

SUMMARY
Although the function of the extracellular region of programmed death ligand 1 (PD-L1) through its interactions with PD-1 on T cells is well studied, little is understood regarding the intracellular domain of PD-L1. Here, we outline a major role for PD-L1 intracellular signaling in the control of dendritic cell (DC) migration from the skin to the draining lymph node (dLN). Using a mutant mouse model, we identify a TSS signaling motif within the intracellular domain of PD-L1. The TSS motif proves critical for chemokine-mediated DC migration to the dLN during inflammation. This loss of DC migration, in the PD-L1 TSS mutant, leads to a significant decline in T cell priming when DC trafficking is required for antigen delivery to the dLN. Finally, the TSS motif is required for chemokine receptor signaling downstream of the Gα subunit of the heterotrimeric G protein complex, ERK phosphorylation, and actin polymerization in DCs.
Graphical Abstract
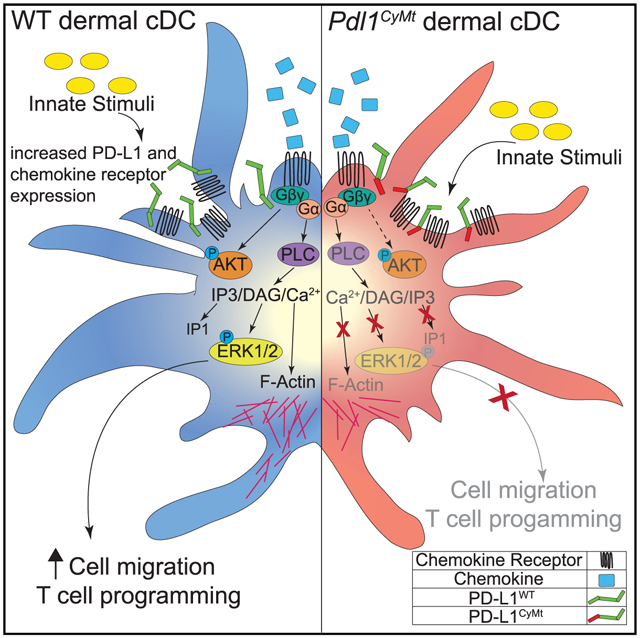
In Brief
Lucas et al. define three residues within the cytoplasmic tail of PD-L1 that are required for proper dendritic cell migration from the skin to the lymph node. These three-amino-acid residues promote chemokine signaling in dendritic cells and productive T cell responses to skin infections.
INTRODUCTION
Programmed death 1 (PD-1)/programmed death ligand 1 (PD-L1) interactions are critical for dampening the immune response to both self and foreign antigens. The signaling of PD-L1 via its cytoplasmic domain, rather than through its interactions with PD-1 via the extracellular domain, has been termed PD-L1 reverse signaling. Recent in vitro evidence has demonstrated that PD-L1 reverse signaling within cancer cells protects the cells from lysis by cytotoxic T cells and interferon (IFN)-induced apoptosis (Azuma et al., 2008; Gato-Cañas et al., 2017; Ghebeh et al., 2010). Although this signaling is beneficial for cancer progression, little is understood about the consequences of PD-L1 reverse signaling in immune cells that express PD-L1 at steady state or in response to infection.
One of the major cell types shown to both express PD-L1 during infection and regulate T cell expansion and differentiation is the dendritic cell (DC). Conventional dendritic cells (cDCs) are separated into two major subsets, cDC1 and cDC2, which develop from a common DC precursor upon expression of FMS-like tyrosine kinase 3 ligand (Flt3L) (Eisenbarth, 2019). These subsets, both lymph node (LN) resident and migratory, are developmentally regulated by specific transcription factors, such as basic leucine zipper transcription factor (Batf3) and interferon regulatory factor (IRF) 8 for cDC1s (Aliberti et al., 2003; Hildner et al., 2008) or IRF4 and Notch for cDC2s (Lewis et al., 2011; Schlitzer et al., 2013); express zbtb46; and can be distinguished as either chemokine XC receptor 1 (XCR1)+ for cDC1s or XCR1− for cDC2s (Bachem et al., 2012; Guilliams et al., 2014). PD-L1 expression in the steady state is highest in cDC2s, whereas PD-L1 expression is induced by both cDC1s and cDC2s (Brown et al., 2019), primarily through IRF1 (Garcia-Diaz et al., 2017; Michalska et al., 2018), during infection. This PD-L1 upregulation occurs concurrently with cDC activation and, for dermal cDCs, is consistent with the time frame in which CCR7 expression is increased and the dermal cDCs begin to migrate to the lymphatic capillaries on their way to the skin draining lymph node (dLN) (Brown et al., 2019; Honda et al., 2003; Ohl et al., 2004; Riol-Blanco et al., 2005; Tiberio et al., 2018). Loss of PD-L1 during infection leads to unchecked T cell proliferation and increased autoimmune T cell responses (Dai et al., 2014; Francisco et al., 2010). Although the T cell-intrinsic role of PD-1 for inhibiting T cell responses has been well explored, little to no effort has been directed at investigating the consequences of PD-L1 reverse signaling in the DCs, despite evidence that PD-L1 has the capacity to alter extracellular regulated kinase (ERK) signaling (Qiu et al., 2018), a requirement for DC migration (Huang et al., 2004; Wilflingseder et al., 2004; Yen et al., 2011).
