

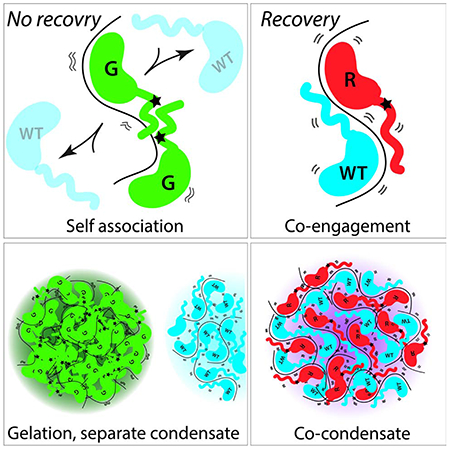
Abstract
The RNA-binding protein Fused in sarcoma (FUS) can form pathogenic inclusions in neurodegenerative diseases like amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia (FTLD). Over 70 mutations in Fus are linked to ALS/FTLD. In patients, all Fus mutations are heterozygous, indicating that the mutant drives disease progression despite the presence of wild-type FUS. Here, we demonstrate that ALS/FTLD-linked FUS mutations in glycine (G) strikingly drive formation of droplets that do not readily interact with wild-type FUS whereas arginine (R) mutants form mixed condensates with wild-type FUS. Remarkably, interactions between wild-type and G mutants are disfavored at the earliest stages of FUS nucleation. In contrast, R mutants physically interact with the wild-type FUS such that wild-type FUS recovers the mutant defects by reducing droplet size and increasing dynamic interactions with RNA. This result suggests disparate molecular mechanisms underlying ALS/FTLD pathogenesis and differing recovery potential depending on the type of mutation.
Keywords: FUS, RNA, miscibility, ALS/FTLD-linked mutation, LLPS, nucleation, arrested dynamics
eTOC blurb
FUS is an RNA binding protein with over seventy mutations linked to the incurable neurodegenerative diseases, ALS/FTLD. Rhine et al demonstrates that FUS mutations in glycine do not associate with the wildtype at all levels while the mutations in arginine interacts with wildtype FUS and thereby recover the mutant defect.
Graphical Abstract
I. Introduction
Protein aggregation is a well-known pathological mechanism for many neurodegenerative diseases (Elbaum-Garfinkle, 2019). In amyotrophic lateral sclerosis (ALS) and frontotemporal lobar dementia (FTLD), which exist on a pathological spectrum, various RNA binding proteins form pathologic inclusions, including TDP-43, FUS, TIA1, TAF15, EWSR1 and hnRNPA1 (Couthouis et al., 2012; DeJesus-Hernandez et al., 2011; Kapeli et al., 2016; Kim et al., 2013; Kwiatkowski et al., 2009; Mackenzie et al., 2017; Rosen et al., 1993; Sreedharan et al., 2008). They possess intrinsically-disordered regions (IDRs), which promote aggregation and are often enriched with repeats of arginines and glycines (Fay et al., 2017; Wang et al., 2018). In addition, they bind RNA, which promotes the formation of phase-separated condensates (Colombrita et al., 2012; Daigle et al., 2013; Niaki et al., 2020; Schwartz et al., 2013). Once formed, the condensates can convert to fibrillar solids, which resemble the pathogenic inclusions observed in post-mortem ALS/FTLD neurons (French et al., 2019; Patel et al., 2015).
FUS is a highly disordered protein known to undergo Liquid-liquid phase-separation (LLPS) (Niaki et al., 2020; Schwartz et al., 2013; Wang et al., 2018). LLPS of FUS involves extensive multivalent interactions between arginine and tyrosine-enriched motifs, which are interspersed with glycines and other amino acids (Martin et al., 2020; Wang et al., 2018; Yoshizawa et al., 2018). While FUS can interact homotypically, RNA promotes FUS-FUS oligomerization in a length-dependent manner (Daigle et al., 2013; Maharana et al., 2018; Niaki et al., 2020; Schwartz et al., 2013). Together, both FUS-FUS and FUS-RNA interactions drive LLPS of FUS.
Mutations can disrupt FUS’s RNA binding and other intermolecular interactions. There are over 70 ALS/FTLD-linked mutations in Fus which are all heterozygous (Deng et al., 2014). They are enriched in IDRs and nuclear localization signal (NLS) (Deng et al., 2014). Mutations in the NLS lead to severe disease pathology due to mislocalization and aggregation (Guo et al., 2018; Hofweber et al., 2018; Qamar et al., 2018; Yoshizawa et al., 2018). By contrast, mutations in the IDRs induce aberrant condensation and defective interactions with RNA (Niaki et al., 2020; Patel et al., 2015). Over half of ALS/FTLD mutations occur at arginine and glycine residues (Deng et al., 2014). We and others have shown that FUS mutations in arginine have defective interactions with RNA and form larger condensates, whereas glycine mutations form solid-like condensates (Niaki et al., 2020; Wang et al., 2018).
Notably, ALS/FTLD progression occurs despite the presence of a healthy wild-type copy of each protein (Kwiatkowski et al., 2009; Mackenzie et al., 2010; Sreedharan et al., 2008). FUS mutations dysregulate normal FUS activities in RNA metabolism, including splicing and RNA trafficking (Daigle et al., 2013; Kabashi et al., 2011; Sun et al., 2015; Wang et al., 2011), but it is unclear if it is due to loss of function of the wildtype or the gain of toxicity due to the mutation (Kamelgarn et al., 2018; Lebedeva et al., 2017; Lopez-Erauskin et al., 2018; Sharma et al., 2016; Shiihashi et al., 2016; Sun et al., 2015). Do the wild-type and mutant protein physically interact, at the molecular level or during condensate formation? If so, does the mutant exert a disruptive effect on wildtype? Or can the wild-type protein temporarily recover the mutant’s defect?
Here, we investigate how wild-type FUS affects mutant FUS RNA binding, oligomerization, condensate formation, and fusion. We sought to dissect the interaction of wild-type and mutant FUS using single-molecule, ensemble, and cellular methods, focusing on four fundamental questions: (1) Do wild-type and mutant FUS interact to form mixed condensates with an RNA scaffold? (2) At what stage does mutant and wildtype FUS engage with or disengage from each other? (3) What is the RNA binding phenotype of wild-type and mutant FUS together? (4) How does this interaction change as mutant FUS matures? We demonstrate that G mutants do not associate with wildtype FUS starting at the earliest stages of FUS-RNA nucleation and condensate formation, whereas R mutants physically interact with wild-type at all stages, resulting in the recovery of the R mutant defect. This recovery effect is reduced if wild-type FUS is added to aged mutant FUS condensates. Moreover, we show by mutational analysis that the molecular defects observed for the ALS/FTLD-linked G156E mutation arise from the position and bulkiness of the amino acid substitution. These results reveal that the accelerated gelation of G mutant may be an important facet of FUS-mediated ALS/FTLD pathogenesis, and the lack of a physical association with wild-type may contribute to a more severe pathology.
R. Results
We expressed, purified, and fluorescently labeled several ALS/FTLD-linked variants of FUS: wild-type (WT), R mutants (R244C, R216C, R521G), and G mutants (G156E, G187S, G399V). The ALS/FTLD lined R and G mutations are located in the QGSY-rich, RGG, and NLS domains of FUS (Corrado et al., 2010; Deng et al., 2014; Kwiatkowski et al., 2009; Mackenzie et al., 2010; Rademakers et al., 2010; Ticozzi et al., 2009). To limit premature oligomerization of FUS during expression and purification, we used mild induction conditions, maintained a low concentration throughout purification, stored in 1 M urea, added a proteolytically removable solubility tag, maltose binding protein (MBP) and only used fresh (<2 weeks old) proteins for experiments (Niaki et al., 2020). FUS was nonspecifically labeled with Cy3- or Cy5-NHS ester to achieve 70-95% labeling efficiency after excess dye was removed through filtration (Figure 1A). Although it was previously reported that Cy3 and Cy5 dyes may cause aberrant LLPS of FUS, we observed minimal changes in droplet morphology (Figure 1B–C) (Riback et al., 2019).
Figure 1: Glycine mutations in FUS lead to separate condensate formation.
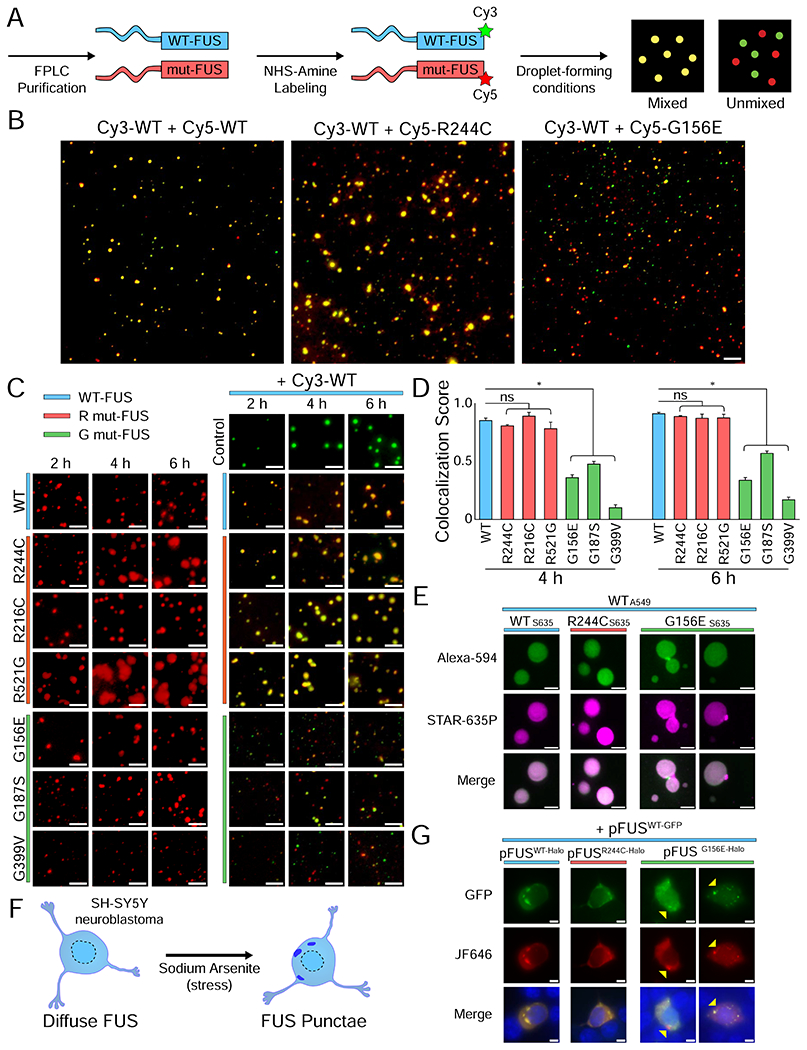
(A) Schematic of purified and labeled FUS preparation for droplet experiments. (B) Wide-field images of 500 nM Cy3-WT FUS with 500 nM Cy5-WT, Cy5-R244C, or Cy5-G156E FUS, and 1 μM unlabeled U40 RNA after 2 h of incubation. Images were acquired in both the Cy3 (green) and Cy5 (red) channels and overlaid using Fiji and Adobe Photoshop CC 2019. The scale bar is 5 μm. (C) Wide-field images of Cy3-WT with Cy5-mutant FUS at 2 h, 4 h, and 6 h. The scale bar is 5 μm. (D) Quantification of (C) by using intensity masking and identifying overlapping droplets. The number of overlapping droplets was divided by the total number of droplets to calculate the colocalization score. Statistical significance was determined using a two-sample proportion z-test with ns = not significant and * = p < 0.05. (E) Stimulated emission depletion (STED) images of Alexa-594-labeled WT FUS with STAR-635P-labeled mutant FUS after 4 h of incubation. Unlabeled FUS was 1 μM concentration whereas labeled FUS was 10 nM. The scale bar is 500 nm. (F) Schematic of SH-SY5Y stress via sodium arsenite addition. (G) Live cell imaging of SH-SY5Y neuroblastoma cells transfected with 200 ng pFUSWT-GFP and 200 ng pFUSmut-Halo and stressed with 0.05 mM sodium arsenite for 1 h. Images show GFP, JF646, and overlay (GFP + JF646 + Hoechst) signal. Yellow arrows indicate granules with only G156E-Halo FUS signal and not WT-GFP FUS.
R.1. G mutant and wild-type FUS form independent condensates
To test whether WT and mutant FUS coalesce together into phase-separated droplets, equimolar ratios of Cy3- and Cy5-FUS were combined under droplet-forming conditions. Briefly, poly-U40 RNA (1 μM) was added to the Cy3- and Cy5-FUS mixture (1 μM) in physiological salt conditions without molecular crowder. The tobacco etch virus (TEV) protease was added to cleave the MBP to initiate condensation. First, we mixed the RNA and Cy3- and Cy5-wild type FUS and observed mixed droplet formation, as expected (Figure 1B–C). The Cy5-labeled R244C mutant also efficiently mixed with Cy3-WT FUS (Figure 1B–C). In striking contrast, the Cy5-G156E mutant did not readily mix with Cy3-WT, forming mostly independent droplets (Figure 1B–C). The same difference was also exhibited in two other ALS/FTLD linked R and G mutations (Figure 1C). Notably, the reduced mixing of G156E with WT FUS was observed with G156E-GFP and WT-mCherry, suggesting that Cy3 and Cy5 labeling is not responsible for generating separate condensates (Figure S1A). Cy5-labeled G mutant FUS also migrates separately from Cy3-WT FUS on electrophoretic mobility shift gels, consistent with the non-mixing condensates (Figure S1B–C). In addition, the least mixed G mutant, G399V, formed multiple oligomeric states devoid of Cy3-WT FUS (Figure S1B–C). The prominent self-association of G mutants with high resistance to physical mixing with WT suggests a gelation process that is inherently different from the LLPS of WT-FUS.
We tracked the overlap between mutant and WT FUS over time by using a custom-built script (STAR Methods, Figure 1D). G mutants remained significantly less colocalized with WT than the R mutants with WT (Figure 1D). We observed a slight increase in the colocalization score from 4 h to 6 h for all G mutants, indicating a small degree of delayed mixing. To test if the RNA-bound form of FUS is responsible for the reduced interaction between G mutant and wild-type FUS, we incubated Cy3-WT and Cy5-G156E FUS without RNA, which still formed independent condensates. Interestingly, when RNA was spiked in after the incubation of the two proteins, we observed increased mixing, indicating that RNA may promote mixing by scaffolding pre-nucleated WT and G mutant FUS (Figure S1D).
