

Abstract
Introduction:
UDP N-acetylglucosamine2-epimerase/N-acetylmannosamine-kinase (GNE) gene mutations can cause mostly autosomal-recessive myopathy with juvenile-onset known as hereditary inclusion-body myopathy (HIBM).
Methods:
We describe a family of a patient showing an unusual HIBM with both vacuolar myopathy and myositis without quadriceps-sparing, hindering diagnosis. We show how genetic testing with functional assays, clinical transcriptome sequencing (RNA-seq) in particular, helped facilitate both the diagnosis and a better understanding of the genotype-phenotype relationship.
Results:
We identified a novel 7.08 kb pathogenic deletion upstream of GNE using array comparative genomic hybridization (aCGH) and a common Val727Met variant. Using RNA-seq, we found only monoallelic (Val727Met-allele) expression, leading to ~50% GNE reduction in muscle. Importantly, α-dystroglycan is hypoglycosylated in the patient muscle, suggesting HIBM could be a “dystroglycanopathy.”
Conclusions:
Our study shows the importance of considering aCGH for GNE-myopathies, and the potential of RNA-seq for faster, definitive molecular diagnosis of unusual myopathies.
Keywords: aCGH, GNE myopathy (HIBM), molecular diagnostics, myositis, next generation sequencing, transcriptome sequencing (RNA-seq)
To enhance molecular diagnostic yield in neuromuscular disorders (NMDs), functional assays downstream of genomic DNA1–4 is recommended by the American College of Medical Genetics and Genomics (ACMG).5 GNE-myopathy (OMIM #605820) or hereditary inclusion-body myopathy (HIBM) is a vacuolar myopathy generally sparing the quadriceps. It is a rare, recessive, inherited degenerative skeletal muscle disorder caused by GNE gene (MIM# 603824; NCBI Gene ID: 10020; NC_000009.12) variants with - early-adult onset.6–8 The GNE enzyme catalyzes the first 2 rate-limiting steps in the biosynthesis of 5-N-acetylneuraminic acid (Neu5Ac)9,10 found as the terminal glycans on various glycoproteins/glycolipids, such as the sarcoglycans and dystroglycan (DG), functioning in variety of cellular pathways.11
Our understanding of the molecular basis of GNE myopathy is unclear.12,13 Here, we describe the lessons learned by using functional genomic approaches to characterize an unusual GNE myopathy in a family with a novel deletion variant in which relative quadriceps sparing in association with both vacuolar myopathy and myositis made diagnosis challenging.
MATERIALS AND METHODS
All protocols were approved by institutional IRB with written consents and are presented here in chronological order. First, trio (patient and parents) exome sequencing and simultaneous patient muscle biopsy immunohistochemistry, and then RNA sequencing of the biopsy, were performed, which prompted us to do array comparative genomic hybridization (aCGH) to identify any deletions/duplications. Immunoblotting was then done using biopsy to identify any glycosylation defect related to the HIBM pathophysiology. Gene ontology-pathway analysis14 led to further understanding of patient muscle glycosylation defects. Molecular-dynamics (MD) simulation of the V727M variant on the GNE kinase-domain crystal-structure was performed. For methods details, see Supplementary Methods, which are available online.
RESULTS
Clinical Phenotype.
The patient was a 21-year-old man, ethnically Indian-Guyanese, with progressive muscle-weakness without any cardiac or respiratory comorbidity. Symptoms began at age 20 years with asymmetric leg pain and weakness, initially with bilateral-foot-drop and mild (Medical Research Council 4/5) weakness in the quadriceps, followed by rapidly progressive and severe bilateral lower extremity distal weakness, additional quadriceps weakness, and upper extremity weakness in the deltoids and the long-finger-flexors. There was no facial or bulbar weakness and tendon reflexes were reduced throughout. Needle electromyography of the quadriceps, tibialis anterior, iliopsoas, and medial gastrocnemius muscles demonstrated abnormal spontaneous activity (fibrillation potentials and positive sharp waves) and early motor unit potential recruitment, compatible with multiple types of myopathies. There was no family history of neuromuscular disease.
Quadriceps Showed Unusual HIBM: Both Vacuolar and Inflammatory Myopathy.
Quadriceps biopsy (Fig. 1) showed increased fiber-size variability with both muscle-fiber necrosis and perivascular inflammation. Prominent rimmed vacuoles were seen on modified Gomori trichrome that were positive by immunohistochemistry for both ubiquitin and TDP-43. There was no lipid or glycogen accumulation. Acid phosphatase stain showed increased lysosomal activity.
