

Many insects host microbiomes with important ecological functions. However, the prevalence of this phenomenon is unclear because in many insect taxa, microbiomes have been studied in only part of the life cycle, if at all. A prominent example is butterflies and moths, in which the composition and functional role of adult-stage microbiomes are largely unknown. We comprehensively characterized microbiomes in adult passion-vine butterflies. Butterfly-associated bacterial communities are generally abundant in guts, consistent within populations, and composed of taxa widely shared among hosts. More closely related butterflies harbor more similar microbiomes, with the most dramatic shift in microbiome composition occurring in tandem with a suite of ecological and life history traits unique to the genus Heliconius. Butterflies are also frequently infected with previously undescribed eukaryotic parasites, which may interact with bacteria in important ways. These findings advance our understanding of butterfly biology and insect-microbe interactions generally.
KEYWORDS: symbiosis, gut bacteria, Lepidoptera, microbiota, parasites
ABSTRACT
Lepidoptera (butterflies and moths) are diverse and ecologically important, yet we know little about how they interact with microbes as adults. Due to metamorphosis, the form and function of their adult-stage microbiomes might be very different from those of microbiomes in the larval stage (caterpillars). We studied adult-stage microbiomes of Heliconius and closely related passion-vine butterflies (Heliconiini), which are an important model system in evolutionary biology. To characterize the structure and dynamics of heliconiine microbiomes, we used field collections of wild butterflies, 16S rRNA gene sequencing, quantitative PCR, and shotgun metagenomics. We found that Heliconius butterflies harbor simple and abundant bacterial communities that are moderately consistent among conspecific individuals and over time. Heliconiine microbiomes also exhibited a strong signal of the host phylogeny, with a major distinction between Heliconius and other butterflies. These patterns were largely driven by differing relative abundances of bacterial phylotypes shared among host species and genera, as opposed to the presence or absence of host-specific phylotypes. We suggest that the phylogenetic structure in heliconiine microbiomes arises from conserved host traits that differentially filter microbes from the environment. While the relative importance of different traits remains unclear, our data indicate that pollen feeding (unique to Heliconius) is not a primary driver. Using shotgun metagenomics, we also discovered trypanosomatids and microsporidia to be prevalent in butterfly guts, raising the possibility of antagonistic interactions between eukaryotic parasites and colocalized gut bacteria. Our discovery of characteristic and phylogenetically structured microbiomes provides a foundation for tests of adult-stage microbiome function, a poorly understood aspect of lepidopteran biology.
IMPORTANCE Many insects host microbiomes with important ecological functions. However, the prevalence of this phenomenon is unclear because in many insect taxa, microbiomes have been studied in only part of the life cycle, if at all. A prominent example is butterflies and moths, in which the composition and functional role of adult-stage microbiomes are largely unknown. We comprehensively characterized microbiomes in adult passion-vine butterflies. Butterfly-associated bacterial communities are generally abundant in guts, consistent within populations, and composed of taxa widely shared among hosts. More closely related butterflies harbor more similar microbiomes, with the most dramatic shift in microbiome composition occurring in tandem with a suite of ecological and life history traits unique to the genus Heliconius. Butterflies are also frequently infected with previously undescribed eukaryotic parasites, which may interact with bacteria in important ways. These findings advance our understanding of butterfly biology and insect-microbe interactions generally.
INTRODUCTION
Insect microbiome research has historically focused on hosts with highly stable, specific, and functionally important microbiomes (1). For example, the obligate nutritional endosymbionts of sap- and blood-feeding insects, which exemplify this scenario, have long served as important model systems. More recently, however, wider microbial explorations have shown that this kind of association does not apply to all insect taxa. In some insect groups, microbiomes are more variable, and the constituent microbes have low or no specificity to their hosts (2, 3). Moreover, the functional importance of these microbiomes is often unclear and possibly variable, low, or nonexistent in some groups (4). It is important to study these insects in order to gain a broader understanding of insect biology and microbiome evolution.
The Lepidoptera (butterflies and moths) are a group in which microbiome form and function have been particularly difficult to resolve. Recent work indicates that larvae (caterpillars) typically harbor microbiomes that are variable, low abundance, and transient (5–9). However, a crucial consideration for Lepidoptera and other insects that undergo complete metamorphosis is that microbial associations may change between life stages (10). In Lepidoptera, larvae and adults often differ in the composition and absolute abundance of their microbiomes (6, 11–15). Beyond this pattern, however, the ecology of adult-stage microbiomes remains poorly understood.
There are three major unknowns about microbiomes of adult Lepidoptera that we sought to address in this study. First, it is unclear how variable microbiomes are among host individuals and taxa and which ecological or evolutionary factors (e.g., host phylogeny) are associated with microbiome variation. Understanding these patterns could give insights into whether microbiomes are important for certain ecological traits, such as feeding, and into the likelihood of host-microbe coevolution. Second, it is unclear whether butterfly-associated microbes have any specificity to their host species or to higher taxonomic levels; tight specificity is a hallmark of obligate insect-microbe symbioses (3). Previous studies of adult Lepidoptera microbiomes have largely used 97% operational taxonomic units (OTUs) based on short fragments of the 16S rRNA gene. OTUs can encompass substantial strain-level diversity (16), hindering analyses of specificity. Third, the full diversity of eukaryotic microbes that may be associated with adult Lepidoptera has not been explored, as previous studies have exclusively focused on bacteria or fungi.
Our study focuses on neotropical passion-vine butterflies (Nymphalidae: Heliconiini). This tribe, which includes the genus Heliconius, is a foundational system in evolutionary biology with over 150 years of scientific study (17). Given the scientific relevance of heliconiines (18) and the myriad roles that microbes can play in animal biology (19), it is important to know the structure and function of heliconiine-associated microbiomes. Heliconiine butterflies are also a potentially useful system for the field of host-microbe interactions given the wealth of genomic data available (20) and the ability to rear and genetically manipulate members of this group (21). A few amplicon sequencing-based microbiome studies have included adult heliconiines, but these were limited to a small number of individuals and species (11, 22, 23). Consequently, we lack even basic information about the microbes harbored by heliconiine butterflies and how they may vary across individuals and the host phylogeny.
In addition to the practical advantages mentioned above, heliconiines are also an interesting system to address how host ecological traits evolve in concert with microbiomes. Within the Heliconiini, the genus Heliconius exhibits unique feeding and life history traits relative to other genera: larvae feed on young passion-vine shoots and leaves as opposed to old foliage, adults of most species (besides Heliconius aoede) feed on pollen as well as nectar, and they have distinctive defensive chemistry and a greatly extended adult-stage life span of several months (18, 24). These traits could influence and be influenced by microbiomes in many ways. For example, we hypothesized that pollen feeding might be associated with a distinctive microbiome, either because it is a source of microbes (25) or nutrients that shift resident microbiome composition (26) or because the trait itself depends on metabolic contributions from novel microbes (11). A useful starting point to uncovering these kinds of interactions is to examine whether Heliconius butterflies have microbiomes that are internally consistent yet distinct from those of other heliconiine genera. If so, then we can zero in on the specific ecological and evolutionary drivers of variation and the potential functional roles of microbiomes in Heliconius biology.
