

Abstract
Tuberculosis is an infectious disease caused by the bacterium Mycobacterium tuberculosis (Mtb). Mtb protein tyrosine phosphatase B (mPTPB) is a virulence factor required for Mtb survival in host macrophages. Consequently, mPTPB represents an exciting target for tuberculosis treatment. Here, we identified N-phenyl oxamic acid as a highly potent and selective monoacid-based phosphotyrosine mimetic for mPTPB inhibition. SAR studies on the initial hit, compound 4 (IC50 = 257 nM), resulted in several highly potent inhibitors with IC50 values lower than 20 nM for mPTPB. Among them, compound 4t showed a Ki of 2.7 nM for mPTPB with over 4500-fold preference over 25 mammalian PTPs. Kinetic, molecular docking, and site-directed mutagenesis analyses confirmed these compounds as active site-directed reversible inhibitors of mPTPB. These inhibitors can reverse the altered host cell immune responses induced by the bacterial phosphatase. Furthermore, the inhibitors possess molecular weights <400 Da, log D7.4 < 2.5, topological polar surface area < 75, ligand efficiency > 0.43, and good aqueous solubility and metabolic stability, thus offering excellent starting points for further therapeutic development.
Graphical Abstract

INTRODUCTION
Protein tyrosine phosphatases (PTPs) counterbalance the activity of protein tyrosine kinases, and consequently, these two enzyme families have a central role in determining the status of protein tyrosine phosphorylation inside the cell.1 The phosphorylation of a protein can alter its enzymatic activity, subcellular localization, macromolecular interactions, stability, and ultimately control normal cellular homeostasis and disease processes.2 Dysregulation of PTPs is associated with a multitude of diseases, and many members of the PTP family have been recognized as potential therapeutic targets.3 Potent and selective inhibitors of PTPs are essential for interrogating the biological function of the PTPs and they may ultimately be developed into valuable therapeutics in the treatment of several pathological human conditions, including cancer, autoimmune disorders, and infectious diseases.4-7
Tuberculosis (TB), an infectious disease caused by the bacteria called Mycobacterium tuberculosis (Mtb),8 is one of the top 10 causes of human death worldwide.9 In 2017, an estimated 10 million people became ill with TB and 1.6 million people died of TB (including 0.3 million people with HIV-associated TB). It is also estimated that about one-quarter of the world’s population has latent TB (people have been infected by TB but are not ill (yet) with the disease). The major obstacle in the treatment of TB is antibiotic resistance. Some of the common causes of drug resistance are lack of compliance from patients, inadequate diagnosis and/or drug regimen, mutation/gene transfer, irregular drug supply, and poor drug quality with low bioavailability.10 The rapid emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) TB demands the development of new therapeutic agents with novel molecular targets and mechanisms of actions.11 Antivirulence strategies are now emerging as an alternative therapeutic approach to combat antibiotic resistance in a number of microbial infections including TB.12-14 Mtb protein tyrosine phosphatase B (mPTPB) is a virulence factor that is secreted into the host macrophages.15 mPTPB is critical for the survival of Mtb and persistence of the infection inside the macrophages of animal models. Deletion of mPTPB has no effect on the growth of the pathogen itself but reduces the intracellular survival of Mtb in infected macrophages and reduces the bacterial load in a guinea pig model of TB infection.16,17 As mPTPB is secreted by Mtb into the cytoplasm of macrophages, we previously elucidated the role of mPTPB in host cell biology using Raw264.7 mouse macrophages stably expressing mPTPB.18 Treatment of the Raw264.7 cells with IFN-γ induces the activation of the MAP kinases ERK1/2, p38, and JNK. Moreover, IFN-γ stimulation also leads to increased production of interleukin-6 (IL-6), a proinflammatory cytokine secreted by the macrophage that is involved in upregulating microbicidal activity and initiates the immune response to Mtb. Expression of mPTPB in activated macrophages subverts the innate immune responses by blocking the ERK1/2 and p38 kinase-mediated IL-6 production and prevents macrophage cell death through activation of the AKT pathway, both of which are essential for Mtb to overcome the host defense mechanisms.18 Inhibition of mPTPB with small-molecule inhibitors can reverse the altered host immune responses induced by the bacterial phosphatase and impair the survival of MDR-TB in human macrophages and reduce infection burden in guinea pig models.18-23 Selective inhibition of mPTPB also increases the intracellular killing efficacy of first-line antibiotics rifampicin and isoniazid, indicating their suitability for combination therapies.19,20 These results provide important proof-of-concept for the notion that specific inhibitors of mPTPB may serve as effective anti-TB agents. Targeting the virulent mPTPB is expected to specifically undermine pathogen–host interactions without adverse effects on bacterial growth, therefore exerting less selective pressure for drug resistance.24 More importantly, the lack of a human orthologue (minimal side effects on the host) makes mPTPB an attractive drug target for specific treatment of TB. Moreover, mPTPB inhibitors function within the host macrophage cytosol and mechanistically do not overlap with the existing anti-TB agents. Consequently, mPTPB inhibitors, when used together with the current anti-TB agents, may reduce the lengthy treatment of >6 months. Finally, because mPTPB acts outside of the bacterium, mPTPB inhibitors are not required to cross the thick, hydrophobic, and waxy mycobacterial cell wall, which presents a significant challenge for the efficient delivery of traditional antibacterial agents.25,26
The design and synthesis of inhibitors for PTPs with optimal potency, selectivity, and pharmacological properties remain a challenging endeavor mostly because of the highly conserved and positively charged nature of PTP active sites. To enable the engagement with the positively charged active-site pockets, numerous negatively charged functional group-containing compounds such as carboxylic acids, salicylic acids, sulfamic acids, α-sulfophenylacetic amide (SPAA), and 2-oxalylamino benzoic acid (OBA) have been reported as nonhydrolyzable phosphotyrosine (pTyr) mimetics for the inhibition of various members of the PTP family.5,27,28 OBA was reported as a competitive, reversible inhibitor of protein-tyrosine phosphatase 1B (PTP1B) and cocrystal structures of this chemical class were also described (Figure 1).29 OBA was further optimized by structure-based design to generate several highly potent PTP1B inhibitors, such as compounds II, III, and IV (Figure 1).30-32 Although potent (Ki values in the nM range), the high polar surface area (PSA), molecular mass, and charge of these compounds make further optimization difficult.33,34 Hartshorn and co-workers identified compound V as a PTP1B inhibitor using a high-throughput X-ray crystallography technique.35 This compound showed an IC50 of 86 μM against PTP1B, and no selectivity studies or further modifications of V were reported by the authors. Grundner et al. reported (oxalylamino methylene)-thiophene sulfonamide (OMTS, VI), as a competitive inhibitor for mPTPB. Compound VI showed an IC50 of 440 ± 50 nM and over 60-fold selectivity for mPTPB over six human PTPs, although no cellular activity data were provided for VI.36
Figure 1.
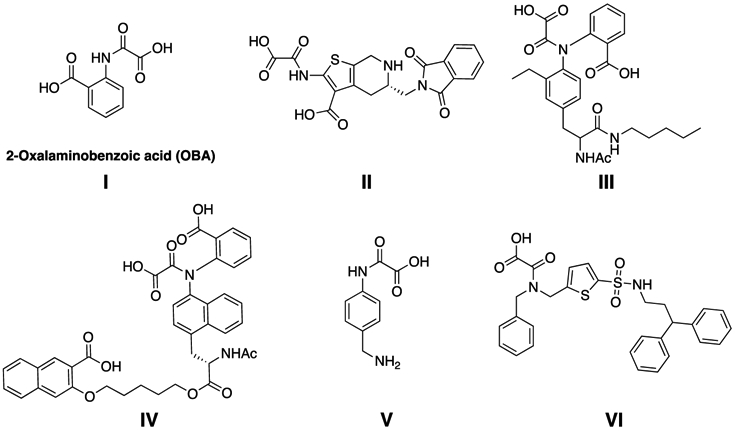
Previously reported oxamic acid-based pTyr mimetics for PTP inhibition.
We envisioned that selective and cell-permeable PTP active site-directed inhibitors should possess smaller molecular weight (<400 Da) and no more than one negative charge. To this end, we designed singly charged N-aryl oxamic acid derivatives as pTyr mimetics for active site-directed inhibition of mPTPB. This research led to the development of several highly potent and selective N-aryl oxamic acid analogues for mPTPB inhibition. Among them, inhibitor 4t exhibited a Ki of 2.7 nM for mPTPB and over 4500-fold selectivity over a panel of 25 mammalian PTPs. Importantly, the inhibitors reported here show molecular weights < 400 Da, log D7.4 < 2.5, ligand efficiency (LE) > 0.43, and good aqueous solubility and microsomal stability. In addition, the inhibitor efficiently blocks mPTPB-mediated signaling in mouse macrophages, further validating the concept that small-molecule mPTPB inhibitors may be developed to treat tuberculosis. The work further demonstrates that it is feasible to obtain active site-directed, polar, but cell-permeable PTP inhibitors.
RESULTS AND DISCUSSION
Design and Synthesis of mPTPB Inhibitors.
To identify the best N-aryl oxamic acid-based pTyr mimetics for mPTPB, we first prepared phenyl-, biphenyl-, phenylethynyl-, alkynyl phenyl-, and quinoline-containing oxamic acids (Figure 2). Compounds 1–8 were prepared by a reaction between the corresponding aryl amine and methyl chlorooxoacetate in a DIPEA/tetrahydrofuran (THF) reaction followed by mild hydrolysis using 1 N KOH/THF (1:1 v/v) solution. Our efforts to synthesize other bicyclic (naphthyl, benzoxazole, and benzothiazole)-based oxamic acids resulted in compounds with poor solubility profiles likely because of a higher degree of molecular planarity.37 Half-inhibitory concentration values (IC50) of 1–8 were measured in a p-nitrophenyl phosphate (pNPP) assay against mPTPB, PTP1B, and SHP2 phosphatases.
Figure 2.
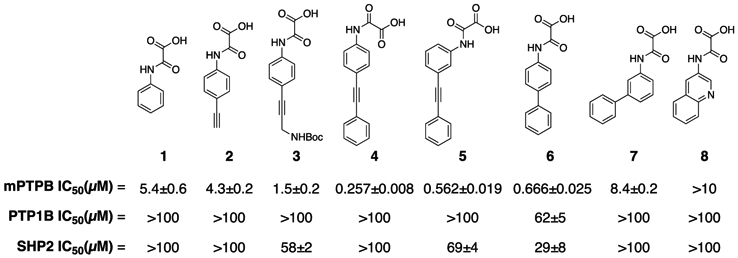
IC50 values of various oxamic acid derivatives against mPTPB, PTP1B, and SHP2.
As shown in Figure 2, the phenylethynyl-containing analogues (4 and 5) demonstrated superior inhibition and selectivity for mPTPB in comparison with the biphenyl, alkyl, or bicyclic analogues (2, 3, 6, 7, and 8). Importantly, para derivative 4 showed twofold more potency compared to the meta derivative 5, and a similar trend was observed in the case of compounds 6 and 7. These results suggest the preference for para substitution over meta for mPTPB inhibition. Consequently, we prepared several analogues of compound 4 to further improve its activity against mPTPB. Based on the commercial availability of the starting materials (aryl halides and aryl alkynes), we used two different synthetic strategies to prepare analogues of 4. In the first strategy, we treated 4-ethynyl or 3-ethynyl aniline with methyl chlorooxoacetate in a DIPEA/THF reaction to yield the corresponding oxamate ester. Subsequently, the oxamate ester was treated with various aryl halides in a Sonogashira coupling reaction using Pd(PPh3)Cl2, CuI, Na2CO3, and N,N-dimethylformamide (DMF) to provide phenylethynyl oxamate esters in good yields.38 A mild hydrolysis of these esters using 1 N KOH/THF furnished the final compounds in excellent yields (Scheme 1A). In the second strategy, we treated 4-iodoaniline with methyl chlorooxoacetate in a DIPEA/THF reaction to yield methyl 2-((4-iodophenyl)amino)-2-oxoacetate (Scheme 1B). The Sonogashira coupling reaction of the ester with alkynes followed by mild hydrolysis provided final compounds in excellent yields.
Scheme 1.
