxes in A-A″, highlighting the RCs (Pav, green) and fusome (Adducin, purple) in a wild-type testis at three different stages of development: mitotic (zone 1), post-mitotic (zone 2) and elongated spermatids (zone 3). Insets in C-C″ highlight one RC. (D-D″,I-I″) Zone 2* highlights the growing ends of spermatid tails from the same ROI, but different z plane, as zone 2. (F-F″) Testes with α-Spec RNAi driven by nos-Gal4 lack Adducin staining at the fusome, but testis morphology appears unaffected. (G-J″) Detailed images of the regions marked in F showing Adducin staining at the membrane rather than in a fusome pattern, while Pav::GFP remained localized to the RCs. Insets in H-H″ highlight one RC. Scale bars: 150 µm in A-A″,F-F″; 20 μm in B-E″,G-J″; 1 µm in insets in C-C″,H-H″.
We also examined nos> and bam>hts RNAi testes stained with either Adducin or α-Spectrin. In nos>hts RNAi testes, the fusome was effectively disrupted in the mitotic region but Adducin- and α-Spectrin-containing fusome fragments were present outside of the zone of nos expression, in zones 2 and 3 (data not shown). In bam>hts RNAi testes, fusome morphology in zone 1 was indistinguishable from control testes. In zones 2 and 3, Adducin-containing fusome fragments were observed in addition to Adducin at the growing ends of spermatid tails (Fig. S7D). Similarly, we observed α-Spectrin-labeled fusome-like fragments in zones 2 and 3 in bam>hts RNAi and at the growing ends of spermatid tails (data not shown). Surprisingly, despite the absence of an intact fusome, overall testis morphology appeared normal (Fig. 5F).
As nos>aSpec RNAi produced a more complete knockdown of the fusome, we used this genotype to assess the effect of fusome disruption. We examined Pav::GFP in nos>ɑSpec RNAi testes to determine whether fusome disruption had an effect on RC formation. We found that fusome disruption did not affect accumulation of Pav::GFP to RCs (compare Fig. 5C′ with H′), and that the diameters of the RCs in wild-type and fusome-compromised testes were not statistically different (P=0.98); RCs were the same diameter, ∼1.6 µm, in both wild type (n=300) and nos>ɑSpec RNAi (n=201) throughout spermatogenesis. However, 11.1% of RCs in the nos>ɑSpec RNAi spermatogonia and primary spermatocytes appeared morphologically abnormal (not round; Fig. 5H, inset) or collapsed (having no obvious lumen; Fig. S8), suggesting that the fusome may play a role in RC stability.
To assess the extent of fusome disruption at greater resolution, we performed electron microscopy on htsΔG or ɑSpec RNAi testes driven by nos-Gal4. RCs were easily identifiable in cross-section by their electron-dense plasma membranes (Fig. 6A-D, Fig. S9A-C). In wild-type testes, the fusome appeared as a region of ribosome- and mitochondria-free cytoplasm that extended through the RCs (n=12) (Fig. 6A,A′, purple shading in A′) and between connecting cells. We also observed ER-like vesicles frequently embedded in the fusome, similar to electron micrographs of Drosophila ovaries (Lin et al., 1994). In nos>ɑSpec (n=12; Fig. 6C,D) and htsΔG (n=5; Fig. S9C) samples, we did not observe the characteristic ribosome-free cytoplasmic compartment within RC lumens.
Fig. 6.

Ribosome density reveals lack of fusomes in α-Spectrin RNAi testes. (A) EM of Pav::GFP testis revealed electron-dense RCs surrounding a fusome. (A′) False coloring of A highlights electron-dense RCs (green), plasma membrane (black), ER-like vesicles (yellow arrowhead) and a ribosome-free fusome area (purple). (B) Area outlined in A′ showing locations used for quantification of ribosome density in E. (C) EM of two cells connected by a RC in a α-Spec RNAi testis. (C′) False coloring of C highlighting RCs (green) and a plasma membrane (black) but no discernible fusome structure. (D) Area outlined in C′ shows locations used for quantification of ribosome density in E in the α-Spec RNAi testis. (E) Quantification of ribosome density shown as a split violin plot of standard deviation of pixel intensity values from regions of either RC (blue) or non-RC (orange) cytoplasmic compartments in controls or α-Spec RNAi testes.
To quantify the loss of the fusome in RNAi cells, we measured the ribosome density in cytoplasm within the RC lumen compared with cytoplasm outside of the RC lumen. To do this, we obtained the standard deviation (s.d.) of the mean pixel intensity from three non-overlapping 40 µm2 regions-of-interest (ROI) in the RC lumen (Fig. 6B,D; ‘RC’, blue) or cytoplasm distant from the RC lumen (Fig. 6B,D; ‘non-RC’, orange) in electron micrographs (see Fig. S10 for ROIs). We averaged the three values corresponding to each cellular compartment. In the cytoplasm of control RCs, the mean±s.d. was 22.30±3.5, and in the non-RC cytoplasm the value was 27.53±4.2 (P=0.0083, two-tailed Student's t-test) (Fig. 6E). The higher s.d. in the non-RC cytoplasm represents a broader distribution of pixel intensity values, which corresponds to the presence of electron-dense ribosomes. In contrast, the lower SD in the RC lumen cytoplasm corresponds to the observed reduction in electron-dense ribosomes that marks the fusome. We performed these same analyses in nos>ɑSpec RNAi RCs and observed that the s.d. values of RC and non-RC cytoplasm were not statistically different with average s.d. values of 26.32±4.1 and 26.24±4.2, respectively (Fig. 6E, P=0.96).