Upon immunization or infection, DCs must migrate through the extracellular matrix (ECM) via amoeboid movement to the lymphatic capillaries, guided by C-C motif chemokine ligand 21 (CCL21) secreted from the lymphatics (Lämmermann et al., 2008; Schumann et al., 2010). C-C motif chemokine receptor 7 (CCR7) is a G protein-coupled receptor that is upregulated by DCs during infection and binds to both secreted and surface CCL21. This CCL21-activated CCR7 leads to mitogen-activated protein kinase (MAPK)/ERK phosphorylation, phosphatidylinositol 3-kinase (PI3K) activation, cell division control protein 42 (Cdc42) activation via guanine nucleotide exchange factors (GEFs), Janus kinase (JAK) activation, Ca2+ mobilization, and actin polymerization (Hauser and Legler, 2016). Once the DCs undergo CCL21/CCR7-mediated signaling events and have reached the lymphatic capillaries, the DCs interact with the lymphatics through integrins and CCL21 at transmigratory cups (Muto et al., 2014). Following an active actin remodeling process, which is required for successful intravasation into the lymphatic capillaries, the DCs crawl along the luminal surface of the lymphatics (Jackson, 2019). At this stage, interactions between lymphatic endothelial cells (LECs) and DCs are maintained primarily through integrin-intracellular adhesion molecule 1 (ICAM1) interactions (Nitschké et al., 2012). The DCs are guided by lymphatic-derived chemotactic CCL21 gradients until they reach the downstream lymphatic collectors, in which they are propelled to the LN by lymphatic flow. These processes are required for presentation of skin pathogens in the LN (Chu et al., 2011; Dejani et al., 2016; Johansen et al., 2008; Manolova et al., 2008; Tomura et al., 2014), as well as the acquisition of archived antigens retained by LECs (Kedl et al., 2017; Tamburini et al., 2014).
Here, we demonstrate that PD-L1 reverse signaling is required for dermal DC migration during an active immune response. We show that loss of PD-L1 severely reduces DC migration from the skin to the dLN during immunization or infection. Loss of dermal DC migration in the absence of PD-L1 leads to reduced cytotoxic T cell responses to bacterial infection of the dermis. This is in contrast to T cell priming of soluble antigens, which directly access the dLN and do not require dermal DC migration for T cell priming. Here, we define a region within the cytoplasmic domain of PD-L1 and demonstrate the requirement of this region of PD-L1 in DC migration. We discovered three amino acids within the cytoplasmic tail of PD-L1 were critically important for CCR7 signaling and thereby significantly impeded CCL21-mediated G protein activation, downstream ERK phosphorylation, and actin polymerization. Altogether, these data identify a role for PD-L1 in mediating chemokine signaling events and DC migration during dermal infection and identify a signaling motif in the PD-L1 cytoplasmic domain responsible for these events.
RESULTS
Pdl1−/− Mice Have Fewer DCs in the LN Immediately following Subcutaneous poly(I:C) Injection
In our studies looking at PD-L1 signaling in response to type 1 IFN, we discovered that PD-L1 was required for LEC division and survival in the LN following poly(I:C) subcutaneous injection (Lucas et al., 2018). Because immune cells can also upregulate PD-L1 in response to type 1 IFN (Bazhin et al., 2018; Brown et al., 2019), and because poly(I:C) injection leads to a strong induction of type 1 IFN, we asked whether there were differences in number or frequency in immune cells in wild-type (WT) and Pdl1−/− mice either before or 1 day after intradermal or subcutaneous poly(I:C) injection. Upon examination of LNs at baseline, using established markers (Brown et al., 2019) (Figure 1A), we observed no differences in the number or frequency of any cell type we evaluated, including DCs, monocytes, T cells, or B cells, between WT and Pdl1−/− mice (Figure 1; Figure S1). One day after poly(I:C) subcutaneous or intradermal injection, we found that both cDC1s, as defined by XCR1+, and cDC2s, as defined as XCR1− and CD11b+, significantly upregulated PD-L1 expression (Figures 1A and 1B). We also found a significant decrease of both cDC1s and cDC2s in the dLN in the Pdl1−/− mice compared with WT (Figure 1C; Figure S1A). This was in contrast to total CD11c− CD11b+ cells (Figure 1D; Figure S1B) and Ly6C-high monocytes (Figure S1C), which were unchanged in frequency or number after poly(I:C) injection in Pdl1−/− mice compared with WT mice. The frequency and number of Langerhans cells were also unchanged in the Pdl1−/− mice compared with WT mice (Figure S1D), either at baseline or following poly(I:C) injection. This is not surprising, because Langerhans cells do not enter the LN until ~72 h after stimulation (Bursch et al., 2007; Kissenpfennig et al., 2005) and therefore could not be sufficiently evaluated at the 24 h time point. Furthermore, total LN cellularity, as well as CD4 T cells, CD8 T cells, or B cells, was unchanged after poly(I:C) injection in Pdl1−/− mice compared with WT mice (Figures S1E and S1F). To better evaluate whether this decline of DCs in the dLN was caused by a deficit in the number of DCs in the skin that are recruited to the LN after poly(I:C) injection (Kilgore et al., 2020), we quantified the frequency and number of DCs in the skin at the site of intradermal injection (PBS or poly(I:C)). We did not find significant differences in the number of DCs in the skin in either WT or Pdl1−/− mice given a PBS injection; however, after poly(I:C) injection, we found a significant decrease in WT DCs in the skin that was not observed in the Pdl1−/− mice (Figure 1E; Figure S1G). Thus, Pdl1−/− DCs appear to be unable to properly increase in the dLN and decline in the skin following intradermal or subcutaneous poly(I:C) injection.