FIGURE 1.
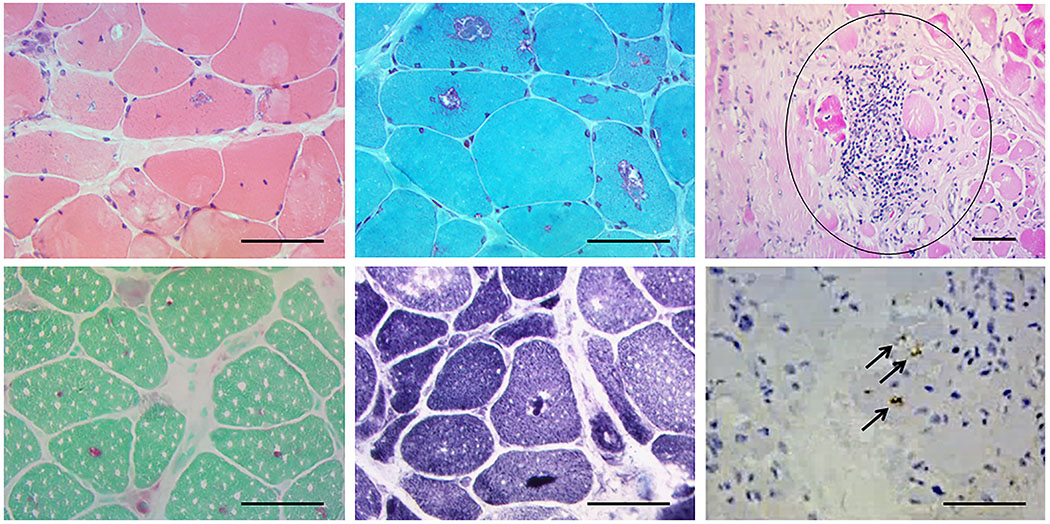
Muscle biopsy showing central and sub-sarcolemmal vacuoles in hemotoxylin and eosin (H&E) (top panel left), which demonstrates red “rimming” with modified Gomori trichrome (top center panel). An example of inflammation is seen in the top right panel (circled). Bottom panel: Acid phosphatase (left) and NADH (center) showing positive material within the vacuoles and vacuoles are stained positive for ubiquitin (black arrows) (right). Scale bar = 50 μm. Additionally, the connective tissue was mildly increased. Atrophic fibers were round and pyknotic nuclear clumps were not seen, and the biopsy showed a moderate number of fibers with internalized nuclei. Regenerating fibers were not seen. The following stains were normal: cytochrome oxidase (COX), myosin ATPase (normal distribution of fiber types), Oil red O, periodic acid–Schiff (PAS), phosphorylase, Congo red. Neither muscle fiber-type grouping, nor type specific atrophy was seen.
Exome and RNA Sequencing Revealed Monoallelic Expression of V727M Allele.
A known “likely pathogenic” heterozygous missense variant (c.2179C>T (p.V727M)) (Supplementary Fig. S1; Supplementary Table S1) was identified in GNE (rs121908627; allele frequency of 0.0141) prevalent in South East Asian populations.6,15–21 Exome analysis did not identify a second variant resulting in no molecular diagnosis. RNA sequencing using target muscle biopsy revealed the presence of only the GNE V727M allele (Fig. 2A) suggesting mono-allelic expression. Absence of transcription from the alternate chromosome could be due to a deletion/duplication not detected by ES. Thus, we performed aCGH using patient genomic DNA.
FIGURE 2.