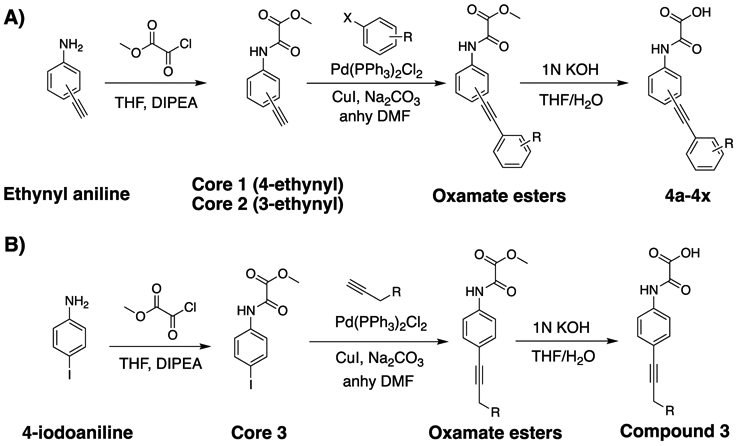
Strategies for the Synthesis of mPTPB Inhibitors. (A) Synthesis of Phenylethynyl Oxamic Acids from Ethynylanilines. (B) Synthesis of Phenylethynyl Oxamic Acids from 4-Iodoaniline
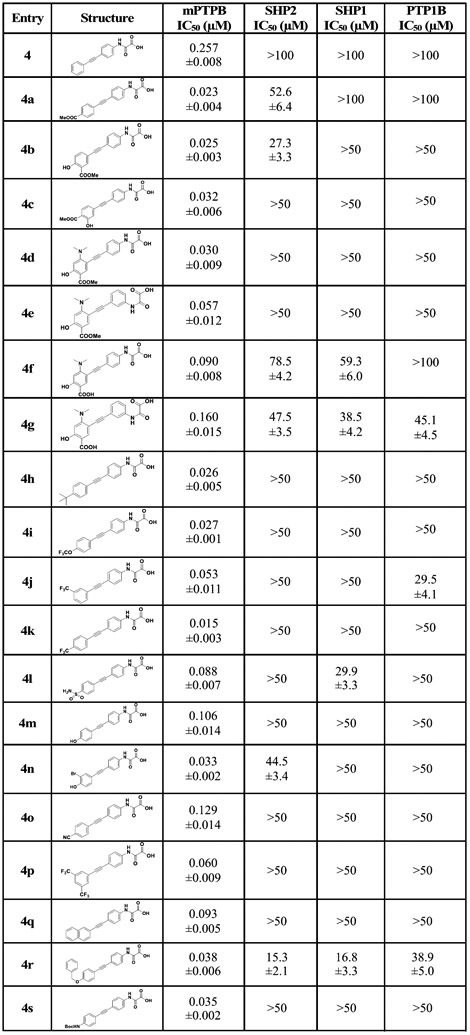
We first introduced several substituents that vary in size and polarity on the distal phenyl ring of 4, to improve the binding interactions with mPTPB active-site residues. As shown in Table 1, the introduction of substituents such as COOMe, OH, and NMe2 (4a–4d) provided inhibitors with over 10-fold improved binding affinity compared to the parent compound 4. The double- and triple-substituted compounds showed similar or less activity against mPTPB compared to the single-substituted analogues. Importantly, compounds 4f and 4g with an ionizable functional group COOH demonstrated ~three-fold decrease in mPTPB binding affinity compared to 4d and 4e with nonionizable functional groups. Compounds 4e and 4g with aryl alkyne substitution meta to the oxalamide group showed approximately twofold less activity compared to their para counterparts, 4d and 4f, respectively. This is consistent with the results observed in the case of compounds 4 and 5 (Figure 2). Interestingly, changing the position of CF3 from meta (4j) to para (4k) improved the IC50 by 3.5-fold. In addition, 4p with two CF3 groups showed no improvement in affinity compared to 4j with only one CF3. Thus, we evaluated several substituents only at the para position. Replacing the CF3 group with tBu, OCF3, or NHBoc resulted in twofold less-potent inhibitors (4h, 4i, and 4s). Inhibitors with polar functional groups such as OH, COOH, SO2NH2, and CN (4f, 4l, 4m, 4n, and 4o) showed weak activity against mPTPB (Table 1). Compounds with naphthalene (4q) or benzyloxybenzene (4r) in place of the distal phenyl ring in 4 displayed poor solubility and selectivity (Table 1).
Table 1.
IC50 Values of Compounds 4, 4a–4s against a Panel of PTPs
 |
After obtaining the potent inhibitor 4k with an IC50 of 15 nM for mPTPB, we next sought to further improve its activity through the installation of additional substituents in the distal phenyl ring. Halogens, mainly the lighter ones fluorine and chlorine, are widely used in medicinal chemistry.39 Compounds containing chlorine, bromine, or iodine can form interactions of the type R─X⋯Y─R (halogen bond), where X is the halogen (acts as a Lewis acid) and Y can be any electron-donor moiety. In protein–ligand environments, halogen bonds can be formed between a halogen atom in the ligand and any nearby Lewis base in the protein, such as backbone carbonyl oxygen.40 Additionally, halogen bonds can be formed with groups present in the side chains, such as −OH (Ser, Thr, and Tyr), −COOH (Asp and Glu), sulfur (Cys and Met), nitrogen (His), and the π surfaces (Tyr, Phe, Try, and His).41 Moreover, halogens such as fluorine are known to alter the physicochemical properties of compounds and increase the metabolic stability of drug molecules.42,43 To this end, we synthesized 4t, 4u, and 4v with chlorine, bromine, and fluorine atoms meta to the CF3 in 4k, respectively. This resulted in compounds with improved affinity (Table 2). Analogues 4t and 4u demonstrated ~twofold improvement in binding compared to 4k, which also represents a 44-fold increase in activity compared to parent compound 4 (Table 1). Although both 4t and 4u showed similar affinity toward mPTPB, 4t showed superior aqueous solubility, Log D, c Log P, and selectivity (Table 7). On the other hand, nitrile-containing analogue, 4w demonstrated a decrease in potency compared to 4k.
Table 2.
IC50 Values of 4k Analogues against a Panel of PTPs
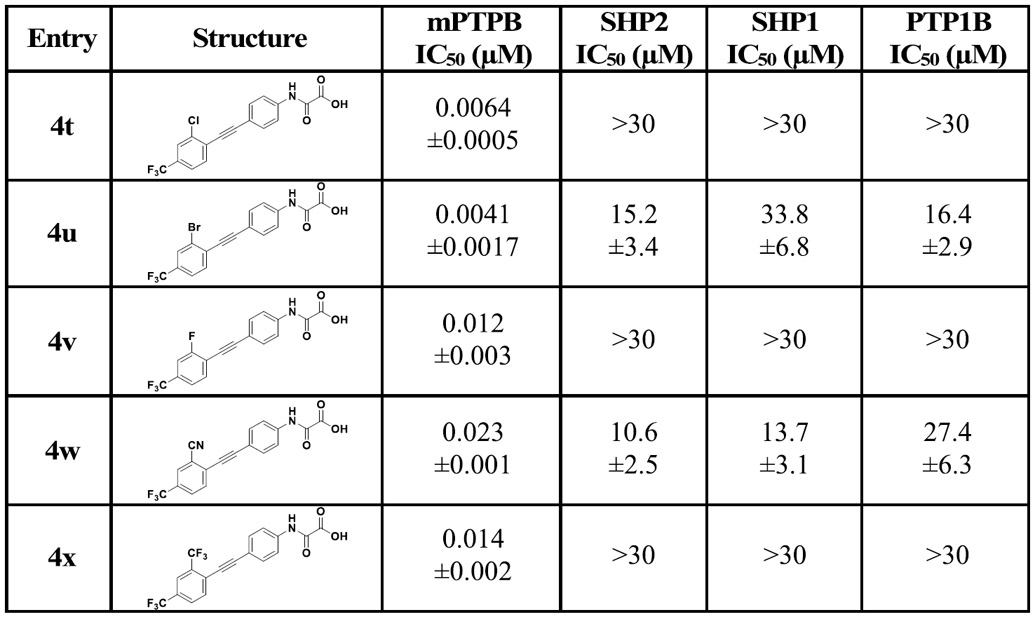 |
Table 7.
Calculated and Determined Physicochemical Properties of the Selected Compounds
| compound | MWa | tPSAb | c Log Pc | Log Dd | aq. sol (μM)e | LEf | LLEg | microsome stabilityh |
|---|---|---|---|---|---|---|---|---|
| 4b | 339 | 112.9 | 1.96 | 1.94 | 38.6 | 0.42 | 5.64 | NT |
| 4h | 321 | 66.4 | 3.50 | 2.32 | 18.4 | 0.43 | 5.26 | 32 ± 2 |
| 4k | 333 | 66.4 | 2.69 | 1.97 | 22.4 | 0.44 | 5.85 | 43 ± 3 |
| 4t | 368 | 66.4 | 3.29 | 1.84 | 67.3 | 0.45 | 6.31 | 52 ± 4 |
| 4u | 412 | 66.4 | 3.42 | 2.28 | 24.6 | 0.46 | 6.12 | 55 ± 1 |
| 4v | 351 | 66.4 | 2.78 | 1.81 | 33.5 | 0.43 | 6.11 | 64 ± 3 |
Molecular weight.
Topological PSA (A2).
Molinspiration calculated Log P.
Log D was experimentally determined using the shake flask method (n-octanol and pH 7.4 PBS).
Kinetic solubilities measured in pH 7.4 PBS buffer.
LE calculated using the equation: LE = −1.37 Å × log(potency(M))/heavy atom count.
Lipophilic LE calculated using the equation: LLE = −log(potency (M)) − Log D.
Mouse liver microsomes, half-life in minutes (reactions were run in duplicate). NT = not tested.
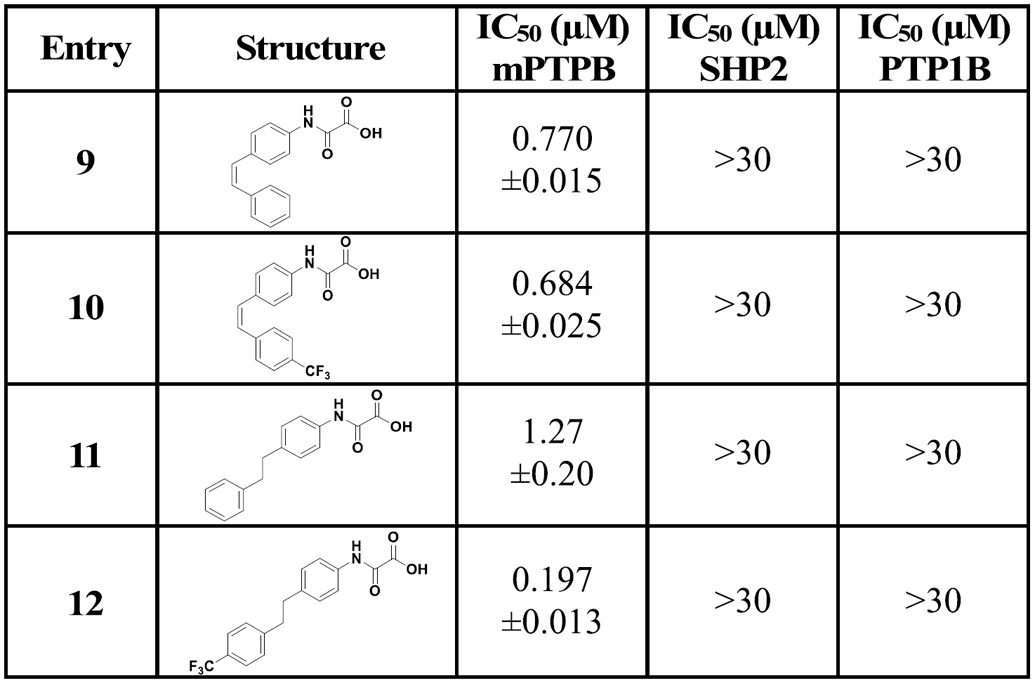
Finally, to evaluate the importance of the triple bond, we prepared compounds with a double bond or a single bond using the hydrogenation reaction (Table 3). To synthesize compounds 9 and 10, we used the Lindlar catalyst (5% Pd/BaSO4, quinoline, and H2 gas). On the other hand, we used 10% Pd/C, methanol, and H2 gas to synthesize the single bond-containing analogues 11 and 12 (Scheme 2). As expected, the potency of the double bond- or single bond-containing derivatives dramatically decreased. Compounds 9 and 10 showed 3- and 45-fold less potency compared to their corresponding alkyne analogues 4 and 4k, respectively. Similarly, compounds 11 and 12 showed 5- and 13-fold less activity compared to 4 and 4k, respectively. These observations indicate the importance of rigidity and directionality of the triple-bonded substitutions at the para position of the N-aryloxamic acid core for optimal interaction with mPTPB.
Table 3.
IC50 Values of Alkene- and Alkane-Containing Analogues of 4 and 4k against mPTPB, SHP2, and PTP1B
 |
Scheme 2.

Synthesis of Alkene- and Alkane-Based Derivatives of 4 and 4k
Inhibitor Selectivity Studies.
In order to enable pharmacological assessment of mPTPB as a novel anti-TB target, the specificity of the mPTPB inhibitor is of utmost importance. Compounds 4g, 4k, and 4t were subjected to specificity profiling for mPTPB versus a large panel of PTPs, including the bacterial PTPs, mycobacterium protein tyrosine phosphatase A (mPTPA) (the only other PTP in the Mtb genome)8 and YopH from Yersinia; the nonreceptor PTPs, SHP1, SHP2, PTP1B, TC-PTP, MEG2, HePTP, STEP, FAP1, and LYP; the receptor-like PTPs, CD45, PTPσ, PTPα, PTPμ, PTPγ, and PTPε; and the dual-specificity phosphatases Laforin, VHR, MKP3, MKP5, and Cdc14A. Compound 4g was subjected to specificity profiling for mPTPB versus a panel of over 25 PTPs. This compound showed no inhibition against the majority of PTPs even at 50 μM, except SHP1, SHP2, PTP1B, LMWPTP, and PTPβ (Table 4). On the other hand, when screened at 30 μM, compounds 4k and 4t showed no inhibition against PTPs tested, suggesting selectivity greater than 2000- and 4500-fold for mPTPB over other PTPs, respectively.
Table 4.
Specificity of 4g, 4k, and 4t for mPTPB over a Large Panel of PTPs
| PTP | 4g IC50 (∝M) | 4k IC50 (∝M) | 4t IC50 (∝M) |
|---|---|---|---|
| mPTPB | 0.160 | 0.015 | 0.0064 |
| mPTPA | >50 | >50 | >30 |
| SHP1 | 38.5 | >50 | >30 |
| SHP2 | 47.5 | >50 | >30 |
| PTP1B | 45.1 | >50 | >30 |
| TC-PTP | >50 | >50 | >30 |
| LYP | >50 | >50 | >30 |
| HePTP | >50 | >50 | >30 |
| FAP-1 | >50 | >30 | >30 |
| DEP-1 | >50 | >30 | >30 |
| Laforin | >50 | >30 | >30 |
| PTP-MEG2 | >50 | >30 | >30 |
| MKP3 | >50 | >30 | >30 |
| MKP5 | >50 | >30 | >30 |
| YopH | >50 | >30 | >30 |
| LMPTP-1 | 34.5 | >30 | >30 |
| PTP-PEST | >50 | >30 | >30 |
| CD45 | >50 | >30 | >30 |
| CDC14A | >50 | >30 | >30 |
| STEP | >50 | >30 | >30 |
| VHR | >50 | >30 | >30 |
| PTPσ | >50 | >30 | >30 |
| PTPα | >50 | >30 | >30 |
| PTPβ | 11.5 | >30 | >30 |
| PTP∝ | >50 | >30 | >30 |
| PTPγ | >50 | >30 | >30 |
| PTPε | >50 | >30 | >30 |
The mode of mPTPB inhibition by these compounds was determined by steady-state kinetic analyses, varying the substrate pNPP and inhibitor concentrations. Lineweaver–Burk plots of these inhibitors revealed them as classic competitive inhibitors, affecting the apparent Km value, while Vmax was unchanged (Figure 3). This observation is consistent with the expectation that these compounds bind to the active site of mPTPB because of the pTyr mimetic properties of the N-aryl oxamic acid moiety. Compounds 4t, 4u, and 4v showed inhibition constant (Ki) values of 2.7 ± 0.2, 1.2 ± 0.1, and 4.9 ± 0.6 nM, respectively. To rule out the possibility of promiscuous inhibition, we conducted IC50 experiments for 4p and 4v with and without 0.01% (v/v) of Triton X-100 in the buffer and time-dependent inhibition (enzyme and inhibitor were preincubated for 30 min prior to IC50 determination).44,45 These experiments showed no significant difference in the IC50 values of the inhibitors tested, suggesting them as valid active site-directed inhibitors for mPTPB.