Figure 1. Loss of PD-L1 Leads to a Reduction in the Number of Dendritic Cells (DCs) in the Draining Lymph Node (dLN) following Poly(I:C).

(A) Flow cytometry gating strategy for conventional DC subsets and myeloid cells in the LN from WT and Pdl1−/− mice. PD-L1 expression shown is from naive mice and mice 24 h following poly(I:C) injection.
(B) Quantification of PD-L1 expression on WT conventional dendritic cell (cDC) 1 and cDC2 at baseline and 24 h following poly(I:C), as well as Pdl1−/− DCs.
(C) Number of cDC1 and cDC2 in the dLN of WT and Pdl1−/− before and 24 h after intradermal poly(I:C) injection (10 μg/mouse). Data are shown from axillary and brachial LNs combined.
(D) Number of CD11b+ CD11c− cells in the dLN of WT and Pdl1−/− mice. Data are shown from axillary and brachial LNs combined.
(E) Number and percentage of CD11c-high cells in the skin from the site of injection of WT and Pdl1−/− mice based on weight.
For (B–E), n = 3 mice per group. Data are shown from one experiment that was repeated four times with similar results. Statistical analysis was done using a one-way ANOVA. *p < 0.05; **p < 0.01; ****p < 0.0001; n.s., p > 0.05. Error bars indicate standard error of the mean. See also Figure S1.
PD-L1 Expression Is Necessary for Dermal DC Migration to the dLN
We next asked whether the reason we found fewer DCs in the dLN and more DCs in the skin after poly(I:C) injection in the Pdl1−/− mice was caused by a loss of dermal DC migration. We chose to use a bone marrow chimeric system to avoid PD-L1 reverse signaling in non-hematopoietic LECs during immunization/infection, which could potentially confound our results (Lucas et al., 2018). We lethally irradiated Pdl1−/− and WT mice with 1,000 rads and allowed reconstitution with WT or Pdl1−/− bone marrow for 12 weeks. It is possible to separate DC populations in the LN into migratory and resident using major histocompatibility complex class II (MHC class II) and CD11c expression. However, MHC class II expression is upregulated on all DCs 24 h following poly(I:C) injection (Figure S1H), as noted previously (Merad et al., 2013), making these two populations indistinguishable. Therefore, following reconstitution, we measured trafficking of dermal DCs from the skin to the dLN using a fluorescein isothiocyanate (FITC) painting assay. In this assay, FITC acquired by dermal DCs is used to identify migratory DCs, in which the DCs in the skin acquire the FITC before migrating to the skin dLN (Macatonia et al., 1986, 1987). In this way, we were able to specifically identify DCs that migrated from the site of injection to the dLN. We performed this assay with or without intradermal poly(I:C) injection (Figure 2A). Although we found 24 h after FITC painting that both WT and Pdl1−/− DCs migrated to the dLN, we saw a dramatic increase in DC migration following poly(I:C) injection in WT mice that was not replicated either in the total Pdl1−/− or when the hematopoietic compartment was Pdl1−/− (Figures 2B and 2C). Importantly, the fold increase in the number of DCs migrated in the WT mice following poly(I:C) was about 2-fold, which was similar to the fold decrease in the number of DCs in the skin at the same time point (Figure 1E). We confirmed that the DCs recruited to the dLN were Pdl1−/− or WT by flow cytometry (Figure 2D). We also observed a defect in CD80 upregulation on the Pdl1−/− dermal DCs (Figure 2E), as well as CD86, but did not see a similar impact on CD40 or MHC class II (Figures S2A and S2B). Because a loss of dermal DC migration has not been reported in the Pdl1−/− mice, and to determine whether the defect was intrinsic to DCs, we confirmed these findings using a co-transfer of bone marrow-derived dendritic cells (BMDCs) from WT or Pdl1−/− mice into WT mice. Mice were injected with a 1:1 ratio of congenically different and differentially labeled (carboxyfluorescein succinimidyl ester [CFSE] or violet proliferation dye [VPD]) BMDCs. 24 h after poly(I:C) injection, BMDCs were transferred into the footpad of mice, and 20 h after transfer, the frequency and number of BMDCs in the dLN was quantified (Figure 2F). We found a significant decrease in the ratio of Pdl1−/−:WT BMDCs in the dLN (Figures 2G and 2H) and a decrease in the number of migrated Pdl1−/− BMDCs compared with WT (Figure 2I). Upon digestion of the skin from the injection site (Figure S2C), we found a significant increase in the ratio of Pdl1−/−:WT BMDCs in the skin (Figures S2D and S2E). To determine whether the differences we observed resulted from increased death of the Pdl1−/− BMDCs, we evaluated viability and found no differences (Figure S2F). We confirmed that the BMDCs had similar expression of CD11c and that the Pdl1−/− BMDCs were devoid of PD-L1 expression before injection (Figures S2G and S2H). These findings demonstrate that expression of PD-L1 has a direct effect on DC migration from the skin to the dLN following a poly(I:C) injection. Furthermore, these findings demonstrate PD-L1 deficiency results in an accumulation of DCs in the skin rather than a loss of Pdl1−/− DCs in the dLN caused by increased death.
Figure 2. Loss of PD-L1 Impairs Dermal Dendritic Cell (dDC) Migration following Poly(I:C).