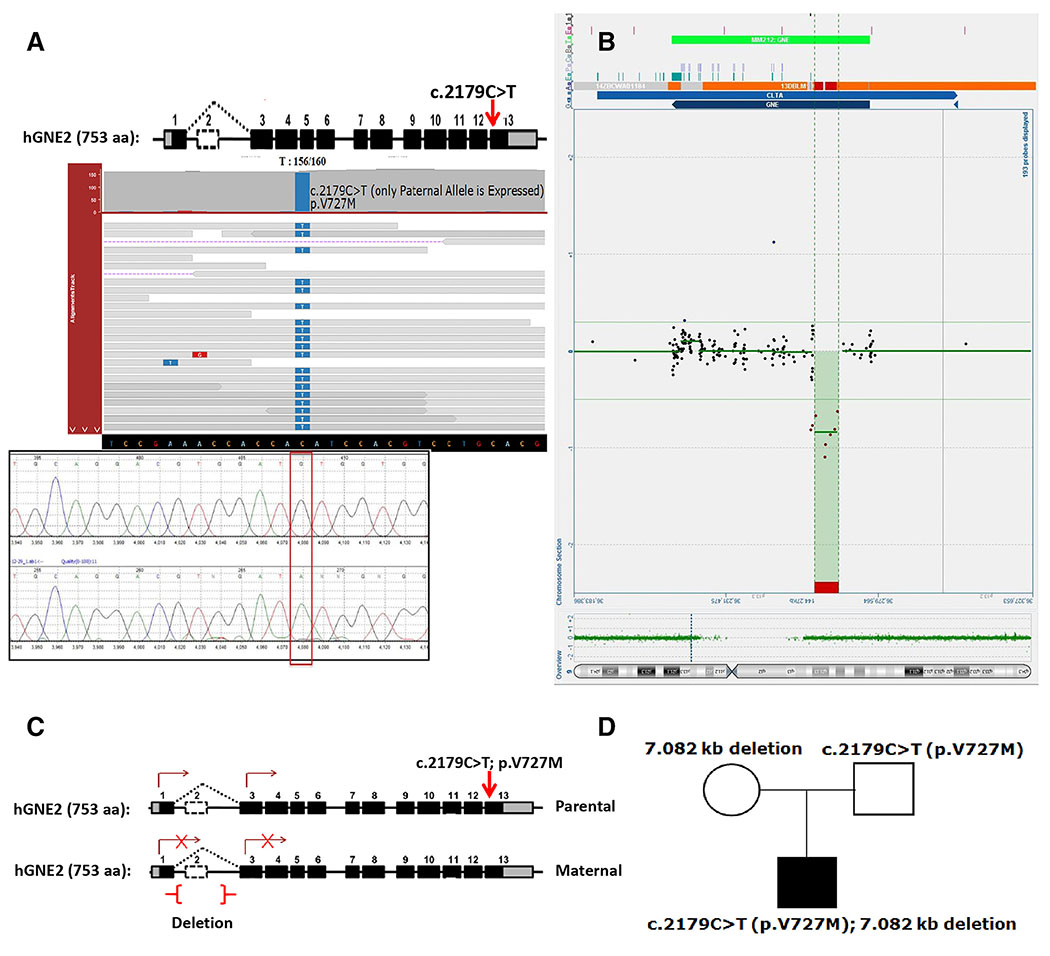
(A) Integrated Genomics Viewer (IGV) pile up of RNA-sequencing showing monoallelic expression of GNE gene with only the allele harboring c.2179C>T:G>A (p.V727M) missense “likely pathogenic” variant expressed. The red arrow indicates the position of the V727M variant in exon 13 of the GNE gene. Sanger sequencing confirmation was performed on cDNA showing monoallelic expression as shown below. (B) aCGH signal showing a deletion upstream of the GNE gene with genomic breakpoints at nucleotide positions g.36,259,402 and 36,266,483 was detected in this individual (SCV000599234). This deletion is 7.08 kb in size and encompasses the untranslated exon 2 of the hGNE2 transcript but upstream of the hGNE1 transcript of the GNE gene. (C,D) Exome sequencing and later aCGH of trios reveal that monoallelic expression was due to expression of only the paternal allele of GNE in the proband.
aCGH Identified Novel Deletion Encompassing Exon2 of hGNE2, Upstream of hGNE1.
Recently, a ~11.3-kb deletion encompassing exon 2 was found in a patient along with a single V727M variant.22 In our study, aCGH revealed a novel 7.08 kb deletion (g.36,259,402 to g.36,266,483) (SCV000599234) upstream of the GNE gene (different from deletions identified in Zhu et al.23) (Fig. 2B). This reports a novel compound-heterozygous variant combination (Supplementary Table S1) of a large deletion upstream of GNE in trans with the missense V727M, which causes the mono-allelic expression.
Gene Expression Analysis Showed 50% Reduced GNE Expression.
Cluster analysis of 274 NMD-associated genes (Table S2) from next generation sequencing-based transcriptome sequencing (RNA-seq) data showed separate clustering of the patient’s vastus lateralis (VL) muscle samples from the controls’ VL muscles (Supplementary Fig. S2). GNE expression was reduced by ~50% (Fig. 3A). A total of 89 NMD-associated genes were differentially expressed and several key extracellular matrix (ECM) genes, namely collagen fiber and laminin genes (COL6A1/A2/A3/12A1, LAMA2) were up-regulated (Table S3).