Here, we addressed the following main questions. (i) How variable are heliconiine microbiomes among host individuals, species, and genera and across the heliconiine phylogeny? (ii) Are host traits, such as pollen feeding, associated with microbiome variation? (iii) How host specific are butterfly-associated microbes? At the same time, we sought to facilitate future microbiome research on heliconiine butterflies by answering some additional questions. (iv) How are microbes distributed within the butterfly body? (v) How abundant are these microbes? (vi) How reliable is amplicon sequencing of 16S rRNA genes for studying butterfly-associated bacterial communities? (vii) Are there eukaryotic microbes that we might be missing with targeted amplicon sequencing?
We collected 214 wild adult butterflies, representing 23 species and subspecies of Heliconiini, and characterized their microbiomes by 16S rRNA gene sequencing. While many adult Lepidoptera microbiome studies have used reared, captive specimens (6, 12, 13, 22, 27), these may give a biased picture of microbial community structure in the wild (11). We also used shotgun metagenomic sequencing on a subset of butterflies to provide both an untargeted assessment of microbial diversity (including eukaryotes) and a finer-resolution picture of microbial specificity than is available from amplicon sequencing of short 16S rRNA gene regions. Furthermore, we used quantitative PCR (qPCR) to estimate the absolute abundance of bacteria across different heliconiine taxa and tissue types. Our work illustrates the ecological and evolutionary dynamics of microbiomes in adult heliconiine butterflies and advances our general understanding of Lepidoptera-microbe interactions.
RESULTS
Adult heliconiine butterflies host whole-body microbiomes that are typically low in diversity and evenness. Across all individuals, the median exact sequence variant (ESV) richness was 26, of which only 11 accounted for 95% or more of the sequences. The composition of these communities is also reasonably consistent within host species. For example, within our most deeply sampled population (Heliconius erato demophoon in Gamboa, Panama; n = 23), a median of 84% of the 16S rRNA gene reads obtained from a given individual’s microbiome belonged to a core set of 10 bacterial genera (see Fig. S1 in the supplemental material). Some of these genera are present in roughly similar relative abundances across individuals and across our two sampling years (Fig. S1). Applying the above-mentioned calculation to all heliconiine species from which five or more individuals were sequenced, we found that this intraspecific consistency extends beyond H. erato demophoon. For most host species, over 80% of the constituent individuals’ microbiome reads belonged to the 10 most abundant bacterial genera for that species (median = 88%; range = 77 to 94%).
Microbiomes from whole, homogenized butterfly bodies are mainly composed of gut-associated taxa. Isolated guts are similar to conspecific whole-body microbiomes in their bacterial community profiles (Fig. S1), with many bacterial genera occurring at similar relative abundances in guts and head/thorax tissue (Table S1). Some bacteria differed between whole-body and gut samples, such as Wolbachia, which usually inhabits reproductive tissues (28) (Fig. S1). Likewise, in both Heliconius and other heliconiine genera, the relative abundance of Acinetobacter was ∼10-fold higher in head/thorax samples than in guts (Table S1). In contrast, Orbus, Enterobacter, Asaia, and some other dominant bacterial genera are enriched in gut tissue (Table S1).
We used metagenomes (n = 15) to examine the accuracy of amplicon sequencing for describing bacterial community composition in our broader sample set. Interindividual microbiome variation was highly correlated between the amplicon-based data set and the metagenomic data set (Mantel r = 0.63; P < 0.001). Furthermore, for six of the most abundant bacterial genera, relative abundances in amplicon libraries were highly predictive of relative abundances in metagenomes (Fig. S2). These results support the use of amplicon sequencing for evaluating bacterial community composition in adult heliconiine butterflies.
Adult Heliconius butterflies generally harbor a high absolute abundance of gut bacteria, although titers sometimes varied substantially even among conspecific individuals collected from the same location (Fig. 1A). On average, Heliconius harbored significantly higher (by ∼5.5-fold) gut bacterial titers than species belonging to other passion-vine butterfly genera (P = 0.039). Bacterial communities associated with head and thorax tissue did not significantly differ in titer between Heliconius and other genera (P = 0.38) (Fig. S3).
FIG 1.
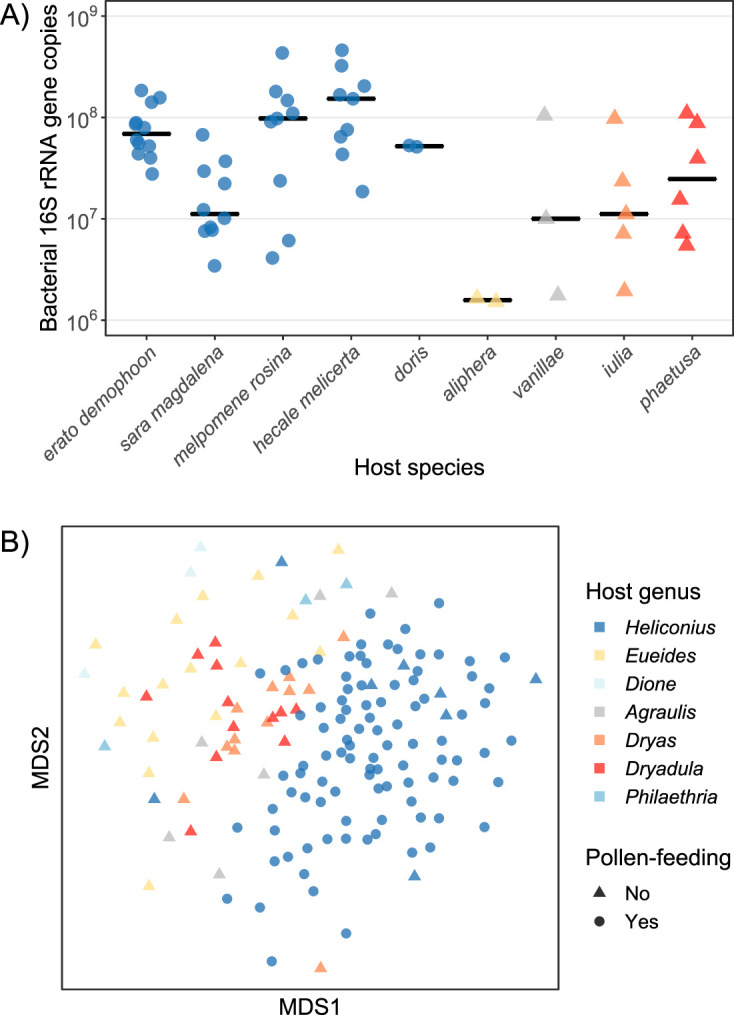
(A) Adult Heliconius butterflies have high, although often variable, titers of gut bacteria. Shown are quantitative PCR-derived estimates of bacterial abundances in terms of the number of bacterial 16S rRNA gene copies per individual gut. These individuals were collected from Gamboa, Panama, in 2016 (n = 42 Heliconius and 16 other Heliconiini). (B) Heliconius butterflies host distinct adult-stage bacterial communities compared with related genera. Shown is a nonmetric multidimensional scaling (MDS) ordination of microbiome variation (Bray-Curtis dissimilarities) among all whole-body samples collected at various sites in Panama and Ecuador in 2014 (n = 104 Heliconius and 52 other Heliconiini). With the exception of H. aoede (dark blue triangles), pollen feeding is exclusive to, and ubiquitous within, Heliconius butterflies.