(A) Experimental design for (B)–(E), in which black and white lines indicate chimerism.
(B) Representative flow plots showing FITC+ dDCs (CD11c+, MHC2hi, and B220−) in dLNs of WT and Pdl1−/− chimeric mice.
(C) Number of FITC+ dDCs in the dLN 24 h after intradermal poly(I:C).
(D) PD-L1 expression on FITC+ dDCs in the dLN.
(E) CD80 expression on FITC+ dDCs in the dLN.
(F) Experimental design for (G)–(I). WT and Pdl1−/− bone marrow-derived dendritic cells (BMDCs) co-injected in the footpad of WT mice 24 h after poly(I:C) (5 μg/footpad).
(G) Representative flow plots of CD11c+ cells before transfer and from the popliteal dLN 24 h after transfer.
(H) Ratio of Pdl1−/− BMDCs to WT BMDCs found in the dLN 24 h after transfer into WT hosts.
(I) Number of WT and Pdl1−/− BMDCs in the dLN 24 h after transfer.
For (B–E), data are shown from axillary and brachial LNs combined, n = 5–7 mice in data shown, repeated four times with similar results. For (G–I), data are shown from two popliteal LNs, n = 4 mice in data shown, repeated three times with similar results. Statistical analysis was done using a one-way ANOVA (C–E), an unpaired Student’s t test (H), or a paired Student’s t test (I). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Error bars indicate standard error of the mean. See also Figure S2.
Mutation of Three Amino Acids within Exon 6 of PD-L1 in C57BL/6 Mice
The evidence outlined earlier demonstrates a role for PD-L1 in directing DC migration to the dLN during an immune response. It has previously been shown that PD-L1 controls LEC division and survival during an immune response (Lucas et al., 2018) and that the DTSSK domain may regulate PD-L1 reverse signaling, in cancer cells in vitro, following IFNβ (Gato-Cañas et al., 2017). To determine whether the threonine-serine-serine (TSS) region is a motif that controls DC migration, we mutated the three amino acids, TSS, within exon 6 of PD-L1 to alanine-alanine-alanine (AAA) using CRISPR-Cas9 mutagenesis in C57BL/6 embryos (Figure 3A). Guide design was done using CRISPR and Broad Institute sgRNA Design software, both of which have been refined to better identify off-target events (Doench et al., 2016; Haeussler et al., 2016). Results from each software were compared, and the guide that performed best using both algorithms was chosen. Guide activity was verified by incubating guide RNA (gRNA) and Cas9 protein with a PCR product containing the target sequence and comparing the ratio of cut to uncut PCR product (Figure S3A). After verifying candidate guide activity, we microinjected the gRNA, Cas9 protein, and the single-stranded oligonucleotide template into B6 zygotes and transferred the embryos into pseudo-pregnant recipients. Two litters, totaling 11 pups, were born. Genotyping was performed by PCR amplification using primers flanking the target sequence followed by restriction enzyme digest for a novel restriction enzyme, Pst1 (CTGCAG), introduced into the CD274 DAAAK allele by silent mutations (Figure 3A; Figure S3B). Of the pups born, 2 showed evidence of non-homologous end joining (NHEJ) that produced large enough insertions or deletions (indels) to be observed upon gel electrophoresis. Of the remaining 9 mice, 1 mouse appeared homozygous (lane 8) and the other 8 mice appeared to have both the modification (277 bp band) and a WT allele (363 bp band) or an allele with small indels that cannot be easily visualized by gel electrophoresis (96 bp) (Figure S3B). Five of these putative founder mice were then bred to C57BL/6 WT mice. Based on litter size, F1 mice from founders 7, 8, and 11 were sequenced and confirmed to have the desired modification. The five off targets were PCR amplified and sequenced for founder 8. There was no evidence of off-target activity for these five putative off targets (Table S1). Founder 8 was bred, and all experiments were performed with this founder line. When evaluating the spleen and LNs, we did not observe significant differences between the mice carrying the mutation and the WT mice. When evaluating morphology via hematoxylin and eosin (H&E) staining of spleens and LNs, we did see a trend for increased size, but not the number of periarterial lymphatic sheaths (PALS) in the spleen, and we saw no difference in the number of B cell follicles in the LN (Figures S3C and S3D). We also quantified total cellularity and immune cell composition and found no differences between mutant and WT mice (Figure S3E). Upon characterization of PD-L1 expression, we found no significant differences at baseline when compared with WT (Figures S3F and S3G). For simplicity, this mouse strain will be called Pdl1 cytoplasmic mutant (Pdl1CyMt) for the remainder of this manuscript.
Figure 3. Creation of a Mouse Model with a Three-Amino-Acid Mutation in the Cytoplasmic Domain of PD-L1.

(A) Design of the guide RNA (underlined sequence) for CRISPR-Cas9 mutagenesis. The lowercase red letters indicate nucleotide changes leading to the amino acid replacements indicated. A recognition sequence for Pst1 (CTGCAG) was also introduced. The box indicates the PAM (protospacer adjacent motif) sequence.
(B) Total number of cDC1s and cDC2s in WT and Pdl1CyMt mice as in Figure 1A. n = 3 mice each in data shown, repeated three times with similar results.
(C) Number of FITC+ DCs migrated to the dLN as in Figure 2C. n = 4–5 mice in data shown, data combined from two independent experiments.