We next asked whether these mixed droplets are homogenous or if they contain substructures (Gasior et al., 2019; Protter et al., 2018) that cannot be resolved by wide-field microscopy. We used stimulated emission depletion (STED) microscopy to image FUS droplets at 4-5-fold higher resolution than wide-field microscopy. To accommodate the far-red depletion beam of our STED setup, we used labeled FUS with Alexa 594 and STAR 635P, which did not alter the condensate formation or morphology (Figure S1E). STED-imaged R244C-WT and WT-WT droplets had evenly-distributed color, indicating homogenous mixing within the droplets (Figure 1E). By contrast, Alexa-594-WT mixed with STAR 635P-G156E FUS revealed numerous incomplete fusion events at the 4 h time point (Figure 1E).
To determine whether mutant and wild-type FUS interacted in vivo, we performed a multicolor co-transfection experiment with pFUSWT-GFP and pFUSmut-halo. A diffuse FUS localization was achieved by transfecting SH-SY5Y neuroblastoma cells with a low concentration of FUS plasmid (Shelkovnikova et al., 2014; Takanashi and Yamaguchi, 2014) (Figure S1F–G). FUS condensation into cytoplasmic granules was then triggered by addition of 0.05 mM sodium arsenite (Figure 1F–G, Figure S1F–G). R244C and G156E FUS formed granules without adding sodium arsenite (Figure S1H). Consistent with the in vitro results, the co-transfection of pFUSWT-GFP with either pFUSWT-Halo or pFUSR244C-Halo led to the formation of mixed cytoplasmic granules while a small population of G156E-only granules was observed with pFUSG156E-Halo (Figure 1G). Altogether, these results indicate that G mutants, due to accelerated gelation, may form independent condensates in cells and in vitro (Harmon et al., 2017).
R.2. WT and G mutant exclude each other from engaging with the same RNA
We next asked how early the segregation of the two proteins occurs during condensation. To this end, we developed a single-molecule dual-color nucleation assay in which color-coded WT and mutant FUS binding on an RNA scaffold was observed in real-time via total internal reflection (TIRF) microscopy (Figure 2A). Unlabeled partially-duplexed U50 RNA was tethered to the single-molecule surface by biotin-NeutrAvidin linkage, and a TEV-cleaved mixture of Cy3- and/or Cy5-labeled FUS was flowed in while recording the single-molecule movies (Joo and Ha, 2012a, b; Sarkar and Myong, 2018). As individual molecules of color-coded FUS engaged with RNA, fluorescence intensity increased in a stepwise manner (Figure 2A–B). We interpreted single steps as arising from single FUS molecules binding to a single RNA based on our 80-90% labeling efficiency of FUS (see Methods). Decreases in fluorescence intensity can be interpreted as dissociation since photobleaching is minimized by the imaging buffer. WT and mutants exhibit similar affinity to RNA as measured previously by EMSA and here by fluorescence anisotropy (Figure S2A–F) (Niaki et al., 2020). We confirmed FUS-RNA binding specificity devoid of surface interaction (Figure S2G–M).
Figure 2: Arginine mutations enhance oligomerization of single FUS molecules.
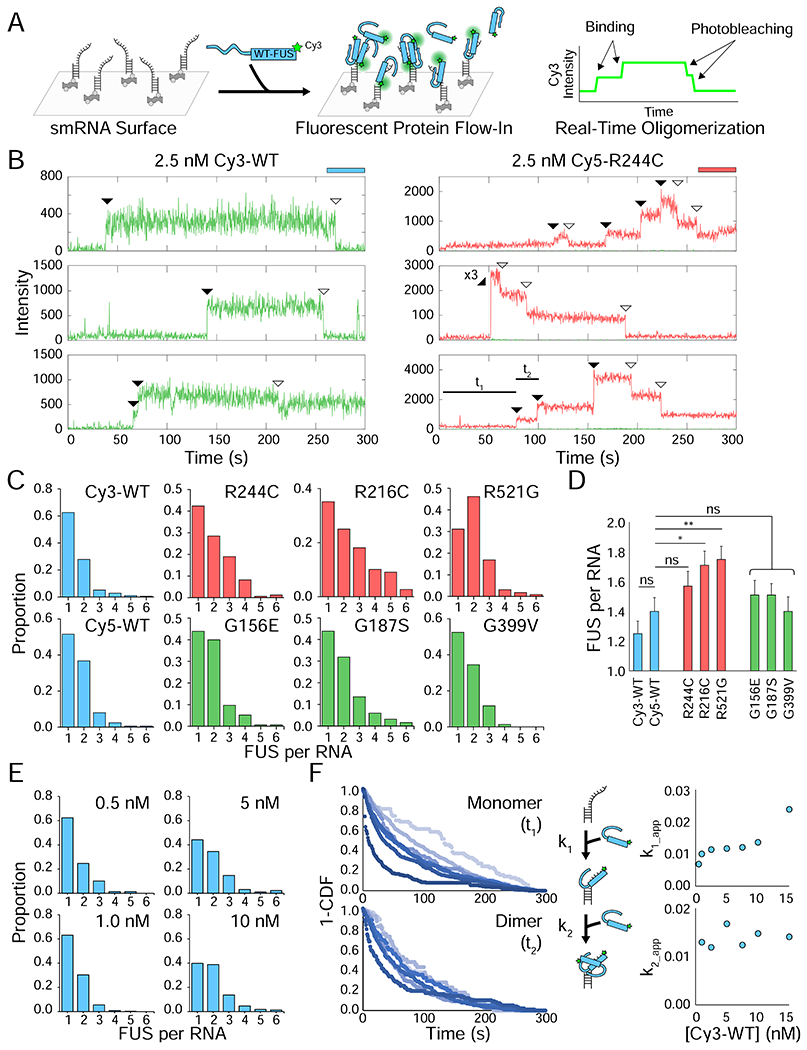
(A) Schematic of the one-color nucleation experiment in which FUS is flowed onto an RNA surface in real-time. (B) Sample single-molecule traces of either 2.5 nM Cy3-WT or 2.5 nM Cy5-R244C FUS interacting with RNA. The Cy3 (green) and Cy5 (red) intensity is shown over time at each spot. Flow occurs 6 s after the start of the video. Shaded arrows indicate binding events and unshaded arrows indicate photobleaching events. The dwell times used to quantify kinetics, t1 and t2, are indicated on the last trace to show how kinetics were determined. (C) Frequency histograms depicting the number of stable FUS binding events per RNA molecule for different FUS variants. All FUS were flowed at 2.5 nM, and all FUS were Cy5-labeled except Cy3-WT. (D) Average FUS count per RNA obtained by fitting a Poisson distribution to histograms from (C). Error is standard error ((λ/n)0.5). Statistics were calculated using a two-sample Poisson distribution test where ns = not significant, * = p < 0.05, and ** = p < 0.01. (E) Same as (C) but with 0.5, 1, 5, and 10 nM Cy3-WT FUS instead of 2.5 nM Cy3-WT. (F) 1-CDF plots for the two-step dimerization reaction on RNA (t1 for monomer, t2 for dimer) for Cy3-WT FUS concentrations of 0.5 (lightest blue), 1, 2.5, 5, 7.5, 10, and 15 nM (darkest blue). The apparent k1 and k2 rates for each concentration were plotted; the standard deviation for each point is smaller than the size of the icons, so they are not shown.
First, we tested one-color nucleation of FUS, which revealed the oligomerization status of FUS on single RNA molecules and the kinetics of FUS oligomerization. Upon flowing in 2.5 nM Cy5-FUS to the single-molecule surface, R mutants on average accommodated more FUS per RNA (1.6-1.8) than either WT FUS (1.2-1.4) or G mutants (1.4-1.6), which is consistent with our previous reports showing that R mutants form large condensates (Figure 2B–D) (Niaki et al., 2020). Higher concentration of Cy3-WT FUS resulted in increased oligomerization of FUS on RNA (Figure 2E). We measured dwell times corresponding to initial binding (t1) and successive binding (t2), to determine k1 and k2. Unlike in k1, we observed no FUS concentration dependence for k2 which may indicate that there is a rate limiting step in the monomer bound state, for example, a conformational rearrangement of the monomer required for the subsequent binding of another unit. It may also be due to the limited concentration of <10 nM, which is the highest concentration allowed by this assay due to high background signal (Figure 2F).
To test the association between WT and mutant FUS, we applied the alternating laser excitation of color-coded FUS variants (Kapanidis et al., 2015) from which stable (>10 s) binding events were captured. Cy3- and Cy5-labeled FUS (2.5 nM each) were coincubated and flowed together to a single molecule chamber containing unlabeled RNA on the surface. The fluorescence intensity was tracked for both fluorophores over time (Figure 3A–B, Figure S3A–B). Single-molecule traces showed binding events of one or both fluorophores (Figure 3B, Figure S3A–B). More than 100 traces for each condition were categorized based on the number of Cy3- and/or Cy5-binding events and binned into a two-dimensional heatmap. In this plotting scheme, the middle of the heatmap represents heterotypic binding events (Cy3 and Cy5 together) while the perimeter of the heatmap represents homotypic binding events (Cy3 or Cy5 alone) (Figure 3C). Monomer-bound RNAs were excluded from the heatmaps. Three mixed samples were compared: WT-WT, G mutant-WT, and R mutant-WT. G mutant-WT FUS mixtures displayed a significantly higher fraction of homotypic traces (i.e self-interacting, perimeter-enriched heatmaps) than the other conditions, indicating that G mutants and WT did not readily interact even at the single-molecule level (Figure 3C–D). Accordingly, the fraction of homotypic binding events (expressed as colocalization score) for G mutants was substantially lower than for R mutants and WT (Figure 3D). Taken together, G mutants disfavor physically associating with WT from the very initial stages of nucleation when one, two, or three proteins start interacting on an RNA scaffold (Figure 1). To test this hypothesis further, we determined the number of unsuccessful (<1-2 s) Cy5-WT or Cy5-mutant binding attempts after an initial Cy3-WT binding event (see Figure 3B for an example). Indeed, Cy5-G mutants had significantly more transient, unsuccessful interactions with Cy3-WT compared to the WT or R mutant mixture (Figure 3E). This result indicates that G mutants may have a disrupted conformation that is dynamically arrested and unable to accommodate interactions with WT (Foffi et al., 2005).
Figure 3: Two-color single-molecule nucleation of FUS shows reduced interactions between G mutant and wild-type FUS.
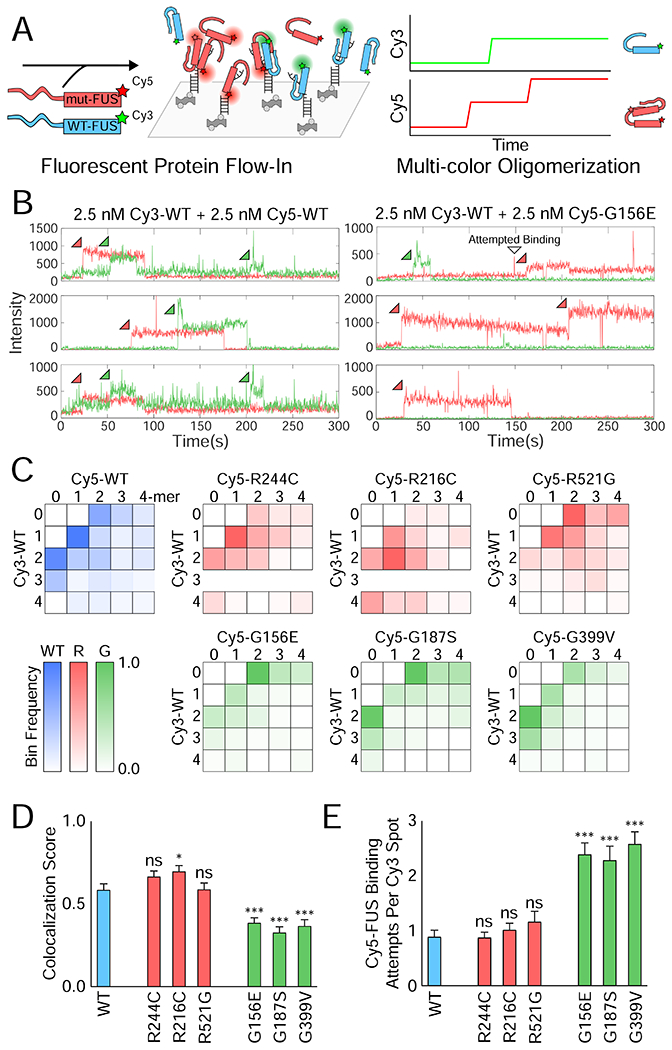
(A) Schematic depicting two-color FUS nucleation on a single-molecule RNA surface. (B) Example single-molecule traces showing Cy3 (green) and Cy5 (red) intensity over time for the green and red excitation of the single-molecule surface. Sample traces are shown for 2.5 nM Cy3-WT FUS with either 2.5 nM Cy5-WT or Cy5-G156E FUS simultaneously flowed into the flow cell. (C) Heatmaps of the normalized frequency of each combination of FUS multimers in reactions of 2.5 nM Cy3-WT and 2.5 nM Cy5-mutant FUS. For instance, the 1-1 bin corresponds to the frequency of traces with a monomer of Cy5-mutant and a monomer of Cy3-WT on that RNA molecule. More transparent bins denote a lower proportion of overall traces falling into the indicated bin (bottom left). (D) The FUS colocalization fraction calculated from (C) by dividing the number of traces with some combination of both FUS molecules (the interior of the heatmap) by the total number of 2-mer+ traces. Error was calculated from the proportion and the number of traces, and significance was determined using a two-sample proportion z-test where ns = not significant, * = p < 0.05, and *** = p < 0.001. (E) Quantification of the average number of binding attempts by Cy5-mutant FUS on traces that had Cy3-WT FUS bind first. Error bars denote SEM. Significance was calculated using a two-tailed two-sample Student’s t-test with *** = p < 0.001. Degrees of freedom were calculated using the Satterthwaite two-sample approximation.
Despite the Cy5-R mutants exhibiting a higher oligomerization status in one-color experiments, co-incubating Cy3-WT with either G or R mutants did not increase the oligomerization state of WT (Figure S3C–D).
R.3. WT recovers the R244C mutant defect by increasing dynamic RNP interactions and reducing condensate size
The physical interaction between R mutant and WT FUS sparked an intriguing question: could WT FUS ameliorate the RNA binding and LLPS defects observed in ALS/FTLD-linked R mutant? The four prominent defects seen in R mutants include (1) static interaction with RNA, (2) high aggregation propensity, (3) large size and (4) loss of fluidity in condensates. First, we employed a previously established single-molecule (sm) FRET assay to probe FUS-RNA interaction (Sarkar and Myong, 2018). WT FUS exhibits a highly dynamic interaction with RNA compared to the more static interaction induced by R244C and G156E FUS (Niaki et al., 2020).