Figure 3.

Compound 4t is a reversible and competitive inhibitor of mPTPB with pNPP as a substrate. Lineweaver–Burk plot for 4t-mediated mPTPB inhibition. Compound 4t concentrations were 0 (●), 10 (○), and 20 (▼) nM, respectively. The Ki value of 2.7 ± 0.2 nM was determined from three independent measurements.
Molecular Docking Studies.
To understand the potential interactions between these inhibitors and mPTPB, we conducted molecular docking studies using Glide.46 The active site structures of PTPs are positively charged, which facilitates their interaction with negatively charged molecules, such as pTyr mimetics with high affinity (Figure 4A). All PTPs share a conserved active-site signature motif of HCX5R, in the catalytic domain of about 240 amino acids. The phosphate-binding loop (P-loop) harbors the catalytic cysteine (Cys160 in mPTPB) within the HCX5R motif. The P-loop sequence of HCFAGKDR is unique to mPTPB and differentiates this enzyme from other members of the PTP family. A comparison of P-loop residues of mPTPB with various members of the PTP superfamily is shown in Figure 4B,C.
Figure 4.

Comparison of structural features of mPTPB with other human PTPs. (A) Electrostatic potential is calculated using APBS and is mapped onto the molecular surface of the mPTPB structure from the PDB code: 2OZ5 (using PyMOL).64 The active site of mPTPB is highly positively charged (blue region). (B) Comparison of P-loop residues of mPTPB with various other members of the PTP family. (C) Overlay of P-loop residues of mPTPB and human PTP1B. mPTPB residues were colored in red (PDB code: 2OZ5) and human PTP1B residues were colored in green (PDB code: 1XBO).
Compared to its mammalian PTP counterparts, mPTPB has a relatively deeper and broader active site, which is consistent with its additional phosphoinositide activity.47 Based on the docking model, the polar oxamic acid head group occupies the P-loop region (blue, Figure 5A) with various polar interactions and the hydrophobic distal phenyl ring bearing CF3 fills the hydrophobic region (red, Figure 5A). As predicted by the modeling work, inhibitor 4t binds to the active-site pocket of mPTPB and makes key hydrogen-bonding interactions with P-loop residues, Phe161, Ala162, Lys164, Asp165, and Arg166 (Figure 5B). The oxalamide group forms an intricate network of hydrogen bonds to Asp165, Arg166, and the backbone of Phe161, Gly-163, and Asp165. In mPTPB, Phe161 is adjacent to the catalytic cysteine making hydrophobic contacts with the phenyl ring in 4t, whereas the human PTPs such as PTP1B, SHP1, SHP2, TC-PTP, and HePTP have a Ser in this position. Most importantly, three of the P-loop amino acids Phe161, Lys164, and Asp165 that make H-bonding interactions with 4t are not present in other mammalian PTPs (Figure 4B).36 The internal alkyne provides the required space between the two aromatic rings, which allows the distal benzene ring to make hydrophobic interactions with Phe98. In addition, the fluorine atoms in CF3 of 4t are making favorable interactions with a hydrophobic pocket formed by residues Ile203, Met206, and Ile207. Hence, the high affinity of 4k, 4t, 4u, and 4v for mPTPB is likely attributed to the proposed hydrophobic interactions of the fluorine atoms in CF3 with the hydrophobic pocket in addition to the polar interactions between the oxamic acid moiety and the P-loop residues. Thus, these molecules achieve their high potency and selectivity by exploiting the nonconserved regions in both hydrophilic and hydrophobic regions in the mPTPB active site. The abovementioned findings suggest that these compounds are expected to show specificity toward mPTPB over other PTPs in addition to the ones screened here (Table 4).
Figure 5.

Molecular docking models of 4t with mPTPB (PDB code: 2OZ5). (A) Binding model of 4t with surface representation of mPTPB. The polar P-loop residues are shown in blue and the hydrophobic region is shown in red.64 (B) Detailed interactions of 4t with mPTPB. The hydrogen-bonding interactions are shown using red lines.
Mutagenesis of Key Binding Residues in mPTPB.
To further validate the binding mode, we mutated residues that are predicted to make interactions with 4t (Figure 5B). Molecular docking studies predicted the hydrogen bonding interactions between the oxamic acid moiety and the Phe161, Lys164, and Asp165 residues, which are unique to mPTPB (Figure 4B). On the other hand, the PhCF3 group is suggested to form hydrophobic interactions with the side chains of residues Ile203, Met206, and Ile207. To ascertain the importance of the unique P-loop residues in binding 4t, we substituted residues Phe161, K164, and Asp165 to their mammalian PTP counterparts, namely, Ser, Gly, and Ile, respectively. To increase the distance between the PhCF3 and the sec-butyl side chain of Ile203 and Ile207 in order to disrupt this hydrophobic interaction, we mutated both residues to alanine (methyl side chain). To further disrupt this pocket, we also replaced Ile207 by a lysine, which represents a hydrophobic to hydrophilic residue conversion. The wild-type and all of the mutant mPTPB were expressed in Escherichia coli and purified to homogeneity. As shown in Table 5, the kinetic parameters of kcat and Km for I203A, I207A, and I207K are comparable to those of the wild-type mPTPB, indicating that these mutations do not perturb the overall enzyme structure and function. Consistent with the importance of the P-loop in PTP catalysis, the K164I and D165G mutants exhibit no measurable phosphatase activity. Interestingly, although the kcat for F161S is 14.6-fold lower than that of the wild-type enzyme, the Km values are similar for both the wild-type mPTPB and F161S.
Table 5.
Kinetic Parameters of the Wild-Type and Mutant mPTPBs with pNPP as a Substratea
| enzyme | Km (in mM) | kcat (s−1) |
|---|---|---|
| wild type | 3.58 ± 0.11 | 4.37 ± 0.12 |
| I203A | 2.73 ± 0.09 | 1.61 ± 0.10 |
| I207A | 4.85 ± 0.17 | 4.94 ± 0.19 |
| I207K | 4.53 ± 0.05 | 1.96 ± 0.02 |
| F161S | 4.47 ± 0.27 | 0.30 ± 0.02 |
| K164I | NA | NA |
| D165G | NA | NA |
Kinetics measured at pH = 7.0, in 50.0 mM 3,3-dimethylglutarate, 1 mM EDTA, 0.15 M NaCl buffer at room temperature. NA = showed no measurable phosphatase activity.
We then determined the impact of altering mPTPB residues Ile203, Ile207, and Phe161 on the IC50 values of compounds 4k, 4t, 4u, and 4v. Mutant I203A showed a 3–6-fold decrease in binding affinity, while I207A demonstrated 13–18-fold decrease in inhibition for compounds 4k, 4t, 4u, and 4v as compared to the wild type (Table 6). The decrease in affinity was in the expected range as we have shown that addition of the CF3 group increased the affinity of our core compound by 18-fold (compound 4 vs 4k). This indicates that the CF3 group likely fits in the abovementioned hydrophobic pocket and hence increases the binding affinity of the inhibitor series derived from 4k. Not to our surprise for mutant I207K, the loss in affinity was even higher, around 41–166-fold for compounds 4k, 4t, 4u, and 4v, which is in line with the extent of the structural perturbation at this position. Consistent with the importance of P-loop residues in binding the oxamic acid moiety, the IC50 values of F161S for compounds 4k, 4t, 4u, and 4v are 570–1800-fold higher than those of the wild-type mPTPB. Taken together, these observations support the proposed binding mode for compound 4t and its close analogues and indicate that compounds 4k, 4t, 4u, and 4v indeed bind to the mPTPB active site.
Table 6.
Comparison of IC50 Data for 4k, 4t, 4u, and 4v against Wild-Type and Mutant mPTPB
| entry | wild-type IC50 (μM) | I203A IC50 (μM) | I207A IC50 (μM) | I207K IC50 (μM) | F161S IC50 (μM) |
|---|---|---|---|---|---|
| 4k | 0.015 ± 0.003 | 0.043 ± 0.003 | 0.190 ± 0.007 | 0.627 ± 0.021 | 8.5 ± 1.1 |
| 4t | 0.0064 ± 0.0005 | 0.025 ± 0.001 | 0.091 ± 0.001 | 0.374 ± 0.036 | 5.4 ± 0.9 |
| 4u | 0.0041 ± 0.0017 | 0.021 ± 0.002 | 0.075 ± 0.002 | 0.684 ± 0.071 | 7.4 ± 1.1 |
| 4v | 0.012 ± 0.003 | 0.040 ± 0.002 | 0.161 ± 0.005 | 0.698 ± 0.065 | 9.1 ± 1.2 |
Physicochemical Properties.
Given the excellent potency and selectivity of these inhibitors, we next set out to measure or calculate the physicochemical properties for selected compounds. Physicochemical properties such as topological PSA (tPSA), c Log P, Log D, and aqueous solubility of 4b, 4h, 4k, 4t, 4u, and 4v are shown in Table 7. All these compounds showed moderate aqueous solubility in the range of 18–68 μM [pH = 7.4 in phosphate-buffered saline (PBS) buffer]. Hopkins et al. proposed LE as a method for comparing molecules according to their average binding energy per heavy atom.48 It is a powerful tool for assessing the quality of ligand molecules, as larger molecules tend to show better potency because of the larger number of interactions but may not necessarily be the most efficient. Hence, smaller but more efficient molecules are needed as they have a higher probability of successfully advancing in the lead optimization process. Lipophilic LE (LLE) is an estimate of the specificity of a molecule in binding to the target relative to partitioning into n-octanol.49 The proposed acceptable values of LE and LLE for drug candidates are LE > 0.3 and LLE > 5.50 All the compounds shown in Table 7 show LE values of >0.42 and LLE values of >5.3. Importantly, these compounds also follow the GSK 4/400 rule (higher risks of off-target interactions and toxicity, if MW > 400 and c Log P > 4), suggesting that they could serve as potential candidates for drug development.
An ideal compound for the mouse in vivo studies should be soluble, have good absorption, and must exhibit metabolic stability. A sufficient oral dosage must survive first-pass clearance through the liver before reaching its biological target. The majority of drugs/xenobiotics undergo phase I metabolism, mediated by the cytochrome P450 (CYP450) family of heme-thiolate proteins predominantly in the liver. Although mouse liver microsomal (MLM) stability studies are not a perfect substitute for in vivo metabolic clearance studies, they serve as an initial cell-based model system that can correlate well with human liver microsomal (HLM) stability and in vivo activity in mice.51,52 To this end, compounds 4h, 4k, 4t, 4u, and 4v were allowed to react with mouse liver microsomes and the fraction of the remaining parent compound over time was determined. Compound 4h with a tert-butyl was the least stable as measured by the half-life compared to all other compounds with CF3. Incorporation of fluorine into drug candidates generally improves their potency, bioavailability, and metabolic stability. In addition, fluorinated molecules are, in general, nontoxic and mimic the corresponding nonfluorinated analogues in their stereoelectronic properties, so the drug’s affinity for the receptor either remains the same or, in some cases, increases. The improved metabolic stability of CF3-containing 4k, 4t, 4u, and 4v compared to 4h, partly because of the stronger C─F bond compared to C─H (116 and 99 kcal mol−1, respectively), makes the C─F bond less sensitive to metabolic degradation. Importantly, the analysis of cofactor-absent microsome reactions suggests that all these analogues are stable at 37 °C for 2 h. Therefore, the loss of compounds tested here is attributed only to oxidative phase I metabolism. The high microsomal stability of these compounds could be exploited pharmacologically.
Cell Cytotoxicity and Permeability Studies.
Before conducting the cell permeability and cellular activity studies, we first tested the cytotoxicity of our best compounds 4t and 4s against mouse embryonic fibroblasts (MEF cells). Cell viability was measured using tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell proliferation assay. Compounds 4s and 4t did not exhibit any significant cytotoxicity when treated against MEF cells even at as high as 25 μM concentration. Cell permeability is the major hurdle in the development of active site-directed PTP inhibitors. The majority of the pTyr mimetics reported, carrying one or more negative charges for binding with the positively charged PTP active site, are deficient in cell membrane permeability, which limits further advancement of such compounds as drug candidates. We treated Raw264.7 cells with compounds 4k, 4t, 4u, and 4v and determined the cell permeability potential of our compounds using a previously described LC─MS-based method.53,54 The cellular bioavailability studies indicated that these compounds can easily be taken up by Raw264.7 cells (Supporting Information).
Cellular Activity of Compound 4t in Blocking mPTPB-Mediated Signaling.
After evaluating the cell permeability of these compounds in Raw264.7 cells, we proceeded to investigate compound 4t’s cellular efficacy. We have previously demonstrated that once inside the host macrophage, mPTPB activates Akt signaling and blocks ERK1/2 and p38 activation to prevent macrophage apoptosis and attenuate IFN-γ stimulation-mediated IL-6 production.18,21 Because mPTPB functions within the macrophage, Raw264.7 mouse macrophage cell lines stably expressing mPTPB (pcN-HA-mPTPB) or vector (pcN-HA) control were used to investigate the role of mPTPB in host cell biology.18 Using these systems, we previously established that mPTPB decreases ERK1/2 (T202/Y204) and p38 (T180/Y182) phosphorylation and enhances AKT (S473) phosphorylation. Phosphorylation of these sites directly correlates with kinase activation which is a surrogate for kinase activity measurement. Therefore, mPTPB inhibitors are expected to increase ERK1/2 and p38 phosphorylation and reduce AKT phosphorylation.
We showed that in the vector control Raw 264.7 cells, INF-γ stimulation led to an increase in pAKT, pERK1/2, and pp38 levels by 4.3-, 4.1-, and 3.4-fold, respectively (Figure 6, lane 1 vs lane 2). Compared to the vector control cells, the level of INF-γ-stimulated pAKT (7.2-fold over basal) was further elevated in Raw 264.7 cells expressing mPTPB, while the pERK1/2 and pp38 levels (1.5- and 2.4-fold over basal) were suppressed upon INF-γ stimulation (Figure 6, lane 7 and lane 8). These findings are consistent with previous observations18,21-23 that mPTPB promotes AKT activation and attenuates ERK1/2 and p38 activity.