Heliconius butterflies also harbor compositionally distinct whole-body bacterial communities compared with other passion-vine butterfly genera (Fig. 1B). Microbiomes clustered by host genera to various degrees depending on the distance metric used. The effect was strongest with taxonomic or phylogenetic metrics that incorporate information on the relative abundances of ESVs (Bray-Curtis, R2 = 0.13 and P = 0.001; weighted UniFrac, R2 = 0.18 and P = 0.001) as opposed to purely presence/absence-based metrics (Jaccard, R2 = 0.11 and P = 0.001; unweighted UniFrac, R2 = 0.10 and P = 0.001). This separation of Heliconius microbiomes from those of related butterfly genera is not fully explicable by adult-stage diet, as individuals of the non-pollen-feeding species Heliconius aoede mostly clustered with pollen-feeding Heliconius species (Fig. 1B). An analysis of species-by-species Bray-Curtis dissimilarities confirmed that H. aoede microbiomes are not uniquely distinct from those of pollen-feeding Heliconius species (Fig. S4).
As suggested by the host genus-level clustering, we found that variation in butterfly microbiome composition was associated with host relatedness (phylogenetic distance). More closely related butterfly lineages harbored more similar microbiomes (Bray-Curtis, Mantel r = 0.40 and P = 0.01), and the topologies of the butterfly phylogeny and the dendrogram of microbiome similarity were moderately congruent (Fig. 2). This pattern was still statistically significant when microbiome variation was measured using weighted UniFrac distances (Mantel r = 0.31; P = 0.03) but not when using unweighted UniFrac (Mantel r = 0.03; P = 0.41) or Jaccard (Mantel r = 0.19; P = 0.08) distances, which do not incorporate information on relative abundance.
FIG 2.

Variation in heliconiine butterfly microbiomes is correlated with host relatedness. Shown is the concordance between the host phylogeny (from reference 37) and a dendrogram representation of microbiome variation among species (Bray-Curtis dissimilarities). Here, all nodes have been rotated to maximize tip matching. Note, however, that sets of parallel lines connecting tips between the phylogeny and the dendrogram do not always signify congruent branching structure.
We then determined which bacterial taxa contribute to the observed taxonomic (Fig. 1B) and phylogenetic (Fig. 2) structuring in overall community composition. The dominant bacterial genera Enterobacter, Orbus, and Entomomonas/Pseudomonas were proportionally more abundant in Heliconius than in the non-pollen-feeding butterfly genera, while Asaia and Apibacter showed the opposite pattern (Fig. 3). None of these bacterial genera, however, were exclusively restricted to one host feeding guild, genus, or species, and their relative abundances were occasionally highly variable (Fig. 3). We also analyzed the data at the ESV level, including all individual ESVs that were reasonably abundant within at least one host species (≥5% within-species average). We did not find evidence for prevalent host species- or genus-restricted ESVs (Fig. 4). Most ESVs are present, albeit with different relative abundances, across host genus and species boundaries.
FIG 3.

Dominant bacterial genera are largely shared among Heliconius and other butterflies, although some are differentially abundant. Shown are the relative abundances of the top 10 bacterial genera, ranked by mean abundance, in whole-body microbiomes. Dots indicate replicate individuals, and black bars indicate median proportions within a host species. Starred bacterial genera differed significantly in relative abundance between Heliconius and non-Heliconius butterflies (P < 0.05 after FDR correction). The arrangement of host species on the x axis corresponds to the phylogeny shown in Fig. 4. Note that Enterobacter here includes sequences originally assigned as Klebsiella and some other closely related Enterobacteriaceae genera (see Materials and Methods).
FIG 4.

Most bacterial exact sequence variants (ESVs) are not host species or host genus specific. Shown are all ESVs in the data set that had a ≥5% mean relative abundance across conspecific individuals for one or more host species. For each ESV, the species-level mean relative abundance in whole-body samples, after log transformation, is indicated by the color of the cells (white, not detected in that species). Note that bacterial genera (labeled at the top) contained various numbers of ESVs that met the above-mentioned prevalence threshold.
Amplicon-derived ESVs are limited in their ability to resolve bacterial strains as they represent only a short region of the 16S rRNA gene (here, ∼250 bp). To test for potential host specificity at a finer level of resolution, we obtained near-full-length 16S rRNA gene sequences from the bacterium Orbus, which is highly prevalent across gut and whole-body samples (Fig. 3; Fig. S1) and almost exclusively composed of a single ESV (Fig. 4). These sequences were reconstructed from the 12 metagenomes in which Orbus was sufficiently abundant. A phylogeny based on these sequences and other Orbaceae shows that butterfly-associated Orbus bacteria form a unique clade (Fig. 5). Host phylogenetic or geographic structure was not evident within this clade. In fact, many of the Panamanian heliconiine butterflies harbored Orbus bacteria that have 16S rRNA gene sequences that are identical or nearly identical to that of an Orbus strain isolated from an East Asian butterfly, Sasakia charonda (29).
FIG 5.

Maximum likelihood phylogenetic reconstruction of the bacterial family Orbaceae (Gammaproteobacteria: Pasteurellales). Some host taxonomic structure is apparent at the order level (i.e., Lepidoptera, beetles, bees, and flies) but not within butterflies. 16S rRNA gene sequences from Agraulis, Dryadula, Dryas, and Heliconius were assembled from short metagenomic reads. Other 16S rRNA gene sequences are from GenBank. Branch support values are shown next to the nodes. Haemophilus influenzae (Pasteurellaceae) was used as the outgroup.
We also used shotgun metagenomes to search for microbial eukaryotes in butterfly gut samples and found that microsporidia and trypanosomatids were prevalent (27% and 47% of the 15 individual samples analyzed, respectively) (Fig. S5). Fungi and other microeukaryotic taxa were only sporadically detected and at very low relative abundances. When trypanosomatids were detected in a given sample, we also sometimes detected 16S rRNA gene reads in the metagenomic data classified as Kinetoplastibacterium, an obligate bacterial endosymbiont of certain trypanosomatids (30). Kinetoplastibacterium was not detected without its corresponding trypanosomatid host (Fig. S5). This observation led us to reexamine the larger, amplicon-based bacterial data set. We found that 9% of Heliconius individuals and 4% of individuals belonging to other heliconiine genera were infected with a trypanosomatid, as inferred by the presence of Kinetoplastibacterium. These proportions were not significantly different (P = 0.28 by Fisher’s exact test). They are also likely underestimates given the above-mentioned prevalence of trypanosomatids among metagenomes (47%) and the potential for false negatives; in five metagenomes, trypanosomatids were detected without Kinetoplastibacterium (Fig. S5).
DISCUSSION
We found that adult-stage microbiomes in heliconiine butterflies are generally similar in composition among conspecific individuals and over time and are also abundant within gut tissue (especially in Heliconius). Our estimates of total bacterial abundances (via qPCR), in parallel with those reported previously (23), support the hypothesis that bacteria actively colonize adult butterflies as opposed to passing through the gut transiently. These features contrast with the typical situation in lepidopteran larvae (5), reinforcing the idea that insect-microbe associations can differ strongly between life stages (10).
Although stable relative to larvae, dominant butterfly-associated microbes exhibit high levels of interindividual variability compared with obligate nutritional endosymbionts or gut microbes of some other insect groups. For example, in honey bees, >98% of sequences belong to a honey bee-specific set of 5 to 9 bacterial species (31). We found that ∼80 to 90% of sequences in a given butterfly species’ microbiome belonged to a set of 10 core bacterial genera. This contrast suggests that microbiomes are more facultative for butterflies or that there is greater functional redundancy among butterfly-associated microbes.