FIGURE 3.
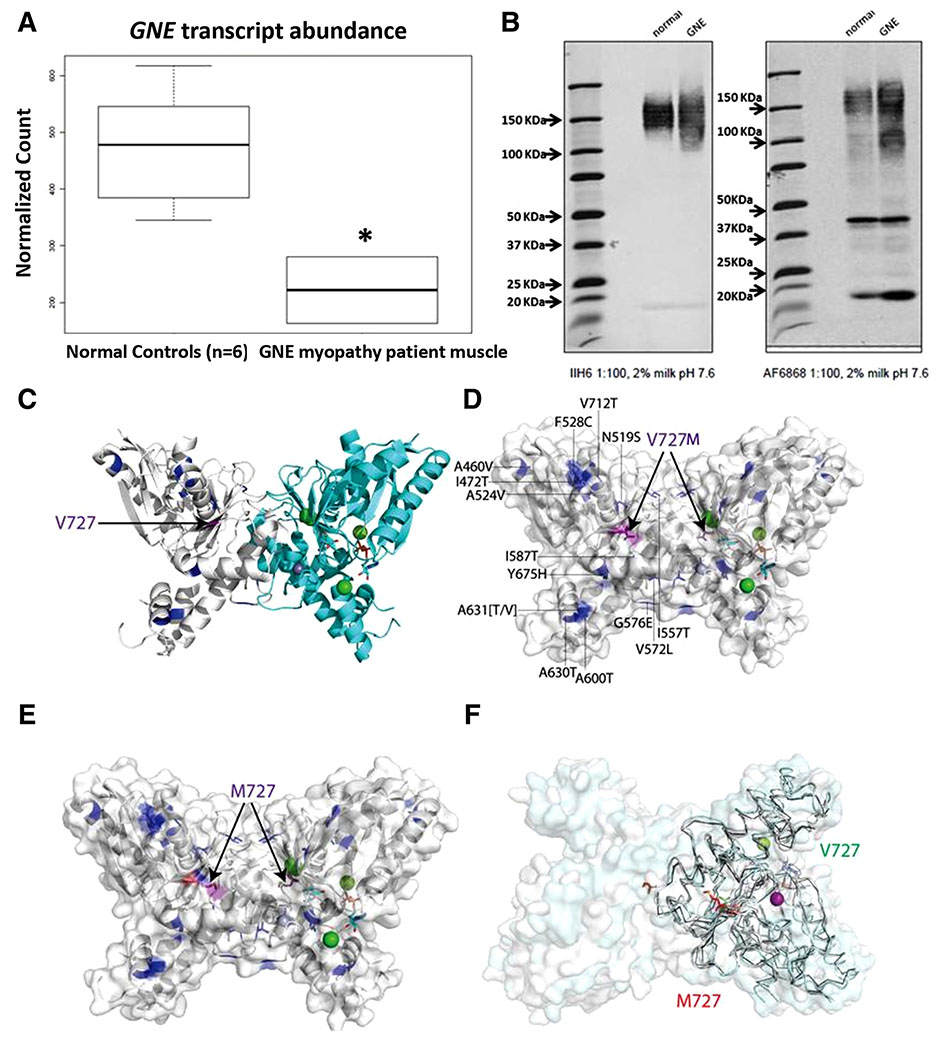
(A) Approximately 50% lower-expression (P < 0.05) of GNE in GNE myopathy patient muscle compared with that in 6 control normal muscle biopsies. (B) IIH6-antibody against glycosylated-α-DG shows hypoglycosylation (lighter signal, broader smear) of α-DG in patient muscle compared with control. AF6868 against core α-DG, β-DG shows different α-DG smearing patterns in patient muscle compared with control. Two predominant staining areas are 150 kDa and 100 kDa in patient sample. Roughly same β-DG-fragment expression at ~43 kDa is seen in both patient and control muscles. (C) HIBM-causing point mutations (blue, purple sticks) mapped onto GNE-N-acetylmannosamine-kinase-domain (PDB ID: 2YHY); N-acetylmannosamine-kinase-dimer backbone shown as cartoon. Protomers are colored white and cyan, associated divalent ions as spheres (chloride ions: green, zinc ion: lavender). Active site bound ADP and N-acetylmannosamine are shown as sticks on the right hand side GNE protomer. HIBM-causing residues are mapped on the left hand side GNE protomer. The V727M (V696M) mutation (purple sticks), other HIBM mutations (blue sticks). (D) Valine-727, and other HIBM mutations, are shown on both dimer subunits. (E) The Valine-727 sidechain is replaced by a Methionine residue on both dimer subunits and colored in Corey, Pauling, Koltun scheme (oxygen in red and sulfur in yellow). (F) The rotamer is changed upon replacing V727 (green sticks) by a methionine (red sticks) in MD simulations.