Figure 6.
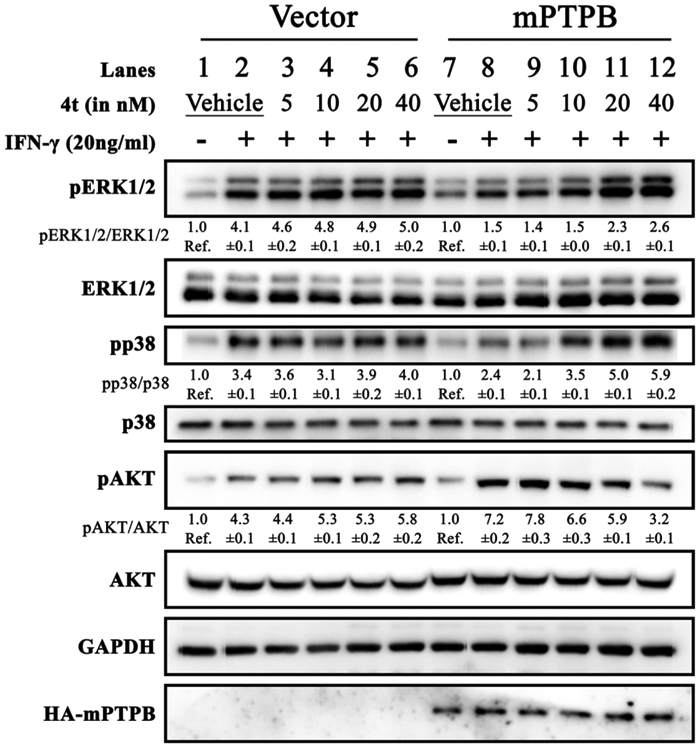
Cellular efficacy of the mPTPB inhibitor 4t. Raw 264.7 cells were transfected with pcN-HA control vector (lanes 1–6) or pcN-HA-mPTPB (lanes 7–12) to create stable cell lines.18 Raw264.7 cells expressing mPTPB exhibited decreased IFN-γ-stimulated ERK1/2 and p38 activation and increased AKT activity compared to control vector cells. Cellular activity of compound 4t was evaluated by treating it with both cell lines. Compound 4t was able to block mPTPB-mediated cellular signaling by restoring ERK1/2 and p38 activity and inhibiting AKT activation by mPTPB, whereas no effect on control vector-transfected cells was observed as shown by the blots above.
As expected, treatment of compound 4t to the Raw 264.7 vector cells upon IFN-γ stimulation (Figure 6, lanes 3–6) elicited no significant change in the pAKT, pERK1/2, and pp38 levels compared to the vehicle-treated cells (Figure 6, lane 2). This indicates that compound 4t does not interfere with these signaling pathways inside the cells. In contrast, treatment of IFN-γ-stimulated mPTPB expressing Raw 264.7 cells with 5–40 nM compound 4t resulted in a dose-dependent reduction of pAKT and the corresponding increase in pERK1/2 and pp38 (Figure 6, lanes 9–12). These results are similar to those described in previous studies with several structurally unrelated small-molecule mPTPB inhibitors in cell-based assays18,21-23,55 and strongly suggest that the observed cellular effects of 4t in macrophages are indeed from specific inhibition of mPTPB. Importantly, the finding that 4t’s cellular activity tracked very well with its IC50 value measured with purified recombinant mPTPB in biochemical assays further supports the excellent cell permeability of compound 4t. Together, these results indicate that 4t is highly specific and efficient in blocking mPTPB activity inside the cell.
CONCLUSIONS
Given the importance of PTPs in regulating cellular signaling and homeostasis, there is increasing interest in developing PTP-based therapeutics for a wide range of diseases including cancer, diabetes, autoimmune disorders, and infectious diseases.3 As a key virulence factor for Mtb survival within host macrophages, mPTPB has garnered substantial attention as a novel anti-TB target. Importantly, the absence of a human orthologue for mPTPB makes it a highly attractive target for the development of novel anti-TB candidates. However, the acquisition of highly potent, specific, and efficacious PTP inhibitors with properties suitable for in vivo experiments and clinical translation has proven to be difficult primarily because of the highly conserved and positively charged nature of the PTP active sites. We have shown that through fragment-based approaches, we can transform negatively charged pTyr mimetics such as salicylic acids and α-sulfophenylacetic amide into highly potent and selective active site-directed PTP inhibitors with robust in vivo efficacy.5 Nevertheless, the design of the minimally charged, stable, and high-affinity nonhydrolyzable pTyr surrogate has generally met with limited success and this presents a major challenge to the development of novel therapeutics based on the PTPs.
Here, we identified N-phenyl oxamic acid as a highly potent and selective monoacid-based pTyr mimetic for mPTPB inhibition. We then conducted SAR studies by varying the substituents on the phenyl ring and discovered the 4-phenylethynyl-containing analogue (compound 4) as the best core structure for further modification. Several mono-, di-, and trisubstituted analogues of compound 4 were synthesized; among them, compound 4t showed a Ki of 2.7 nM for mPTPB with >4500-fold preference over 25 PTPs. Kinetic, molecular docking, and site-directed mutagenesis analyses confirmed these compounds as active site-directed reversible inhibitors of mPTPB. Importantly, these N-phenyl oxamic acid inhibitors penetrate cell membranes and inhibit mPTPB inside the cells. Additionally, these oxamic acid-based inhibitors are capable of reversing the altered host cell immune responses induced by the bacterial phosphatase. Furthermore, the reported mPTPB inhibitors possess highly compact structures with molecular weights <400 Da, c Log P < 4, Log D7.4 < 2.5, and LE > 0.42 and with favorable drug-like properties.56-58 Collectively, the results indicate that the N-phenyl oxamic acid pharmacophore is sufficiently polar to bind the PTP active site yet remains capable of efficiently crossing cell membranes, offering PTP inhibitors with both high affinity and selectivity and excellent cellular efficacy. The results also provide another example of developing minimally charged, highly potent, and selective active site-directed PTP inhibitors by exploiting the specific structural features of the active sites of different PTPs. Our work not only offers further opportunities to evaluate mPTPB inhibition as a tuberculosis treatment but also stimulates interest in targeting other mPTPB orthologues, which are present in over 50 human pathogens, including Listeria monocytogenes.59,60 Finally, among the compounds identified in this study, 4t merits further therapeutic development by virtue of its low nanomolar mPTPB inhibitory potency, outstanding selectivity over other PTPs, low molecular weight, good aqueous solubility, superior metabolic stability, and efficacious cellular activity. Furthermore, 4t can also serve as a molecular probe in unraveling the role of mPTPB in normal physiology and in diseases, in addition to the potential therapeutic value.
EXPERIMENTAL SECTION
General Synthetic Procedures and Reagents.
Unless otherwise noted, all reagents were purchased from commercial suppliers and used without further purification. Thin-layer chromatography was performed using glass precoated Merck silica gel 60 F254 plates. Column chromatography was performed using KP-SIL silica gel (Biotage, USA), and flash column chromatography was performed on Biotage prepacked columns using the automated flash chromatography system Biotage Isolera One. Organic solvents were evaporated using rotary evaporation at 40–45 °C. The 1H- and 13C NMR spectra were recorded on a Bruker AVANCE 500 MHz spectrometer using CDCl3 or dimethyl sulfoxide (DMSO) (d6) as the solvent. Chemical shifts are expressed in ppm (δ scale) and referenced to the residual protonated solvent. Peak multiplicities are reported using the following abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), or br (broad singlet). Low-resolution mass spectra and purity data were obtained using an Agilent Technologies 6470 series, triple quadrupole LC─MS. The purity of all final tested compounds was determined to be >95% (UV, λ = 254 nm). High-resolution mass analysis was performed on an Agilent 6550 iFunnel Q-TOF mass LC─MS. Recombinant mouse IFN-γ was purchased from PeproTech Inc. Anti-ERK1/2 (catalog#4696), antiphospho-ERK1/2 (catalog#9101), anti-p38 (catalog#9212), antiphospho-p38 (catalog#9211s), anti-AKT (catalog#2920s), and antiphospho Akt473 (catalog#9271s) antibodies were purchased from Cell Signaling. Anti-HA (catalog#SC-7392) and anti-GAPDH (catalog#SC-59541) antibodies were purchased from Santa Cruz. pNPP was purchased from Thermo Scientific (catalog#PI34045).
Synthesis of Oxanilic Acid (1).
To a stirred solution of aniline (300 mg, 3.22 mmol) and N,N-diisopropyl ethylamine (1.11 mL, 6.44 mmol) in 15 mL of anhydrous CH2Cl2, methyl chlorooxoacetate (327 μL, 3.54 mmol) was added dropwise. The resulting solution was stirred at room temperature under N2 gas for 30 min. The reaction mixture was washed with 15 mL of DI water and 15 mL of brine. The organic layer was dried over anhydrous Na2SO4 and evaporated in vacuo. Crude ester was subjected to hydrolysis using 12 mL of 1 N KOH/THF (1:1 v/v) at room temperature for 1 h. After completion, THF was evaporated, the aqueous layer was acidified to pH ~2 using 3 N HCL, and the resulting solid was filtered. Column chromatographic purification of the residue using silica and 10% methanol in dichloromethane (DCM) as an eluent afforded the product as a white solid (490 mg, 92% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.68 (s, 1H), δ 7.74 (d, J = 7.5 Hz, 2H), δ 7.33 (t, J = 7.5 Hz, 2H), δ 7.11 (t, J = 7.5 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 162.6, 157.3, 138.1, 129.2, 125.0, 120.8; mass spectra (ESI) m/z: 164 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C8H6NO3, 164.0348; found, 164.0354.
Synthesis of Methyl 2-((4-ethynylphenyl)amino)-2-oxoacetate (Core 1).
To an ice-cold solution of 4-ethynylphenylamine (4.0 g, 34.14 mmol) and N,N-diisopropyl ethylamine (11.82 mL, 68.29 mmol) in 100 mL of anhydrous CH2Cl2, methyl chlorooxoacetate (3.46 mL, 37.56 mmol) was added dropwise under a N2 atmosphere. The resulting solution was stirred at room temperature for 1 h. The reaction mixture was washed with 100 mL of DI water and 100 mL of brine. The organic layer was dried over anhydrous Na2SO4 and evaporated in vacuo. The product was then purified by column chromatography on silica gel using a mixture of 2:8 ethylacetate/hexanes as an eluent. White powder; (6.2 g, 96% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.94 (s, 1H), δ 7.77 (d, J = 9.0 Hz, 2H), δ 7.45 (d, J = 9.0 Hz, 2H), δ 4.11 (s, 1H), δ 3.84 (s, 3H); 13C NMR (DMSO-d6, 125 MHz): δ 161.3, 155.7, 138.5, 132.8, 120.8, 118.2, 83.7, 80.9, 53.7. Mass spectra (ESI) m/z: 202 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C11H9NO3, 202.0504; found, 202.0510.
Synthesis of Methyl 2-((3-ethynylphenyl)amino)-2-oxoacetate (Core 2).
To an ice-cold solution of 3-ethynylphenylamine (2.0 g, 17.07 mmol) and N,N-diisopropyl ethylamine (5.91 mL, 34.14 mmol) in 50 mL of anhydrous CH2Cl2, methyl chlorooxoacetate (1.73 mL, 18.78 mmol) was added dropwise under a N2 atmosphere. The resulting solution was stirred at room temperature for 1 h. The reaction mixture was washed with 50 mL of DI water and 50 mL of brine. The organic layer was dried over anhydrous Na2SO4 and evaporated in vacuo. The product was then purified by column chromatography on silica gel eluted with a mixture of 3:7 ethylacetate/hexanes. Off-white powder; (3.0 g, 93% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.88 (s, 1H), δ 7.90 (s, 1H), δ 7.75–7.77 (m, 1H) δ 7.35 (t, J = 8.0 Hz, 1H), δ 7.24 (d, J = 7.5 Hz, 1H), δ 4.18 (s, 1H), δ 3.84 (s, 3H); 13C NMR (DMSO-d6, 125 MHz): δ 161.3, 155.8, 138.2, 129.7, 128.4, 123.7, 122.5, 121.6, 83.6, 81.3, 53.7. Mass spectra (ESI) m/z: 202 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C11H9NO3, 202.0504; found, 202.0510.
Synthesis of Methyl 2-((4-iodophenyl)amino)-2-oxoacetate (Core 3).
To an ice-cold solution of 4-iodoaniline (3.0 g, 13.7 mmol) and N,N-diisopropyl ethylamine (3.0 mL, 20.55 mmol) in 60 mL of anhydrous CH2Cl2, methyl chlorooxoacetate (1.5 mL, 16.44 mmol) was added dropwise under a N2 atmosphere. The resulting solution was stirred at room temperature for 1 h. The reaction mixture was washed with 60 mL of DI water and 60 mL of brine. The organic layer was dried over anhydrous Na2SO4 and evaporated in vacuo. The product was then purified by column chromatography on silica gel eluted with a mixture of 3:7 ethylacetate/hexanes. White powder; (4.0 g, 96% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.88 (s, 1H), δ 7.68 (d, J = 8.5 Hz, 2H), δ 7.57 (d, J = 8.5 Hz, 2H), δ 3.83 (s, 3H); 13C NMR (DMSO-d6, 125 MHz): δ 161.3, 155.7, 137.9, 137.8, 123.0, 89.4, 53.7. Mass spectra (ESI) m/z: 304 (M − H)−.
Synthesis of 2-((4-Ethynylphenyl)amino)-2-oxoacetic Acid (2).
A stirred solution of core 1 (100 mg) in 8 mL of 1 N KOH/THF (1:1 v/v) was stirred at room temperature for 1 h. After completion, THF was completely evaporated, the aqueous layer was acidified to pH ~2 using 3 N HCL, and the resulting solid was filtered to yield pure compound 2 as an off-white solid (90 mg, 95% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.85 (s, 1H), δ 7.78 (d, J = 8.5 Hz, 2H), δ 7.44 (d, J = 8.5 Hz, 2H), δ 4.11 (s, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 162.4, 157.5, 138.7, 132.8, 120.6, 117.9, 83.8, 80.9. Mass spectra (ESI) m/z: 188 (M − H)−.