To test the recovery effect in the context of RNA-FUS interaction, we performed the smFRET measurement in which increasing concentrations of WT FUS (up to 1 μM) was titrated to mutant FUS (fixed at 250 nM). Surprisingly, 50 nM of WT FUS mixed into 250 nM mutant was sufficient to recover the dynamic FRET fluctuations for R244C but not for G156E (Figure 4B–D). Such recovery was observed for the G mutant only when equimolar or excess concentrations of WT FUS was added (Figure 4B–C). The dynamic fraction for R mutant increased as a function of WT FUS concentration whereas it remained low for G156E FUS until WT FUS was in excess of G156E FUS (Figure 4D). This selective recovery effect likely arises from the physical association between the R mutant and WT FUS, which is disfavored for the dynamically arrested G mutant and WT FUS (Figure 3).
Figure 4: Defective single-molecule FRET signature of G156E-FUS is ineffectively recovered by WT FUS.
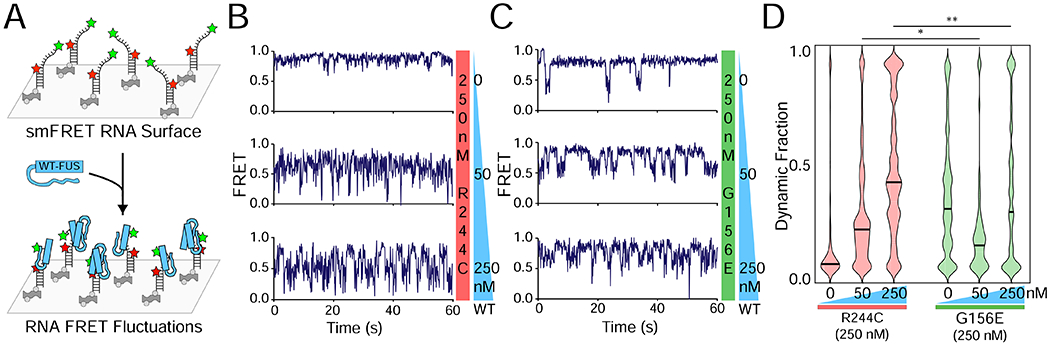
(A) Schematic of FUS binding to single RNAs in smFRET measurements. (B) Sample FRET efficiency traces for FRET-labeled pdU50 RNA with 250 nM R244C and increasing WT concentrations (0 nM, 50 nM, and 250 nM). (C) Same as (B) but with G156E. (D) Violin plot of the dynamic fraction of >100 traces for each condition in (B) and (C). The dynamic fraction represents the proportion of the trace with rapid FRET fluctuations (see Methods). The black bar indicates the median dynamic fraction for each condition. Significance was calculated using a two-tailed two-sample Student’s t-test with ns = not significant, * = p < 0.05, and ** = p < 0.01. Degrees of freedom were calculated using the Satterthwaite two-sample approximation.
We next asked if this recovery observed at the single-molecule level extends to the scale of condensates visualized by fluorescently labeled RNA formed with unlabeled protein. Fluorescence recovery after photobleaching (FRAP) and droplet size were both monitored as droplets matured (Figure 5A). When equimolar WT FUS (500 nM) was mixed with R244C FUS (500 nM), droplet size was dramatically reduced compared to the R244C (1 μM) condensates (Figure 5B, D). Strikingly, large (>10 μm2) droplets of R244C-RNA droplets completely disappeared (Figure 5D). All G mutants were the same size as WT (Figure 5E, Figure S4A–B).
Figure 5: G156E-FUS exerts a delayed disruptive effect on WT FUS RNA droplet fluidity.
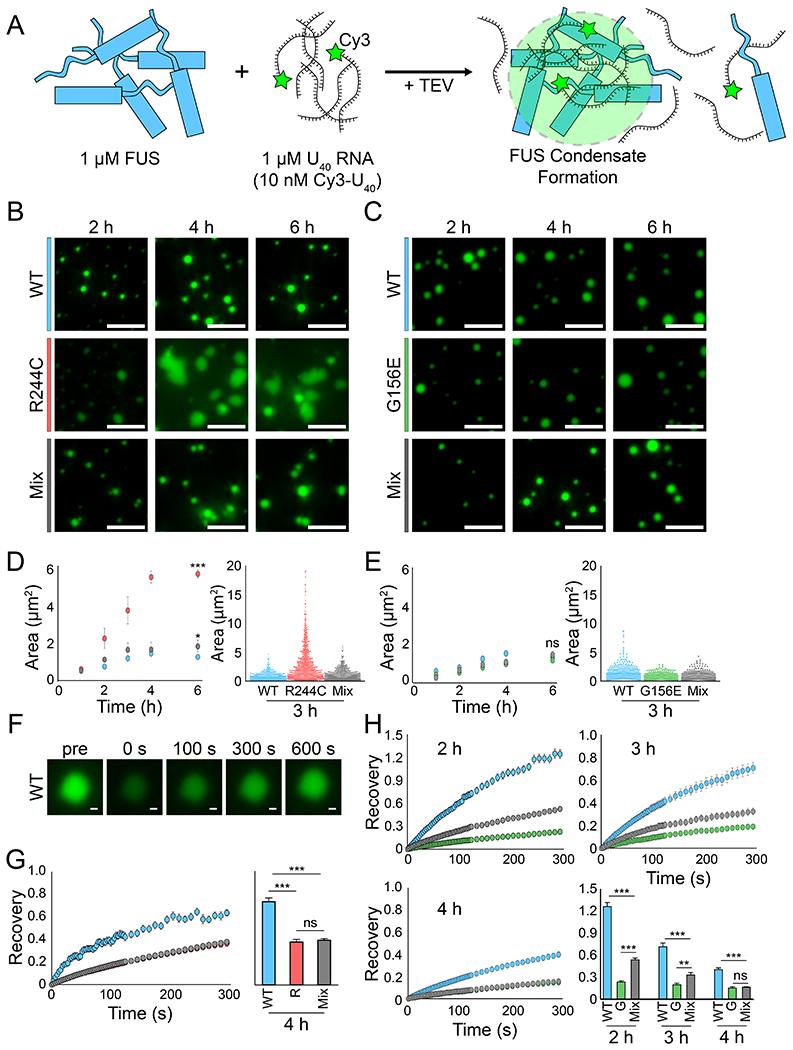
(A) Schematic detailing the experimental setup in which 1 μM FUS is combined with 1 μM (1:100 Cy3-labeled:unlabeled) U40 RNA to form droplets. (B) Representative wide-field images of WT, R244C, and WT/R244C mixed (500 nM each) droplets at 2 h, 4 h, and 6 h. The scale bar is 5 μm. (C) Same as (B) but with G156E and WT/G156E mixed droplets instead. (D) Area quantification (WT = blue, R244C = red, mix = gray) of the droplets from (B) via intensity thresholding and ROI auto-selection. Left, scatter plot of area versus time. Error is standard deviation. Right, dot plot of all droplet areas for the 3 h time point. (E) Same as (D) but with G156E (green). (F) Representative fluorescence recovery after photobleaching (FRAP) images from WT FUS at the 3 h time point for one droplet. The scale bar is 0.5 μm. (G) Left, fluorescence recovery for WT, R244C, and WT/R244C mixed droplets at 4 h over 300 s. Error is SEM with n = 8. Right, bar plot of recovery at the 300 s time point. (H) Same as (G) but for G156E and WT/G156E mixed droplets at 2 h, 3 h, and 4 h. All statistics for this figure are calculated using a two-tailed two-sample Student’s t-test with ns = not significant, * = p < 0.05, ** = p < 0.01, and *** = p < 0.001. Degrees of freedom were calculated using the Satterthwaite two-sample approximation.
FRAP was performed by photobleaching (405 nm laser) RNA or FUS (Figure 5F). Our values for RNA FRAP closely mirrored the FRAP values for protein, in agreement with previous studies (Patel et al., 2015; Wang et al., 2018). The G156E droplets displayed a slower recovery as expected (Figure S4C). The co-condensate of R244C mixed with WT FUS had moderately reduced FRAP compared to WT FUS (Figure 5G). The lower fluidity of the R244C/WT mix may represent incomplete or partial recovery of the R244C by the WT FUS or altered condensate formation under heterotypic conditions. The other R mutants had no differences in FRAP compared to wild-type in either homotypic or heterotypic conditions (Figure S4D).
The FRAP for WT/G156E mix shifted to a G156E-like recovery over the course of 2-4 h (Figure 5H). At 2 h, the mixed population showed an average fluorescence recovery between homotypic WT and G156E droplets (Figure 5H). Despite the low physical association between G mutant and WT, an undetectable amount of the G mutant (Figure 1B–D) may integrate into WT droplets and reduce the RNA diffusion of the entire condensate. Such effect became more prominent as seen by further diminished FRAP concomitant with the increase in G156E/WT mixing over time (Figure 1C–E, 5H). Similar result was obtained for G187S and G399V co-condensates with WT (Figure S4D). In summary, WT was effective in reducing the R244C condensate size to WT levels, but was not able to recover the fluidity of the resulting condensates. The G mutants’ low, gel-like fluidity was not recovered by the WT, consistent with the lack of recovery observed at the single-molecule level.
R.4. The G156E mutation does not affect FUS’s Csat
We next asked whether the gel-like properties of the G156E mutation could be explained by changes in the phase separation propensity. Csat is the concentration threshold above which LLPS occurs; arginine and tyrosine residues control the Csat through reversible intermolecular crosslinks whereas glycine-like residues do not influence Csat (Choi et al., 2020b; Harmon et al., 2017; Wang et al., 2018). Using a turbidity assay, we measured condensate formation as a function of RNA and FUS concentrations for WT, R244C, and G156E FUS to construct phase diagrams. As expected, R244C FUS had a lower Csat, which is consistent with our previous results (Figure S5A–B) (Niaki et al., 2020). Nevertheless, G156E’s Csat closely matched WT, suggesting that the mutation does not substantially impact the phase separation propensity of FUS (Figure S5A). Instead, G156E alters the physical properties of the condensates by promoting gelation that lead to dynamically arrested condensates with limited internal diffusion (Foffi et al., 2005).
R.5. A large structural disruption at the Gly-156 residue drive ALS/FTLD mutant behavior
Glycine is the smallest amino acid with a side chain of a single hydrogen atom, predicted to increase the fluidity of FUS droplets as a spacer between bulky residues like tyrosine, arginine, and proline (Choi et al., 2020b; Harmon et al., 2017; Wang et al., 2018). Based on the dramatic reductions in droplet fluidity, reduced interactions with WT, and lack of WT-mediated recovery of the G mutants (Figures 1, 3–5), we asked if these changes arise from the loss of glycine or the gain of glutamic acid in G156E, which causes aggressive form of ALS and is one of the few FUS mutations also linked with FTLD (Deng et al., 2014).
To this end, we designed four non-ALS/FTLD-linked FUS mutation at the Gly-156 position: G156A, which is small and uncharged like glycine; G156D, which has the same negative charge but is one carbon shorter than glutamic acid; G156Q, which resembles glutamic acid’s size and structure but not its charge; and G156P, which also does not have steric hindrance at the R group (Figure 6A). The non-ALS/FTLD-linked G156A, G156D, and G156P FUS all recapitulated the rapid FRET fluctuations of WT-FUS whereas G156Q resembled that of G156E FUS, displaying static high FRET with intermittent FRET fluctuations. (Figure 6B). Overall, G156A, G156D, and G156P all showed a moderate level of dynamic status whereas G156Q resembled the static G156E smFRET signature (Figure 6B). Thus, the bulkiness of the mutation at this residue appears to drive the disrupted RNA-FUS interaction of G156E.
Figure 6: ALS/FTLD-linked G156E mutation disrupts the LLPS of FUS due to a bulky amino acid substitution.
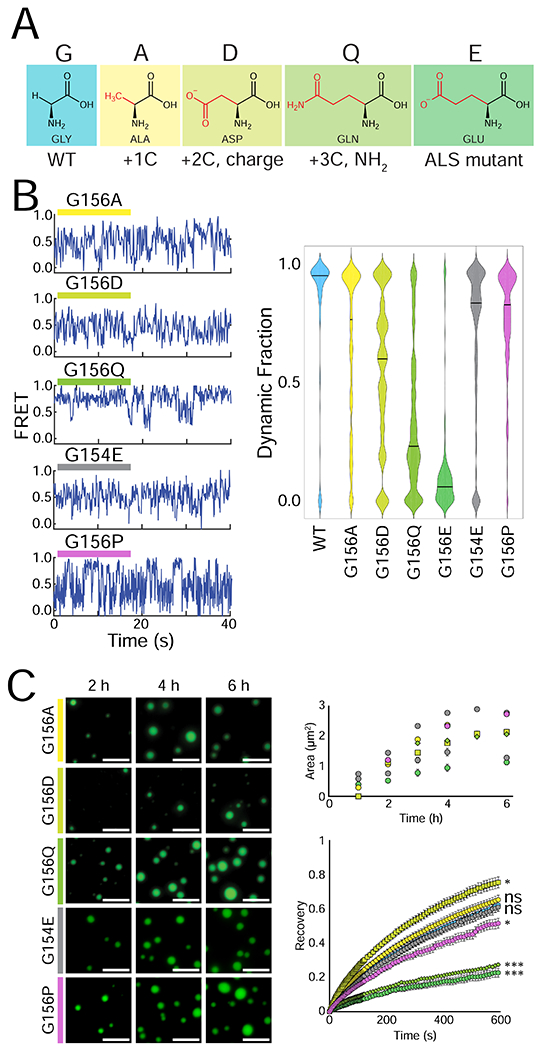
(A) Structures of the different amino acids introduced at position 156. (B) Representative smFRET traces of 500 nM G156A, G156D, G156Q, G154E, and G156P FUS with 50 pM FRET-labeled pdU50 RNA. The dynamic fraction of >100 molecules was quantified and is visualized with a violin plot. (C) Wide-field images of 1 μM G156A, G156D, G156Q, G154E and G156P FUS with 1 μM U40 RNA and TEV protease at 2 h, 4 h, and 6 h. The fluorescent signal is 10 nM U40 RNA, and the scale bar is 5 μm. Droplet area was quantified by intensity thresholding and is plotted over time, and droplet fluorescence recovery after photobleaching is shown for the 4 h timepoint: G156E = green circle, G156A = yellow circle, G156D = yellow-green square, G156Q = lime diamond, G154E = gray circle, and G156P = purple circle. Significance was calculated using a two-tailed two-sample Student’s t-test with ns = not significant, * = p < 0.05, ** = p < 0.01, and *** = p < 0.001. Degrees of freedom were calculated using the Satterthwaite two-sample approximation.
The droplet properties of G156A, G156D, G156P, and G156Q FUS alone were tested in the same way as before, with labeled RNA. The droplet size and morphology of all Gly-156 mutant FUS were similar to WT and G156E (Figure 6C). However, the FRAP results mirrored the pattern observed with smFRET: G156Q had low droplet fluidity like G156E whereas G156A, G156D, and G156P displayed high, WT-FUS-like droplet recovery (Figure 5F, 6D). In color-coded condensates, Cy5-labeled G156A readily mixed with Cy3-labeled WT FUS as evident in colocalization score similar to Cy3-WT/Cy5-WT (Figure S5C–D). Surprisingly, G156Q was also mixed with wild-type, indicating a key difference between G156E and G156Q despite their other molecular similarities (Figure S5C–D). Overall, the FUS-RNA interaction and droplet fluidity of G156Q closely resembled the ALS-linked G156E FUS, whereas G156A and G156D followed the patterns of WT FUS. This result signifies that the bulky size of E in G156E, rather than loss of G is primarily responsible for the accelerated gelation of the G156E mutant.