Fusome disruption has minimal impact on male fertility
We evaluated the effect of fusome disruption on male fertility by comparing wild-type fertility rates to males with α-Spec or hts RNAi. We detected little discernible effect on male fertility in males lacking an intact fusome (Fig. S11A). This was unexpected as previous work had reported male sterility caused by the hts1103 mutation (Wilson, 2005). Upon testing, we found that male fertility of three hts alleles was dramatically reduced (Fig. S11B). Two hts alleles, hts1103 and hts1, displayed declining progeny counts over a 14-day period, whereas htsΔG and htsW532X produced very few progeny throughout the same time-frame (Fig. S11C). Given the marked reduction in fertility rate in hts mutants, but negligible effect of germline-specific inhibition of hts expression, it is possible that the sterility of hts mutant males may be caused by a fusome-independent function of Adducin. Alternatively, as previously mentioned, in both α-Spectrin and Hts knockdown testes, Adducin localization to fusome fragments in the post-mitotic and meiotic zones (zones 2 and 3) as well as elongating spermatid tails may suggest that any remaining Adducin protein is sufficient for normal fertility, despite overt fusome disruption.
The results of our examination of germline-specific RNAi directed to structural components of the fusome, when taken together, demonstrate that intact fusomes are not necessary for spermatogenesis in normal, unchallenged, conditions. This is in stark contrast to Drosophila oogenesis, where disruption of fusomes results in oogenesis arrest and sterility (Huynh and St Johnston, 2004).
The fusome is not necessary for movement of GFP between cells
Studies in Drosophila oogenesis demonstrated a requirement for the fusome in the sharing of cytoplasmic information among the germ cells within a cyst (Yue and Spradling, 1992). However, as the fusome was not required for male fertility, we explored whether disruption of the fusome had an impact on protein movement through the RCs during spermatogenesis. We activated PA-GFP in α-Spec RNAi testes and monitored the movement of cytoplasmic GFP (Fig. 7, Movies 8, 9). After photoactivation in a single cell, PA-GFP moved to neighboring cells in 2-, 4- and 16-cell cysts, just as in wild-type testes, and on a similar time scale (n=93) (Fig. 7A-F). Similarly, movement of PA-GFP through the RCs in post-meiotic cells was not impacted by disruption of the fusome (Fig. 7G-I, Movie 10). Moreover, FLIP analysis in a fusome knockdown background showed that, as in wild-type testes, GFP::Oda moved between cells, whereas GFP::CaM did not (data not shown). To determine whether the rate of GFP diffusion between cells was affected by fusome disruption, we analyzed the diffusion of GFP from one cell into another. From 16-cell cysts, we isolated groups of two cells connected by a single RC in which we could measure the diffusion of total GFP from one cell directly into another cell in the presence of the fusome versus in fusome knockdown cells (Fig. S12).The initial rate of diffusion was slightly faster in fusome knockdown cells (Fig. S12), suggesting the possibility that the fusome moderates the rate of cytoplasm exchange between cells. In summary, our data suggest the fusome is not necessary to mediate transport between the cells in a cyst during spermatogenesis although it may modulate the rate of diffusion between cells.
Fig. 7.

PA-GFP moves through RCs despite knockdown of fusome components. (A-F) α-Spec RNAi driven by nos-Gal4 in two- and four-cell spermatogonia (A-C) and bam-Gal4 in 16-cell spermatocyte cysts (D-F; red outlines) did not affect movement of GFP through RCs (marked with Pav::GFP). PA-GFP was activated in one cell (yellow outline) and moved through the RCs to other cells within that cyst (white outline). (G-I) PA-GFP movement occurred through RCs in elongated spermatids, even after disruption of the fusome with α-Spec RNAi. Scale bars: 10 μm.
DISCUSSION
Interconnectivity is a major feature during Drosophila spermatogenesis, involving at least three types of intra-cyst cellular connections: ring canals, the fusome and lateral perforations in post-meiotic spermatid tails. Our data could support hypotheses that these cells maintain extensive connectivity for quality control under stress (Lu and Yamashita, 2017), synchronizing signals that govern cell cycling (Fuller, 1993; Gärthner et al., 2014; Huckins and Oakberg, 1978; Ren and Russell, 1991) and sharing of X- or Y-linked genes products in post-meiotic cells (Braun et al., 1989). We have demonstrated that RCs are open channels for protein sharing throughout all the stages of spermatogenesis, and that GFP and some endogenous proteins travel readily between connected cells. Under normal laboratory conditions, our data show the fusome is not required for spermatogenesis as compromising it does not impair RC formation, overall testis morphology or male fertility. The fusome may function instead in response to abnormal conditions – a synchronous ‘all-or-none’ cell death response of spermatogonial cysts in response to DNA damage involves the fusome (Lu and Yamashita, 2017).
This work represents the first extensive evidence that RCs mediate the sharing of cytoplasmic components throughout the entire process of Drosophila spermatogenesis, highlighting their role in cell-cell communication and protein sharing in differentiating male germline cells. Using live-imaging approaches, we documented diffusion of GFP and endogenous proteins through RCs in all stages, including post-meiotically. In post-meiotic haploid spermatid bundles, ring canals are located at the opposite end of the cell from the nuclei, yet we observed rapid GFP diffusion along the length of bundles, and spreading to neighboring cells, primarily through RCs. We cannot rule out protein sharing via lateral perforations between spermatid tails; however, tracking of GFP indicates the faster route relies on RCs (Fig. 4 and Fig. S3).