We also discovered that Heliconius butterfly microbiomes are distinct from those of other heliconiine genera in terms of overall community structure (Fig. 1B), relative abundances of specific bacterial genera (Fig. 3), and total numbers of gut bacteria (Fig. 1A). This finding opens the possibility that one or more of the ecological traits specific to Heliconius influence, and may be influenced by, the microbiome. As these traits evolved in tandem (18), it is difficult to disentangle their potential links to microbes, but our analysis of the species Heliconius aoede suggests that pollen feeding is not a primary driver. H. aoede does not pollen feed (32, 33), yet its microbiomes were not uniquely distinct from pollen-feeding Heliconius species (Fig. 1B; see also Fig. S4 in the supplemental material). This result contrasts with previous work on butterflies finding a strong association between adult-stage feeding ecology (nectivory versus frugivory) and adult gut microbiomes (23).
Larval feeding ecology, however, warrants further investigation as a potential driver of variation in adult gut microbiomes. Microbiomes of butterfly larvae and adults are not fully decoupled: in H. erato, some adult-stage microbes appear to be carried over from the larval stage (11). All Heliconius species (including H. aoede) feed on young passion-vine foliage, while other heliconiine genera feed on mature foliage (18). Related variation in diet-derived microbes, or in host processes that determine which microbes persist through metamorphosis (10, 34), could potentially underlie variation in adult-stage microbiomes.
In addition to the clear separation of Heliconius from other heliconiine genera (Fig. 1B), there was also a strong signal of the host phylogeny in heliconiine butterfly microbiomes (Fig. 2). The strength of this signal was comparable to those observed in gut microbiomes of mammals (35) and other host groups (36). This result is also in agreement with a recent survey of neotropical butterflies, in which microbiomes were found to be phylogenetically structured across a broad diversity of hosts (six different families) (23). Thus, even in a comparatively young radiation such as the one studied here (37), host phylogenetic history is clearly an important factor shaping the composition of adult butterfly microbiomes. The question then becomes, how has this pattern (also known as phylosymbiosis [38]) arisen, and what does it signify?
There are at least two, non-mutually exclusive processes that can lead to phylosymbiosis: host-microbe codiversification and contemporary host filtering of environmental microbes (36, 39). In the former process, there is a shared evolutionary history of host and microbial lineages. This does not necessarily apply to the latter process, which occurs when host traits that influence microbial colonization, such as diet preference, are conserved to some degree across the host phylogeny. We suggest that codiversification is unlikely in this system given the broad distribution of bacterial phylotypes across host species and genera (discussed below). Rather, host filtering of environmental microbes likely explains phylogenetic structure in heliconiine microbiomes, as is the case in a wide variety of other animal groups (36). A priority for future work on butterflies is to identify the specific traits underlying host filtering. In Heliconiini, two candidates are larval host plant use (i.e., Passiflora species identity and age of tissue consumed) and adult foraging behavior (especially whether pollen is collected and from which plant species), both of which exhibit phylogenetic signal (18, 40, 41).
Bacterial community-level variation among butterfly genera and across the phylogeny was largely driven by shifts in the relative abundance of shared bacterial genera and ESVs as opposed to the presence or absence of host-specific bacterial taxa (Fig. 3 and 4). Moreover, a finer-resolution analysis of the nearly ubiquitous bacterium Orbus did not find evidence for specificity within butterflies (Fig. 5). This pattern is notable in part because it weighs against the codiversification model described above and in part because it provides a contrast to the high degree of host specificity documented in a number of other insect-microbe symbioses (42–45). While not highly host specific, at least some of the dominant bacteria in butterflies appear to be insect specialists as opposed to cosmopolitan environmental taxa. For example, phylogenetic evidence supports a general insect association for Entomomonas (46), Apibacter (47), and Orbus (Fig. 5) (48). The ecology of these groups is poorly understood; one important unknown is how they are transmitted among butterflies, which may be via flowers (49) or other shared resources.
Shotgun metagenomes allowed us to corroborate the amplicon-based bacterial data (Fig. S2) and led to the discovery of putative eukaryotic parasites (trypanosomatids and microsporidia) in gut tissue of many adult heliconiine butterflies (Fig. S5). Beyond fungi (12, 23), which do not appear to be abundant in heliconiines, microeukaryotes are almost unknown from adult-stage Lepidoptera, with the exception of the neogregarine Ophryocystis elektroscirrha in monarch butterflies (50). Given their potential interactions with gut bacteria and relevance to host fitness, more targeted analyses of butterfly-associated microeukaryote diversity are clearly warranted.
While the effects of endosymbionts such as Wolbachia and Spiroplasma have been documented (51), a major open question is what functional role gut microbes play in the biology of adult heliconiines and Lepidoptera generally (52). In Heliconius, our hypothesis that microbes mediate pollen feeding is not strongly supported. To directly influence pollen digestion, which occurs extraorally using saliva exuded from the proboscis (53), microbes would likely need to inhabit the proboscis or salivary gland. Yet we saw no clear signal of elevated microbial abundances or unique microbial taxa in these tissues in Heliconius compared with non-pollen-feeding butterflies (Fig. S3). Endogenous pollen digestion mechanisms (33, 54, 55) may be sufficient.
Other microbial roles in host nutrition are possible, although a recent experiment on the butterfly Speyeria mormonia did not find evidence for nutrition-related functions (56). We suggest that colonization resistance (i.e., protection from pathogens and parasites), which is a feature common to many symbioses (39, 57), could be a primary ecological function of adult butterfly gut microbiomes. We found that heliconiines are frequently infected by trypanosomatids and microsporidia as well as Serratia species (Fig. 4), which are common opportunistic bacterial pathogens in insects (58, 59). Adult butterflies may be similar to pyrrhocorid bugs, tsetse flies, and social bees, in which gut bacteria provide an important layer of defense against related parasites and pathogens (60–64). Importantly, colonization resistance can readily evolve even in symbioses lacking strong host-microbe specificity (65), such as that between heliconiines and their adult-stage microbiomes.
Conclusions.
This study provides an in-depth characterization of Heliconius and other passion-vine butterfly microbiomes, adding a new dimension to a classic model system in evolutionary biology. The characteristics of adult-stage microbiomes that we report contrast with those of larval Lepidoptera, emphasizing that holometabolous insects are able to interact with microbes in very different ways across life stages (10). However, many Lepidoptera do not feed or even have a digestive tract as adults (66), and we do not yet know whether patterns from heliconiines or other butterflies are generalizable to these other groups. We further suggest that heliconiines may serve as a useful system for exploring how animal ecology and life history relate to microbes. Heliconius butterflies host phylogenetically structured microbiomes that are markedly distinct from those of their close relatives, and this finding sets the stage for experiments to test the specific host traits that may be involved. Finally, we uncovered novel microeukaryote diversity in butterflies and hypothesize that these microeukaryotes are parasites that interact antagonistically with gut bacteria. Further research on adult-stage microbiomes will help advance our understanding of both insect-microbiome evolution and the biology of Lepidoptera, a diverse group of considerable ecological, societal, and scientific importance (66, 67).
MATERIALS AND METHODS
Field collections.