Furthermore, we asked whether the 156 position is important for driving the mutant defects of G156E. To test this, we created a G154E variant, which is not ALS-linked (Deng et al., 2014; Mackenzie et al., 2010). Unlike the ALS-linked G156E mutation, the G154E protein variant displayed WT-like characteristics seen by dynamics in smFRET measurement and high fluidity in droplets (Figure 6B–C). This result reveals that the position of the mutation potentially plays an important role in giving rise to molecular defect.
R.6. Aged mutant FUS exerts a disruptive effect on WT FUS
FUS and FUS-RNA droplets solidify over time, mirroring the solid-like aggregates observed in patients (Kwiatkowski et al., 2009; Niaki et al., 2020; Patel et al., 2015; Vance et al., 2009; Wang et al., 2018). This maturation process occurs over the course of several hours in vitro and is greatly accelerated in mutants as evidenced by the decrease in mobile fraction of condensate measured by FRAP (Figure 5F–H). In vivo, mutant FUS mislocalizes to the cytoplasm (Guo et al., 2018; Hofweber et al., 2018; Takanashi and Yamaguchi, 2014), increasing the propensity to coalesce with other proteins due to the low RNA:protein ratio (Maharana et al., 2018). Cellular stress can drive WT FUS to the cytoplasm (Guo et al., 2018; Hofweber et al., 2018; Takanashi and Yamaguchi, 2014), increasing the interaction frequency between WT and mutant FUS, which may have formed into higher order complex (Daigle et al., 2013; Guo et al., 2018; Patel et al., 2015; Shiihashi et al., 2016; Sun et al., 2015; Vance et al., 2013). Therefore, we asked if WT can recover pre-formed mutant FUS condensates (Figure 7A).
Figure 7: Mutant FUS traps new WT FUS that enters mutant condensates.
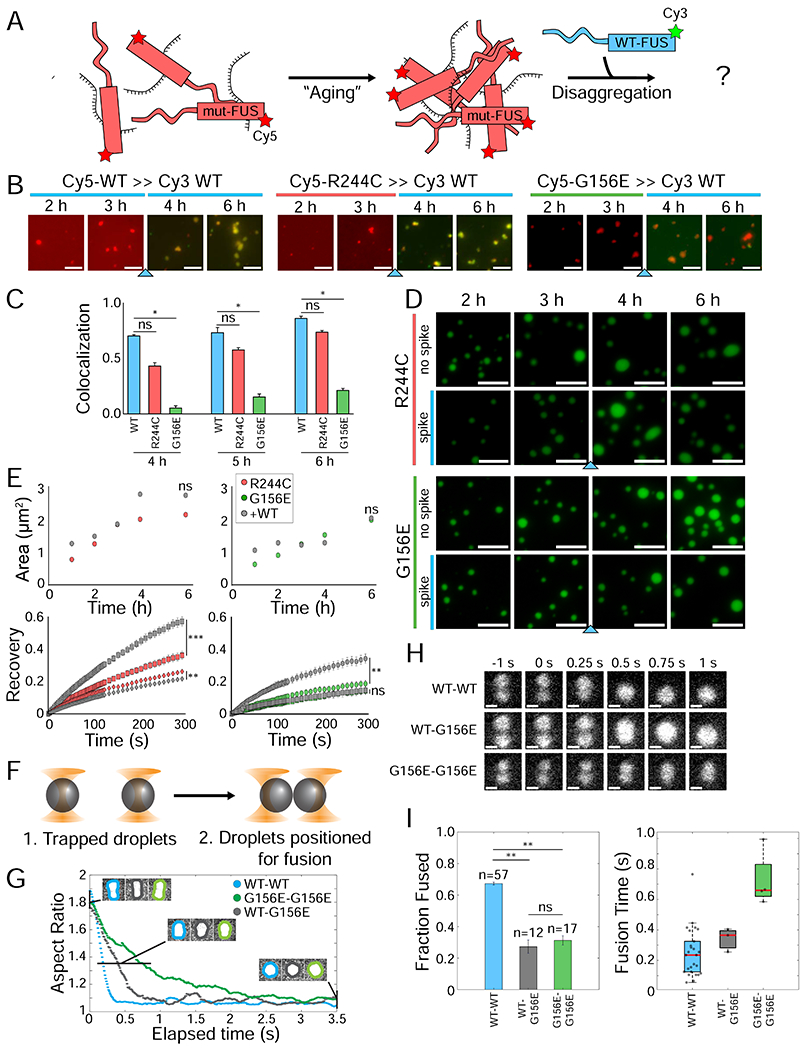
(A) Schematic showing how mutant FUS is pre-aged in the absence of WT FUS. (B) Wide-field images of Cy5-labeled WT, R244C, and G156E FUS that are pre-aged at 1 μM concentration with 1 μM unlabeled U40 RNA for 3 h. Following the addition of Cy3-WT FUS (blue arrow), both Cy3-WT and Cy5-mutant FUS are 500 nM each with 1 μM total RNA. Images immediately following the addition of Cy3-WT are shown at 0 min, 10 min, 20 min, and 30 min. The scale bar is 5 μm. (C) Quantification of colocalization in (B). (D) Wide-field images of unlabeled R244C and G156E FUS are incubated under droplet-forming conditions alone at 1 μM concentration for 3 h. Droplets are visualized by 10 nM Cy3-labeled U40. Following addition of unlabeled WT (blue arrow), each FUS is at 500 nM concentration. (E) Droplet area and fluorescence recovery for mutant alone (R244C = red, G156E = green) and mutant with WT spike (gray). For FRAP curves, recovery is shown at 3 h (circles), 4 h (squares), and 6 h (diamonds). Statistical significance was calculated for the final time point for each spiked reaction compared to the mutant alone. (F) Schematic of the optical trap experiment, in which individual droplets are immobilized and brought in close proximity until spontaneous fusion occurs. (G) Representative plots of droplet pair aspect ratio over time for WT-WT, WT-G156E, and G156-G156E. (H) Still Cy3 fluorescence images from sample optical tweezer droplet fusions of pre-aged WT-WT, WT-G156E, and G156E-G156E droplet pairs. The scale bar is 1 μm. (I) Quantification of the fraction of successful droplet fusions and of the fusion time for WT-WT, WT-G156E, G156E-G156E. Fusion times were estimated by determining the aspect ratio for each frame of the videos from (G) and plotting over time. All statistics for this figure are calculated using a two-tailed two-sample Student’s t-test with ns = not significant, * = p < 0.05, ** = p < 0.01, and *** = p < 0.001. Degrees of freedom were calculated using the Satterthwaite two-sample approximation.
To assess if the colocalization behaviors observed in Figure 1 changed after mutant FUS aging, we mixed 3 hr pre-aged Cy5-mutant FUS with Cy3-WT FUS (Figure 7B). The control, Cy3-WT and Cy5-WT quickly coalesced, forming mixed droplets within 10-20 minutes. Cy3-WT applied to pre-aged Cy5-G156E remained unmixed, similar to our earlier results (Figures 1B–D, 7B). The addition of Cy3-WT to the pre-aged Cy5-R244C gradually mixed over the course of 1 h, which was slightly slower than for WT-WT mixing (Figure 7B–C).
We next monitored how the fluidity of pre-aged mutant FUS condensates were altered by WT addition. Mutant FUS was pre-incubated for 3 hours independently, to which fresh WT FUS was added (Figure 7A). Fluorescence recovery for both R244C and G156E increased following the WT spike, but further droplet maturation led back to low mutant-like recovery (Figure 7D–E). Droplet area was unaffected by WT FUS (Figure 7E). Notably, WT FUS does not effectively disassemble mutant FUS droplets, a phenomenon that we observed with known disaggregases such as Kapβ2 (Guo et al., 2018; Hofweber et al., 2018; Kroschwald et al., 2017; Yoshizawa et al., 2018) (Figure S6A–B). RNase A can likewise disassemble FUS-RNA droplets (Figure S6C).
Finally, the fusion kinetics of pre-aged FUS condensates were tested using dual trap optical tweezers outfitted with a confocal fluorescent microscope (see Methods). The homotypic (WT-WT and G156E-G156E) and heterotypic (WT-G156E) droplet fusions were performed by trapping a pair of droplets with fluorescent Cy3-labeled RNA in the dual traps and bringing them in close proximity (until the Gaussian tails of fluorescence intensities overlapped) to promote droplet fusion (Figure 7F). For heterotypic fusions, WT and G156E mutant FUS condensates were formed and flowed through separate microfluidic channels (Figure S7A). The droplet fusion kinetics were then extracted by determining the aspect ratio of the fusing condensate over time (Figure 7G–H, also see Methods). Homotypic WT-WT fusions occurred ~70% of the time, and the fusion had a characteristic fusion half-time, τ1/2, of ~0.22 s (Figure 7G–I). By contrast, both the heterotypic WT-G156E and homotypic G156E-G156E fusion events only occurred in ~30% of cases, in agreement with the decelerated mixing characteristic of the G mutant (Figure 7I). Intriguingly, in all but one case, the unfused droplets remained attached and could not be pulled apart by the optical traps (Figure S7B–C), which suggests the existence of an attractive interaction similar to that observed in STED imaging (Figure 1E). Of the fusion events, homotypic G156E-G156E condensates fused significantly slower than WT-WT, with the τ1/2 of ~0.66 s (Figure 7I). Heterotypic WT-G156E fusion events were only slightly slower, suggesting that the G156E condensate dominates the fusion kinetic, likely due to the higher viscosity (Figure 7I). Together, this provides further evidence about the inherently selective interaction between WT and G156E FUS.
D. Discussion
In this work, we demonstrate that three ALS/FTLD-linked point mutations in FUS glycine residues form dynamically arrested condensates that remain distinctive and do not mix with wild-type FUS over long timescales. There exists a three-way interplay of homotypic interactions among mutant proteins, homotypic interactions among wild-type FUS proteins, and heterotypic interactions among mutant and wild-type proteins, all in the context of RNA The preference for homotypic interactions exerts control over the timescales of fusion events and the mixing of condensates formed by the mutant and wild-type proteins. Importantly, the preference for homotypic interactions is evident on multiple length scales, including the single-molecule, condensate, and cellular levels. The three disease-linked glycine mutations are located in different domains throughout FUS (Deng et al., 2014), and we find that the bulky glutamic acid substitution at Gly-156 (ALS/FTLD-linked) – but not Gly-154 (not ALS/FTLD-linked) – drives the formation of dynamically arrested condensates (Figure 6). All three G mutants preferentially nucleate on RNA without wild-type FUS (Figure 3), thus giving rise to independent droplets that do not efficiently mix with wild-type FUS condensates (Figure 1). However, the G mutant condensates can eventually incorporate wild-type FUS after 4-6 hours (Figure 1D), as STED imaging reveals complete mixing within mature condensates (Figure 1G). Surprisingly, the wild-type FUS proteins that eventually interact with the G mutant condensates also become dynamically arrested, as evidenced by the drastic reduction in RNA exchange measured by FRAP (Figure 5). By contrast, arginine mutations interact extensively with wild-type at all levels, including the single molecule level in which wild-type and R mutant FUS molecules bind together on single RNA molecules (Figure 3). Remarkably, such physical association leads to a recovery effect by recovering dynamic RNA-protein interactions (Figure 4) and reducing condensate size (Figure 5). Nevertheless, addition of wild-type FUS to pre-aged mutant FUS cannot effectively recover the mutant condensate (Figure 7).
How do we explain the physical and chemical basis underlying the G mutant behavior? The stickers-and-spacers model describes the behavior of associative biopolymers such as multivalent protein and RNA molecules, which might provide mechanistic explanation for our observations regarding G mutants and wild-type FUS (Choi et al., 2020b). In this model, stickers engage in reversible physical crosslinks formed by a hierarchy of directional non-covalent interactions (Choi et al., 2020a; Choi et al., 2020b; Martin et al., 2020; Peran and Mittag, 2020; Wang et al., 2018). The number and strength of these physical crosslinks control how associative biopolymers undergo phase separation and percolation (or gelation) above Csat (saturation concentration) and Cperc (percolation threshold), respectively. Csat and Cperc are usually coupled with one another, and the effective solvation volumes of spacers determine whether percolation is aided by phase separation or not (Choi et al., 2020b; Harmon et al., 2017). If Cperc and the concentration of the dense, droplet phase (Cdense) are similar, then phase separation is achieved through the formation and breakage of crosslinks at equilibrium. However, mutations that lower Cperc without changing Cdense will induce rapid percolation (gelation), leading to the dynamical arrest as we observed in our RNA condensates (Foffi et al., 2005; Zeng et al., 2020). We propose that these physical concepts may explain our observations of the G mutants with wild-type FUS.
A plausible scenario is that the change of glycine to glutamate at position 156 lowers Cperc by replacing a spacer residue with a bulky amino acid. The effects of a single substitution can become extended in the context of a protein-protein network, whereby G mutants make favorable crosslinks with other G mutants and not with wild-type. In agreement, large-scale replacement of glycine residues in FUS decreased the fluidity of the resulting condensates (Wang et al., 2018), highlighting the role of glycines as important spacers that lower gelation propensity. Indeed, we and others have shown that ALS/FTLD-linked point mutations in glycine residues are also sufficient to reduce fluidity (Niaki et al., 2020; Patel et al., 2015). It is worth noting that glycine residues constitute approximately 30% of amino acids in FUS (152 out of 526), and mutations at over 20 of them are associated with ALS/FTLD, reflecting an extremely high level of glycine enrichment and the critical role played by glycine in FUS (Deng et al., 2014). The G156E mutation in FUS likely promotes aberrant crosslinking and thereby lowers Cperc, leading to dynamically arrested phase separation without altering Csat or Cdense (Choi et al., 2020b) (Zeng et al., 2020) (Foffi et al., 2005; Harmon et al., 2017; Ruter et al., 2020). Indeed, we observe that the G156E mutation has a similar Csat to wild-type FUS (Figure S5A), strongly suggesting that Cperc is decoupled from Csat by the disease-linked mutation. The unique gelation of G mutants leads to the unsuccessful coengagement of wild-type and G mutant to the same RNA at the single-molecule level (Figure 3E), the slow exchange of RNA molecules in condensates (Figure 5H), and reduced droplet fusion (Figure 7F–I). By contrast, wild-type condensates have high fluidity and likely engage in many transient interactions before eventually hardening into a more solidified state as shown before (Patel et al., 2015). We further postulate that spacers within the wild-type protein are more solvated and are therefore incompatible to interact with the G mutant protein. Our observations of dynamical arrest are also concordant with similar findings reported for model peptide-RNA systems (Boeynaems et al., 2019).