(D) PD-L1 expression (by mean fluorescence intensity [MFI]) on FITC+ DCs in the dLN.
(E) CD80 expression (MFI) on FITC+ DCs in the dLN.
(F) Representative flow plots of transferred WT and Pdl1CyMt BMDCs before transfer and in the dLN (after transfer) as in Figure 2G.
(G) Quantification of (F) showing the ratio of Pdl1CyMt BMDCs to WT BMDCs as in Figure 2H.
(H) Number of WT and Pdl1CyMt BMDCs in the dLN 24 h after transfer.
For (B–E), data are shown from axillary and brachial LNs combined. For (D) and (E), n = 3 mice in data shown, repeated two times with similar results. For (F–H), data are shown from popliteal LNs, n = 6 mice in data shown; data are combined from three different experiments. Statistical analysis was done using a one-way ANOVA (B–D), an unpaired Student’s t test (G), or a paired Student’sttest (H). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Error bars indicate standard error of the mean. See also Figures S3 and S4 and Table S1.
Pdl1CyMt Mice Have Defects in DC Migration
Once we confirmed the Pdl1CyMt mouse strain was viable, fertile, and normal in size with no overt physical or behavior abnormalities, we believed this would be a useful model for determining whether this cytoplasmic region of PD-L1 was required for DC migration. Unlike the Pdl1−/− strain, the extracellular domain of PD-L1 is intact and expressed in the Pdl1CyMt mice; thus, any differences we found will be independent of PD-1 signaling, because PD-1 will still receive signals from PD-L1. We first evaluated differences in DC frequency and number in naive or Pdl1CyMt mice. As in Figure 1, we saw no differences in DC number or frequency in Pdl1CyMt mice compared with WT in either the LN or the skin at baseline. However, similar to Pdl1−/− mice, we saw significantly fewer cDC1 and cDC2 subsets arriving in the dLN 24 h after poly(I:C) injection (Figure 3B). We next employed the FITC painting assay, and 20–24 h following poly(I:C) injection and FITC painting of the skin, we observed robust DC migration to the LN following poly(I:C) in WT mice, but not in Pdl1CyMt mice (Figure 3C). Upon evaluation of PD-L1, we observed no differences in PD-L1 upregulation or in CD80, CD86, CD40, or MHC class II upregulation by DCs from either WT or Pdl1CyMt mice after poly(I:C) (Figures 3D and 3E; Figure S4A). This was important, because we demonstrate (Figure 2E), like others (Talay et al., 2009), that CD80 expression by Pdl1−/− DCs is diminished. This is likely caused by PD-L1 interactions with CD80 in cis that are required for blocking CD80 binding to CD28 (Sugiura et al., 2019; Zhao et al., 2019). This also indicates that the decrease in migration observed in Pdl1−/− or Pdl1CyMt mice is likely independent of the defects in the upregulation of CD80 or CD86 that we saw in the Pdl1−/− mice (Figure 2E; Figure S2B). As in Figure 2, we confirmed these findings were a result of an intrinsic defect in DCs by co-transferring WT and Pdl1CyMt BMDCs after poly(I:C) injection into WT mice. 24 h after transfer, we found a significant decrease in Pdl1CyMt BMDCs in the dLN (popliteal) and a significant increase in Pdl1CyMt BMDCs in the skin at the injection site (foot) (Figures 3F-3H; Figure S4B). We again evaluated the viability of the transferred BMDCs at the site of injection and in the dLN and found no difference in cell death in the Pdl1CyMt BMDCs compared with WT (Figure S4C). We found no differences in expression of PD-L1, CD11c, CD80, CD86, and CD40 in the BMDCs transferred into the mice (Figure S4D). These data demonstrate that an intracellular signaling cascade, which requires the threonine and serine residues at positions 277–279, mutated in the Pdl1CyMt mice is required for DC migration and demonstrates a signaling function for this region of PD-L1.
Pdl1CyMt Mice Do Not Efficiently Prime CD8 T Cells when DC Migration Is Required
Because we had seen defects in DC migration, we next asked whether there were differences in T cell responses. In bacterial skin infections, DC migration to the LN is of critical importance, because most bacteria (400–2,000 nm) are too large to pass through the lymphatic capillaries (Manolova et al., 2008). However, protein immunization (30–50 kDa) and virus given subcutaneously pass through the lymphatics directly to the dLN, in which LN-resident DCs can present the antigen and mount an optimal T cell response (Gerner et al., 2017; Loo et al., 2017). We immunized mice with ovalbumin (ova)/poly(I:C)/αCD40, a soluble antigen immunization regimen that does not require CD4 T cell help (Edwards et al., 2013) or DC trafficking from the skin to dLN. Seven days, later we measured antigen-specific CD8 T cell responses in the popliteal dLN using H2-Kb SIINFEKL tetramers, gating on CD8+ and CD3+ cells (Figure S5A). We found no significant difference in soluble antigen-specific T cell responses to ova/poly(I:C)/αCD40 in Pdl1−/− or Pdl1CyMt mice compared with WT mice (Figure 4A). We next infected mice with Listeria monocytogenes-expressing ovalbumin (LM-ova), an infection that normally results in robust type 1 IFN production by monocytes and macrophages even after intradermal infection (Solodova et al., 2011). Indeed, we found that LM-ova given intradermally to WT mice initiated a robust antigen-specific CD8 T cell response in the dLN 7 days after infection (Figure 4B). This was in contrast to the Pdl1−/− or Pdl1CyMt, in which we observed a significant deficit in T cell priming (Figure 4B). We asked whether the defect in endogenous T cell responses was caused by defective PD-L1 back-signaling into T cells, as recently described (Diskin et al., 2020). To do this, we evaluated WT OT1s transferred into Pdl1−/− or Pdl1CyMt mice (Figure S5B). We found a similar loss in OT1 responses as evaluated by OT1 division following WT OT1 transfer into either Pdl1−/− or Pdl1CyMt mice (Figure S5B), suggesting a defect in T cell priming even when T cells were WT. These findings demonstrate that the defects we found in T cell priming in the Pdl1−/− or Pdl1CyMt mice were not caused by T cell-intrinsic factors.