Interestingly, protein size does not appear to correlate with its ability to move into neighboring cells. Within the time frame of our live imaging experiments, proteins ranging from 28 to 69 kDa moved through RCs, while many proteins in the same range of molecular mass did not (Table 1). These results are similar to our assessment of movement across the smaller RCs connecting somatic follicle cells in Drosophila egg chambers (Airoldi et al., 2011). An unexpected difference is that GFP-tagged Calmodulin (CaM), a 17 kDa calcium-binding messenger protein, moved freely through ovarian follicle cell RCs, but not through testis RCs. Previous work has shown differences in CaM diffusion rates depending on whether CaM is bound to other complexes or immobile structures (Sanabria et al., 2008). The ability of proteins to diffuse between cells may be dependent on whether they are associated with a larger complex, an organelle or the cytoskeleton. This could suggest that CaM in the testis, but not in the ovarian follicle cells, is part of a larger complex.
Our work supports the long-held idea that male RCs are required to allow post-meiotic sharing of X- or Y-linked gene products between haploid cells. Although active biosynthesis of crucial mRNAs and proteins occurs during the 3-day growth phase of the primary spermatocytes prior to meiosis (Fuller, 1993), a subset of genes, called ‘cup’ and ‘comet’ genes based on the localization of mRNAs clustered at one end of spermatid bundles, is transcribed only after meiosis during spermatid tail elongation (Barreau et al., 2008a,b; Jandura et al., 2017). The timing of cup and comet gene expression suggests that they function during spermatid development. Mutants of one comet gene, scotti, which is involved in spermatid individualization, are male sterile (Barreau et al., 2008a,b). As scotti heterozygotes are fertile, products made in haploid spermatids with the wild-type allele likely spread to spermatids with the mutant allele (White-Cooper, 2010). Although this equilibration could be mediated by lateral perforations between spermatid tails (Fabrizio et al., 1998; Tokuyasu et al., 1972), our work suggests the most efficient path between cells is through RCs. This would be especially crucial for products of post-meiotically expressed X-linked genes, such as r-cup and p-cup. Our evidence of protein exchange through RCs even during post-meiotic stages of spermatogenesis demonstrates that haploid cells are indeed able to share gene products with their neighbors.
In addition to RCs, Drosophila male germline cells contain fusomes, which provide another type of connectivity between cells. We dramatically disrupted fusomes by RNAi inhibition of structural components and found negligible effects on fertility. Furthermore, the rate of movement of GFP between cells lacking fusomes was unchanged, suggesting that fusomes are not needed to either promote or prevent protein movement through RCs under normal conditions. Discovering that compromising fusomes has little effect on spermatogenesis was unexpected as disruption of fusomes in ovaries causes egg chamber arrest and female sterility (Lin et al., 1994; Yan et al., 2014). The composition of fusomes is also different in males and females: although they share some cytoskeletal proteins (α-Spectrin, Adducin and Ankyrin), female fusomes are richer in endoplasmic reticulum (ER) membranes and ER proteins (De Cuevas et al., 1997; Hime et al., 1996; Lighthouse et al., 2008; Lu et al., 2017; Snapp et al., 2004; Yamashita, 2018).
The striking functional difference between male and female fusomes may reflect the role of the fusome in breaking symmetry early in oogenesis. Whereas male fusomes persist throughout spermatogenesis (Hime et al., 1996), female fusomes are present only in the cystoblast stage of development, where they have been implicated in mitotic spindle orientation, cell cycle control and oocyte specification (De Cuevas and Spradling, 1998; Deng and Lin, 1997; Lilly et al., 2000; Lin et al., 1994; Lin and Spradling, 1995; McGrail and Hays, 1997; Yamashita, 2018). Female fusome disassembly begins immediately after the completion of mitotic divisions in the ovary. As the fusome fades, a polarized microtubule cytoskeleton forms in its place that promotes oocyte fate for one of the 16 sibling cells, with the rest becoming nurse cells (Grieder et al., 2000; McGrail and Hays, 1997; Theurkauf et al., 1993). In contrast, all 16 post-mitotic cells of male cysts proceed to meiosis and produce 64 sperm, so a structural mechanism is not needed to mediate different cell fates. In other words, one stem cell daughter in the female produces one egg, and in the male, one stem cell daughter produces 64 sperm. Perhaps removal of the female fusome is necessary to ensure the production of one egg per stem cell division. Although there is evidence that male fusomes participate in mitotic spindle alignment (Miyauchi et al., 2013), we can now conclude that fusomes are not essential for either mitosis or meiosis during spermatogenesis, at least under normal conditions. They may, however, have a role in responding to abnormal conditions, as seen in the presence of DNA damage in 16-cell spermatogonial cysts (Lu and Yamashita, 2017).
Our live-imaging data support a function for RCs during spermatogenesis in mediating cytoplasmic content sharing between the cells in a cyst; however, more could be learned by manipulating RC function. Our attempts at RC disruption have focused on RNAi of RC proteins, but these efforts led to cytokinesis defects rather than a RC-specific phenotype. To progress, new tools must be developed to disrupt RC function in vivo, perhaps by occlusion or targeted disruption, to study the functional consequences of RC loss during spermatogenesis. Disruption of the RCs in this manner could provide additional evidence for RC involvement in cell-cycle synchronization, maintenance of overall cyst health and sharing of post-meiotic gene products.