The dynamically arrested model posits that fusion of condensates will occur, albeit over a longer timescale. This is indeed what we observe. Using STED microscopy we find that wild-type and G mutant proteins eventually reach a well-mixed state (Figure 1E); further, we observe that mixed condensates initially have an average fluidity (Figure 5H), and that wild-type and mutant condensates can occasionally fuse in optical tweezers measurements (Figure 7F–I). Over timescales of several hours, even the wildtype condensate undergoes gradual hardening and gelation, though the sticker-sticker, sticker-spacer, and spacer-spacer contacts are likely different from the G mutant. Perhaps this aged and hardened state of the wild-type FUS makes the intermolecular or inter-condensate interactions less selective, i.e., more favorable for engaging with different aberrant conformers of the aged G mutants. We note that dynamical arrest is distinct from fibrillization, which has been studied more extensively in the context of neurodegeneration (Elbaum-Garfinkle, 2019). In cells, additional factors such as ATP-dependent helicases, protein chaperones, heat shock proteins or other RNA binding proteins may buffer G mutant gelation, allowing heterogeneous interactions with wild-type FUS (Hondele et al., 2019; Maharana et al., 2018). DEAD/H-box helicases in particular might be adept at fluidizing dynamically arrested RNA condensates, as observed previously (Hondele et al., 2019). Indeed, we find that the addition of RNA to separate condensates aids the coalescence of wild-type and mutant FUS in vitro (Figure S1D). Other cellular proteins and RNAs may likewise act as scaffolds to enable wild-type-G mutant mixing. Our in vitro results suggest that this may also lead to accelerated hardening since the mixed condensates converge to G mutant’s gel-like properties (Figures 5H and 7E).
Thus, the initial exclusion of wild-type FUS from G mutant FUS condensates is likely not thermodynamic immiscibility; rather, there is a decelerated interaction between wild-type and mutant that is driven by G mutant’s rapid gelation. Immiscibility arises in unique circumstances that entail differences in condensate surface tension, RNA composition, or post-translational modifications (Feric et al., 2016; Gibson et al., 2019; Langdon et al., 2018). Feric et al. demonstrate that subcompartments in nucleoli emerge from immiscible phase separation driven by a relative difference in surface tension. Interestingly, the RNA binding domain is responsible for the formation of immiscible condensates (Feric et al., 2016). In the fungus Ashbya gossypii, Whi3 proteins form immiscible condensates, governed by the interacting RNAs, CLN3 and BNI1, which exhibit differing shapes (Langdon et al., 2018). In nucleosomes, acetylated and non-acetylated chromatin form distinct compartments that touch one another without content mixing (Gibson et al., 2019). Importantly, all of these condensates display liquid-like properties with differing fluidities (Feric et al., 2016; Gibson et al., 2019; Langdon et al., 2018; Zhang et al., 2015). Because of the decreased Cperc of FUS G mutants, the liquid-like properties of wild-type FUS are supplanted by the arrested dynamics of the mutant (Boeynaems et al., 2019; Foffi et al., 2005).
Arginine mutants do not have these altered interactions with wild-type FUS. Instead, we observe that R mutants readily interact with wild-type FUS at the single-molecule, condensate, and cellular levels (Figures 1 and 3). This is likely because the loss of the sticker residue does not decrease Cperc; in our previous results and in this study, the R244C mutation decreases Csat (Figure S5A) (Niaki et al., 2020). Although the global loss of stickers increases Csat (Wang et al., 2018), we suggest that the loss of a single sticker residue may simply allow alternative crosslinks that are stronger and thus decrease Csat. When R mutants interact with wild-type FUS, the wild-type protein may inhibit these aberrant crosslinks, which would be consistent with our single-molecule and condensate data showing that R mutant condensate-and RNA-interaction properties can be recovered by adding wild-type FUS (Figures 4 and 5). However, in pre-aging experiments, these aberrant sticker-sticker interactions may be locked into position and unable to be recovered by wild-type FUS as we observed in Figure 7.
These results demonstrate that mutant FUS LLPS is altered simply by adding wild-type FUS. In cells, FUS interacts with many binding partners (Kamelgarn et al., 2016; Reber et al., 2019), and these proteins and RNAs may further modulate its ability to undergo LLPS and the properties of the resulting condensates (Maharana et al., 2018). Many granules are known to constantly exchange protein and RNA constituents (Alexander et al., 2018; Khong et al., 2017; Moon et al., 2019; Youn et al., 2019), providing ample opportunities for heterotypic interactions such as the wild-type-mutant interaction we test here (Cid-Samper et al., 2018; Langdon et al., 2018; Niaki et al., 2020; Shevtsov and Dundr, 2011; Shiina, 2019). Previously, FUS has been shown to recruit other client proteins into its in vitro condensates through extensive sticker-sticker crosslinks (Wang et al., 2018). The complex RNP network that exists in dynamic equilibrium within cells consists of many binary and multivalent interactions that may tune FUS and FUS mutant properties (Rhine et al., 2020).
What are the lost and gained interactions that drive ALS/FTLD-linked FUS variants to enter a pathogenic fate? Although pulldown studies have not identified significant differences between FUS and FUS mutant interactomes (Reber et al., 2019), RNA metabolism such as splicing outcomes are compromised by FUS mutations (Sun et al., 2015). Two common pathogenic features found in ALS/FTLD patients’ neurons are the aggregated and mislocalized FUS along with other RNA binding proteins (Kwiatkowski et al., 2009; Vance et al., 2009). Our study provides a mechanism by which a single point mutation at three glycine residues alters the protein properties to exclude wild-type FUS. The single-molecule platform we have devised in this report is uniquely primed for probing and screening the fundamental interactions that seed nucleation for LLPS. For FUS, linking the mutant FUS and RNAs that modulate its LLPS is important for understanding how ALS/FTLD progress in the cell – and for how to develop treatments that are effective against mutant FUS gelation.
Limitations
The molecular features pertaining to the ALS/FTLD associated mutants of FUS that we present in this work may only partially depict the cellular defect found in neurodegeneration. The stickers-and-spacers model may not fully explain our observation of G mutant forming separate condensate from the wildtype FUS. It is plausible that the single G mutant induces local fibril that manifest into aberrant RNA binding and conformational abnormality.
STAR Methods section
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sua Myong (smyong1@jhu.edu).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
Single Molecule FRET data acquisition and analysis package can all be obtained freely from Center for Physics of Living Cells’ website (https://cplc.illinois.edu/software/).
Matlab code from this manuscript can be downloaded from Github: (https://github.com/Myong-Lab). IDL (http://www.exelisvis.co.uk/ProductsServices/IDL.aspx), MATLAB (https://www.mathworks.com/), Adobe (https://www.adobe.com/products/photoshop.html), and Prism7 (https://www.graphpad.com/scientific-software/prism/) software with academic or individual licenses can be obtained from their respective software companies. ImageJ is an open-source program available from the NIH (https://imagej.nih.gov/ij/). The violin plot MATLAB code was available online through MATLAB file exchange (https://www.mathworks.com/matlabcentral/fileexchange/45134-violin-plot).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
E. coli Culturing
All expression plasmids were transformed into either competent BL21 E. coli with Lysogeny broth plus 50 mg/L kanamycin sulfate (E. coli expression vectors) or competent NEB Turbo E. coli with Lysogeny broth (LB) plus 100 mg/L ampicillin (mammalian expression vectors). Bacteria were grown overnight in LB plus antibiotic, and afterwards were added 1:1 to 50% (v/v) glycerol to generate frozen stocks that were stored at -80 °C until used for DNA or protein preparation as described below.
Mammalian Cell Culture
Female human SH-SY5Y neuroblastoma cells (ATCC) were grown at 37 °C with 5% CO2 in a DMEM solution supplemented with 10% FBS, 2 mM glutamate, 100 μg/mL Pen-Strep, 0.15% sodium bicarbonate, and 1 mM sodium pyruvate. Cell lines were periodically tested with a mycoplasma-detecting test. A 10 cm2 plate with 10 mL DMEM was used to grow and passage cells, and cells were washed twice with 1X distilled PBS before trypsinizing with 0.05% trypsin. The trypsin was neutralized by 1:1 addition of DMEM. Cells were transferred to 15 mL conical vials and pelleted at 1500 x g for 5 min and resuspended in fresh DMEM for 1:20-1:50 dilutions into new plates. For long-term storage, SH-SY5Y cells were trypsinized and DMSO was added to a final concentration of 5% (v/v). Cells were first frozen at -80 °C before being transferred to liquid nitrogen for long-term storage.
METHOD DETAILS
Purification of WT FUS and ALS FUS Variants
Plasmid Preparation
E. coli expression vectors for many FUS variants (WT, WT-GFP, LCD-only, RBD-only, G156E, G187S, R216C, R244C, G399V, and R521G) were generated by Genscript (Piscataway, NJ) for our previous work with FUS (Niaki et al., 2020). Expression vectors for WT-mCherry, G154E, G156A, G156D, G156Q, G156E-GFP, and R244C-GFP were created by site-directed mutagenesis of the WT FUS vector by Genscript and/or synthesis of the fluorescent tag. Each expression vector is designed with a maltose binding protein (MBP) tag and a 6xHis tag fused the N-terminus of the FUS open reading frame, which was codon-optimized for E. coli expression. A tobacco etch virus (TEV) recognition motif was included between the poly-His tag and the FUS N-terminus.
Mammalian expression vectors for FUS were synthesized for WT FUS by Genscript. For the GFP constructs, both GFP and mammalian codon-optimized FUS were synthesized and cloned into a pcDNA3.1 vector. For the Halo constructs, a previously-generated EF1-Halo plasmid was used as a target for FUS subcloning from the GFP construct by Genscript (Li et al., 2016). Site-directed mutagenesis was then performed to introduce the R244C and G156E mutations into the FUS-Halo vector.
FUS Purification
FUS was purified as described previously (Niaki et al., 2020). Briefly, the FUS expression vectors were transformed into BL21 (DE3) competent E. coli. cells. A starter 5 mL E. coli culture was grown in LB with 50 mg/L Kanamycin sulfate overnight at 37 °C with 200 rpm shaking. LB (0.5 L – 2 L) was then inoculated with this starter culture in the morning. After further incubation at 37 °C with shaking to an OD600 of approximately 0.4 (~ 2-3 h), IPTG was added to a final concentration of 0.25 mM. Bacteria were further incubated at 30 °C for 2 h. Cultures were then pelleted in a JA-14 rotor at 5000 rpm (3825 × g) at 4 °C in large centrifuge bottles. The cell pellets were consolidated and either stored at -80 °C or immediately used for purification.
FUS was purified via Nickel affinity column chromatography on an FPLC system. Cell pellets were resuspended in FUS Lysis Buffer (1 M KCl, 1 M Urea, 50 mM Tris-HCl pH 7.4, 10 mM imidazole, 1.5 mM 2-mercaptoethanol, 1% (v/v) IGEPAL-CA630, 5% (v/v) glycerol, one-half tablet of EDTA-free cOmplete protease inhibitor tablets, and 5 mg RNase A) on ice. The resuspended cells were lysed via sonication for 6 min with 8 s pulses at 20% amplitude. Lysate debris was pelleted by centrifuging in a JA-20 rotor at 14000 rpm (23645 × g) at 4 °C in small centrifuge bottles. The supernatant fraction was collected and filtered through 0.22 μm filter attached to a 30 mL Luer-lock syringe into a new 50 mL conical vial. FUS was then purified using a 5 mL His-Trap column (GE Healthcare) on an AKTA pure 25 M FPLC system (GE Healthcare). The lysate was loaded via a 50 mL Superloop, and the column was washed for 10 column volumes with FUS Binding Buffer (1 M KCl, 1 M Urea, 50 mM Tris-HCl pH 7.4, 10 mM imidazole, 1.5 mM 2-mercaptoethanol, 5% (v/v) glycerol). FUS was eluted with a linear gradient of FUS Binding Buffer to FUS Elution Buffer (1 M KCl, 1 M Urea, 50 mM Tris-HCl pH 7.4, 500 mM imidazole, 1.5 mM 2-mercaptoethanol) over the course of 25 2-mL fractions. FUS generally eluted in fractions 15 and 16, and fraction 15 was used to perform all experiments. Purified FUS was immediately diluted 1:1 with 50% (v/v) glycerol and stored at 4 °C. The FUS concentration was determined by a Nanodrop spectrophotometer using Beer’s Law with εFUS of 139000 M−1 cm−1. All experiments used FUS that was no more than 3 weeks old.
Nickel columns for each protein were stripped and regenerated every 5-10 purifications by incubating in Stripping Buffer (20 mM Na3PO4 pH 7.0, 500 mM NaCl, 50 mM EDTA pH 8.0) for 20 min then 1 M NaOH for 2 h. Nickel (25 mL, 100 mM NiSO4) was flowed at 0.5 mL/min to regenerate the His-Trap column.
Labeling of Purified FUS
FUS was non-specifically labeled using Cy3, Cy5, Alexa 594, and STAR635P NHS esters. First, purified FUS was buffer-exchanged to Labeling Buffer (1 M KCl, 1 M Urea, 1X PBS, 1.5 mM 2-mercaptoethanol, 5% (v/v) glycerol) using two 500 uL Zeba desalting columns to remove Tris, which interferes with amine labeling by NHS esters. Zeba columns were prepared according to the manufacturer’s instructions by spinning 300 uL of the desired buffer three times at 1500 × g for 2 min in an Eppendorf microcentrifuge. FUS (~10 μM) was then incubated with 200 μM fluorophore (dissolved in DMSO) in 100 mM NaHCO3 for 30 min in the dark at room temperature with gentle rotation. Excess dye was removed by again buffer-exchanging FUS to 20 mM Na3PO4 with two 500 uL Zeba columns. The labeling efficiency of FUS was then determined using a Nanodrop spectrophotometer and Beer’s Law μM dye divided by μM protein), and all labeled FUS used in the following experiments were generally 70-95% labeled. Stored labeled FUS was diluted 1:1 with 2X Labeling Buffer.
RNA Preparation
RNA Synthesis
The following RNAs were synthesized by IDT:
18-mer: 5’-biotin-UGG CGA CGG CAG CGA GGC-3’-amino-modified
18-mer-U50: 5’-amino-modified-U50 GCC UCG CUG CCG UCG CCA-3’
U40: 5’-U40-3’-amino-modified.
Highly-concentrated stocks were diluted in nuclease-free H2O to 1, 10, or 100 μM and stored at −20 °C. Single-use 10 nM aliquots were then diluted in water and stored at −20 °C until used.