The wild adult butterflies used for whole-body microbiome sequencing were collected from seven locations in Panama and Ecuador from May to August 2014 (more detail is provided in the “Collection_localities.txt” file at https://figshare.com/projects/Heliconius_butterfly_microbiomes/70520). Butterflies were euthanized with ethyl acetate and stored in dimethyl sulfoxide (DMSO) after the removal of wings, according to methods reported previously (68). We also stored two DMSO-only blanks to use as negative controls. In June 2016, we collected additional adult butterflies for gut and head/thorax sequencing from Gamboa and Pipeline Road, Panama. For these specimens, we dissected the gut (hindgut, midgut, and the distal ∼1/2 of the foregut) using sterilized tools prior to storage in DMSO. The whole head and thorax (including the proximal foregut) were stored separately. Species or subspecies were identified based on morphology. Butterflies were collected under permit number SC/A-7-11 from Panama’s Autoridad Nacional del Ambiente and permit number 005-13 IC-FAU-DNB/MA from Ecuador’s Ministerio del Ambiente.
Sample processing, qPCR, PCR, and sequencing.
We removed whole bodies and head/thorax samples from DMSO and, after homogenization, used ∼50-mg subsamples of the homogenate for DNA extractions with the MoBio PowerSoil kit according to the manufacturer’s instructions. We added entire guts directly to DNA extraction tubes, in which they were homogenized during the first bead-beating step of the protocol. Two DMSO blanks and 30 DNA extraction blanks were also processed in tandem with the butterfly samples and sequenced. For dissected gut and head/thorax samples, we estimated the total bacterial abundance using quantitative PCR (qPCR) with 16S rRNA gene primers (515F/806R) according to protocols described previously (5).
PCR amplifications (515F/806R primers, V4 region) and 2- by 150-bp Illumina MiSeq sequencing of 16S rRNA genes were performed according to standard Earth Microbiome Project protocols (69). For gut and head/thorax samples of 29 butterfly individuals of nine species, we also attempted PCR amplification with primers that target the internal transcribed spacer (ITS) gene region of fungi (70). However, the amplification success with these fungus-specific primers (as assessed by gel electrophoresis) was very low, suggesting a lack of abundant fungal DNA, which was later corroborated by the shotgun metagenomic data (see Results). DNA extracts from a subset of 15 amplicon-sequenced gut samples were used for shotgun metagenomic sequencing according to an approach described previously (71) with an input DNA concentration of 0.75 ng/μl, Kapa HiFi HotStart ReadyMix, and bead cleanup with AMPure XP beads at a 0.9-fold ratio.
Amplicon data processing.
Amplicons from the 2014 whole-body samples and 2016 gut and head/thorax samples were sequenced on separate runs, demultiplexed using idemp (https://github.com/yhwu/idemp), and combined for further processing. cutadapt (72) was used to remove primer sequences. We then used the DADA2 pipeline (73) to quality filter (with the maximum expected error parameter set to 1) and trim (150-bp forward and 140-bp reverse) reads, infer exact sequence variants (ESVs), merge paired-end reads, and remove chimeras. We classified ESVs using the RDP naive Bayesian classifier algorithm (74) against SILVA training set v.132 (75).
Further data processing and analyses were conducted in R v.3.6.0 (76). We used decontam (77) for prevalence-based identification of putative contaminant ESVs based on 34 negative controls (DMSO and DNA extraction blanks and PCR no-template controls). The median percentage of contaminant sequences across butterfly samples was 0.09%, but two samples had >10% contaminants and were removed from further analysis. ESVs with <100 total sequences across all samples (out of a combined total of 5.6 million sequences) were removed, as were ESVs classified as mitochondrion or chloroplast or bacteria lacking subdomain identification. These ESVs combined typically made up a low proportion of reads from the libraries (median of 2.6% across all samples). We also used these data to correct qPCR-based absolute abundances for nonbacterial amplification. Specifically, we multiplied the proportion of bacteria in sequence libraries by the total number of 16S rRNA gene copies to obtain estimates of bacterial absolute abundance.
As the 16S rRNA gene amplicon libraries were highly variable in read depth across samples, we rarefied all samples to 5,000 sequences, filtering out 13 samples with lower sequence depth. We relabeled Pantoea, Erwinia, Kluyvera, Citrobacter, Klebsiella, and Cronobacter taxonomic assignments to Enterobacter. This step was taken as genera within the Enterobacteriaceae are often polyphyletic and are difficult to resolve from short 16S rRNA gene regions (e.g., see reference 78), and we wanted to avoid spurious separation of ESVs among genera and resulting idiosyncrasies in bacterial genus distributions across butterflies. Another case of ambiguous taxonomy occurred with sequences classified as Pseudomonas by the SILVA reference database, which we label as “Entomomonas/Pseudomonas.” Entomomonas is a recently described genus of apparently insect-specialized Pseudomonadaceae and includes several sequences retrieved from Heliconius genome assemblies (46).
Amplicon and qPCR data analysis.
Beta diversity statistics and plots are based on Bray-Curtis dissimilarities or UniFrac distances (weighted and unweighted). To obtain a bacterial phylogeny for the latter, we used the fragment insertion method (79) to place our ESV sequences into the Greengenes reference tree (80). The butterfly phylogeny was reported previously (37) (TreeBASE identifier Tr77496). Four of the butterfly species in our sample set contained specimens from two distinct subspecies (e.g., Heliconius sara sara and Heliconius sara magdalena). To include these in the species-level host phylogeny, we inserted subspecies tips halfway along the terminal branches to their sister subspecies. Here, we refer to these subspecies as “species” for simplicity.
To test for host phylogenetic signal in microbiomes, we used Mantel tests with 9,999 permutations to calculate the correlation between microbial community dissimilarities/distances and host phylogenetic distances (36). Intraspecific variation in microbiomes was handled by averaging the pairwise dissimilarities/distances between all individuals of one species and all individuals of another species. We used the phytools package (81) to visualize concordance between topologies of the host phylogeny and a dendrogram of bacterial community dissimilarities. Nodes were rotated with the “cophylo” function in phytools to maximize tip matching between the two trees.
Differences in overall community composition between host genera were tested by permutational multivariate analysis of variance (PERMANOVA) as implemented in the vegan package (82). Using the “betadisper” function, we corroborated that significant test results were due to host genus-level differences in location and not dispersion (83). We used a nonparametric statistical test (Wilcoxon rank sum) to identify bacterial genera that differed in relative abundance between host taxa or between sample types (gut versus head/thorax) and applied a false discovery rate (FDR) correction to the resulting P values.
We tested whether the total abundances of bacteria differed between Heliconius and other butterfly genera using qPCR data from gut samples and head/thorax samples. Each sample type was analyzed separately using log-transformed counts of bacterial 16S rRNA gene copies. We verified that residuals were approximately normally distributed and used a linear mixed-effects model as implemented in the nlme package (84), treating host genus (Heliconius versus others) as a fixed effect and host species as a random effect.
Metagenome data processing and analysis.