Synthesis of 2-((4-(3-((tert-Butoxycarbonyl)amino)prop-1-yn-1-yl)phenyl)amino)-2-oxoacetic Acid (3).
To a stirred solution of methyl 2-((4-iodophenyl)amino)-2-oxoacetate (400 mg, 1.0 equiv) in 5 mL of anhydrous DMF, tert-butyl prop-2-yn-1-ylcarbamate (224 mg, 1.1 equiv), sodium carbonate (278 mg, 2.0 equiv), Pd(PPh3)2Cl2 (5 mol %), and copper iodide (5 mol %) were added sequentially. The resulting mixture was stirred at room temperature under a N2 atmosphere. Reactions were monitored by LC─MS. After completion of the reaction, DMF was completely removed using high-vac. The residue was dissolved in 70 mL of ethyl acetate and washed with 50 mL of DI water. The organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated. Crude ester was subjected to hydrolysis using 1 N KOH/THF (1:1 v/v) at room temperature for 1 h. After completion, THF was evaporated, the aqueous layer was acidified to pH ~3 using cold 1 N HCL, and the resulting solid was filtered. Compound 3 was purified by column chromatography using a mixture of 1:9 methanol/DCM as an eluent on silica gel (stationary phase) as an off-white powder (351 mg, 84% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.59 (s, 1H), δ 7.76 (d, J = 8.5 Hz, 2H), 7.33 (d, J = 8.5 Hz, 2H), 3.94 (d, J = 5.5 Hz, 2H), δ 1.38 (s, 9H); 13C NMR (DMSO-d6, 125 MHz): δ 162.8, 160.4, 155.8, 138.8, 132.3, 120.2, 117.9, 87.4, 81.9, 78.7, 30.6, 28.7; mass spectra (ESI) m/z: 317 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C16H17N2O5, 317.1132; found, 317.1134.
General Procedure for the Synthesis of Compounds 4, 4a–4x, and 5.
To a stirred solution of methyl 2-((4-ethynylphenyl)-amino)-2-oxoacetate (200 mg, 1.0 equiv) in 3 mL of anhydrous DMF, aryl iodides (1.1 equiv), sodium carbonate (2.0 equiv), Pd(PPh3)2Cl2 (5 mol %), and copper iodide (5 mol %) were added sequentially. The resulting mixture was stirred at room temperature under a N2 atmosphere. Reactions were monitored by LC─MS. After completion of the reaction, DMF was completely removed using high-vac. The residue was dissolved in 50 mL of ethyl acetate and washed with 40 mL of DI water. The organic layer was dried over anhydrous Na2SO4 and the solvent was evaporated. Crude ester was subjected to hydrolysis using 1 N KOH/THF (1:1 v/v) at room temperature for 1 h. After completion, THF was evaporated, the aqueous layer was acidified to pH ~2 using 3 N HCL, and the resulting solid was filtered. Products were then purified by column chromatography on silica gel eluted with a mixture of 1:9 methanol/DCM.
General Procedure for Preparation of Compounds 9 and 10.
To a stirred solution of 4 or 4k (50 mg) in 4 mL of THF/methanol (1:1 v/v) were added 5 mg of 5% Pd/BaSO4 and 50 μL of quinoline and then the mixture was allowed to stir for 12 h at room temperature under a H2 atmosphere. After completion of the reaction (monitored by LC─MS), the mixture was filtered through Celite pad, and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography using the 1:9 methanol/DCM mixture as an eluent to furnish compounds 9 and 10 in >70% yields.
General Procedure for Preparation of Compounds 11 and 12.
To a stirred solution of 4 or 4k (50 mg) in ethanol (4 mL), 5 mg of 10% Pd/C was added, and the mixture was allowed to stir for 4 h at room temperature under a H2 atmosphere. After completion of the reaction (monitored by LC─MS), the mixture was filtered through Celite pad, and the filtrate was concentrated under reduced pressure. The residue was purified by column chromatography using the 1:9 methanol/DCM mixture as an eluent to furnish compounds 11 and 12 in >90% yields.
2-Oxo-2-((4-(phenylethynyl)phenyl)amino)acetic Acid (4).
White solid; (235 mg, 90% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.62 (s, 1H), δ 7.82 (d, J = 8.5 Hz, 2H), δ 7.48–7.53 (m, 4H), δ 7.38–7.41 (m, 3H); 13C NMR (DMSO-d6, 125 MHz): δ 160.7, 139.2, 132.2, 131.7, 129.2, 129.1, 122.9, 120.2, 117.7, 89.9, 89.2. Mass spectra (ESI) m/z: 264 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C16H10NO3, 264.0661; found, 264.0670.
2-((4-((4-(Methoxycarbonyl)phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4a).
White powder; (249 mg, 78% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.58 (s, 1H), δ 7.95 (d, J = 8.5 Hz, 2H), δ 7.86 (d, J = 9.0 Hz, 2H), δ 7.65 (d, J = 8.5 Hz, 2H), δ 7.52 (d, J = 8.5 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 166.1, 163.8, 162.7, 139.8, 132.6, 131.9, 129.9, 129.5, 127.8, 120.1, 116.9, 93.2, 88.4, 52.8. Mass spectra (ESI) m/z: 322 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C18H12NO5, 322.0715; found, 322.0712.
2-((4-((4-Hydroxy-3-(methoxycarbonyl)phenyl)ethynyl)-phenyl)amino)-2-oxoacetic Acid (4b).
Pale-yellow solid; (238 mg, 71% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.79 (s, 1H), δ 10.70 (s, 1H), δ 7.87 (d, J = 2.0 Hz, 1H), δ 7.81 (d, J = 7.0 Hz, 2H), δ 7.62 (d, J = 8.5 Hz, 1H), δ 7.49 (d, J = 9.0 Hz, 2H), δ 7.01 (d, J = 8.5 Hz, 1H), 3.87 (s, 3H); 13C NMR (DMSO-d6, 125 MHz): δ 168.4, 162.5, 160.1, 158.3, 138.6, 138.2, 133.7, 132.3, 120.5, 118.7, 118.3, 114.7, 113.8, 88.7, 88.4. Mass spectra (ESI) m/z: 338 (M − H)−. HRMS (ESI-TOF, [M + H]+) m/z: calcd for C18H14NO6, 340.0815; found, 340.0815.
2-((4-((3-Hydroxy-4-(methoxycarbonyl)phenyl)ethynyl)-phenyl)amino)-2-oxoacetic Acid (4c).
Pale-yellow solid; (240 mg, 72% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.87 (s, 1H), δ 10.57 (s, 1H), δ 7.87 (br, 2H), δ 7.76 (d, J = 8.5 Hz, 1H), δ 7.55 (d, J = 6.5 Hz, 2H), δ 7.10 (s, 1H), δ 7.06 (d, J = 8.5 Hz, 1H), δ 3.87 (s, 3H); 13C NMR (DMSO-d6, 125 MHz): δ 168.8, 159.9, 132.8, 130.9, 129.5, 122.7, 120.6, 120.0, 117.6, 113.9, 92.7, 88.4, 53.0. Mass spectra (ESI) m/z: 338 (M − H)−. HRMS (ESI-TOF, [M + H]+) m/z: calcd for C18H14NO6, 340.0815; found, 340.0815.
2-((4-((2-(Dimethylamino)-4-hydroxy-5-(methoxycarbonyl)-phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4d).
Pale-yellow solid; (264 mg, 70% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.71 (s, 1H), δ 7.85 (br, 1H), δ 7.78 (s, 1H), δ 7.59 (s, 1H), δ 7.48 (s, 1H), δ 6.29 (s, 1H), δ 3.84 (s, 3H), δ 3.08 (s, 6H); 13C NMR (DMSO-d6, 125 MHz): δ 169.0, 161.9, 158.9, 137.2, 133.7, 131.7, 120.8, 119.3, 103.9, 103.5, 103.0, 92.7, 88.8, 52.6. Mass spectra (ESI) m/z: 381 (M − H)−. HRMS (ESI-TOF, [M + H]+) m/z: calcd for C20H19N2O6, 383.1237; found, 383.1236.
2-((3-((2-(Dimethylamino)-4-hydroxy-5-(methoxycarbonyl)-phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4e).
White powder; (259 mg, 69% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.72 (s, 1H), δ 7.94 (s, 1H), δ 7.93 (s, 1H), δ 7.75 (d, J = 8.5 Hz, 1H), δ 7.35 (t, J = 8.0 Hz, 1H), δ 7.23 (d, J = 10.0 Hz, 1H), δ 6.30 (s, 1H), δ 3.84 (s, 3H), δ 3.09 (s, 6H); 13C NMR (DMSO-d6, 125 MHz): δ 168.9, 162.5, 162.1, 158.9, 158.2, 138.6, 137.4, 129.7, 126.9, 123.7, 122.4, 120.5, 104.0, 103.2, 103.0, 92.5, 89.2, 52.5; mass spectra (ESI) m/z: 381 (M − H)−. HRMS (ESI-TOF, [M + H]+) m/z: calcd for C20H19N2O6, 383.1237; found, 383.1236.
5-((4-(Carboxyformamido)phenyl)ethynyl)-4-(dimethylamino)-2-hydroxybenzoic Acid (4f).
Off-white solid; (245 mg, 68% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.81 (s, 1H), δ 7.81 (d, J = 7.5 Hz, 2H), δ 7.76 (s, 1H), δ 7.46 (d, J = 8.0 Hz, 2H), δ 6.27 (s, 1H), δ 3.06 (s, 6H); 13C NMR (DMSO-d6, 125 MHz): δ 171.6, 162.9, 159.0, 137.4, 133.6, 131.6, 120.7, 119.3, 104.7, 103.3, 103.0, 92.5, 89.1, 42.6; mass spectra (ESI) m/z: 367 (M − H)−. HRMS (ESI-TOF, [M + H]+) m/z: calcd for C19H17N2O6, 369.1081; found, 369.1081.
5-((3-(Carboxyformamido)phenyl)ethynyl)-4-(dimethylamino)-2-hydroxybenzoic Acid (4g).
Off-white solid; (238 mg, 66% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.57 (s, 1H), δ 7.95 (s, 1H), δ 7.78 (s, 1H), δ 7.75 (d, J = 7.5 Hz, 1H), δ 7.33 (t, J = 7.5 Hz, 1H), δ 7.20 (d, J = 7.5 Hz, 1H), δ 6.26 (s, 1H), δ 3.04 (s, 6H); 13C NMR (DMSO-d6, 125 MHz): δ 171.7, 163.4, 158.9, 138.9, 137.4, 129.6, 126.6, 123.9, 122.2, 120.2, 106.4, 103.2, 102.8, 92.3, 89.7, 42.7. Mass spectra (ESi) m/z: 367 (M − H)−. HRMS (ESI-TOF, [M + H]+) m/z: calcd for C19H17N2O6, 369.1081; found, 369.1081.
2-((4-((4-(tert-Butyl)phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4h).
Pale-yellow solid; (259 mg, 82% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.30 (s, 1H), δ 7.80 (d, J = 9.0 Hz, 2H) δ 7.42 (q, J = 9.0, 8.0 Hz, 6H), δ 1.27 (s, 9H); 13C NMR (DMSO-d6, 125 MHz): δ 164.8, 162.7, 151.6, 139.9, 132.3, 131.5, 126.0, 120.1, 119.5, 116.9, 89.6, 88.9, 35.0, 31.4. Mass spectra (ESI) m/z: 320 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C20H18NO3, 320.1281; found, 320.1280.
2-Oxo-2-((4-((4-(trifluoromethoxy)phenyl)ethynyl)phenyl)amino)acetic Acid (4i).
Pale-yellow powder; (268 mg, 78% yield). 1H NMR (DMSO-d6 500 MHz): δ 10.69 (s, 1H), δ 7.83 (d, J = 8.5 Hz, 2H): δ 7.65 (d, J = 8.5 Hz, 2H), δ 7.51 (d, J = 8.5 Hz, 2H), δ 7.40 (d, J = 8.0 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.6, 159.9, 148.5, 139.4, 133.8, 132.5, 122.3, 121.84, 120.3, 117.4, 90.9, 87.8. Mass spectra (ESI) m/z: 348 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C17H9F3NO4, 348.0485; found, 348.0482.
2-Oxo-2-((4-((3-(trifluoromethyl)phenyl)ethynyl)phenyl)amino)acetic Acid (4j).
White solid; (267 mg, 81% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.89 (s, 1H), δ 7.82–7.87 (m, 4H), δ 7.74 (d, J = 8.0 Hz, 1H), δ 7.64 (t, J = 8.0 Hz, 1H), δ 7.56 (d, J = 8.5 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 1162.4, 157.6, 139.0, 135.6, 132.7, 130.5, 130.2, 129.9, 129.7, 128.2, 125.6, 125.3, 124.0, 123.1, 120.7, 117.8, 91.4, 87.9. Mass spectra (ESI) m/z: 332 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C17H9F3NO3, 332.0534; found, 332.0532.
2-Oxo-2-((4-((4-(trifluoromethyl)phenyl)ethynyl)phenyl)amino)acetic Acid (4k).
Off-white solid; (263 mg, 80% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.91 (s, 1H), δ 7.85 (d, J = 8.5 Hz, 2H), δ 7.75 (dd, J = 15.0, 9.0 Hz, 4H), δ 7.57 (t, J = 7.0 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.3, 157.5, 139.1, 132.7, 132.5, 129.3, 129.1, 128.8, 128.6, 127.2, 126.1, 125.5, 123.4, 120.7, 117.7, 92.3, 88.1. Mass spectra (ESI) m/z: 332 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C17H9F3NO3, 332.0534; found, 332.0532.
2-Oxo-2-((4-((4-sulfamoylphenyl)ethynyl)phenyl)amino)acetic Acid (4l).
Off-white solid; (226 mg, 67% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.79 (s, 1H), δ 7.82–7.86 (m, 2H), δ 7.71 (d, J = 8.5 Hz, 1H), δ 7.53–7.62 (m, 3H), δ 7.45 (s, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 162.5, 159.0, 144.1, 139.4, 133.6, 132.7, 132.5, 132.2, 132.0, 131.9, 129.3, 129.2, 126.5, 126.3, 120.5, 117.4, 92.3, 88.3. Mass spectra (ESI) m/z: 343 (M − H)− 367 (M + Na)+. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C16H12N2O5S, 343.0388; found, 343.0391.