MATERIALS AND METHODS
Drosophila strains
The following Drosophila lines were generously provided by the referenced authors: 20XUAS-mC3PA-GFP and 10XUAS-GFP (Pfeiffer et al., 2012), bam-Gal4 (Chen and McKearin, 2003), hts1(Yue and Spradling, 1992), htsW532X (a gift from Trudy Schüpbach, Princeton University, USA), and htsΔG (Koundakjian et al., 2004). The following FlyTrap lines were used: GFP::CaM (YC0069LE), eIF4A::GFP (YC0001) and GFP::Oda (YD0523) (Buszczak et al., 2007; Lowe et al., 2014; Nagarkar-Jaiswal et al., 2015; Quiñones-Coello et al., 2007). nos-Gal4 (stock 7303), α-Spectrin shRNA (stock 56932), hts1103 (stock 10989), hts shRNA (stock 35421), Df(2R)BSC135/CyO (stock 9423), GFP::Clu (stock 6842), GFP::eIF4E1 (stock 50858), GFP::CG32701 (stock 50839), GFP::Lost (stock 6832), Mito::GFP (stock 8442), Pdcd4::GFP (stock 38446), GFP::Sgg (stock 50887), GFP::Kra (stock 50873), GFP::Men-B (stock 50854) and GFP::βTub56D (stock 50867) were obtained from the Bloomington Drosophila Stock Center. All animals were raised at room temperature or in a 25°C incubator. See Table S1 for more information.
Construction of transgenes and generation of transgenic lines
To visualize RC components at endogenous levels, we recombineered GFP into a BAC containing the Pavarotti (Pav) gene at the C-terminus (BAC ID 322-102N3) to create Pav::GFP. This BAC contains the entire pav locus on a 21 kb genomic fragment (chr3L:4,229,286…4,250,505, FlyBase release 6). Briefly, we used a two-step BAC recombineering protocol to first insert a Kanamycin resistance cassette (Wang et al., 2006) that was subsequently replaced by HA::GFP::FLAG through streptomycin selection. The final plasmid was injected by Rainbow Transgenic Flies into stock 24872 for insertion at the attP-3B site on chr2L. See Table S1 for more information.
Immunocytochemistry
Testes were dissected in IMADS buffer (ionically matched Drosophila saline) (Singleton and Woodruff, 1994) and fixed for 10 min in 4% paraformaldehyde in PBT (phosphate-buffered saline with 0.3% Triton X-100 and 0.5% BSA). Fixed tissue was washed in PBT and incubated with anti-Hts1B1 [1:50, Developmental Studies Hybridoma Bank (DSHB) (Zaccai and Lipshitz, 1996)] or anti-ɑSpec 3A9 (1:50, DSHB; Dubreuil et al., 1987). Secondary antibodies used were goat anti-mouse conjugated to Alexa-568 (1:500, Invitrogen). Samples were washed in PBT and mounted on slides in Aqua PolyMount (Polysciences). Samples were imaged with a Leica SP8 confocal microscope and a 40×1.3 NA oil-immersion objective lens and analyzed as maximum z-projections. See Table S1 for more information.
Photoactivation of PA-GFP
Live testes expressing Pav::GFP and PA-GFP with or without α-Spec shRNA driven by a nos-Gal4 or bam-Gal4 driver were dissected in a small drop of IMADS on a coverslip. Testes were gently scored to release both spermatogonia and spermatocytes, and break the muscle to prevent muscle contraction and prevent the testes from shifting during imaging. A slide was placed over the coverslip squashing the testes and extra buffer was wicked away using a Kimwipe. The slide was sealed with VALAP (equal parts Vaseline, lanolin, and paraffin) and imaged within 15 min. Photoactivation and subsequent live imaging of PA-GFP was accomplished on an inverted Leica SP8 confocal microscope and a 40×1.3 NA oil-immersion objective lens using the FRAP mode. Photoactivation was accomplished with 30 scan iterations of 405 nm light over regions of interest. Activated GFP was observed by capturing a single z-slice using 488 nm excitation every 30 s for ∼10 min.
Male fertility assay
Fertility of the fusome-less males was assessed by pairing a single male with three CantonS or w1118 virgin females. These males were shifted to new vials with fresh females every 2 days for 14 days and the total number of adult progeny was counted to determine fertility.
Transmission electron microscopy
For analysis by EM, ∼20 testis samples were fixed in 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 30 min at room temperature followed by 1 h at 4°C. The samples were rinsed in buffer then post-fixed in 1% osmium tetroxide and en bloc stained in 2% aqueous uranyl acetate for 1 h. Tissue was rinsed and dehydrated in an ethanol series followed by epon resin (Embed812 Electron Microscopy Science) infiltration, oriented and baked overnight at 60°C. Hardened blocks were cut using a Leica UltraCut UCT. Sections (60 nm) were collected on formvar/carbon-coated grids and contrast stained using 2% uranyl acetate and lead citrate. Samples were viewed using FEI Tencai Biotwin TEM at 80Kv. Images were taken using a Morada CCD and iTEM (Olympus) software.