For 15 gut samples, we obtained shotgun metagenomic data to complement the bacterial 16S rRNA gene amplicon data set. We quality filtered these reads with sickle (85) and trimmed adapters with cutadapt (72). We then used Bowtie 2 (86) to filter out reads matching a given sample’s corresponding host species’ genome, obtained from Lepbase (20). The two Dryadula phaetusa metagenomes were mapped to a genome of the sister species Dryas iulia as no Dryadula genome was available. Since there was a high proportion of host-derived reads, we focused here on describing microbial diversity using rRNA gene reads present in the metagenomes. With the host-filtered reads, we used phyloFlash (87) to find and classify eukaryotic and bacterial small-subunit (SSU) rRNA reads. Bacterial community compositions were compared between the amplicon and shotgun metagenomic data sets using a Mantel test. We also used phyloFlash to assemble 16S rRNA genes from the 150-bp shotgun reads. These longer sequences allowed us to estimate the phylogeny of Orbus, the dominant bacterium in these 15 samples. Orbus sequences were aligned with MUSCLE (88), curated with Gblocks (89), and used for maximum likelihood reconstruction with the phylogeny.fr implementation (90) of PhyML (91).
Data availability.
The amplicon data, metadata, and R code are available from figshare (https://figshare.com/projects/Heliconius_butterfly_microbiomes/70520). Metagenomes are available from MG-RAST (project accession number MGP89563).
Supplementary Material
ACKNOWLEDGMENTS
This material is based on work supported by the Cooperative Institute for Research in Environmental Sciences and the Ecology and Evolutionary Biology Department at the University of Colorado at Boulder; a National Science Foundation graduate research fellowship (1144083); and a postdoctoral fellowship from the USDA National Institute of Food and Agriculture (2018-08156) to T.J.H. This work was further supported by grants from the Simons Foundation (429440) and by funding from the Smithsonian Tropical Research Institute.
We thank Jessica Henley for assistance with sequencing, the Smithsonian Tropical Research Institute staff for administrative support, and the Panamanian and Ecuadorian authorities for permission to collect butterflies. We also acknowledge the reviewers for helpful comments that improved the manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Buchner P. 1965. Endosymbiosis of animals with plant microorganisms. John Wiley & Sons, New York, NY. [Google Scholar]
- 2.Jones RT, Sanchez LG, Fierer N. 2013. A cross-taxon analysis of insect-associated bacterial diversity. PLoS One 8:e61218. doi: 10.1371/journal.pone.0061218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engel P, Moran NA. 2013. The gut microbiota of insects—diversity in structure and function. FEMS Microbiol Rev 37:699–735. doi: 10.1111/1574-6976.12025. [DOI] [PubMed] [Google Scholar]
- 4.Hammer TJ, Sanders JG, Fierer N. 2019. Not all animals need a microbiome. FEMS Microbiol Lett 366:fnz117. doi: 10.1093/femsle/fnz117. [DOI] [PubMed] [Google Scholar]
- 5.Hammer TJ, Janzen DH, Hallwachs W, Jaffe SP, Fierer N. 2017. Caterpillars lack a resident gut microbiome. Proc Natl Acad Sci U S A 114:9641–9646. doi: 10.1073/pnas.1707186114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staudacher H, Kaltenpoth M, Breeuwer JAJ, Menken SBJ, Heckel DG, Groot AT. 2016. Variability of bacterial communities in the moth Heliothis virescens indicates transient association with the host. PLoS One 11:e0154514. doi: 10.1371/journal.pone.0154514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Priya NG, Ojha A, Kajla MK, Raj A, Rajagopal R. 2012. Host plant induced variation in gut bacteria of Helicoverpa armigera. PLoS One 7:e30768. doi: 10.1371/journal.pone.0030768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitaker M, Pierce N, Salzman S, Kaltenpoth M, Sanders J. 2016. Microbial communities of lycaenid butterflies do not correlate with larval diet. Front Microbiol 7:1920. doi: 10.3389/fmicb.2016.01920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phalnikar K, Kunte K, Agashe D. 2019. Disrupting butterfly caterpillar microbiomes does not impact their survival and development. Proc Biol Sci 286:20192438. doi: 10.1098/rspb.2019.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hammer TJ, Moran NA. 2019. Links between metamorphosis and symbiosis in holometabolous insects. Philos Trans R Soc Lond B Biol Sci 374:20190068. doi: 10.1098/rstb.2019.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hammer TJ, McMillan WO, Fierer N. 2014. Metamorphosis of a butterfly-associated bacterial community. PLoS One 9:e86995. doi: 10.1371/journal.pone.0086995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison JG, Urruty DM, Forister ML. 2016. An exploration of the fungal assemblage in each life history stage of the butterfly, Lycaeides melissa (Lycaenidae), as well as its host plant Astragalus canadensis (Fabaceae). Fungal Ecol 22:10–16. doi: 10.1016/j.funeco.2016.02.001. [DOI] [Google Scholar]
- 13.Paniagua Voirol LR, Weinhold A, Johnston PR, Fatouros NE, Hilker M. 2020. Legacy of a butterfly’s parental microbiome in offspring performance. Appl Environ Microbiol 86:e00596-20. doi: 10.1128/AEM.00596-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Phalnikar K, Kunte K, Agashe D. 2018. Dietary and developmental shifts in butterfly-associated bacterial communities. R Soc Open Sci 5:171559. doi: 10.1098/rsos.171559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Sun S, Yang X, Cheng J, Wei H, Li Z, Michaud JP, Liu X. 2020. Variability of gut microbiota across the life cycle of Grapholita molesta (Lepidoptera: Tortricidae). Front Microbiol 11:1366. doi: 10.3389/fmicb.2020.01366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chase AB, Karaoz U, Brodie EL, Gomez-Lunar Z, Martiny AC, Martiny JBH. 2017. Microdiversity of an abundant terrestrial bacterium encompasses extensive variation in ecologically relevant traits. mBio 8:e01809-17. doi: 10.1128/mBio.01809-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bates HW. 1862. Contributions to an insect fauna of the Amazon Valley. Lepidoptera: Heliconidae. Trans Linn Soc Lond 23:495–566. doi: 10.1111/j.1096-3642.1860.tb00146.x. [DOI] [Google Scholar]
- 18.Jiggins CD. 2016. The ecology and evolution of Heliconius butterflies. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 19.McFall-Ngai M, Hadfield MG, Bosch TCG, Carey HV, Domazet-Lošo T, Douglas AE, Dubilier N, Eberl G, Fukami T, Gilbert SF, Hentschel U, King N, Kjelleberg S, Knoll AH, Kremer N, Mazmanian SK, Metcalf JL, Nealson K, Pierce NE, Rawls JF, Reid A, Ruby EG, Rumpho M, Sanders JG, Tautz D, Wernegreen JJ. 2013. Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci U S A 110:3229–3236. doi: 10.1073/pnas.1218525110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Challis RJ, Kumar S, Dasmahapatra KK, Jiggins CD, Blaxter M. 2016. Lepbase: the Lepidopteran genome database. bioRxiv 10.1101/056994. [DOI]