2-((4-((4-Hydroxyphenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4m).
White solid; (230 mg, 83% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.78 (s, 1H), δ 7.80 (d, J = 8.5 Hz, 2H), δ 7.45 (d, J = 8.5 Hz, 2H), δ 7.34 (d, J = 8.5 Hz, 2H), δ 6.78 (d, J = 8.5 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.5, 158.5, 158.1, 138.2, 133.4, 132.1, 120.56, 119.0, 116.2, 113.0, 90.1, 87.7; mass spectra (ESI) m/z: 280 (M − H)−. HRMS (ESI-TOF, [M + H]+) m/z: calcd for C16H12NO4, 282.0761; found, 282.0759.
2-((4-((3-Bromo-4-hydroxyphenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4n).
Pale-yellow powder; (261 mg, 74% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.83 (s, 1H), δ 7.81 (d, J = 8.5 Hz, 2H), δ 7.64 (d, J = 2.0 Hz, 1H), δ 7.47 (d, J = 8.5 Hz, 2H), δ 7.35 (dd, J = 8.5, 2.0 Hz, 1H), δ 6.96 (d, J = 8.5 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 162.5, 157.7, 155.3, 138.4, 136.0, 132.5, 132.2, 120.6, 118.6, 116.9, 114.7, 109.7, 88.7, 88.4; mass spectra (ESI) m/z: 358 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C16H9NO4, 357.9715; found, 357.9727.
2-((4-((4-Cyanophenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4o).
White powder; (226 mg, 79% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.92 (s, 1H), δ 7.86 (d, J = 8.5 Hz, 4H), δ 7.70 (d, J = 8.5 Hz, 2H), δ 7.57 (d, J = 8.0 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.5, 157.5, 139.3, 133.1, 132.8, 132.5, 127.8, 120.7, 118.9, 117.6, 111.3, 93.9, 88.2. Mass spectra (ESI) m/z: 289 (M − H)−. HRMS (ESI-TOF, [M + H]+) m/z: calcd for C17H11N2O3, 291.0764; found, 291.0764.
2-((4-((3,5-Bis(trifluoromethyl)phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4p).
Off-white solid; (305 mg, 77% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.53 (s, 1H), δ 8.22 (s, 2H) δ 8.09 (s, 1H), δ 7.87 (d, J = 8.5 Hz, 2H), δ 7.55 (d, J = 8.5 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.8, 162.7, 140.4, 132.8, 132.2, 131.7, 131.4, 131.2, 130.9, 125.8, 124.5, 122.3, 122.1, 119.9, 116.0, 93.7, 86.3. Mass spectra (ESI) m/z: 400 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C18H8F6NO3, 400.0408; found, 400.0413.
2-((4-(Naphthalen-2-ylethynyl)phenyl)amino)-2-oxoacetic Acid (4q).
Off-white solid; (249 mg, 80% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.70 (s, 1H), δ 8.15 (s, 1H), δ 7.92–7.95 (m, 3H), δ 7.85 (d, J = 8.5 Hz, 2H), δ 7.54–7.59 (m, 5H); 13C NMR (DMSO-d6, 125 MHz): δ 162.5, 139.3, 133.1, 132.8, 132.5, 131.5, 128.8, 128.5, 128.2, 127.5, 127.3, 120.3, 117.8, 90.3, 89.6. Mass spectra (ESI) m/z: 314 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C20H12NO3, 314.0811; found, 314.0812.
2-((4-((4-(Benzyloxy)phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4r).
White powder; (245 mg, 67% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.69 (s, 1H), δ 7.84 (br, 2H), δ 7.55 (d, J = 8.0 Hz, 2H), δ 7.43–7.47 (m, 5H), δ 7.38 (t, J = 8.5 Hz, 2H), δ 7.33 (t, J = 7.5 Hz, 2H), δ 7.03 (d, J = 8.5 Hz, 2H), δ 5.12 (s, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 159.0, 137.2, 133.6, 133.3, 132.2, 128.9, 128.4, 128.3, 120.4, 118.4, 115.7, 115.1, 89.4, 88.6, 69.8. Mass spectra (ESI) m/z: 370 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C23H16NO4, 370.1076; found, 370.1076.
2-((4-((4-((tert-Butoxycarbonyl)amino)phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4s).
Pale-yellow solid; (275 mg, 73% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.80 (s, 1H), δ 9.57 (s, 1H), δ 7.81 (d, J = 8.5 Hz, 2H) δ 7.40–7.50 (m, 6H), δ 1.46 (s, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.5, 158.2, 153.0, 140.5, 138.4, 132.3, 132.2, 120.6, 118.7, 118.4, 115.9, 89.8, 88.6, 79.9, 28.5. Mass spectra (ESI) m/z: 379 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C21H19N2O5, 379.1294; found, 379.1300.
2-((4-((2-Chloro-4-(trifluoromethyl)phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4t).
Off-white powder; (280 mg, 77% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.78 (s, 1H), δ 8.00 (s, 1H), δ 7.87 (t, J = 9.0 Hz, 3H), δ 7.74 (d, J = 8.5 Hz, 1H), δ 7.57 (d, J = 8.5 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.5, 159.6, 139.9, 135.7, 134.5, 132.8, 130.3, 130.1, 126.9, 126.7, 124.7, 122.5, 120.4, 116.7, 97.7, 84.9. Mass spectra (ESI) m/z: 366 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C17H7ClF3NO3, 366.0145; found, 366.0143.
2-((4-((2-Bromo-4-(trifluoromethyl)phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4u).
Off-white powder; (302 mg, 74% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.65 (s, 1H), δ 8.12 (s, 1H), δ 7.82–7.88 (m, 3H), δ 7.78 (d, J = 8.5 Hz, 1H), δ 7.56 (d, J = 8.5 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.6, 161.0, 140.2, 134.3, 132.8, 129.6, 129.2, 125.6, 125.1, 120.2, 116.4, 97.2, 86.9. Mass spectra (ESI) m/z: 410 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C17H8BrF3NO3, 409.9640; found, 409.9636.
2-((4-((2-Fluoro-4-(trifluoromethyl)phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4v).
White solid; (292 mg, 84% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.76 (s, 1H), δ 7.87 (d, J = 9.0 Hz, 2H) δ 7.79 (t, J = 8.0 Hz, 1H), δ 7.70 (d, J = 11.5 Hz, 1H), δ 7.55 (d, J = 8.5 Hz, 3H); 13C NMR (DMSO-d6, 125 MHz): δ 162.6, 160.2, 159.7, 158.1, 139.8, 132.8, 129.8, 129.7, 128.4, 128.2, 123.9, 121.8, 120.4, 120.0, 119.8, 116.7, 93.8, 86.9. Mass spectra (ESI) m/z: 350 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C17H8F4NO3, 350.0435; found, 350.0442.
2-((4-((2-Cyano-4-(trifluoromethyl)phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4w).
Pale-yellow solid; (250 mg, 71% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.78 (s, 1H), δ 8.41 (s, 1H) δ 8.07 (d, J = 8.5 Hz, 1H), δ 7.94 (d, J =11.5 Hz, 1H), δ 7.90 (d, J = 8.5 Hz, 2H), δ 7.58 (d, J = 8.5 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.6, 160.1, 140.4, 133.4, 133.1, 130.7, 130.4, 129.8, 129.5, 129.3, 129.0, 124.5, 122.4, 120.5, 116.8, 115.9, 115.4, 98.7, 85.0; mass spectra (ESI) m/z: 357 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C18H8F3N2O3, 357.0487; found, 357.0493.
2-((4-((2,4-Bis(trifluoromethyl)phenyl)ethynyl)phenyl)amino)-2-oxoacetic Acid (4x).
White powder; (275 mg, 73% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.87 (s, 1H), δ 8.10 (s, 2H), δ 8.01 (d, J = 8.5 Hz, 1H), δ 7.88 (d, J = 8.5 Hz, 2H), δ 7.55 (d, J = 8.5 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.4, 158.6, 139.9, 135.4, 132.8, 130.1, 125.2, 123.6, 120.7, 116.7, 98.3, 84.2; Mass spectra (ESI) m/z: 400 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C18H8F6NO3, 400.0408; found, 400.0415.
2-Oxo-2-((3-(phenylethynyl)phenyl)amino)acetic Acid (5).
Off-white solid; (229 mg, 88% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.73 (s, 1H), δ 8.00 (s, 1H), δ 7.74–7.77 (m, 1H), δ 7.54.7.56 (m, 2H), δ 7.39–7.43 (d, 3H), δ 7.37 (d, J = 7.5 Hz, 2H), δ 7.29 (d, J = 7.5 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 162.6, 158.6, 138.7, 131.9, 129.7, 129.4, 129.3, 127.7, 122.9, 122.6, 121.1, 89.7, 89.6. Mass spectra (ESI) m/z: 264 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C16H10NO3, 264.0661; found, 264.0670.
Synthesis of 2-([1,1′-Biphenyl]-4-ylamino)-2-oxoacetic Acid (6).
To a stirred solution of 4-biphenylamine (300 mg, 1.77 mmol) and N,N-diisopropyl ethylamine (613 μL, 3.55 mmol) in 20 mL of anhydrous CH2Cl2 was added methyl chlorooxoacetate (180 μL, 1.95 mmol) dropwise under a N2 atmosphere. The resulting solution was stirred at room temperature for 30 min. The reaction mixture was washed with 20 mL of DI water and 20 mL of brine. The organic layer was dried over anhydrous Na2SO4 and evaporated in vacuo. Crude ester was subjected to hydrolysis using 15 mL of 1 N KOH/THF (1:1 v/v) at room temperature for 1 h. After completion, THF was evaporated, the aqueous layer was acidified to pH ~2 using 3 N HCL, and the resulting solid was filtered. Products were then purified by column chromatography on silica gel eluted with a mixture of 1:9 methanol/DCM. White solid; (374 mg, 87% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.80 (s, 1H), δ 7.86 (d, J = 8.5 Hz, 2H), δ 7.63–7.66 (m, 4H), δ 7.43 (t, J = 7.5 Hz, 2H), δ 7.32 (t, J = 7.5 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 162.6, 157.4, 139.9, 137.6, 136.6, 129.4, 127.7, 127.4, 126.8, 121.1. Mass spectra (ESI) m/z: 240 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C14H10NO3, 240.0661; found, 240.0669.
Synthesis of 2-([1,1′-Biphenyl]-3-ylamino)-2-oxoacetic Acid (7).
To an ice-cold solution of 3-biphenylamine (300 mg, 1.77 mmol) and N,N-diisopropyl ethylamine (613 μL, 3.55 mmol) in 20 mL of anhydrous CH2Cl2 was added methyl chlorooxoacetate (180 μL, 1.95 mmol) dropwise under a N2 atmosphere. The resulting solution was stirred at room temperature for 30 min. The reaction mixture was washed with 20 mL of DI water and 20 mL of brine. The organic layer was dried over anhydrous Na2SO4 and evaporated in vacuo. Crude ester was subjected to hydrolysis using 15 mL of 1 N KOH/THF (1:1 v/v) at room temperature for 1 h. After completion, THF was evaporated, the aqueous layer was acidified to pH ~2 using 3 N HCL, and the resulting solid was filtered. Products were then purified by column chromatography on silica gel eluted with a mixture of 1:9 methanol/DCM. White powder; (360 mg, 84% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.75 (s, 1H), δ 8.08 (s, 1H), δ 7.78 (dt, J = 7.0, 2.0 Hz, 2H), δ 7.61 (d, J = 9.5 Hz, 1H), δ 7.41–7.48 (m, 4H), δ 7.36 (t, J = 7.0 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 162.6, 157.7, 141.2, 140.4, 138.8, 129.8, 129.5, 128.1, 127.1, 123.3, 119.7, 119.1. Mass spectra (ESI) m/z: 240 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C14H10NO3, 240.0661; found, 240.0669.
Synthesis of 2-Oxo-2-(quinolin-3-ylamino)acetic Acid (8).
To a stirred solution of 3-aminoquinoline (400 mg, 2.77 mmol) and N,N-diisopropyl ethylamine (960 μL, 5.55 mmol) in 25 mL of anhydrous CH2Cl2 was added methyl chlorooxoacetate (281 μL, 3.05 mmol) dropwise under a N2 atmosphere. The resulting solution was stirred at room temperature for 30 min. The reaction mixture was washed with 25 mL of DI water and 25 mL of brine. The organic layer was dried over anhydrous Na2SO4 and evaporated in vacuo. Crude ester was subjected to hydrolysis using 20 mL of 1 N KOH/THF (1:1 v/v) at room temperature for 1 h. After completion, THF was evaporated, the aqueous layer was acidified to pH ~2 using 3 N HCL, and the resulting solid was filtered. Compound 7 was then purified by column chromatography on silica gel eluted with a mixture of 1:9 methanol/DCM. Yellow powder; (450 mg, 75% yield). 1H NMR (DMSO-d6, 500 MHz): δ 11.35 (s, 1H), δ 9.25 (s, 1H), δ 8.97 (s, 1H), δ 8.07 (t, J = 7.0 Hz, 2H), δ 7.78 (t, J = 7.0 Hz, 1H), δ 7.67 (t, J = 7.0 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 161.9, 158.0, 143.8, 142.2, 132.3, 130.4, 128.7, 128.5, 128.2, 127.1, 126.7. Mass spectra (ESI) m/z: 215 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C11H7N2O3, 215.0457; found, 215.0464.
(Z)-2-Oxo-2-((4-styrylphenyl)amino)acetic Acid (9).
Off-white powder; (37 mg, 74% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.67 (s, 1H), δ 7.64 (d, 8.5 Hz, 2H), δ 7.24 (d, 8.5 Hz, 2H), δ 7.19–7.22 (m, 3H), δ 7.16 (d, J = 8.5 Hz, 1H), δ 6.57 (s, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.6, 157.9, 137.4, 137.3, 133.3, 130.1, 129.9, 129.4, 129.0, 128.9, 128.8, 128.7, 127.7, 120.4. Mass spectra (ESI): m/z: 266 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C17H12NO3, 266.0817; found, 266.0826.