Fluorescence loss in photobleaching (FLIP)
FLIP of UAS-GFP and all GFP-traps was conducted using a Leica SP8 microscope. 16- or 64-cell cysts were dissected out of the testis in a manner as described above. Microscope pinhole size was set to 7 to generate visible bleaching of GFP using the following sequence: (one pre-bleach, 30 iterations of bleaching, one post-bleach)×48 for 1 h of imaging while repositioning the sample as necessary to account for drift. GFP bleaching and single z-slice image capture was performed using a 488 nm laser. All images were processed using FIJI. To quantify fluorescence loss, a region of interest (ROI) was drawn around the entire bleached area of a cyst and the mean pixel intensity for this region was measured for the duration of the movie using Time Series Analyzer (FIJI). Similarly, a ROI was drawn around the remainder of the cyst, outside the bleached region, and the mean pixel intensities were measured. These values were plotted against the mean pixel values from a ROI of similar size in a neighboring control cell to control for loss of signal due to generalized photobleaching. Raw values of the mean pixel intensities were exported into GraphPad Prism to generate representative graphs of each GFP protein.
Quantification of fusome knockdown
Knockdown of the fusome was quantitatively assessed from electron micrographs of control (Pav::GFP, w1118 or nos-Gal4; n=12) and nos>aSpec RNAi (n=12) testes. Three non-overlapping ROIs of 40 μm2 were assigned to the RC lumen between or immediately adjacent to the electron dense plasma membrane. Similarly, three non-overlapping ROIs of 40 μm2 were assigned to measure the cytoplasm distant from the RC lumen. From the resulting histograms, the standard deviation was measured and used as a proxy for ribosome density. The standard deviations from each cytoplasmic compartment (RC or non-RC) were averaged to account for differences in staining across preparations. Measurements were exported into R Studio for further analysis and data visualization. Statistical significance was determined by a two-tailed Student's t-test.
Quantitative analysis of GFP movement
Movement of GFP fluorescence, both in PA-GFP and FLIP experiments, was assessed by recording fluorescence intensities in experimental and non-activated control cells over the course of the movies. Average pixel intensity values from a 1256 pixel2 region of interest (ROI) in the cytoplasm of either the donor or acceptor cell(s) were measured using the Time Series Analyzer in FIJI. In cysts with more than one acceptor cell, all cells in the focal plane were averaged to generate a single trace. Fluorescence values were normalized by subtracting the average fluorescence intensity of an ROI of the same size from two adjacent, non-photoactivated cells. Measurements were exported into Excel for further analysis and GraphPad Prism for data visualization.
Measurements of GFP movement in elongated spermatids were acquired using the Time Series Analyzer in FIJI. Six 1256 pixel2 ROIs were assigned as described in Fig. 4M and the mean pixel values of each ROI were plotted as a function of time. Raw values were exported into Excel for further analysis and GraphPad Prism for data visualization. A total of three spermatid bundles were analyzed and plotted individually because of differing levels of GFP fluorescence.
To quantitatively assess rates of movement of PA-GFP between cells in wild-type and fusome knockdown testes, a Bruker Opterra II Swept Field Microscope was used, with a 60× water immersion objective lens. Wild-type (w1118) and bam>ɑSpec RNAi testes were scored and mounted as described above. PA-GFP was activated by a single iteration of 405 nm light in one z-plane and movies were captured in a 15-20 slice z-stack encompassing the cyst every 10 s for a total of 10 min. Maximum intensity projections were generated in FIJI, and the total fluorescence of PA-GFP in the activated and recipient cell was measured at each time point. For comparison purposes, we quantified only movies in which PA-GFP was activated within a single spermatocyte cell and diffused into one other recipient cell. Although the cells imaged in these experiments were from 16-cell cysts, PA-GFP diffusion usually was restricted to one to five other cells, indicating that the tissue-scoring preparation used may have caused cell clusters to become dissociated from the rest of the cyst. The PA-GFP relative fluorescence units (RFU) between the activated and the recipient cells were summed and normalized to 1. The mean RFU of PA-GFP in the recipient cell (as a fraction of total RFU between the two cells) for wild-type and fusome knockdown conditions was plotted over time (n=9 for wild-type; n=11 for fusome knockdown). A nonlinear regression was used to fit the data to a one-phase exponential association model using the following equation:
The best-fit curve was plotted alongside the mean RFU data points for each condition (see Fig. S12). Comparison of fits was performed to check for statistically significant differences in the best-fit values between wild-type and fusome knockdown fits.
Supplementary Material
Acknowledgements
We thank Peter McLean for his help in recombineering the Pav BAC transgene; Allison James for her help screening RNAi lines; Charlotte Killiam for help with statistical analysis; Ronit Wilk, Jack Hu and Henry Krause for help with post-meiotic movement experiments; Julie Brill for helpful conversations about lateral perforations in spermatid tails; Tian Xu and Kaelyn Sumigray for providing access to the Leica SP8 confocal microscope; and Morven Graham and the Yale Center for Cellular and Molecular Imaging for help with EM imaging. Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study. We thank the DRSC/TRiP Functional Genomics Resources at Harvard Medical School for providing transgenic RNAi fly stocks used in this study.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: R.S.K., K.L.P., A.M.H., L.C.; Methodology: R.S.K., K.L.P., K.M.M., K.M.A., L.C.; Formal analysis: R.S.K., K.L.P., K.M.M.; Investigation: R.S.K., K.L.P., K.M.M., K.M.A.; Resources: A.M.H., L.C.; Writing - original draft: R.S.K.; Writing - review & editing: R.S.K., K.L.P., L.C.; Visualization: R.S.K., K.L.P., K.M.M.; Supervision: L.C.; Project administration: L.C.; Funding acquisition: L.C.