Gene Ontology Pathway Analysis.
Gene ontology-based gene set enrichment (GO-Pathway) analysis on the differentially-regulated 89 genes (Supplementary Fig. S3; Supplementary Table S4) identified major enriched “pathways,” “cellular compartments,” and “molecular functions.” The common biology is a predicted effect on of protein and lipid glycosylation affecting the cytoskeleton-intracellular matrix and ECM cross-talk through sarcolemmal proteins, important for the sarcomere integrity.
α-DG Hypo-glycosylation in Muscle Resembling Congenital Muscular Dystrophy.
Using IIH6 antibody,24 we detected significant hypo-glycosylation of α-DG in the patient skeletal muscle compared with normal control (Fig. 3B, left; band intensity values: 182,500 vs. 466,900) with a lighter and a broader smear from 110–180 kDa compared with a strong signal for normal muscle at 130–180 kDa. Using AF6868 antibody24 the normal muscle showed a band at 150 kDa and β-DG at ~43 kDa, similar to the patient muscle with equivalent β-DG fragment with the exception of a second smear at 90–100 kDa (Fig. 3B, right).
No Substantial Structural Change on MD Simulation of V727M on GNE Kinase-Domain
Mutation mapping and MD simulation suggests that all known missense variants in kinase domain are away from the active site, and there is no significant structural change due to only the V727M change in the kinase domain (Fig. 3C–F). But subtle fold changes can be observed (Fig. 3D,F) when V727 and M727 structures are overlaid. Subtle fold changes in concert with another variant can cause a substantial difference in functional output.
DISCUSSION
Our study provides important insights for molecular diagnostic approaches to understand the pathological and molecular nature of unusual myopathies. We report here a family having a patient with a novel upstream promoter-region large deletion in the GNE gene, which abolishes expression of the respective allele. Previous reports showed that patients with compound heterozygous variants in both epimerase and kinase GNE mdomains manifest more severe phenotypes than those with both variants in 1 domain,25 suggesting that mild pathogenicity of missense variants in each domain needed for more disease severity. Although V727M pathogenicity is uncertain given its relatively high prevalence in South Asians, the most parsimonious conclusion given many other similar reports is that this compound heterozygous state contributes to the pathology.
Generally, inflammation is not associated with HIBM, in which quadriceps muscles are relatively spared of any rimmed vacuolar pathology compared with other muscle types, as seen in studies of smaller numbers of individuals with inclusion-body myositis (IBM)22 and in larger study cohorts.25 The presence of both inflammation and rimmed vacuoles in the quadriceps muscle of this patient is not characteristic of either primary inflammatory or rimmed vacuolar myopathies. The second causal variant was inferred from the combination of aCGH and RNA-seq that definitively diagnosed the case as GNE-related myopathy, and led to identification of multiple gene expression perturbations. This study shows the involvement of quadriceps muscle directly with both rimmed vacuoles and inflammation, unlike previous reports of individual cases and cohorts,22,23,26,27 enhancing the molecular-pathological spectrum of GNE-myopathy that is important to understand for patient stratification in clinical trials.
Previously, Zhu et al.23 showed that large promoter region deletions in GNE are common in already clinically diagnosed GNE-myopathy patients, and Garland et al.22 showed that a combination of such deletions and a V727M missense variant causes wa more severe reduction in GNE expression than the combination of V727M and another missense variant. Here, we show that such variant combinations are associated with unique GNE-related myopathy pathology and the clinical/molecular diagnostic hurdles faced. Consequently, it is likely that the combination of reduced transcription due to promoter region deletion and possible V727M-induced subtle altered kinase activity is required for the unique HIBM-like symptoms. Further functional studies are needed to classify the pathogenicity of V727M.