RNA Labeling
RNA (10 or 100 μM) was combined with 0.2 mg NHS-conjugated fluorophores (Cy3 or Cy5) and 100 μM NaHCO3 and labeled overnight at room temperate in the dark with gentle rotation. After the labeling reaction finished, NaCl and ice-cold ethanol were added to 300 mM and 70% (v/v), respectively. RNA was stored at −20 °C for thirty minutes and then pelleted at 21000 × g for 30 min at 4 °C. The supernatant fraction was carefully removed and the pellet was washed with 70% (v/v) ethanol. The RNA pellet was resuspended in 10 mM Tris pH 7.4 and pelleted again as described above. After the second round of washing, the RNA pellet was air-dried for 20 min in the dark and resuspended in T50 buffer (10 mM Tris pH 7.4, 50 mM NaCl). RNA concentration was then calculated using a Nanodrop spectrophotometer with the appropriate extinction coefficient for the RNA and fluorophore.
RNA Annealing
Complementary RNAs (i.e. constructs 1 and 2 from RNA Synthesis above.) were annealed by combining the RNAs in a 1:1 ratio in T50 buffer. The RNA mixture was then heated to 85 °C for 2 min followed by stepwise 1 °C/min cooling to room temperature.
FUS-RNA Phase-Separation
Phase-Separation Reactions
All FUS phase-separation reactions occurred with the 6×His and MBP solubility tag cleaved and with RNA as previously described (Niaki et al., 2020; Sarkar and Myong, 2018). Unlabeled FUS was buffer-exchanged into 20 mM Na3PO4 pH 7.4 using Amicon filters with a molecular weight cutoff of 50 kDa. This was achieved by loading protein onto the top of the column, centrifuging per the manufacturer’s instructions (21000 × g for 3 min), and diluting the remaining protein with 20 mM Na3PO4 pH 7.4 successively. Using Amicon filters avoided premature aggregation observed through small-scale dialysis and other methods while still removing a sufficient amount of the high-salt, high-urea storage buffer. Labeled FUS was used immediately after the final buffer-exchange into 20 mM Na3PO4 pH 7.4 with Zeba desalting columns.
FUS (1 μM) was combined with 1 μM unlabeled U40 RNA and 10 nM Cy3-labeled U40 in 1X Cleavage Buffer (50 mM Tris pH 7.4, 100 mM NaCl, 1 mM EDTA pH 8, 1 mM DTT). TEV (5 U) was added to start the reaction by cleaving the 6xHis and MBP solubility tags. Cy3-labeled RNA was not used for reactions with labeled FUS. Reactions were carried out in 8-well Nunc chambers. For reactions with WT and mutant FUS together, each FUS was added to a final concentration of 500 nM for a total FUS concentration of 1 μM. For reactions in which one FUS was incubated alone before addition of another FUS at a later time point, the initial reaction proceeded with 1 μM mutant FUS at a lower volume before addition of WT FUS to a final concentration of 500 nM each at a 1:1 mixing ratio. Each droplet experiment was performed in with three separate batches of purified protein.
Droplets were imaged every 1 h on a Nikon Ti Eclipse wide-field microscope equipped with a 100x oil-immersion objective. An EMCCD camera was used to capture images in the Cy3 (555 nm) and Cy5 (637 nm) channels. For reactions with both Cy3- and Cy5-labeled FUS, Cy5-labeled FUS was briefly photobleached (~2 s) with high laser intensity to allow visualization of the Cy3 FUS, which underwent FRET with the Cy5 FUS in the droplets. For videos, the Cy3- and Cy5-channels were imaged every 10 s for 30 min.
In Vitro Stimulated Emission Depletion Imaging
Two-color Stimulated Emission Depletion (STED) imaging was performed using a previously-described setup (Han and Ha, 2015; Ma and Ha, 2019). Unlabeled WT and mutant FUS (500 nM each) were combined with 10 nM Alexa 594-labeled WT FUS and 10 nM STAR 635P-labeled mutant FUS in Cleavage Buffer. U40 RNA and TEV were added to start the reaction. Droplets were imaged on a custom-built 100X oil-immersion objective STED setup with 600/620 and 655/670 excitation/emission pathways. A STED laser of 760-780 nm was used for depletion. To overcome the partial overlap of the emission spectra, selective excitation and linear unmixing was used to deconvolute the channels (Han and Ha, 2015).
Fluorescence Recovery After Photobleaching
FRAP experiments were performed on the same Nikon Ti Eclipse wide-field microscope described in Phase Separation Reactions above. A 50 mW 405 nm bleaching laser and a Brucker Galvano mirror scanner were used to bleach small regions-of-interest containing single droplets. Droplets were bleached with 50% laser intensity for 5 ms per pixel with each ROI consisting of a 10-20-pixel diameter circle. Cy3 fluorescence was measured twice before bleaching, and successively for 10 min after bleaching at 3 s intervals for the first 2 min and 10 s intervals for the remaining 8 min. Eight droplets were bleached per video.
Cell Culture Experiments
SH-SY5Y cells were passaged into 4-well Nunc chambers with 500 uL DMEM mixture two days before imaging. The FUS mammalian expression vectors (200 ng each plasmid; see Experimental Model and Subject Details above) were transfected into SH-SY5Y cells one day before imaging by following the manufacturer’s instructions for the Lipofectamine 3000 kit. Following one additional day of growth, cells were prepared for imaging by adding Hoechst 33432 to a final concentration of 1X and, when Halo-tagged expression vectors were used, Janelia Fluor 646 to a final concentration of 25 nM. After a 15-minute incubation at 37 °C, cells were imaged on a Nikon Ti Eclipse wide-field microscope as described for FUS droplets above (see Phase Separation Reactions above). To stress cells, NaAsO2 was added to a final concentration of 0.05 mM 1 h before imaging. Cell culture experiments were performed in triplicate.
Electrophoretic Mobility Shift Assay
Three-color protein-RNA interactions were resolved on 6% retardation gels. Alexa 488-labeled U50 RNA (1 nM) was added to increasing concentrations of Cy3- and Cy5-labeled FUS (100 nM, 500 nM, and 1 μM). Binding reactions were incubated at room temperature in the dark for 45 min in Binding Buffer (50 mM Tris-HCl pH 7.5, 100 mM KCl, 100 μg/mL BSA, 100 mM 2-mercaptoethanol, 2 mM MgCl2). Before loading, Loading Buffer (final concentration of bromophenol blue, sucrose) was added to the reactions. The 6% retardation gels were electrophoresed for 1.5 h at 100 V and imaged on a Typhoon 5 fluorescent gel imager.
Single-Molecule Total Internal Reflection Microscopy
Single-Molecule Nucleation Experiments
Low-density biotin PEG-passivated slides were purchased from the Slide Production Core for Microscopy at Johns Hopkins Medical Institute, where they were prepared as described previously (Joo and Ha, 2012a; Sarkar and Myong, 2018). Slides were stored at −20 °C, and were thawed to room temperature approximately 15 min prior to assembly. Individual lanes were created using double-sided tape. The coverslip was glued to the slide using the double-sided tape and quick-drying epoxy. Flow chambers were constructed using P200 tips as buffer reservoirs and epoxy molds for syringe connections.
The prism-type TIRF microscope setup that was used for these experiments has been described previously (Joo and Ha, 2012b). Nucleation assays were performed at room temperature. First, 1 mg/mL NeutrAvidin was flowed onto the single-molecule surface and incubated for five minutes. Following a T50 buffer wash, 50 pM pdU50 was flowed onto the surface. For two-color nucleation experiments, unlabeled pdU50 was used. For single-color Cy3- or Cy5-labeled FUS experiments, pdU50 RNA with Cy5- or Cy3-labeled 18-mer (construct 1 from RNA Synthesis above). were used, respectively. Following another T50 buffer wash, Imaging Buffer (20 mM Tris-HCl pH 7.4, 100 mM KCl, 0.5% (w/v) glucose, 1 mg/mL glucose oxidase, 1.8 U/mL catalase, ~10 mM Trolox, 2 U TEV, 4 U RNase Inhibitor) was flowed using a syringe pump at 1 mL/min flow rate. Protein was diluted in Imaging Buffer immediately prior to flowing onto the surface, which was synchronized with the start of the video with a 6 s delay. Flow videos were acquired using 100 ms exposure time with maximum gain for 300 s. For two-color flow experiments, laser excitation was alternated between Cy3 and Cy5 every 1 s (10 frames). For one-color flow experiments, laser excitation alternated for one cycle to visualize labeled RNA during the delayed flow period followed by constant excitation of the protein label channel.
For control experiments, protein was flowed onto the single-molecule surface without the RNA addition step.
FRET Experiments
FRET experiments were carried out essentially as described in Single-Molecule Nucleation Experiments. (Niaki et al., 2020; Sarkar and Myong, 2018), except for the following changes: (1) P200 reservoirs and epoxy molds were not used for most smFRET experiments, (2) FRET-labeled pdU50 was used instead of unlabeled or singly-labeled pdU50, (3) unlabeled FUS was equilibrated with labeled RNA for ~30 min before imaging, and (4) the surface was illuminated with the Cy3 laser only.
Fluorescence Anisotropy
Anisotropy reactions were carried out in Binding Buffer (see Electrophoretic Mobility Shift Assays above). for 1 h in Thermo Scientific Nunc MicroWell 96-well microplates. FUS concentration was varied from the micromolar to the picomolar range and Cy3-U40 concentration was held constant at 10 nM. Fluorescence polarization was measured by a Tecan Spark 10M plate reader with fluorescent excitation at 563 nm and emission at 615 nm (20 nm bandwidth).
Phase Diagrams
Phase diagrams were determined using a turbidity assay in which varying concentrations of FUS were combined with varying concentrations of RNA in LLPS-favorable conditions. Reactions were incubated in the same buffer conditions as the droplet reactions described above (see FUS-RNA Phase Separation) in Thermo Scientific Nunc MicroWell 96-well plates. The A400 value was measured over 4 h every 5 min by a Tecan Spark 10M plate reader, and the absorbance value for each well was visualized using Matlab. In general, we observed that A400 steadily increased from 0.05 to 0.10 for conditions with robust LLPS; wells with a lower FUS concentration had smaller increases. We calculated the percentage turbidity increase for each well compared to the condition with the highest turbidity increase (Figure S5B). We then assayed for droplets for set of conditions (125 nM RNA with varying R244C FUS concentration) in triplicate to determine which percentage cutoff to use for LLPS versus no LLPS. This cutoff was used to map the phase diagram.
Heterotypic Droplet Fusion
Microfluidic Chamber & Droplet Preparation
Droplets were formed as described above in RNase-free 1.5 mL microcentrifuge tubes and transferred to 1 mL syringes (see FUS-RNA Phase Separation above). To prevent mixing and controllably maneuver the WT and G156E droplets, we performed all our experiments in a custom-built microfluidic chamber (Figure S7A) as described previously (Whitley et al., 2017). First, we carefully cleaned a pair of glass coverslips (Fisher Scientific, number 1). Then, using a laser engraver (Universal Laser Systems, VLS2.25) we cut out a pattern in Nescofilm (Karlan) with a design for three laminar-flow channels (Figure S7A). We carefully placed the pattern between glass slides, and moved it onto a heat block until the melted Nescofilm formed a uniform seal. Next, we fit the chamber onto a custom-built bracket, to which we attached Tygon tubing to all the inlets and outlets. Finally, we passivated the surfaces of both the chamber and the tubing with polyethylene glycol (PEG; Laysan Bio, MW 5000) by following a previously described protocol (Comstock et al., 2015; Ha et al., 2002). After each experiment, we washed the chamber with ethanol and water, and dried it with nitrogen. To extend the lifetime of the PEGylated surface we stored the chamber between experiments in the dark at 4° C.
Optical Trapping
To optically trap and monitor fusions of FUS droplets, we used a custom-built instrument which combines three modules: optical tweezers, bright-field and fluorescence microscopy (Mears et al., 2014; Min et al., 2009). The optical tweezers consist of two traps generated by timesharing a single beam generated by a 5-W, 1,064-nm fiber laser (YLR-5-1064-LP, IPG Photonics). To adjust the distance between the trapped droplets, we precisely controlled the trap separation by deflecting the laser with an acousto-optic deflector (DTSXY-250-1064, AA Opto-Electronic).
To avoid any excess oxygen by slowly flushing the chamber with 5 mL of filtered MilliQ water, after which we filled all channels with 1X Cleavage Buffer (see FUS-RNA Phase Separation above). Next, we gently filled the channels with droplet solutions (40 μL at 100-1,000 μL/h), and kept a constant flow rate of 5 μL/h throughout all measurements. We consistently used the top and bottom channels for the WT and G156E solutions, correspondingly.
To minimize the influence of laser-light on droplet integrity, we used the lowest possible trapping power (72 mW), which was sufficient to stably trap the droplets. We controlled the chamber position with a three-axis translational stage (Newport; ESP300) to move the optical traps into channels containing either WT or G156E droplets and to trap one of each (the order of trapping either WT or G156E droplets was alternated for mixed fusions). We moved trapped droplet pairs to the middle, blank channel (10X cleavage buffer, see FUS-RNA Phase Separation above) to record fusions.
Bright-field and Epifluorescence Imaging of Optically-trapped Droplets
We used bright-field imaging to inspect droplet morphology and singularity before and after each fusion attempt. Once we trapped a droplet pair and positioned it in the blank channel, we excited Cy3-labelled RNA within droplets with a 532-nm laser (WorldStar Tech, TECGL-30) in epifluorescence configuration. We set the exposure time to 50 μs and collected fluorescence images of WT-WT, WT-G156E, and G156E-G156E fusions with an EMCCD camera (Andor, iXon3 860 EMCCD) every 0.01 sec (Supplementary Videos 1–3).
To ensure that we did not forcefully merge the droplets by bringing them too close to one another, we plotted the Cy3 fluorescence intensity along the trap separation coordinate in real-time. At the start of each fusion attempt, we kept the two droplets at a distance corresponding to two fully separated Gaussian fluorescence intensity profiles. We decreased the trap separation, reducing the distance between the droplets, until the two Gaussian profiles started to overlap at the tails. We then fixed the trap position and allowed the droplets to attempt to fuse.
Counting Fraction of Successful Fusions
After positioning the droplets in close proximity, we waited for up to ~500 s for the droplets to merge. We followed each fusion attempt with a recording of a bright-field video of the droplet pair released from the traps. We observed two types of events: the droplets either coalesced or remained separate. In case of successful fusion, we observed a larger-sized droplet in one of the traps (Figure S7B, left panel). We confirmed that only one droplet was present by overlapping the traps and pulling them apart in small increments. This single droplet either remained in the same trap or fell into the other trap. In addition, we clearly resolved a single, fused droplet by bright-field imaging when released from the trap (Figure S7C, left panel). Events were scored as unsuccessful if no fusion was observed after ~500 s (Figure S7B, right). In all but one case, we noticed that unfused droplets were attracted to each other and remained tethered when we tried to pull the droplets apart. These unfused droplet dimers could not be separated by increasing the trap distance. Furthermore, we could clearly resolve unfused droplet dimers by bright-field imaging, when released from traps (Figure S7C, right panel).