Funding
This work was supported by the National Institutes of Health (R01 GM043301 and RC1 GM091791 to L.C.). Partial support for predoctoral trainees was provided by National Institutes of Health training grants (T32 GM007223 for R.S.K. and K.M.M.). Support for K.L.P. was provided by the Yale Surdna Venture Fund and the National Institutes of Health (F32 GM136029-01). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at https://dev.biologists.org/lookup/doi/10.1242/dev.190140.supplemental
Peer review history
The peer review history is available online at https://dev.biologists.org/lookup/doi/10.1242/dev.190140.reviewer-comments.pdf
References
- Airoldi S. J., McLean P. F., Shimada Y. and Cooley L. (2011). Intercellular protein movement in syncytial Drosophila follicle cells. J. Cell Sci. 124, 4077-4086. 10.1242/jcs.090456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreau C., Benson E., Gudmannsdottir E., Newton F. and White-Cooper H. (2008a). Post-meiotic transcription in Drosophila testes. Development 135, 1897-1902. 10.1242/dev.021949 [DOI] [PubMed] [Google Scholar]
- Barreau C., Benson E. and White-Cooper H. (2008b). Comet and cup genes in Drosophila spermatogenesis: the first demonstration of post-meiotic transcription. Biochem. Soc. Trans. 36, 540-542. 10.1042/BST0360540 [DOI] [PubMed] [Google Scholar]
- Braun R. E., Behringer R. R., Peschon J. J., Brinster R. L. and Palmiter R. D. (1989). Genetically haploid spermatids are phenotypically diploid. Nature 337, 373-376. 10.1038/337373a0 [DOI] [PubMed] [Google Scholar]
- Brown R. K. and Smith W. L. (1964). Chromosomal studies in ovarian dysgenesis. Trans. N. Engl. Obstet. Gynecol. Soc. 18, 47-54. [PubMed] [Google Scholar]
- Burgos M. H. and Fawcett D. W. (1955). Studies on the fine structure of the mammalian testis. I. Differentiation of the spermatids in the cat (Felis domestica). J. Biophys. Biochem. Cytol. 1, 287-300. 10.1083/jcb.1.4.287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buszczak M., Paterno S., Lighthouse D., Bachman J., Planck J., Owen S., Skora A. D., Nystul T. G., Ohlstein B., Allen A. et al. (2007). The carnegie protein trap library: a versatile tool for drosophila developmental studies. Genetics 175, 1505-1531. 10.1534/genetics.106.065961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. and McKearin D. M. (2003). A discrete transcriptional silencer in the bam gene determines asymmetric division of the Drosophila germline stem cell. Development 130, 1159-1170. 10.1242/dev.00325 [DOI] [PubMed] [Google Scholar]
- De Cuevas M., Lee J. K. and Spradling A. C. (1996). α-spectrin is required for germline cell division and differentiation in the Drosophila ovary. Development 122, 3959-3968. [DOI] [PubMed] [Google Scholar]
- De Cuevas M., Lilly M. and Spradling A. (1997). Germline cyst formation in Drosophila. Annu. Rev. Genet. 31, 405-428. 10.1146/annurev.genet.31.1.405 [DOI] [PubMed] [Google Scholar]
- De Cuevas M. and Spradling A. C. (1998). Morphogenesis of the Drosophila fusome and its implications for oocyte specification. Development 125, 2781-2789. [DOI] [PubMed] [Google Scholar]
- Deng W. and Lin H. (1997). Spectrosomes and fusomes anchor mitotic spindles during asymmetric germ cell divisions and facilitate the formation of a polarized microtubule array for oocyte specification in Drosophila. Dev. Biol. 189, 79-94. 10.1006/dbio.1997.8669 [DOI] [PubMed] [Google Scholar]
- Dubreuil R., Byers T. J., Branton D., Goldstein L. S. and Kiehart D. P. (1987). Drosophilia spectrin. I. Characterization of the purified protein. J. Cell Biol. 105, 2095-2102. 10.1083/jcb.105.5.2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dym M. and Fawcett D. W. (1971). Further observations on the numbers of spermatogonia, spermatocytes, and spermatids connected by intercellular bridges in the mammalian testis. Biol. Reprod. 4, 195-215. 10.1093/biolreprod/4.2.195 [DOI] [PubMed] [Google Scholar]
- Fabrizio J. J., Hime G., Lemmon S. K. and Bazinet C. (1998). Genetic dissection of sperm individualization in Drosophila melanogaster. Development 125, 1833-1843. [DOI] [PubMed] [Google Scholar]
- Fawcett D. W., Ito S. and Slautterback D. (1959). The occurrence of intercellular bridges in groups of cells exhibiting synchronous differentiation. J. Biophys. Biochem. Cytol. 5, 453-460. 10.1083/jcb.5.3.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller M. T. (1993). The Development of Drosophila Melanogaster, 1st edn (ed. Bate M. and Martinez Arias A.) Plainview, New York: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Gärthner S. M. K., Rathke C., Renkawitz-Pohl R. and Awe S. (2014). Ex vivo culture of Drosophila pupal testis and single male germ-line cysts: dissection, imaging, and pharmacological treatment. J. Vis. Exp. 91, e51868 10.3791/51868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum M. P., Iwamori T., Buchold G. M. and Matzuk M. M. (2011). Germ cell intercellular bridges. Cold Spring Harb. Perspect. Biol. 3, 1-18. 10.1101/cshperspect.a005850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieder N. C., de Cuevas M. and Spradling A. C. (2000). The fusome organizes the microtubule network during oocyte differentiation in Drosophila. Development 127, 4253-4264. [DOI] [PubMed] [Google Scholar]