Figure 4. Loss of PD-L1 or Mutation of the PD-L1 Cytoplasmic Domain Impairs T Cell Responses to an Infection Dependent on DC Migration.

(A) Representative flow plots and numbers of ova-specific T cells identified as B220−CD3+CD8+ T cells with a Kb SIINFEKL-specific tetramer (NIH tetramer Core Facility) following subunit immunization in WT, Pdl1−/−, and Pdl1CyMt mice. Mice were immunized with ova (10 μg/injection), poly(I:C) (5 μg/injection), and αCD40 (5 μg/injection) in the flanks and hind footpads. Data are shown from the popliteal and inguinal LNs 7 days post infection (dpi). In the data shown, n = 3 mice per group. The experiment was repeated two times with similar results.
(B) Representative flow plots and numbers of ova-specific T cells, as in (A), following subcutaneous infection with LM-ova. Mice were infected with 1e3 CFU in the hind footpads. Data are shown are from the popliteal LNs 7 dpi. n = 5–6 mice per group. The data shown are combined from two independent experiments.
(C) Representative flow plots and numbers of ova-specific T cells, as in (A) and (B), following intravenous infection with LM-ova. Mice were infected with 2e3 CFU via tail vein injection. Data are shown from the spleens 7 dpi. n = 5–8 mice per group. The data shown are combined from two independent experiments. Statistical analysis was done using a one-way ANOVA. *p < 0.05; **p < 0.01; n.s., p > 0.05. Error bars indicate standard error of the mean. See also Figure S5.
To determine whether the defective T cell response was a systemic difference in the ability of DCs to prime T cells when infected with LM-ova, we next infected mice intravenously with LM-ova. Seven days after intravenous injection of LM-ova, we observed a robust ova-specific CD8 T cell response in Pdl1CyMt or Pdl1−/− mice with levels similar to WT (Figure 4C). Upon quantification of bacterial burden 3 days following infection, we did not find differences in colony-forming units (CFUs) following either subcutaneous or intravenous infection with LM-ova (Figure S5C). These findings indicate that differences in antigen-specific T cell responses following subcutaneous infection that we observed in the Pdl1−/− or Pdl1CyMt mice did not result from a difference in the ability of LM-ova to initially colonize the mice. To confirm that the differences we saw in CD8 T cell priming following dermal listeria injection were caused by defective DC recruitment to the LN, not differences in antigen processing or presentation capability, we again isolated BMDCs from WT, Pdl1CyMt, or Pdl1−/− mice. After maturation, BMDCs were given either DQ-ova, which fluoresces only during proteolytic degradation, or ova and then co-cultured with WT OT1 T cells. We found no differences among WT, Pdl1CyMt, and Pdl1−/− BMDCs in either DQ-ova fluorescence or OT1 division (Figures S5D and S5E). These findings suggest that the Pdl1CyMt or Pdl1−/− mice have a significant defect in initiating the adaptive response only when DC trafficking is required. However, when soluble antigen arrives in the LN directly, eliminating the requirement for DC trafficking, LN-resident Pdl1CyMt or Pdl1−/− DCs can efficiently prime CD8 T cells.
Loss of PD-L1 Intracellular Signals Causes Defective CCR7-Mediated Chemotaxis
Based on the data presented earlier, we next investigated the mechanism behind the defective ability for DCs to migrate in the Pdl1CyMt and Pdl1−/− mice. Although other chemokines play important roles in DC migration, CCR7 is required for homing of antigen-bearing DCs from the skin to the LN (Jackson, 2019; Ohl et al., 2004). However, in addition to chemokine-mediated migration, S1P1R signaling has been shown to be necessary for DC migration via a distinct but overlapping signaling mechanism (Czeloth et al., 2005). To address whether chemokine or S1P1R-mediated DC migration was affected by either loss or mutation of Pdl1 (Pdl1CyMt), we used an in vitro transwell assay, in which CCL21, CCL19, CXLC12, or S1P was placed in the bottom chamber and BMDCs from WT, Pdl1−/−, or Pdl1CyMt mice on the top chamber, with 5 μm pore filter inserts between wells (Figure 5A). Although WT BMDCs activated with poly(I:C), LPS, or tumor necrosis factor alpha (TNF-α)/PGE2 migrated efficiently through the 5 μm pores in response to CCL21, neither the Pdl1−/− nor the Pdl1CyMt BMDCs migrated through the 5 μm pores more than BMDCs of the same genotype without CCL21 in the bottom well (Figure 5B). To determine whether this was specific to CCR7, we evaluated DCs activated with either LPS or TNF-α/PGE2, which was described to be critical for S1P-mediated migration (Czeloth et al., 2005). We found similar numbers of migrated DCs from WT, Pdl1−/−, and Pdl1CyMt in the presence of S1P following TNF-α/PGE2 activation, but not LPS activation (Figure 5C) as previously described (Czeloth et al., 2005). Similarly, we observed a defect in Pdl1−/− and Pdl1CyMt BMDC migration compared with WT BMDCs in response to CCL21, but not S1P, at 4 h after addition of the chemoattractant to the bottom well (Figures 5B-5D). We next evaluated whether the defect observed in the Pdl1−/− and Pdl1CyMt BMDCs was specific to CCL21 or was a more general defect in chemokine signaling. We found a similar loss of migration to CCL19, which also signals through CCR7 (Figure 5E) (Yoshida et al., 1997), as well as a defect in response to CXCL12, which signals through the receptor CXCR4 (Figure 5D) (Bleul et al., 1996). Finally, to confirm that the differences observed in migration were not specific to granulocyte-macrophage colony-stimulating factor (GM-CSF)-derived BMDCs, we cultured bone marrow in the presence of FLT3L to generate FLT3L-derived BMDCs and then repeated the transwell assay with CCL21 and S1P. We found a similar defect in the ability of Pdl1−/− and Pdl1CyMt BMDCs to migrate to CCL21 but no impairment in responses to S1P (Figure 5F).