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of Phase Separation Images
For image processing, Nikon images were first converted from the Nikon image format (.nd2) to TIFF images for each channel using Fiji. Each channel was then further processed in Adobe Photoshop by adjusting the contrast and exposure to limit background and better visualize droplets. Multi-channel images were merged into one Photoshop document and exported as overlaid or individual channel TIFF images.
Colocalization between Cy3- and Cy5-labeled FUS was quantified using a customized Matlab script. First, a binary mask was generated for droplets in both the Cy3 and Cy5 channels. The masks were overlaid to identify droplets present in both channels. The number of overlapping droplets was divided by the total number of droplets (Cy3 droplets plus Cy5 droplets minus overlapping droplets) to determine the colocalization score. Significance was calculated using a two-sample proportion z-test.
Analysis of FRAP Videos
FRAP videos were processed using a custom Matlab 2015b code as described previously (Niaki et al., 2020). Briefly, 8 droplets were selected for photobleaching by drawing circular ROIs; these droplets usually ranged in size from 1-5 μm2, depending on the condition. The droplet intensity before after photobleaching was determined and normalized for each ROI with the initial intensity equal to 1 and the frame after photobleaching equal to 0. Background intensity was subtracted, and a reference ROI was used to correct for photobleaching caused by Cy3 excitation. Drift in the xy-plane was also corrected using a reference droplet. We occasionally observe FRAP values above 1.0 because the droplets continue to grow over the 600 s video, especially at the 1-2 h timepoints. Generally, Welch’s t-test was used to test the significance of the FRAP values for the final time point of each pairwise comparison.
Cell Image Processing
Images were processed as described above for droplet images (see Analysis of Phase Separation Images above).
Analysis of EMSA Images
Images were processed with Fiji and Adobe Photoshop essentially as described for droplet images (see Analysis of Phase Separation Images above). Intensity plots were generated using the “Plot Profile” function on Fiji and normalizing the intensity for each channel. The Pearon’s correlation coefficient for the Cy3 and Cy5 channels was calculated using R Studio.
Analysis of Single-Molecule Traces
Single-molecule nucleation and FRET traces were analyzed with IDL mapping scripts and a suite of customized Matlab scripts (Li et al., 2016). A mapping file generated from a control bead slide was first used to map spots between the Cy3 and Cy5 channels with the correct offset. Mapped traces were then analyzed using Matlab scripts that plotted the Cy3 and Cy5 intensities for each spot over time. For alternating-excitation traces, the Cy3 and Cy5 excitations were de-interleaved and plotted separately. For FRET traces, FRET was calculated and plotted as described previously. The dynamic fraction of each trace was calculated by manually identifying portions with FRET fluctuations. These values were then plotted using a freely-available violin plot Matlab code.
Kinetics were determined by indicating each binding event (t1, t2, etc.) on single-molecule nucleation traces. Binding events were considered “real” if they lasted >10 s, and a poisson distribution was fitted to each single-color nucleation histogram. The preflow time (6 s) was subtracted from the initial binding event. Cumulative distribution frequency plots were generated from the dwell times for each condition as described previously (Zhuang et al., 2000). Single-exponent decays were fit for each 1-CDF plot using Matlab’s built-in curve fitting application, and standard deviations were calculated from the 95% confidence interval of the exponential fit. The apparent kon values were equal to the exponential term.
Analysis of Two-Color Nucleation Traces
Cy3- and Cy5-binding events were manually identified for >100 traces for each two-color condition. In general, a binding event was considered legitimate if the protein remained bound and fluorescent for at least 10 s. We also considered the number of photobleaching events to accurately determine the number of binding events. Because we did not observe unbinding in smFRET experiments, we assumed that all FUS remained bound and that all decreases in fluorescence intensity were due to photobleaching, not unbinding. However, we cannot exclude the possibility that higher-order multimers may dissociate from the RNA as our FRET assay is not sensitive enough to discern differences between higher-order multimers. Once the number of binding events was identified, traces were manually binned into the two-dimensional heatmaps, and the proportion in each heatmap was calculated from the total number of RNAs with two or more proteins bound. Because RNAs with only one protein bound could not be considered mixed or unmixed, we excluded these bins from our heatmaps and from the colocalization calculations. The colocalization score was calculated by dividing the number of molecules in the interior of the heatmap by the total number of molecules. Error was determined using the proportion, and significance was calculated using Welch’s t-test.
Attempted binding events were identified as transient (<1 s) increases in intensity that occurred after an initial Cy3-WT binding event but before a successful Cy5-mutant binding event (for an example, See Figure 3B). The average and error of attempted binding events was determined from the distribution of each condition. Welch’s t-test was performed to calculate significance.
Anisotropy Binding Isotherms
Anisotropy was calculated automatically by the Tecan software via the following equation: r = 1000 × (Ip − Ie) / (Ip + 2 Ie) where Ip is parallel fluorescence intensity and Ie is perpendicular fluorescence intensity. Binding isotherms were plotted and fitted using GraphPad Prism 7. Error is reported as the error of the fitted KD value for three replicates, and Welch’s t-test was used for pairwise comparisons of fitted KD values.
Quantification of the Number of Successful Fusion Events
We calculated the fraction of successful fusions, F.F. = Nfused/Ntotal, where Nfused is the number of successful fusions, and Ntotal is the total number of all fusion attempts. The average and standard deviation for each condition was determined, and Welch’s t-test was performed to test the significance.
Quantification of Fusion Time, τ1/2, from Fluorescence Movies
We processed the fluorescence recordings with custom MATLAB (MATLAB R2019b) code which used standard functions for image processing (Figure S7D). The field of view was 128 × 128 pixels or 102.4 × 102.4 μm. For all steps, we processed each .tiff frame individually. Firstly, we converted the images to ‘uint16’ (function: cast), and adjusted the pixel intensity to saturate the bottom and top 1% of all values (function: imadjust) (Figure S7D, panel 1). Then, we de-noised the image by creating a morphological structuring element in a shape of a 1-pixel disk (functions: strep and imopen) (Figure S7D, panel 2), and adjusted the pixel intensity once again (functions: greythresh and imadjust) (Figure S7D, panel 3). Next, we detected edges of the droplets; the most reliable method for edge detection involved finding local maxima in the gradient in pixel intensities calculated using the derivative of a Gaussian filter (function: edge, method: ‘Canny’, degree: 0.5) (Figure S7D, panel 4). We then closed disconnected edges (function: imclose), filled the objects (function: imfill, method: ‘holes’) and cleaned the image of small areas of background-intensity pixels in the background (function: bwmorph, method: ‘majority’). Finally, we determined the droplet centroid, major and minor axes, and radii by fitting the objects to an ellipse (function: regionprops) (Figure S7D, panel 5).
We calculated the aspect ratio, A.R. = lmajor/lminor, where lmajor and lminor are the major and minor axes. When two objects were detected, corresponding to spatially nonoverlapping droplets, we summed the two aspect ratios.
We determined the fusion time τ1/2 for different fusion events from the time at which fusion reached half completion (Figure S7E). We systematically identified the start of each event using changepoint analysis (Beausang et al., 2011). The end of fusion was defined by fitting a line to a stable baseline in aspect ratio of a merged droplet.
Supplementary Material
Supplemental Video 1 (related to Figure 7 and STAR Methods): An example wild-type-wild-type FUS-RNA droplet fusion.
Supplemental Video 2 (related to Figure 7 and STAR Methods): An example wild-type-G156E FUS-RNA droplet fusion.
Supplemental Video 3 (related to Figure 7 and STAR Methods): An example G156E-G156E FUS-RNA droplet fusion.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. Coli BL21(DE3) Chemically Competent Cells | Millipore Sigma | CMC0014-20X40UL |
| NEB® Turbo Competent E. coli (High Efficiency) | New England Biolabs | C2984H |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AcTEV Protease | Fisher Scientific | 12-575-015 |
| Ribonuclease A from bovine pancreas | Millipore Sigma | R6513 |
| cOmplete™ Protease Inhibitor Cocktail | Roche | 11697498001 |
| Glucose Oxidase from Aspergillus niger | Sigma-Aldrich | G2133 |
| Catalase from bovine liver | Sigma-Aldrich | C3155 |
| IGEPAL® CA-630 | Sigma-Aldrich | I8896 |
| His-Trap FF Crude, 5 mL | GE Healthcare | 17528601 |
| Cy3 NHS Ester | GE Healthcare | PA13101 |
| Cy5 NHS Ester | GE Healthcare | PA15100 |
| Alexa Fluor™ 594 NHS Ester (Succinimidyl Ester) | Thermo Scientific | A20004 |
| Abberior STAR 635P | Abberior | ST635P |
| Janelia Fluor® 646 HaloTag® Ligand | Promega | GA112A |
| RNase Inhibitor, Murine | New England Biolabs | M0314L |
| Lipofectamine™ 3000 Transfection Reagent | Thermo Scientific | L3000001 |
| Sodium (meta)arsenite (>90%) | Millipore Sigma | S7400-100G |
| Experimental Models: Cell Lines | ||
| Human: SH-SY5Y | ATCC | CRL-2266 |
| Oligonucleotides | ||
| Biotin-18mer: 5’- /biotin/rUrGrG rCrGrA rCrGrG rCrArG rCrGrA rGrGrC/3AmMO/ -3’ | Niaki et al., 2020; IDT | N/A |
| U50-18mer: 5’- /5AmMC6/rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrG rCrCrU rCrGrC rUrGrC rCrGrU rCrGrC rCrA -3’ | Niaki et al., 2020; IDT | N/A |
| U40: 5’- rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rUrUrU rU/3AmMO/ -3’ | Niaki et al., 2020; IDT | N/A |
| Recombinant DNA | ||
| pTHMT/FUSWT (encoding 6xHis-MBP-FUSWT) | Niaki et al., 2020 | N/A |
| pTHMT/FUSR244C (encoding 6xHis-MBP-FUSR244C) | Niaki et al., 2020 | N/A |
| pTHMT/FUSR216C (encoding 6xHis-MBP-FUSR216C) | Niaki et al., 2020 | N/A |
| pTHMT/FUSR521G (encoding 6xHis-MBP-FUSR521G) | Niaki et al., 2020 | N/A |
| pTHMT/FUSG156E (encoding 6xHis-MBP-FUSG156E) | Niaki et al., 2020 | N/A |
| pTHMT/FUSG187S (encoding 6xHis-MBP-FUSG187S) | Niaki et al., 2020 | N/A |
| pTHMT/FUSG399V(encoding 6xHis-MBP-FUSG399V) | Niaki et al., 2020 | N/A |
| pTHMT/FUSRBD(encoding 6xHis-MBP-FUSRBD) | Genscript | N/A |
| pTHMT/FUSLCD(encoding 6xHis-MBP-FUSLCD) | Genscript | N/A |
| pTHMT/FUSG156A(encoding 6xHis-MBP-FUSG156A) | Genscript | N/A |
| pTHMT/FUSG156D(encoding 6xHis-MBP-FUSG156D) | Genscript | N/A |
| pTHMT/FUSG156Q(encoding 6xHis-MBP-FUSG156Q) | Genscript | N/A |
| pTHMT/FUSG154E(encoding 6xHis-MBP-FUSG154E) | Genscript | N/A |
| pTHMT/FUSG156P(encoding 6xHis-MBP-FUSG154E) | Genscript | N/A |
| pTHMT/FUSWT-EGFP (encoding 6xHis-MBP-FUSWT-EGFP) | Niaki et al., 2020 | N/A |
| pTHMT/FUSWT-mCherry (encoding 6xHis-MBP-FUSWT-mCherry) | Genscript | N/A |
| pTHMT/FUSR244C-EGFP (encoding 6xHis-MBP-FUSR244C-EGFP) | Genscript | N/A |
| pTHMT/FUSG156E-EGFP (encoding 6xHis-MBP-FUSG156E-EGFP) | Genscript | N/A |
| pFUSWT-GFP_pcDNA3.1(+)-C-eGFP | Genscript | N/A |
| pFUSWT-Halo_piggybac-EF1-Halo | Li et al., 2016; Genscript | N/A |
| pFUSR244C-Halo_piggybac-EF1-Halo | Li et al., 2016; Genscript | N/A |
| pFUSG156E-Halo_piggybac-EF1-Halo | Li et al., 2016; Genscript | N/A |
| Software and Algorithms | ||
| MATLAB and IDL scripts | https://physics.illinois.edu/cplc/software/; This manuscript | |
| NIS-Elements Ar Package | Nikon Inc. | |
| Adobe Photoshop CC | Adobe (https://www.adobe.com/products/photoshop.html) | |
| Prism 7 | GraphPad (https://www.graphpad.com/scientific-software/prism/) | |
| ImageJ (Fiji) | NIH (https://imagej.nih.gov/ij/) | |
| RStudio | RStudio (https://rstudio.com/products/rstudio/download/) | |
| Other | ||
| Nunc™ Lab-Tek™ Chambered Coverglass | Thermo Scientific | 155361 |
| DNA Retardation Gels (6%) | Invitrogen | EC63655BOX |
| Amicon Ultra-0.5 Centrifugal Filter Unit | Millipore | UFC501096 |
| Zeba™ Spin Desalting Columns, 7K MWCO, 0.5 mL | Thermo Scientific | 89883 |
Highlights.
ALS/FTLD-linked FUS mutations in glycine cause decelerated mixing with wild-type FUS
G mutant does not associate with wild-type FUS, leading to separate condensates
R mutant interacts and condenses with wild-type FUS, thereby recovering the mutant defect
The position and bulkiness of the G156E is critical for encoding the mutant properties
Acknowledgements
We thank Rohit Pappu for help interpreting our results and formulating a mechanistic basis of our observations, Olivia Yang and Dmitriy Bobrovinikov for help with data analysis, Laura Ganser for proofreading the manuscript, and the rest of the Ha-Myong labs for technical support and advice. We are also grateful to Charlotte Fare and Jim Shorter, who kindly provided purified Karyopherin-β2, Anthony K. Leung, who provided the SH-SY5Y cell line, and Jee Min Kim and Carl Wu, who gifted JF646 fluorophore. We acknowledge funding support from 1-F31-NS113439 (to K.R.), 1-T32-GM007231-45 (to K.R., J.L., and K.F.C.), 1-RF1-NS113636-01 (to S.M.), 1-R01-GM122569 (to T.H.), 1-R01-GM120353 (to Y.C.), the National Science Foundation PHY-1430124 via the Center for Physics of Living Cells (to T.H., Y.C., and S.M.), the Howard Hughes Medical Institute (to T.H.), and the NSF Research Experience for Undergraduates (to C.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare that they do not have any competing interests.