As per ACMG guidelines,5 because the deletion variant causes a 50% reduction in GNE gene expression, we clinically classify the variant as “pathogenic.” This potentially results in a significant reduction in key sarcolemmal protein α-DG glycosylation and aberrant expression of core α-DG and β-DG (Fig. 3B), which along with altered expression of genes and pathways found in GO-pathway analysis could explain the muscle wasting and weakness. Disrupted glycan metabolism and glycosyl transferase likely explains hypoglycosylation of α-DG, potentially causing de-regulation of the actin cytoskeleton, cell cortex, sarcolemma, T-tubule, and ECM (Supplementary Fig. S3).
The nature of reduced α-DG glycosylation and overexpression of β-DG fragment is a hallmark of congenital muscular dystrophies (CMDs),28 found also by Huizing et al.29 in GNE-related HIBM. Our study contributes to an emerging literature suggesting that GNE-related myopathy shares molecular signatures of “dystroglycanopathy” similar to CMDs, with glycosylation-defect-related muscle wasting and weakness as the primary cause and an inflammatory response as a secondary effect. The muscle structural degeneration in HIBM resembling CMD is possibly due to inability of the sarcolemmal machinery to protect the sarcomere from the load of ECM proteome dysregulation. Overall, functional assays suggest a GNE-associated inherent core muscle glycosylation defect as the cause for this unusual GNE-related myopathy.
Importantly, this study shows the power of using aCGH, RNA-seq and focused functional assays on target muscle tissue following clinical/pathological clues for improving diagnostic efficiency and timeliness in the evaluation of undiagnosed myopathies. We believe that this approach will be broadly applicable to the diagnosis of NMDs, and will thus harness the advances in clinical genomics and developing precision therapies.
Supplementary Material
Acknowledgments
This work was also supported by Paul D. Wellstone Muscular Dystrophy Cooperative Research Center Grant (1U54NS053672)
Funding: This work was supported by the Muscular Dystrophy Association grant MDA578400 to S.C. and grant MDA418496 to M.H.
We thank the reported patient and his family, and the normal individuals from whom normal control muscle biopsies were used, for participating in this study. This work was also supported by Paul D. Wellstone Muscular Dystrophy Cooperative Research Center Grant (1U54NS053672). K.P.C. is an investigator of the Howard Hughes Medical Institute. Ethical Publication Statement: The authors confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. Part of this work has been presented before in the following conferences: American Society of Human Genetics meeting 2016 Vancouver, American Academy of Neurology conference 2017, in Boston, and American College of Medical Genetics and Genomics Annual Meeting 2017, in Phoenix. Data Deposition and Access: Whole-exome sequencing data are not publicly available because consent could not be obtained. The deletion variant associated with the phenotype has been submitted to ClinVar under accession number SCV000599234 (https://www.ncbi.nlm.nih.gov/clinvar/) and will be available after publication.
Footnotes
Additional supporting information may be found in the online version of this article.
Conflicts of Interest: None of the authors has any conflict of interest to disclose.
REFERENCES
- 1.Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med 2017;9:eaa15209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lappalainen T, Sammeth M, Friedlander MR, t Hoen PA, Monlong J, Rivas MA, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013;501:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakravorty S, Hegde M. Gene and variant annotation for Mendelian disorders in the era of advanced sequencing technologies. Annu Rev Genomics Hum Genet 2017;18:229–256. [DOI] [PubMed] [Google Scholar]
- 4.Chakravorty S, Hegde M. Clinical utility of transcriptome sequencing: toward a better diagnosis for mendelian disorders. Clin Chem 2018;64:882–884. [DOI] [PubMed] [Google Scholar]
- 5.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet 2001;29:83–87. [DOI] [PubMed] [Google Scholar]
- 7.Huizing M, Krasnewich DM. Hereditary inclusion body myopathy: a decade of progress. Biochim Biophys Acta 2009;1792:881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huizing M, Malicdan MCV, Krasnewich DM, Manoli I, Carrillo-Carrasco N. GNE myopathy In: Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson KM, Mitchell G, editors. The online metabolic and molecular bases of inherited disease. New York, NY: The McGraw-Hill Companies, Inc; 2014. [Google Scholar]