Figure 5. Loss of PD-L1 or Mutation of the PD-L1 Cytoplasmic Domain Impairs DC Response to CCL21.

(A) Experimental outline for (B–F).
(B) Number of BMDCs migrated across a transwell membrane in response to CCL21 placed in the bottom well (1 μg/mL, 12 h). BMDCs were activated with poly(I:C) (1 μg/mL, 4 h), LPS (200 ng/mL, 4 h), or TNF-α/PGE2 (30 ng/mL, 1 μg/mL, 48 h), and the number of BMDCs that migrated across the membrane was counted. The dashed line indicates the number migrated without chemokine in the bottom well.
(C) Number of BMDCs migrated across a transwell membrane to S1P placed in the bottom well (500 nM,12 h). BMDCs were activated with LPS or TNF-α/PEG2 as in (A). The dashed line indicates the number migrated without S1P in the bottom well.
(D) Number of BMDCs migrated across a transwell membrane after 4 h in response to CCL21 (1 μg/mL), CXCL12 (1 μg/mL), or S1P (500 nM) placed in the bottom well. BMDCs were activated with LPS (200 ng/mL, 4 h, for CCL21 and CXCL12 migration) or TNF-α/PEG2 (30 ng/mL, 1 μg/mL, 48 h for S1P migration), and the number of BMDCs that migrated across the membrane was counted. The dashed line indicates the number migrated without chemokine in the bottom well.
(E) Number of BMDCs migrated across a transwell membrane in response to a CCL19 gradient (1 μg/mL). BMDCs were activated with LPS (200 ng/mL, 4 h) and allowed to migrate for 12 h. Data are shown with n = 3 technical replicates per group.
(F) Number of FLT3L-derived BMDCs migrated across a transwell membrane after 4 h in response to CCL21 (1 μg/mL) or S1P (500 nM) placed in the bottom well. BMDCs were BMDCs were activated with LPS (200 ng/mL, 4 h, for CCL21 migration) or TNF-α/PEG2 (30 ng/mL, 1 μg/mL, 48 h for S1P migration).
(G) Representative flow plots and quantification of CCR7 surface expression on LPS-matured BMDCs. Fluorescence minus one (FMO) was performed with all antibodies except anti-CCR7. n = 3 technical replicates per group. The experiment was repeated two times with similar results.
For (B–F), n = 2–3 technical replicates per group. The data shown are combined from two independent experiments. Statistical analysis was done using one-way ANOVA. **p < 0.01; ***p < 0.001; ****p < 0.0001; n.s., p > 0.05. Error bars indicate standard error of the mean. See also Figure S6.
We then evaluated surface expression of CCR7 on the BMDCs to determine whether loss of receptor expression was causing the defect we observed in migration toward CCL21. Intriguingly, WT, Pdl1−/−, and Pdl1CyMt BMDCs all had similar surface expression of CCR7 following LPS activation (Figure 5G), and all genotypes were able to induce CCR7 mRNA expression over naive following LPS activation (Figure S6A). Similarly, when we evaluated CCR7 expression on cDC1s and cDC2s from mice, we found no differences in CCR7 expression (Figure S6B). These data demonstrate that the deficiency in migration in the Pdl1CyMt or Pdl1−/− mice is caused by the inability of chemokine receptors to initiate downstream signaling events that lead to DC migration to chemokines, not the regulation of CCR7 expression or S1P1R signaling.
Loss of PD-L1 Intracellular Signals Leads to Loss of Signaling through CCR7
Others have published that STAT3 phosphorylation is increased in Pdl1−/− cancer cells (Diskin et al., 2020; Gato-Cañas et al., 2017), so we first asked whether STAT3 phosphorylation was increased in CCL21-treated BMDCs. Although we saw a significant increase in STAT3 phosphorylation in LPS-activated Pdl1CyMt BMDCs compared with WT BMDCs without CCL21, this did not change with CCL21 treatment (Figure S6C). This is consistent with data in cancer cells, suggesting type 1 IFN in Pdl1−/− melanoma cells results in increased STAT3 phosphorylation (Gato-Cañas et al., 2017). However, because our findings suggest a signaling mechanism that is downstream of chemokine/chemokine receptor interactions, and because STAT3 phosphorylation does not occur as a consequence of these interactions (Figure S6C), we looked into regulatory proteins downstream of CCR7 signaling that are common to other chemokine receptors. Upon binding of CCR7, both CCL19 and CCL21 induce similar signaling cascades (Yanagawa and Onoé, 2002). Chemokine receptors, including CCR7, require G protein activation. The Gα subunit leads to phospholipase C (PLC)-mediated activation of calcium flux, actin polymerization, and MAPK signaling events. In addition, PI3K activation occurs via the Gγβ subunits, leading to downstream protein kinase B (AKT) activation (Figure 6A) (Hauser and Legler, 2016).