REFERENCES
- Alexander EJ, Ghanbari Niaki A, Zhang T, Sarkar J, Liu Y, Nirujogi RS, Pandey A, Myong S, and Wang J (2018). Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proc Natl Acad Sci U S A 115, E11485–E11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausang JF, Goldman YE, and Nelson PC (2011). Changepoint analysis for single-molecule polarized total internal reflection fluorescence microscopy experiments. Methods Enzymol 487, 431–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Holehouse AS, Weinhardt V, Kovacs D, Van Lindt J, Larabell C, Van Den Bosch L, Das R, Tompa PS, Pappu RV, et al. (2019). Spontaneous driving forces give rise to protein-RNA condensates with coexisting phases and complex material properties. Proc Natl Acad Sci U S A 116, 7889–7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J-M, Hyman AA, and Pappu RV (2020a). Generalized models for bond percolation transitions of associative polymers. In arXiv e-prints, pp. arXiv:2004.03278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JM, Holehouse AS, and Pappu RV (2020b). Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu Rev Biophys 49, 107–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid-Samper F, Gelabert-Baldrich M, Lang B, Lorenzo-Gotor N, Ponti RD, Severijnen L, Bolognesi B, Gelpi E, Hukema RK, Botta-Orfila T, et al. (2018). An Integrative Study of Protein-RNA Condensates Identifies Scaffolding RNAs and Reveals Players in Fragile X-Associated Tremor/Ataxia Syndrome. Cell Rep 25, 3422–3434 e3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, Silani V, and Ratti A (2012). TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J Biol Chem 287, 15635–15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comstock MJ, Whitley KD, Jia H, Sokoloski J, Lohman TM, Ha T, and Chemla YR (2015). Protein structure. Direct observation of structure-function relationship in a nucleic acid-processing enzyme. Science 348, 352–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrado L, Del Bo R, Castellotti B, Ratti A, Cereda C, Penco S, Soraru G, Carlomagno Y, Ghezzi S, Pensato V, et al. (2010). Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J Med Genet 47, 190–194. [DOI] [PubMed] [Google Scholar]
- Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Mojsilovic-Petrovic J, Panossian S, et al. (2012). Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet 21, 2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle JG, Lanson NA Jr., Smith RB, Casci I, Maltare A, Monaghan J, Nichols CD, Kryndushkin D, Shewmaker F, and Pandey UB (2013). RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum Mol Genet 22, 1193–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Gao K, and Jankovic J (2014). The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol 10, 337–348. [DOI] [PubMed] [Google Scholar]
- Elbaum-Garfinkle S (2019). Matter over mind: Liquid phase separation and neurodegeneration. J Biol Chem 294, 7160–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay MM, Anderson PJ, and Ivanov P (2017). ALS/FTD-Associated C9ORF72 Repeat RNA Promotes Phase Transitions In Vitro and in Cells. Cell Rep 21, 3573–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feric M, Vaidya N, Harmon TS, Mitrea DM, Zhu L, Richardson TM, Kriwacki RW, Pappu RV, and Brangwynne CP (2016). Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 165, 1686–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foffi G, De Michele C, Sciortino F, and Tartaglia P (2005). Arrested phase separation in a short-ranged attractive colloidal system: a numerical study. J Chem Phys 122, 224903. [DOI] [PubMed] [Google Scholar]
- French RL, Grese ZR, Aligireddy H, Dhavale DD, Reeb AN, Kedia N, Kotzbauer PT, Bieschke J, and Ayala YM (2019). Detection of TAR DNA-binding protein 43 (TDP-43) oligomers as initial intermediate species during aggregate formation. J Biol Chem 294, 6696–6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasior K, Zhao J, McLaughlin G, Forest MG, Gladfelter AS, and Newby J (2019). Partial demixing of RNA-protein complexes leads to intradroplet patterning in phase-separated biological condensates. Phys Rev E 99, 012411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, and Rosen MK (2019). Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484 e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O’Donovan K, Fare CM, Diaz Z, Singh N, et al. (2018). Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 173, 677–692 e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T, Rasnik I, Cheng W, Babcock HP, Gauss GH, Lohman TM, and Chu S (2002). Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature 419, 638–641. [DOI] [PubMed] [Google Scholar]
- Han KY, and Ha T (2015). Dual-color three-dimensional STED microscopy with a single high-repetition-rate laser. Opt Lett 40, 2653–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon TS, Holehouse AS, Rosen MK, and Pappu RV (2017). Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofweber M, Hutten S, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, Ruepp MD, Simons M, Niessing D, Madl T, et al. (2018). Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 173, 706–719 e713. [DOI] [PubMed] [Google Scholar]
- Hondele M, Sachdev R, Heinrich S, Wang J, Vallotton P, Fontoura BMA, and Weis K (2019). DEAD-box ATPases are global regulators of phase-separated organelles. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo C, and Ha T (2012a). Preparing sample chambers for single-molecule FRET. Cold Spring Harb Protoc 2012, 1104–1108. [DOI] [PubMed] [Google Scholar]
- Joo C, and Ha T (2012b). Prism-type total internal reflection microscopy for single-molecule FRET. Cold Spring Harb Protoc 2012. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Bercier V, Lissouba A, Liao M, Brustein E, Rouleau GA, and Drapeau P (2011). FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet 7, e1002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamelgarn M, Chen J, Kuang L, Arenas A, Zhai J, Zhu H, and Gal J (2016). Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim Biophys Acta 1862, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamelgarn M, Chen J, Kuang L, Jin H, Kasarskis EJ, and Zhu H (2018). ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc Natl Acad Sci U S A 115, E11904–E11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapanidis A, Majumdar D, Heilemann M, Nir E, and Weiss S (2015). Sample Preparation and Data Acquisition for mus-ALEX. Cold Spring Harb Protoc 2015, 1029–1031. [DOI] [PubMed] [Google Scholar]
- Kapeli K, Pratt GA, Vu AQ, Hutt KR, Martinez FJ, Sundararaman B, Batra R, Freese P, Lambert NJ, Huelga SC, et al. (2016). Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses. Nat Commun 7, 12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, and Parker R (2017). The Stress Granule Transcriptome Reveals Principles of mRNA Accumulation in Stress Granules. Mol Cell 68, 808–820 e805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S, Maharana S, and Simon A (2017). Hexanediol: a chemical probe to investigate the material properties of membrane-less compartments. Matters. [Google Scholar]
- Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- Langdon EM, Qiu Y, Ghanbari Niaki A, McLaughlin GA, Weidmann CA, Gerbich TM, Smith JA, Crutchley JM, Termini CM, Weeks KM, et al. (2018). mRNA structure determines specificity of a polyQ-driven phase separation. Science 360, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva S, de Jesus Domingues AM, Butter F, and Ketting RF (2017). Characterization of genetic loss-of-function of Fus in zebrafish. RNA Biol 14, 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu H, Dong P, Li D, Legant WR, Grimm JB, Lavis LD, Betzig E, Tjian R, and Liu Z (2016). Real-time imaging of Huntingtin aggregates diverting target search and gene transcription. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Erauskin J, Tadokoro T, Baughn MW, Myers B, McAlonis-Downes M, Chillon-Marinas C, Asiaban JN, Artates J, Bui AT, Vetto AP, et al. (2018). ALS/FTD-Linked Mutation in FUS Suppresses Intra-axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron 100, 816–830 e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, and Ha T (2019). Fight against background noise in stimulated emission depletion nanoscopy. Phys Biol 16, 051002. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, et al. (2017). TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 95, 808–816 e809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R, and Neumann M (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9, 995–1007. [DOI] [PubMed] [Google Scholar]
- Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I, Bickle M, Rizk S, Guillen-Boixet J, Franzmann TM, et al. (2018). rNa buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EW, Holehouse AS, Peran I, Farag M, Incicco JJ, Bremer A, Grace CR, Soranno A, Pappu RV, and Mittag T (2020). Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 367, 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears PJ, Koirala S, Rao CV, Golding I, and Chemla YR (2014). Escherichia coli swimming is robust against variations in flagellar number. Elife 3, e01916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min TL, Mears PJ, Chubiz LM, Rao CV, Golding I, and Chemla YR (2009). High-resolution, long-term characterization of bacterial motility using optical tweezers. Nat Methods 6, 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SL, Morisaki T, Khong A, Lyon K, Parker R, and Stasevich TJ (2019). Multicolour single-molecule tracking of mRNA interactions with RNP granules. Nat Cell Biol 21, 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niaki AG, Sarkar J, Cai X, Rhine K, Vidaurre V, Guy B, Hurst M, Lee JC, Koh HR, Guo L, et al. (2020). Loss of Dynamic RNA Interaction and Aberrant Phase Separation Induced by Two Distinct Types of ALS/FTD-Linked FUS Mutations. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. (2015). A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 162, 1066–1077. [DOI] [PubMed] [Google Scholar]
- Peran I, and Mittag T (2020). Molecular structure in biomolecular condensates. Curr Opin Struct Biol 60, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protter DSW, Rao BS, Van Treeck B, Lin Y, Mizoue L, Rosen MK, and Parker R (2018). Intrinsically Disordered Regions Can Contribute Promiscuous Interactions to RNP Granule Assembly. Cell Rep 22, 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qamar S, Wang G, Randle SJ, Ruggeri FS, Varela JA, Lin JQ, Phillips EC, Miyashita A, Williams D, Strohl F, et al. (2018). FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-pi Interactions. Cell 173, 720–734 e715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Stewart H, Dejesus-Hernandez M, Krieger C, Graff-Radford N, Fabros M, Briemberg H, Cashman N, Eisen A, and Mackenzie IR (2010). Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve 42, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reber S, Lindsay H, Devoy A, Jutzi D, Mechtersheimer J, Domanski M, Mühlemann O, Barabino SML, and Ruepp M-D (2019). The phase separation-dependent FUS interactome reveals nuclear and cytoplasmic function of liquid-liquid phase separation. bioRxiv, 806158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhine K, Vidaurre V, and Myong S (2020). RNA Droplets. Annu Rev Biophys 49, 247–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riback JA, Bowman MA, Zmyslowski AM, Plaxco KW, Clark PL, and Sosnick TR (2019). Commonly used FRET fluorophores promote collapse of an otherwise disordered protein. Proc Natl Acad Sci U S A 116, 8889–8894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. [DOI] [PubMed] [Google Scholar]
- Ruter A, Kuczera S, Gentile L, and Olsson U (2020). Arrested dynamics in a model peptide hydrogel system. Soft Matter 16, 2642–2651. [DOI] [PubMed] [Google Scholar]
- Sarkar J, and Myong S (2018). Single-Molecule and Ensemble Methods to Probe Initial Stages of RNP Granule Assembly. Methods in molecular biology 1814, 325–338. [DOI] [PubMed] [Google Scholar]
- Schwartz JC, Wang X, Podell ER, and Cech TR (2013). RNA seeds higher-order assembly of FUS protein. Cell Rep 5, 918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, and Shneider NA (2016). ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun 7, 10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova TA, Robinson HK, Southcombe JA, Ninkina N, and Buchman VL (2014). Multistep process of FUS aggregation in the cell cytoplasm involves RNA-dependent and RNA-independent mechanisms. Hum Mol Genet 23, 5211–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevtsov SP, and Dundr M (2011). Nucleation of nuclear bodies by RNA. Nat Cell Biol 13, 167–173. [DOI] [PubMed] [Google Scholar]
- Shiihashi G, Ito D, Yagi T, Nihei Y, Ebine T, and Suzuki N (2016). Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain 139, 2380–2394. [DOI] [PubMed] [Google Scholar]
- Shiina N (2019). Liquid- and solid-like RNA granules form through specific scaffold proteins and combine into biphasic granules. J Biol Chem 294, 3532–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Ling SC, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, Li H, Qiu H, Bui A, Yeo GW, et al. (2015). ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nature communications 6, 6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanashi K, and Yamaguchi A (2014). Aggregation of ALS-linked FUS mutant sequesters RNA binding proteins and impairs RNA granules formation. Biochem Biophys Res Commun 452, 600–607. [DOI] [PubMed] [Google Scholar]
- Ticozzi N, Silani V, LeClerc AL, Keagle P, Gellera C, Ratti A, Taroni F, Kwiatkowski TJ Jr., McKenna-Yasek DM, Sapp PC, et al. (2009). Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 73, 1180–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, Urwin H, Manser C, Miller CC, Hortobagyi T, et al. (2013). ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum Mol Genet 22, 2676–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Choi JM, Holehouse AS, Lee HO, Zhang X, Jahnel M, Maharana S, Lemaitre R, Pozniakovsky A, Drechsel D, et al. (2018). A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 174, 688–699 e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Brent JR, Tomlinson A, Shneider NA, and McCabe BD (2011). The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J Clin Invest 121, 4118–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley KD, Comstock MJ, and Chemla YR (2017). High-Resolution Optical Tweezers Combined With Single-Molecule Confocal Microscopy. Methods Enzymol 582, 137–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa T, Ali R, Jiou J, Fung HYJ, Burke KA, Kim SJ, Lin Y, Peeples WB, Saltzberg D, Soniat M, et al. (2018). Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell 173, 693–705 e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JY, Dyakov BJA, Zhang J, Knight JDR, Vernon RM, Forman-Kay JD, and Gingras AC (2019). Properties of Stress Granule and P-Body Proteomes. Mol Cell 76, 286–294. [DOI] [PubMed] [Google Scholar]
- Zeng X, Holehouse AS, Chilkoti A, Mittag T, and Pappu RV (2020). Connecting Coil-to-Globule Transitions to Full Phase Diagrams for Intrinsically Disordered Proteins. Biophys J 119, 402–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Elbaum-Garfinkle S, Langdon EM, Taylor N, Occhipinti P, Bridges AA, Brangwynne CP, and Gladfelter AS (2015). RNA Controls PolyQ Protein Phase Transitions. Mol Cell 60, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Bartley LE, Babcock HP, Russell R, Ha T, Herschlag D, and Chu S (2000). A single-molecule study of RNA catalysis and folding. Science 288, 2048–2051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Video 1 (related to Figure 7 and STAR Methods): An example wild-type-wild-type FUS-RNA droplet fusion.
Supplemental Video 2 (related to Figure 7 and STAR Methods): An example wild-type-G156E FUS-RNA droplet fusion.
Supplemental Video 3 (related to Figure 7 and STAR Methods): An example G156E-G156E FUS-RNA droplet fusion.
Data Availability Statement
Single Molecule FRET data acquisition and analysis package can all be obtained freely from Center for Physics of Living Cells’ website (https://cplc.illinois.edu/software/).
Matlab code from this manuscript can be downloaded from Github: (https://github.com/Myong-Lab). IDL (http://www.exelisvis.co.uk/ProductsServices/IDL.aspx), MATLAB (https://www.mathworks.com/), Adobe (https://www.adobe.com/products/photoshop.html), and Prism7 (https://www.graphpad.com/scientific-software/prism/) software with academic or individual licenses can be obtained from their respective software companies. ImageJ is an open-source program available from the NIH (https://imagej.nih.gov/ij/). The violin plot MATLAB code was available online through MATLAB file exchange (https://www.mathworks.com/matlabcentral/fileexchange/45134-violin-plot).