- 21.Concha C, Wallbank RWR, Hanly JJ, Fenner J, Livraghi L, Rivera ES, Paulo DF, Arias C, Vargas M, Sanjeev M, Morrison C, Tian D, Aguirre P, Ferrara S, Foley J, Pardo-Diaz C, Salazar C, Linares M, Massardo D, Counterman BA, Scott MJ, Jiggins CD, Papa R, Martin A, McMillan WO. 2019. Interplay between developmental flexibility and determinism in the evolution of mimetic Heliconius wing patterns. Curr Biol 29:3996–4009.e4. doi: 10.1016/j.cub.2019.10.010. [DOI] [PubMed] [Google Scholar]
- 22.van Schooten B, Godoy-Vitorino F, McMillan WO, Papa R. 2018. Conserved microbiota among young Heliconius butterfly species. PeerJ 6:e5502. doi: 10.7717/peerj.5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ravenscraft A, Berry M, Hammer T, Peay K, Boggs C. 2019. Structure and function of the bacterial and fungal gut microbiota of neotropical butterflies. Ecol Monogr 89:e01346. doi: 10.1002/ecm.1346. [DOI] [Google Scholar]
- 24.Sculfort O, Castro ECP, Kozak KM, Bak S, Elias M, Nay B, Llaurens V. 2020. Variation of chemical compounds in wild Heliconiini reveals ecological factors involved in the evolution of chemical defenses in mimetic butterflies. Ecol Evol 10:2677–2694. doi: 10.1002/ece3.6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ambika Manirajan B, Ratering S, Rusch V, Schwiertz A, Geissler-Plaum R, Cardinale M, Schnell S. 2016. Bacterial microbiota associated with flower pollen is influenced by pollination type, and shows a high degree of diversity and species-specificity: the bacterial microbiota of flower pollen. Environ Microbiol 18:5161–5174. doi: 10.1111/1462-2920.13524. [DOI] [PubMed] [Google Scholar]
- 26.Kešnerová L, Emery O, Troilo M, Liberti J, Erkosar B, Engel P. 2020. Gut microbiota structure differs between honeybees in winter and summer. ISME J 14:801–814. doi: 10.1038/s41396-019-0568-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kingsley VV. 1972. Persistence of intestinal bacteria in the developmental stages of the monarch butterfly (Danaus plexippus). J Invertebr Pathol 20:51–58. doi: 10.1016/0022-2011(72)90081-X. [DOI] [Google Scholar]
- 28.Saridaki A, Bourtzis K. 2010. Wolbachia: more than just a bug in insects genitals. Curr Opin Microbiol 13:67–72. doi: 10.1016/j.mib.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 29.Kim JY, Lee J, Shin N-R, Yun J-H, Whon TW, Kim M-S, Jung M-J, Roh SW, Hyun D-W, Bae J-W. 2013. Orbus sasakiae sp. nov., a bacterium isolated from the gut of the butterfly Sasakia charonda, and emended description of the genus Orbus. Int J Syst Evol Microbiol 63:1766–1770. doi: 10.1099/ijs.0.041871-0. [DOI] [PubMed] [Google Scholar]
- 30.De Souza W, Motta MCM. 1999. Endosymbiosis in protozoa of the Trypanosomatidae family. FEMS Microbiol Lett 173:1–8. doi: 10.1111/j.1574-6968.1999.tb13477.x. [DOI] [PubMed] [Google Scholar]
- 31.Zheng H, Steele MI, Leonard SP, Motta EVS, Moran NA. 2018. Honey bees as models for gut microbiota research. Lab Anim (NY) 47:317–325. doi: 10.1038/s41684-018-0173-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown KS. 1981. The biology of Heliconius and related genera. Annu Rev Entomol 26:427–456. doi: 10.1146/annurev.en.26.010181.002235. [DOI] [Google Scholar]
- 33.Gilbert LE. 1972. Pollen feeding and reproductive biology of Heliconius butterflies. Proc Natl Acad Sci U S A 69:1403–1407. doi: 10.1073/pnas.69.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnston PR, Rolff J. 2015. Host and symbiont jointly control gut microbiota during complete metamorphosis. PLoS Pathog 11:e1005246. doi: 10.1371/journal.ppat.1005246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groussin M, Mazel F, Sanders JG, Smillie CS, Lavergne S, Thuiller W, Alm EJ. 2017. Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat Commun 8:14319. doi: 10.1038/ncomms14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mazel F, Davis KM, Loudon A, Kwong WK, Groussin M, Parfrey LW. 2018. Is host filtering the main driver of phylosymbiosis across the tree of life? mSystems 3:e00097-18. doi: 10.1128/mSystems.00097-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kozak KM, Wahlberg N, Neild AF, Dasmahapatra KK, Mallet J, Jiggins CD. 2015. Multilocus species trees show the recent adaptive radiation of the mimetic Heliconius butterflies. Syst Biol 64:505–524. doi: 10.1093/sysbio/syv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim SJ, Bordenstein SR. 2020. An introduction to phylosymbiosis. Proc Biol Sci 287:20192900. doi: 10.1098/rspb.2019.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moran NA, Ochman H, Hammer TJ. 2019. Evolutionary and ecological consequences of gut microbial communities. Annu Rev Ecol Evol Syst 50:451–475. doi: 10.1146/annurev-ecolsys-110617-062453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Estrada C, Jiggins CD. 2002. Patterns of pollen feeding and habitat preference among Heliconius species. Ecol Entomol 27:448–456. doi: 10.1046/j.1365-2311.2002.00434.x. [DOI] [Google Scholar]
- 41.Boggs CL, Smiley JT, Gilbert LE. 1981. Patterns of pollen exploitation by Heliconius butterflies. Oecologia 48:284–289. doi: 10.1007/BF00347978. [DOI] [PubMed] [Google Scholar]
- 42.Ohkuma M. 2008. Symbioses of flagellates and prokaryotes in the gut of lower termites. Trends Microbiol 16:345–352. doi: 10.1016/j.tim.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 43.Kikuchi Y, Hosokawa T, Nikoh N, Meng X-Y, Kamagata Y, Fukatsu T. 2009. Host-symbiont co-speciation and reductive genome evolution in gut symbiotic bacteria of acanthosomatid stinkbugs. BMC Biol 7:2. doi: 10.1186/1741-7007-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwong WK, Moran NA. 2015. Evolution of host specialization in gut microbes: the bee gut as a model. Gut Microbes 6:214–220. doi: 10.1080/19490976.2015.1047129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaltenpoth M, Roeser-Mueller K, Koehler S, Peterson A, Nechitaylo TY, Stubblefield JW, Herzner G, Seger J, Strohm E. 2014. Partner choice and fidelity stabilize coevolution in a Cretaceous-age defensive symbiosis. Proc Natl Acad Sci U S A 111:6359–6364. doi: 10.1073/pnas.1400457111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Su Q, Zhang X, Li C, Luo S, Zhou X, Zheng H. 2020. Entomomonas moraniae gen. nov., sp. nov., a member of the family Pseudomonadaceae isolated from Asian honey bee gut, possesses a highly reduced genome. Int J Syst Evol Microbiol 70:165–171. doi: 10.1099/ijsem.0.003731. [DOI] [PubMed] [Google Scholar]
- 47.Kwong WK, Moran NA. 2016. Apibacter adventoris gen. nov., sp. nov., a member of the phylum Bacteroidetes isolated from honey bees. Int J Syst Evol Microbiol 66:1323–1329. doi: 10.1099/ijsem.0.000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinson VG, Carpinteyro-Ponce J, Moran NA, Markow TA. 2017. A distinctive and host-restricted gut microbiota in populations of a cactophilic Drosophila species. Appl Environ Microbiol 83:e01551-17. doi: 10.1128/AEM.01551-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graystock P, Goulson D, Hughes WOH. 2015. Parasites in bloom: flowers aid dispersal and transmission of pollinator parasites within and between bee species. Proc Biol Sci 282:20151371. doi: 10.1098/rspb.2015.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altizer SM, Oberhauser KS. 1999. Effects of the protozoan parasite Ophryocystis elektroscirrha on the fitness of monarch butterflies (Danaus plexippus). J Invertebr Pathol 74:76–88. doi: 10.1006/jipa.1999.4853. [DOI] [PubMed] [Google Scholar]