(Z)-2-Oxo-2-((4-(4-(trifluoromethyl)styryl)phenyl)amino)acetic Acid (10).
Off-white powder; (32 mg, 72% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.71 (s, 1H), δ 7.68 (d, J = 8.0 Hz, 2H), δ 7.61 (d, J = 8.0 Hz, 2H), δ 7.41 (d, J = 8.5 Hz, 2H), δ 7.17 (d, J = 8.5 Hz, 2H), δ 6.72 (d, J =12 Hz, 1H), δ 7.64 (d, J =12 Hz, 1H); 13C NMR (DMSO-d6, 125 MHz): δ 162.5, 157.8, 141.7, 137.6, 132.7, 132.1, 129.7, 129.5, 128.6, 125.8, 120.5. Mass spectra (ESI) m/z: 334 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C17H11F3NO3, 334.0691; found, 334.0702.
2-Oxo-2-((4-phenethylphenyl)amino)acetic Acid (11).
White crystals; (47 mg, 93% yield). 1H NMR (MeOH-d4, 500 MHz): δ 7.56 (d, J = 8.5 Hz, 2H), δ 7.21–7.24 (m, 2H), δ 7.12–7.16 (m, 5H), δ 2.88 (s, 4H); 13C NMR (MeOH-d4, 125 MHz): δ 141.5, 138.2, 135.0, 128.5, 128.2, 127.9, 125.5, 119.8, 37.6, 37.2. Mass spectra (ESI) m/z: 268 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C16H15NO3, 268.0968; found, 268.0980.
2-Oxo-2-((4-(4-(trifluoromethyl)phenethyl)phenyl)amino)acetic Acid (12).
White crystals; (45 mg, 90% yield). 1H NMR (DMSO-d6, 500 MHz): δ 10.61 (s, 1H), δ 7.59–7.64 (m, 4H) δ 7.42 (d, J = 8.0 Hz, 2H), δ 7.18 (d, J = 8.5 Hz, 2H), δ 2.93–2.96 (m, 2H), δ 2.84–2.88 (m, 2H); 13C NMR (DMSO-d6, 125 MHz): δ 162.6, 157.2, 146.9, 137.7, 136.1, 129.7, 129.1, 127.5, 127.2, 126.9, 126.7, 126.0, 125.5, 125.4, 123.8, 120.7, 37.0, 36.4. Mass spectra (ESI) m/z: 336 (M − H)−. HRMS (ESI-TOF, [M − H]−) m/z: calcd for C17H14F3NO3, 336.0848; found, 336.0852.
Molecular Modeling Studies.
The molecular modeling studies were performed using the previously published crystal structure of mPTPB in complex with OMTS (PDB code:2OZ5).36 Schrodinger Molecular Modeling Suite 2019-4 (Schrodinger, LLC, New York, NY, 2019) was used for the modeling studies with procedures similar to those described before. We used extra precision Glide (Glide-XP) for our docking studies.61 Briefly, the structures of the protein–ligand complexes were prepared using the protein preparation module, and the ligand binding sites were defined based on the native ligand (OMTS). The inhibitors described in this study were built and prepared for docking using the Ligprep module. Data analyses were performed using the Maestro interface of the software.62
Determination of Log D.
To determine the lipophilicity of our inhibitors, we measured the Log D at pH 7.4 using the shake flask method. An aliquot of 10 mM DMSO stock solutions of test compounds was added into an Eppendorf containing 2 mL of n-octanol and pH = 7.4 PBS (1:1 v/v) to give a final concentration of 100 μM. The tubes were shaken for 8 h and centrifuged at 10,000 rpm for 5 min and then n-octanol and PBS layers were separated. Both layers were analyzed using LC─MS (Agilent Technologies 6470 series, triple quadrupole LC─MS). The ratios of AUC of the peaks were used to calculate the Log D in accordance with the equation
Determination of Kinetic Solubility.
The kinetic solubility of compounds (4b, 4h, 4k, 4t, 4u, and 4v) was measured by diluting 10 μL of 20 mM DMSO stock into 990 μL of PBS buffer at pH = 7.4 (200 μM final inhibitor concentration) in a glass vial at 25 °C. The resulting mixture was stirred at 500 rpm for 90 min and then filtered using a 0.45 μm PVDF membrane filter. After filtration, the concentration of the corresponding compound was determined by HPLC, comparing the AUC obtained with that from a standard solution of the compound. Data represent the mean (±standard deviation, SD) of three separate experiments, performed in triplicate.
MLM Assays.
Mouse (CD-1)-pooled liver microsomes were purchased from Fisher Scientific, IL, USA (0.5 mg/mL, Corning Gentest 3P, cat# 452702), and the assay procedures were followed as previously reported.63 A solution of 1.0 mg of mouse liver microsomes (final concentration) and 1 mM cofactor NADPH (final concentration) in 1 mL of 100 mM (PBS, pH = 7.4) was prewarmed to 37 °C in an Eppendorf tube. Then, 10 μL of a test solution (from 10 mM DMSO stock of the inhibitor) was added to initiate the reaction. Verapamil–HCl was used as a positive control. Another set of control inactive microsome experiments (without cofactor NADPH) were performed. The incubation mixture was kept at 37 °C, and 50 μL aliquots were taken at times of 0, 5, 10, 15, 30, 60, 90, or 120 min. Each of the aliquot was quenched using 200 μL of ice-cold acetonitrile. The mixtures were vigorously vortexed and centrifuged to remove the precipitated protein and the supernatants were analyzed using an Agilent Technologies 6470 series, triple quadrupole LC─MS spectrometer to quantitate the remaining parent compound. The percent of the parent compound remaining was calculated from the formula
IC50 Determination and Ki Determination.
Compounds were tested against mPTPB using the pNPP assay system in a Cary100 UV–vis spectrophotometer by monitoring the increase in absorbance of the product formed, p-nitrophenol (pNP), at 405 nm using a molar extinction coefficient of 18,000 M−1 cm−1.18,21 The reaction was started by the addition of 5 μL of mPTPB into a final volume of 200 μL of the master mix containing pH 7.0, 50.0 mM 3,3-dimethylglutarate, 1 mM EDTA, 0.15 M NaCl buffer, 3.0 mM pNPP (the Km value), and various concentrations of the inhibitor (maximum concentration of 500 nM for 4a to 4x and 10 μM for all other compounds with 1.5-fold dilution and 10 different concentration points). The reaction was quenched after 30 min by the addition of 50 μL of 5 N NaOH. The amount of the product pNP was determined from the absorbance at 405 nm. IC50 values for each compound were determined by fitting the dose–response data into the four-parameter logistic curve (eq 1) model of GraphPad prism 7.02 as follows.
| (1) |
where AI is the absorbance at 405 nm of the sample with the inhibitor; A0 is the absorbance at 405 nm without the inhibitor; and [I] is the concentration of the inhibitor.
For selectivity studies, the catalytic domain of PTPs, including SHP2, SHP1, PTP1B, mPTPA, TCPTP, FAP1, LAR, CD45-D1D2, PTPγ-D1D2, VHR, Cdc14A, LYP, PTP-MEG2, HePTP, PTPα-D1D2, Laforin, DEP-1-D1, MKP3, MKP5, YopH, PTP-PEST, STEP, PTPσ-D1D2, PTPβ-D1, PTPμ-D1, PTPε, and LMWPTP, was expressed and purified from E. coli (BL21). The inhibition assay for these PTPs was performed under the same conditions as mPTPB except using a different pNPP concentration corresponding to the Km of the PTP studied.
The inhibition constants (Ki) for the inhibitors against mPTPB were determined at pH 7.0 and 25 °C. At various fixed concentrations of the inhibitor, the initial rate for a series of pNPP concentrations was measured by following the production of pNP (UV absorbance at 405 nm) as described above, ranging from 0.2- to 5-fold the apparent Km values. The data were fitted using SigmaPlot-Enzyme Kinetics to obtain the inhibition constant and to assess the mode of inhibition.
Cell Permeability Studies.
We determined the intracellular concentrations of our compounds in Raw264.7 cells, using the previously reported protocol by Teuscher et al.53 The calibration curves of compounds are shown in the Supporting Information Figures S49-S51. The input concentration was set to 20 μM. The cell-bound concentration of 4k, 4s, 4t, and 4u at 37 °C was determined after 2 h incubation in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS (Invitrogen), penicillin (50 units/mL), and streptomycin (50 μg/mL). In these experiments, we used our previously reported cellular active mPTPB inhibitor, compound 9, as a positive control.55 All these compounds showed better cellular permeability compared to compound 9 under identical conditions.
Cytotoxicity Studies.
A 90 μL aliquot of MEF cells (3 × 103 cells) was seeded in a 96-well plate and treated with 10 μL aliquots of varying compound concentrations (25 μM maximum concentration) for 72 h. Cell viability was measured using tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay by incubating the cells with MTT for 3 h, dissolving the crystals in DMSO, and measuring the optical density at 490 nm.65
Recombinant Protein Production.
pET28b-mPTPB18 was used to transform into E. coli BL21/DE3 and grown in LB medium containing 50 μg/mL kanamycin at 37 °C to an OD600 of 0.5. Site-directed mutagenesis was performed on wild-type pET28-mPTPB, using Quick Change PCR to generate the mutants I203A, I207A, I207K, F161S, K164I, and D165G. Following the addition of IPTG to a final concentration of 20 μM, the culture was incubated at 20 °C with shaking for additional 16 h. The cells were harvested by centrifugation at 5000 rpm for 5 min at 4 °C. The bacterial cell pellets were resuspended in 20 mM Tris, pH 7.9, 500 mM NaCl, and 5 mM imidazole and were lysed by sonication. Cellular debris was removed by centrifugation at 16,000 rpm for 30 min at 4 °C. The protein was purified from the supernatant using standard procedures of Ni-nitrilotriacetic acid-agarose (Qiagen) affinity purification. The protein eluted from the Ni-NTA column was concentrated with an Amicon Ultra centrifugal filter device (Millipore) and the buffer was changed to 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT. Protein concentration was determined using the Bradford dye binding assay (Bio-Rad) diluted according to the manufacturer’s recommendations with bovine serum albumin as the standard. The purified mPTPB was made to 20% glycerol and stored at −20 °C. The purified proteins were tested using pNPP as a substrate at pH 7.0, in 50.0 mM 3,3-dimethylglutarate, 1 mM EDTA, and 0.15 M NaCl.
Cell Culture and Transfection.
Raw264.7 mouse macrophages were cultured in DMEM supplemented with 10% FBS (Invitrogen), penicillin (50 units/mL), and streptomycin (50 μg/mL) under a humidified atmosphere containing 5% CO2 at 37 °C. mPTPB was subcloned into pcN-HA expression vectors. Raw 264.7 cells were seeded at 40% confluency in an antibiotic-free medium and grown overnight, and HA-tagged mPTPB or pcN-HA empty vectors were transfected into cells by electroporation at 800 μF and 280 V. After 24 h of transfection, 0.5 mg/mL G418 was added to the culture medium. Stable clones were picked after two weeks of selection. PcN-HA vector or mPTPB-transfected Raw264.7 cells were seeded in a 24-well plate at a density of 2 × 104 cells/well as previously reported by our group.55 After 48 h, the cells were treated with the mPTPB inhibitor 4t at different concentrations for 2 h and then stimulated with IFN-γ (20 ng/mL) for 1 h. Cells were then lysed in 1 × SDS sample buffer and the lysates were electrophoresed on a 10% polyacrylamide gel and then transferred to a nitrocellulose membrane and probed with anti-phospho-ERK1/2, anti-phospho-p38, anti-phospho-Akt, anti-ERK1/2, anti-AKT, anti-p38 (Cell Signaling Technology), anti-GAPDH, and anti-HA (Santa Cruz) followed by incubation with horseradish rabbit or mouse peroxidase-conjugated secondary antibodies. The blots were developed by the enhanced chemiluminescence technique using the SuperSignal West Pico Chemiluminescent substrate (Pierce).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Health Grant RO1 CA207288 and the Robert C. and Charlotte Anderson Chair Endowment.
ABBREVIATIONS
- DCM
dichloromethane
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- mPTPA
mycobacterium protein tyrosine phosphatase A
- mPTPB
mycobacterium protein tyrosine phosphatase B
- pTyr
phosphotyrosine
- OBA
2-oxalamino benzoic acid
- pNPP
p-nitrophenyl phosphate
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00302.
1H and 13C NMR spectra for all compounds, LC─MS data for selected compounds, molecular docking structures of 4k and 4t, and calibration curves used for physicochemical property calculations (PDF)
Molecular formula strings for compounds (CSV)
The authors declare no competing financial interest.
Contributor Information
Kasi Viswanatharaju Ruddraraju, Department of Medicinal Chemistry and Molecular Pharmacology, Department of Chemistry, Center for Cancer Research, and Institute for Drug Discovery, Purdue University, West Lafayette, Indiana 4790, United States.
Devesh Aggarwal, Department of Medicinal Chemistry and Molecular Pharmacology, Department of Chemistry, Center for Cancer Research, and Institute for Drug Discovery, Purdue University, West Lafayette, Indiana 4790, United States.
Congwei Niu, Department of Medicinal Chemistry and Molecular Pharmacology, Department of Chemistry, Center for Cancer Research, and Institute for Drug Discovery, Purdue University, West Lafayette, Indiana 4790, United States.
Erica Anne Baker, Department of Medicinal Chemistry and Molecular Pharmacology, Department of Chemistry, Center for Cancer Research, and Institute for Drug Discovery, Purdue University, West Lafayette, Indiana 4790, United States.
Ruo-yu Zhang, Department of Medicinal Chemistry and Molecular Pharmacology, Department of Chemistry, Center for Cancer Research, and Institute for Drug Discovery, Purdue University, West Lafayette, Indiana 4790, United States.
Li Wu, Department of Medicinal Chemistry and Molecular Pharmacology, Department of Chemistry, Center for Cancer Research, and Institute for Drug Discovery, Purdue University, West Lafayette, Indiana 4790, United States.
Zhong-Yin Zhang, Department of Medicinal Chemistry and Molecular Pharmacology, Department of Chemistry, Center for Cancer Research, and Institute for Drug Discovery, Purdue University, West Lafayette, Indiana 4790, United States.