- 9.Varki A Selectins and other mammalian sialic acid-binding lectins. Curr Opin Cell Biol 1992;4:257–266. [DOI] [PubMed] [Google Scholar]
- 10.Varki A Diversity in the sialic acids. Glycobiology 1992;2:25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varki A Sialic acids as ligands in recognition phenomena. FASEB J 1997;11:248–255. [DOI] [PubMed] [Google Scholar]
- 12.Nishino I, Malicdan MC, Murayama K, Nonaka I, Hayashi YK, Noguchi S. Molecular pathomechanism of distal myopathy with rimmed vacuoles. Acta Myol 2005;24:80–83. [PubMed] [Google Scholar]
- 13.Malicdan MC, Noguchi S, Nonaka I, Hayashi YK, Nishino I. A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet 2007;16:2669–2682. [DOI] [PubMed] [Google Scholar]
- 14.Berg AT, Chakravorty S, Koh S, Grinspan ZM, Shellhaas RA, Saneto RP, et al. Why West? Comparisons of clinical, genetic and molecular features of infants with and without spasms. PLoS One 2018;13:e0193599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voermans NC, Guillard M, Doedee R, Lammens M, Huizing M, Padberg GW, et al. Clinical features, lectin staining, and a novel GNE frameshift mutation in hereditary inclusion body myopathy. Clin Neuropathol 2010;29:71–77. [PMC free article] [PubMed] [Google Scholar]
- 16.Nalini A, Gayathri N, Nishino I, Hayashi YK. GNE myopathy in India. Neurol India 2013;61:371–374. [DOI] [PubMed] [Google Scholar]
- 17.Liewluck T, Pho-Iam T, Limwongse C, Thongnoppakhun W, Boonyapisit K, Raksadawan N, et al. Mutation analysis of the GNE gene in distal myopathy with rimmed vacuoles (DMRV) patients in Thailand. Muscle Nerve 2006;34:775–778. [DOI] [PubMed] [Google Scholar]
- 18.Chaouch A, Brennan KM, Hudson J, Longman C, McConville J, Morrison PJ, et al. Two recurrent mutations are associated with GNE myopathy in the North of Britain. J Neurol Neurosurg Psychiatry 2014;85:1359–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishino I, Carrillo-Carrasco N, Argov Z. GNE myopathy: current update and future therapy. J Neurol Neurosurg Psychiatry 2015;86:385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Celeste FV, Vilboux T, Ciccone C, de Dios JK, Malicdan MC, Leoyklang P, et al. Mutation update for GNE gene variants associated with GNE myopathy. Hum Mutat 2014;35:915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garland J, Stephen J, Class B, Gruber A, Ciccone C, Poliak A, et al. Identification of an Alu element-mediated deletion in the promoter region of GNE in siblings with GNE myopathy. Mol Genet Genomic Med 2017;5:410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu W, Mitsuhashi S, Yonekawa T, Noguchi S, Huei JC, Nalini A, et al. Missing genetic variations in GNE myopathy: rearrangement hotspots encompassing 5’UTR and founder allele. J Hum Genet 2017;62: 159–166. [DOI] [PubMed] [Google Scholar]
- 24.Briggs DC, Yoshida-Moriguchi T, Zheng T, Venzke D, Anderson ME, Strazzulli A, et al. Structural basis of laminin binding to the LARGE glycans on dystroglycan. Nat Chem Biol 2016;12:810–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pogoryelova O, Cammish P, Mansbach H, Argov Z, Nishino I, Skrinar A, et al. Phenotypic stratification and genotype-phenotype cor-relation in a heterogeneous, international cohort of GNE myopathy patients: first report from the GNE myopathy Disease Monitoring Program, registry portion. Neuromuscul Disord 2018;28:158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yabe I, Higashi T, Kikuchi S, Sasaki H, Fukazawa T, Yoshida K, et al. GNE mutations causing distal myopathy with rimmed vacuoles with inflammation. Neurology 2003;61:384–386. [DOI] [PubMed] [Google Scholar]
- 27.Argov Z, Eisenberg I, Grabov-Nardini G, Sadeh M, Wirguin I, Soffer D, et al. Hereditary inclusion body myopathy: the Middle Eastern genetic cluster. Neurology 2003;60:1519–1523. [DOI] [PubMed] [Google Scholar]
- 28.Mendell JR, Boue DR, Martin PT. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol 2006;9:427–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huizing M, Rakocevic G, Sparks SE, Mamali I, Shatunov A, Goldfarb L, et al. Hypoglycosylation of alpha-dystroglycan in patients with hereditary IBM due to GNE mutations. Mol Genet Metab 2004;81:196–202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.