- Haglund K., Nezis I. P. and Stenmark H. (2011). Structure and functions of stable intercellular bridges formed by incomplete cytokinesis during development. Commun. Integr. Biol. 4, 1-9. 10.4161/cib.13550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hime G. R., Brill J. A. and Fuller M. T. (1996). Assembly of ring canals in the male germ line from structural components of the contractile ring. J. Cell Sci. 109, 2779-2788. [DOI] [PubMed] [Google Scholar]
- Huckins C. and Oakberg E. F. (1978). Morphological and quantitative analysis of spermatogonia in mouse testes using whole mounted seminiferous tubules. II. The irradiated testes. Anat. Rec. 192, 529-541. 10.1002/ar.1091920407 [DOI] [PubMed] [Google Scholar]
- Huynh J. R. and St Johnston D. (2004). The origin of asymmetry: early polarisation of the Drosophila germline cyst and oocyte. Curr. Biol. 14, 438-449. 10.1016/j.cub.2004.05.040 [DOI] [PubMed] [Google Scholar]
- Jandura A., Hu J., Wilk R. and Krause H. M. (2017). High resolution fluorescent in situ hybridization in Drosophila embryos and tissues using tyramide signal amplification. J. Vis. Exp. 2017, e56281 10.3791/56281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch E. A. and King R. C. (1966). The origin and early differentiation of the egg chamber of Drosophila melanogaster. J. Morphol. 119, 283-303. 10.1002/jmor.1051190303 [DOI] [PubMed] [Google Scholar]
- Koch E. A. and King R. C. (1969). Further studies on the ring canal system of the ovarian cystocytes of Drosophila melanogaster. Zeitschrift für Zellforsch. und Mikroskopische Anat. 102, 129-152. 10.1007/BF00336421 [DOI] [PubMed] [Google Scholar]
- Koundakjian E. J., Cowan D. M., Hardy R. W. and Becker A. H. (2004). The Zuker collection: a resource for the analysis of autosomal gene function in Drosophila melanogaster. Genetics 167, 203-206. 10.1534/genetics.167.1.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- León A. and McKearin D. (1999). Identification of TER94, an AAA ATPase protein, as a Bam-dependent component of the Drosophila fusome. Mol. Biol. Cell 10, 3825-3834. 10.1091/mbc.10.11.3825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lighthouse D. V., Buszczak M. and Spradling A. C. (2008). New components of the Drosophila fusome suggest it plays novel roles in signaling and transport. Dev. Biol. 317, 59-71. 10.1016/j.ydbio.2008.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly M. A., De Cuevas M. and Spradling A. C. (2000). Cyclin A associates with the fusome during germline cyst formation in the Drosophila ovary. Dev. Biol. 218, 53-63. 10.1006/dbio.1999.9570 [DOI] [PubMed] [Google Scholar]
- Lin H. and Spradling A. C. (1995). Fusome asymmetry and oocyte determination in Drosophila. Dev. Genet. 16, 6-12. 10.1002/dvg.1020160104 [DOI] [PubMed] [Google Scholar]
- Lin H., Yue L. and Spradling A. C. (1994). The Drosophila fusome, a germline-specific organelle, contains membrane skeletal proteins and functions in cyst formation. Development 120, 947-956. [DOI] [PubMed] [Google Scholar]
- Lowe N., Rees J. S., Roote J., Ryder E., Armean I. M., Johnson G., Drummond E., Spriggs H., Drummond J., Magbanua J. P. et al. (2014). Analysis of the expression patterns, Subcellular localisations and interaction partners of Drosophila proteins using a pigP protein trap library. Development 141, 3994-4005. 10.1242/dev.111054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K. L. and Yamashita Y. M. (2017). Germ cell connectivity enhances cell death in response to DNA damage in the Drosophila testis. Elife 6, e27960 10.7554/eLife.27960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K., Jensen L., Lei L. and Yamashita Y. M. (2017). Stay connected: a germ cell strategy. Trends Genet. 33, 971-978. 10.1016/j.tig.2017.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrail M. and Hays T. S. (1997). The microtubule motor cytoplasmic dynein is required for spindle orientation during germline cell divisions and oocyte differentiation in Drosophila. Development 124, 2409-2419. [DOI] [PubMed] [Google Scholar]
- McKearin D. and Ohlstein B. (1995). A role for the Drosophila Bag-of-marbles protein in the differentiation of cystoblasts from germline stem cells. Development 121, 2937-2947. [DOI] [PubMed] [Google Scholar]