REFERENCES
- (1).Hunter T A thousand and one protein kinases. Cell 1987, 50, 823–829. [DOI] [PubMed] [Google Scholar]
- (2).Ardito F; Giuliani M; Perrone D; Troiano G; Muzio LL The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med 2017, 40, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).He R.-j.; Yu Z.-h.; Zhang R.-y.; Zhang Z.-y. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol. Sin 2014, 35, 1227–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ruddraraju KV; Zhang Z-Y Covalent inhibition of protein tyrosine phosphatases. Mol. Biosyst 2017, 13, 1257–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zhang Z-Y Drugging the undruggable: therapeutic potential of targeting protein tyrosine phosphatases. Acc. Chem. Res 2017, 50, 122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bialy L; Waldmann H Inhibitors of protein tyrosine phosphatases: Next-generation drugs? Angew. Chem., Int. Ed 2005, 44, 3814–3839. [DOI] [PubMed] [Google Scholar]
- (7).Frankson R; Yu Z-H; Bai Y; Li Q; Zhang R-Y; Zhang Z-Y Therapeutic targeting of oncogenic tyrosine phosphatases. Cancer Res. 2017, 77, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Koul A; Choidas A; Treder M; Tyagi AK; Drlica K; Singh Y; Ullrich A Cloning and characterization of secretory tyrosine phosphatases of Mycobacterium tuberculosis. J. Bacteriol 2000, 182, 5425–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).World Health Organization. Global Tuberculosis Report 2018, 2018.
- (10).Munro SA; Lewin SA; Smith HJ; Engel ME; Fretheim A; Volmink J Patient adherence to tuberculosis treatment: a systematic review of qualitative research. PLoS Med. 2007, 4, No. e238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhang Y The magic bullets and tuberculosis drug targets. Annu. Rev. Pharmacol. Toxicol 2005, 45, 529–564. [DOI] [PubMed] [Google Scholar]
- (12).Rasko DA; Sperandio V Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug. Discov 2010, 9, 117–128. [DOI] [PubMed] [Google Scholar]
- (13).Silva AP; Tabernero L New strategies in fighting TB: targeting Mycobacterium tuberculosis-secreted phosphatases MptpA & MptpB. Future Med. Chem 2010, 2, 1325–1337. [DOI] [PubMed] [Google Scholar]
- (14).Dickey SW; Cheung GYC; Otto M Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug. Discov 2017, 16, 457–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Fanzani L; Porta F; Meneghetti F; Villa S; Gelain A; Lucarelli A; Parisini E Mycobacterium tuberculosis low molecular weight phosphatases (MPtpA and MPtpB): From biological insight to inhibitors. Curr. Med. Chem 2015, 22, 3110–3132. [DOI] [PubMed] [Google Scholar]
- (16).Singh R; Rao V; Shakila H; Gupta R; Khera A; Dhar N; Singh A; Koul A; Singh Y; Naseema M; Narayanan PR; Paramasivan CN; Ramanathan VD; Tyagi AK Disruption of mptpB impairs the ability of Mycobacterium tuberculosis to survive in guinea pigs. Mol. Microbiol 2003, 50, 751–762. [DOI] [PubMed] [Google Scholar]
- (17).Singh R; Singh A; Tyagi AK Deciphering the genes involved in pathogenesis of Mycobacterium tuberculosis. Tuberculosis 2005, 85, 325–335. [DOI] [PubMed] [Google Scholar]
- (18).Zhou B; He Y; Zhang X; Xu J; Luo Y; Wang Y; Franzblau SG; Yang Z; Chan RJ; Liu Y; Zheng J; Zhang Z-Y Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 4573–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Vickers CF; Silva APG; Chakraborty A; Fernandez P; Kurepina N; Saville C; Naranjo Y; Pons M; Schnettger LS; Gutierrez MG; Park S; Kreiswith BN; Perlin DS; Thomas EJ; Cavet JS; Tabernero L Structure-based design of MptpB inhibitors that reduce multidrug-resistant mycobacterium tuberculosis survival and infection burden in vivo. J. Med. Chem 2018, 61, 8337–8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Dutta NK; He R; Pinn ML; He Y; Burrows F; Zhang Z-Y; Karakousis PC Mycobacterial protein tyrosine phosphatases A and B inhibitors augment the bactericidal activity of the standard anti-tuberculosis regimen. ACS Infect. Dis 2016, 2, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).He Y; Xu J; Yu Z-H; Gunawan AM; Wu L; Wang L; Zhang Z-Y Discovery and evaluation of novel inhibitors of mycobacterium protein tyrosine phosphatase B from the 6-Hydroxy-benzofuran-5-carboxylic acid scaffold. J. Med. Chem 2013, 56, 832–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).He R; Yu Z-H; Zhang R-Y; Wu L; Gunawan AM; Zhang Z-Y Cefsulodin inspired potent and selective inhibitors of mPTPB, a virulent phosphatase from mycobacterium tuberculosis. ACS Med. Chem. Lett 2015, 6, 1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zeng L-F; Xu J; He Y; He R; Wu L; Gunawan AM; Zhang Z-Y A facile hydroxyindole carboxylic acid based focused library approach for potent and selective inhibitors of Mycobacterium protein tyrosine phosphatase B. ChemMedChem 2013, 8, 904–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wong D; Chao JD; Av-Gay Y Mycobacterium tuberculosis-secreted phosphatases: from pathogenesis to targets for TB drug development. Trends Microbiol. 2013, 21, 100–109. [DOI] [PubMed] [Google Scholar]
- (25).Jarlier V; Nikaido H Mycobacterial cell wall: structure and role in natural resistance to antibiotics. FEMS Microbiol. Lett 1994, 123, 11–18. [DOI] [PubMed] [Google Scholar]
- (26).Brennan PJ Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 2003, 83, 91–97. [DOI] [PubMed] [Google Scholar]
- (27).He R; Yu Z-H; Zhang R-Y; Wu L; Gunawan AM; Lane BS; Shim JS; Zeng L-F; He Y; Chen L; Wells CD; Liu JO; Zhang Z-Y Exploring the existing drug space for novel pTyr mimetic and SHP2 inhibitors. ACS Med. Chem. Lett 2015, 6, 782–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Combs AP Recent advances in the discovery of competitive protein tyrosine phosphatase 1B inhibitors for the treatment of diabetes, obesity, and cancer. J. Med. Chem 2010, 53, 2333–2344. [DOI] [PubMed] [Google Scholar]
- (29).Andersen HS; Iversen LF; Jeppesen CB; Branner S; Norris K; Rasmussen HB; Møller KB; Møller NPH 2-(oxalylamino)-benzoic acid is a general, competitive inhibitor of protein-tyrosine phosphatases. J. Biol. Chem 2000, 275, 7101–7108. [DOI] [PubMed] [Google Scholar]
- (30).Xin Z; Oost TK; Abad-Zapatero C; Hajduk PJ; Pei Z; Szczepankiewicz BG; Hutchins CW; Ballaron SJ; Stashko MA; Lubben T; Trevillyan JM; Jirousek MR; Liu G Potent, selective inhibitors of protein tyrosine phosphatase 1B. Bioorg. Med. Chem. Lett 2003, 13, 1887–1890. [DOI] [PubMed] [Google Scholar]
- (31).Iversen LF; Andersen HS; Branner S; Mortensen SB; Peters GH; Norris K; Olsen OH; Jeppesen CB; Lundt BF; Ripka W; Møller KB; Møller NPH Structure-based design of a low molecular weight, nonphosphorus, nonpeptide, and highly selective inhibitor of protein-tyrosine phosphatase 1B. J. Biol. Chem 2000, 275, 10300–10307. [DOI] [PubMed] [Google Scholar]
- (32).Szczepankiewicz BG; Liu G; Hajduk PJ; Abad-Zapatero C; Pei Z; Xin Z; Lubben TH; Trevillyan JM; Stashko MA; Ballaron SJ; Liang H; Huang F; Hutchins CW; Fesik SW; Jirousek MR Discovery of a potent, selective protein tyrosine phosphatase 1B inhibitor using a linked-fragment strategy. J. Am. Chem. Soc 2003, 125, 4087–4096. [DOI] [PubMed] [Google Scholar]
- (33).Johnson TO; Ermolieff J; Jirousek MR Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug. Discov 2002, 1, 696–709. [DOI] [PubMed] [Google Scholar]
- (34).Liu G; Xin Z; Pei Z; Hajduk PJ; Abad-Zapatero C; Hutchins CW; Zhao H; Lubben TH; Ballaron SJ; Haasch DL; Kaszubska W; Rondinone CM; Trevillyan JM; Jirousek MR Fragment screening and assembly: a highly efficient approach to a selective and cell active protein tyrosine phosphatase 1B inhibitor. J. Med. Chem 2003, 46, 4232–4235. [DOI] [PubMed] [Google Scholar]
- (35).Hartshorn MJ; Murray CW; Cleasby A; Frederickson M; Tickle IJ; Jhoti H Fragment-based lead discovery using X-ray crystallography. J. Med. Chem 2005, 48, 403–413. [DOI] [PubMed] [Google Scholar]
- (36).Grundner C; Perrin D; Hooft van Huijsduijnen R; Swinnen D; Gonzalez J; Gee CL; Wells TN; Alber T Structural basis for selective inhibition of Mycobacterium tuberculosis protein tyrosine phosphatase PtpB. Structure 2007, 15, 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ishikawa M; Hashimoto Y Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem 2011, 54, 1539–1554. [DOI] [PubMed] [Google Scholar]
- (38).Sonogashira K Development of Pd–Cu catalyzed crosscoupling of terminal acetylenes with sp2 -carbon halides. J. Organomet. Chem 2002, 653, 46–49. [Google Scholar]
- (39).Wilcken R; Zimmermann MO; Lange A; Joerger AC; Boeckler FM Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem 2013, 56, 1363–1388. [DOI] [PubMed] [Google Scholar]
- (40).Lu Y; Wang Y; Zhu W Nonbonding interactions of organic halogens in biological systems: implications for drug discovery and biomolecular design. Phys. Chem. Chem. Phys 2010, 12, 4543–4551. [DOI] [PubMed] [Google Scholar]
- (41).Auffinger P; Hays FA; Westhof E; Ho PS Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 16789–16794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in medicinal chemistry. Chem. Soc. Rev 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]
- (43).Shah P; Westwell AD The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem 2007, 22, 527–540. [DOI] [PubMed] [Google Scholar]
- (44).Irwin JJ; Duan D; Torosyan H; Doak AK; Ziebart KT; Sterling T; Tumanian G; Shoichet BK An aggregation advisor for Ligand Discovery. J. Med. Chem 2015, 58, 7076–7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Aldrich C; Bertozzi C; Georg GI; Kiessling L; Lindsley C; Liotta D; Merz KM; Schepartz A; Wang S The ecstasy and agony of assay interference compounds. ACS Cent. Sci 2017, 3, 143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- (47).Grundner C; Ng H-L; Alber T Mycobacterium tuberculosis protein tyrosine phosphatase PtpB structure reveals a diverged fold and a buried active site. Structure 2005, 13, 1625–1634. [DOI] [PubMed] [Google Scholar]
- (48).Hopkins AL; Groom CR; Alex A Ligand efficiency: a useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- (49).Leeson PD; Springthorpe B The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug. Discov 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- (50).Hopkins AL; Keserü GM; Leeson PD; Rees DC; Reynolds CH The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug. Discov 2014, 13, 105–121. [DOI] [PubMed] [Google Scholar]
- (51).Quintieri L; Fantin M; Palatini P; De Martin S; Rosato A; Caruso M; Geroni C; Floreani M In vitro hepatic conversion of the anticancer agent nemorubicin to its active metabolite PNU-159682 in mice, rats and dogs: a comparison with human liver microsomes. Biochem. Pharmacol 2008, 76, 784–795. [DOI] [PubMed] [Google Scholar]
- (52).Palmer BD; Thompson AM; Sutherland HS; Blaser A; Kmentova I; Franzblau SG; Wan B; Wang Y; Ma Z; Denny WA Synthesis and structure-activity studies of biphenyl analogues of the tuberculosis drug (6S)-2-nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem 2010, 53, 282–294. [DOI] [PubMed] [Google Scholar]
- (53).Teuscher KB; Zhang M; Ji H A versatile method to determine the cellular bioavailability of small-molecule inhibitors. J. Med. Chem 2017, 60, 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Bhat J; Narayan A; Venkatraman J; Chatterji M LC-MS based assay to measure intracellular compound levels in Mycobacterium smegmatis: linking compound levels to cellular potency. J. Microbiol. Methods 2013, 94, 152–158. [DOI] [PubMed] [Google Scholar]
- (55).He R; Yu Z-H; Zhang R-Y; Wu L; Gunawan AM; Zhang Z-Y Cefsulodin inspired potent and selective inhibitors of mPTPB, a virulent phosphatase from mycobacterium tuberculosis. ACS Med. Chem. Lett 2015, 6, 1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Gleeson MP Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem 2008, 51, 817–834. [DOI] [PubMed] [Google Scholar]
- (57).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- (58).Hughes JD; Blagg J; Price DA; Bailey S; Decrescenzo GA; Devraj RV; Ellsworth E; Fobian YM; Gibbs ME; Gilles RW; Greene N; Huang E; Krieger-Burke T; Loesel J; Wager T; Whiteley L; Zhang Y Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett 2008, 18, 4872–4875. [DOI] [PubMed] [Google Scholar]
- (59).Beresford NJ; Saville C; Bennett HJ; Roberts IS; Tabernero L A new family of phosphoinositide phosphatases in microorganisms: identification and biochemical analysis. BMC Genom. 2010, 11, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Kastner R; Dussurget O; Archambaud C; Kernbauer E; Soulat D; Cossart P; Decker T LipA, a tyrosine and lipid phosphatase involved in the virulence of Listeria monocytogenes. Infect. Immun 2011, 79, 2489–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- (62).Schrödinger Release 2019–4; Maestro, Schrödinger, LLC: New York, NY, 2019. [Google Scholar]
- (63).Li D; Zhang X; Ma X; Xu L; Yu J; Gao L; Hu X; Zhang J; Dong X; Li J; Liu T; Zhou Y; Hu Y Development of macrocyclic peptides containing epoxyketone with oral availability as proteasome inhibitors. J. Med. Chem 2018, 61, 9177–9204. [DOI] [PubMed] [Google Scholar]
- (64).The PyMOL Molecular Graphics System. Version 2.0.6; Schrödinger, LLC, 2017. [Google Scholar]
- (65).Mosmann T Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.