- 51.Duplouy A, Hornett EA. 2018. Uncovering the hidden players in Lepidoptera biology: the heritable microbial endosymbionts. PeerJ 6:e4629. doi: 10.7717/peerj.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paniagua Voirol LR, Frago E, Kaltenpoth M, Hilker M, Fatouros NE. 2018. Bacterial symbionts in Lepidoptera: their diversity, transmission, and impact on the host. Front Microbiol 9:556. doi: 10.3389/fmicb.2018.00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eberhard SH, Hikl AL, Boggs CL, Krenn HW. 2009. Saliva or regurgitated nectar? What Heliconius butterflies (Lepidoptera: Nymphalidae) use for pollen feeding. Ann Entomol Soc Am 102:1105–1108. doi: 10.1603/008.102.0619. [DOI] [Google Scholar]
- 54.Harpel D, Cullen DA, Ott SR, Jiggins CD, Walters JR. 2015. Pollen feeding proteomics: salivary proteins of the passion flower butterfly, Heliconius melpomene. Insect Biochem Mol Biol 63:7–13. doi: 10.1016/j.ibmb.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 55.Smith G, Macias-Muñoz A, Briscoe AD. 2016. Gene duplication and gene expression changes play a role in the evolution of candidate pollen feeding genes in Heliconius butterflies. Genome Biol Evol 8:2581–2596. doi: 10.1093/gbe/evw180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ravenscraft A, Kish N, Peay K, Boggs C. 2019. No evidence that gut microbiota impose a net cost on their butterfly host. Mol Ecol 28:2100–2117. doi: 10.1111/mec.15057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kamada N, Chen GY, Inohara N, Núñez G. 2013. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol 14:685–690. doi: 10.1038/ni.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raymann K, Coon KL, Shaffer Z, Salisbury S, Moran NA. 2018. Pathogenicity of Serratia marcescens strains in honey bees. mBio 9:e01649-18. doi: 10.1128/mBio.01649-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grimont F, Grimont PAD. 2006. The genus Serratia, p 219–244. In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (ed), The prokaryotes. Springer, New York, NY. [Google Scholar]
- 60.Onchuru TO, Martinez AJ, Kaltenpoth M. 2018. The cotton stainer’s gut microbiota suppresses infection of a cotransmitted trypanosomatid parasite. Mol Ecol 27:3408–3419. doi: 10.1111/mec.14788. [DOI] [PubMed] [Google Scholar]
- 61.Raymann K, Shaffer Z, Moran NA. 2017. Antibiotic exposure perturbs the gut microbiota and elevates mortality in honeybees. PLoS Biol 15:e2001861. doi: 10.1371/journal.pbio.2001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koch H, Schmid-Hempel P. 2011. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc Natl Acad Sci U S A 108:19288–19292. doi: 10.1073/pnas.1110474108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weiss BL, Maltz MA, Vigneron A, Wu Y, Walter KS, O’Neill MB, Wang J, Aksoy S. 2019. Colonization of the tsetse fly midgut with commensal Kosakonia cowanii Zambiae inhibits trypanosome infection establishment. PLoS Pathog 15:e1007470. doi: 10.1371/journal.ppat.1007470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu Y, Zheng Y, Chen Y, Chen G, Zheng H, Hu F. 2020. Apis cerana gut microbiota contribute to host health though stimulating host immune system and strengthening host resistance to Nosema ceranae. R Soc Open Sci 7:192100. doi: 10.1098/rsos.192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McLaren MR, Callahan BJ. 2020. Pathogen resistance may be the principal evolutionary advantage provided by the microbiome. Philos Trans R Soc Lond B Biol Sci 375:20190592. doi: 10.1098/rstb.2019.0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scoble MJ. 1992. The Lepidoptera. Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 67.Boggs CL, Watt WB, Ehrlich PR. 2003. Butterflies: ecology and evolution taking flight. University of Chicago Press, Chicago, IL. [Google Scholar]
- 68.Hammer TJ, Dickerson JC, Fierer N. 2015. Evidence-based recommendations on storing and handling specimens for analyses of insect microbiota. PeerJ 3:e1190. doi: 10.7717/peerj.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caporaso JG, Ackermann G, Apprill A, Bauer M, Berg-Lyons D, Betley J, Fierer N, Fraser L, Fuhrman JA, Gilbert JA, Gormley N, Humphrey G, Huntley J, Jansson JK, Knight R, Lauber CL, Lozupone CA, McNally S, Needham DM, Owens SM, Parada AE, Parsons R, Smith G, Thompson LR, Thompson L, Turnbaugh PJ, Walters WA, Weber L. 13 April 2018. EMP 16S Illumina amplicon protocol. protocols.io 10.17504/protocols.io.nuudeww. [DOI]
- 70.Gardes M, Bruns TD. 1993. ITS primers with enhanced specificity for basidiomycetes—application to the identification of mycorrhizae and rusts. Mol Ecol 2:113–118. doi: 10.1111/j.1365-294x.1993.tb00005.x. [DOI] [PubMed] [Google Scholar]
- 71.Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R. 2015. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS One 10:e0128036. doi: 10.1371/journal.pone.0128036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 73.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and Web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 77.Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. 2018. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brady C, Cleenwerck I, Venter S, Coutinho T, De Vos P. 2013. Taxonomic evaluation of the genus Enterobacter based on multilocus sequence analysis (MLSA). Syst Appl Microbiol 36:309–319. doi: 10.1016/j.syapm.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 79.Janssen S, McDonald D, Gonzalez A, Navas-Molina JA, Jiang L, Xu ZZ, Winker K, Kado DM, Orwoll E, Manary M, Mirarab S, Knight R. 2018. Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems 3:e00021-18. doi: 10.1128/mSystems.00021-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Revell LJ. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol 3:217–223. doi: 10.1111/j.2041-210X.2011.00169.x. [DOI] [Google Scholar]
- 82.Oksanen J, Kindt R, Legendre P. 2019. vegan: community ecology package. R package version 2.5-6.
- 83.Anderson MJ, Walsh DCI. 2013. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: what null hypothesis are you testing? Ecol Monogr 83:557–574. doi: 10.1890/12-2010.1. [DOI] [Google Scholar]
- 84.Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team. 2019. nlme: linear and nonlinear mixed effects models.
- 85.Joshi NA, Fass JN. 2011. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files. https://github.com/najoshi/sickle.
- 86.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gruber-Vodicka HR, Seah BK, Pruesse E. 2019. phyloFlash—rapid SSU rRNA profiling and targeted assembly from metagenomes. bioRxiv 10.1101/521922. [DOI] [PMC free article] [PubMed]
- 88.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 90.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard J-F, Guindon S, Lefort V, Lescot M, Claverie J-M, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guindon S, Delsuc F, Dufayard JF, Gascuel O. 2009. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol 537:113–137. doi: 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The amplicon data, metadata, and R code are available from figshare (https://figshare.com/projects/Heliconius_butterfly_microbiomes/70520). Metagenomes are available from MG-RAST (project accession number MGP89563).