- McLean P. F. and Cooley L. (2013). Protein equilibration through somatic ring canals in Drosophila. Science 340, 1444-1447. 10.1126/science.1234887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi C., Kitazawa D., Ando I., Hayashi D. and Inoue Y. H. (2013). Orbit/clasp is required for germline cyst formation through its developmental control of fusomes and ring canals in drosophila males. PLoS ONE 8, e58220 10.1371/journal.pone.0058220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales C. R., Lefrancois S., Chennathukuzhi V., El-Alfy M., Wu X., Yang J., Gerton G. L. and Hecht N. B. (2002). A TB-RBP and Ter ATPase complex accompanies specific mRNAs from nuclei through the nuclear pores and into intercellular bridges in mouse male germ cells. Dev. Biol. 246, 480-494. 10.1006/dbio.2002.0679 [DOI] [PubMed] [Google Scholar]
- Nagarkar-Jaiswal S., Lee P. T., Campbell M. E., Chen K., Anguiano-Zarate S., Gutierrez M. C., Busby T., Lin W. W., He Y., Schulze K. L. et al. (2015). A library of MiMICs allows tagging of genes and reversible, spatial and temporal knockdown of proteins in Drosophila. Elife 2015, e05338 10.7554/eLife.05338.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer B. D., Truman J. W. and Rubin G. M. (2012). Using translational enhancers to increase transgene expression in Drosophila. Proc. Natl. Acad. Sci. USA 109, 6626-6631. 10.1073/pnas.1204520109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiñones-Coello A. T., Petrella L. N., Ayers K., Melillo A., Mazzalupo S., Hudson A. M., Wang S., Castiblanco C., Buszczak M., Hoskins R. A. et al. (2007). Exploring strategies for protein trapping in Drosophila. Genetics 175, 1089-1104. 10.1534/genetics.106.065995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H. P. and Russell L. D. (1991). Clonal development of interconnected germ cells in the rat and its relationship to the segmental and subsegmental organization of spermatogenesis. Am. J. Anat. 192, 121-128. 10.1002/aja.1001920203 [DOI] [PubMed] [Google Scholar]
- Robinson D. and Cooley L. (1996). Stable intercellular bridges in development: the cytoskeleton lining the tunnel. Trends Cell Biol. 6, 474-479. 10.1016/0962-8924(96)84945-2 [DOI] [PubMed] [Google Scholar]
- Röper K. (2007). Rtnl1 is enriched in a specialized germline ER that associates with ribonucleoprotein granule components. J. Cell Sci. 120, 1081-1092. 10.1242/jcs.03407 [DOI] [PubMed] [Google Scholar]
- Sanabria H., Digman M. A., Gratton E. and Waxham M. N. (2008). Spatial diffusivity and availability of intracellular calmodulin. Biophys. J. 95, 6002-6015. 10.1529/biophysj.108.138974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton K. and Woodruff R. I. (1994). The osmolarity of adult Drosophila hemolymph and its effect on oocyte-nurse cell electrical polarity. Dev. Biol. 161, 154-167. 10.1006/dbio.1994.1017 [DOI] [PubMed] [Google Scholar]
- Snapp E. L., Iida T., Frescas D., Lippincott-Schwartz J. and Lilly M. A. (2004). The fusome mediates intercellular endoplasmic reticulum connectivity in Drosophila ovarian cysts. Mol. Biol. Cell 15, 4512-4521. 10.1091/mbc.e04-06-0475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurkauf W. E., Alberts B. M., Jan Y. N. and Jongens T. A. (1993). A central role for microtubules in the differentiation of Drosophila oocytes. Development 118, 1169-1180. [DOI] [PubMed] [Google Scholar]
- Tokuyasu K. T., Peacock W. J. and Hardy R. W. (1972). Dynamics of spermiogenesis in Drosophila melanogaster. I. Individualization process. Z. Zellforsch. Mikrosk. Anat. 124, 479-506. 10.1007/BF00335253 [DOI] [PubMed] [Google Scholar]
- Wang J., Sarov M., Rientjes J., Fu J., Hollak H., Kranz H., Xie W., Stewart A. F. and Zhang Y. (2006). An improved recombineering approach by adding RecA to λ red recombination. Mol. Biotechnol. 32, 43-53. 10.1385/MB:32:1:043 [DOI] [PubMed] [Google Scholar]
- White-Cooper H. (2010). Molecular mechanisms of gene regulation during Drosophila spermatogenesis. Reproduction 139, 11-21. 10.1530/REP-09-0083 [DOI] [PubMed] [Google Scholar]
- Wilson P. G. (2005). Centrosome inheritance in the male germ line of Drosophila requires hu-li tai-shao function. Cell Biol. Int. 29, 360-369. 10.1016/j.cellbi.2005.03.002 [DOI] [PubMed] [Google Scholar]
- Yamashita Y. M. (2018). Subcellular specialization and organelle behavior in germ cells. Genetics 208, 19-51. 10.1534/genetics.117.300184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D., Neumüller R. A., Buckner M., Ayers K., Li H., Hu Y., Yang-Zhou D., Pan L., Wang X., Kelley C. et al. (2014). A regulatory network of Drosophila germline stem cell self-renewal. Dev. Cell 28, 459-473. 10.1016/j.devcel.2014.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L. and Spradling A. C. (1992). hu-li tai shao, a gene required for ring canal formation during Drosophila oogenesis, encodes a homolog of adducin. Genes Dev. 6, 2443-2454. [DOI] [PubMed] [Google Scholar]
- Zaccai M. and Lipshitz H. D. (1996). Differential distributions of two adducin-like protein isoforms in the Drosophila ovary and early embryo. Zygote 4, 159-166. 10.1017/S096719940000304X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.