Figure 6. Mutation of the PD-L1 Cytoplasmic Domain Impairs G Protein Activation, ERK Phosphorylation, and F-Actin Polymerization in Response to CCL21.

(A) Diagram showing the signaling pathway of chemokine receptors, including CCR7 and CXCR4. Colored text indicates steps of the pathway interrogated, with red showing steps disrupted by loss of PD-L1 signaling and blue showing steps of the pathway without significant changes.
(B) Quantification of IP-1 concentration in WT and Pdl1CyMt BMDCs following 30 min of CCL21 (1 μg/mL) treatment.
(C) Western blot image using antibodies against ERK1/2, pERK1/2, p38, p-p38, and GAPDH at 0, 1, or 5 min following CCL21 (1 μg/mL) addition to BMDC media. The experiment was repeated twice with similar results.
(D) Quantification of (C), showing fold change over no chemokine.
(E) Representative flow plots of F-actin following 15 s of CCL21 (1 μg/mL) treatment in BMDCs.
(F) Quantification of (E) showing fold change over no chemokine at 15s, 60 s, and 5 min after CCL21. n = 3 technical replicates per group. The data shown are combined from three independent experiments.
(G) Representative flow plots of F-actin following 15 s of S1P (500 nM) treatment in BMDCs.
(H) Quantification of (G) showing fold change over no chemokine. n = 3 technical replicates per group. The data shown are combined from two independent experiments.
Statistical analysis was done using a one-way ANOVA (B and F) or an unpaired Student’s t test (D and H). *p < 0.05; **p < 0.01; ****p < 0.0001; n.s., p > 0.05. Error bars indicate standard error of the mean. See also Figure S6.
To determine how the mutated cytoplasmic domain of PD-L1 affects CCR7 signaling, we interrogated several steps in the CCR7 signaling pathway, indicated by the red (disrupted) and blue (normal) text (Figure 6A). We first looked at inositol monophosphate (IP-1) accumulation in LPS-activated BMDCs following CCL21 treatment. IP-1 is produced downstream of PLC activity and is used as a marker of Gα activation, because PLC is directly downstream of Gα signaling (Liu et al., 2008) (Figure 6A). Although we found a significant increase in IP-1 in WT BMDCs following CCL21, we did not observe a change in IP-1 in Pdl1CyMt BMDCs, indicating that Gα activation is impaired when the cytoplasmic domain of PD-L1 is mutated (Figure 6B). To confirm these findings, we evaluated downstream targets of the Gα and Gβγ subunits. Because MAPKs are activated in response to CCR7 (Thelen, 2001; Wymann et al., 2003), specifically Erk1 and Erk2 (MAPK3 and MAPK1), we next evaluated ERK phosphorylation in WT BMDCs following CCL21 treatment. Consistent with the literature, we found increased phosphorylation of ERK1 and ERK2 following CCL21 treatment of WT BMDCs (Kobayashi et al., 2017; Mendoza et al., 2015; Riol-Blanco et al., 2005). In contrast, we did not see increased phospho-ERK (pERK) 1 or pERK2 in Pdl1CyMt BMDCs (Figures 6C and 6D). This difference was not observed with another MAPK family member, p38, in which phosphorylation decreased equivalently after 5 min between WT and Pdl1CyMt BMDCs (Figures 6C and 6D). We confirmed the difference in ERK1/2 phosphorylation via pERK1/2 flow cytometry on WT and Pdl1CyMt BMDCs and again found no increase in ERK1/2 phosphorylation in the Pdl1CyMt BMDCs in contrast to WT BMDCs (Figure S6D).
Because actin polymerization is also downstream of CCL21/CCR7-mediated Gα activation, requires calcium flux, and can be mediated by ERK signaling to initiate Arp2/3 activity for the generation of branched actin networks (Mendoza et al., 2015), we next evaluated the ability of the BMDCs to polymerize actin in response to CCL21. Based on the preceding data, it was not surprising that the Pdl1CyMt BMDCs did not increase actin polymerization following CCL21 treatment as measured by F-actin intensity, whereas the WT BMDCs increased F-actin almost 2-fold after CCL21 treatment at 15 s (Figures 6E and 6F). We found a similar defect in actin polymerization following CXCL12 treatment (Figure S6E), confirming our earlier transwell results with CXCL12 (Figure S6). This was in contrast to S1P, as we observed actin polymerization occurred normally following 15 s of S1P treatment (Figures 6G and 6H), indicating the defect in F-actin polymerization results from the loss of chemokine signaling. Furthermore, we found that both the intensity of F-actin staining, using phalloidin, and the presence of actin stress fibers appeared to be drastically different in Pdl1CyMt BMDCs 30 min after CCL21 treatment compared with WT (Figures S6F and S6G). We then confirmed that Pdl1CyMt DCs from the LN also had a similar impairment in actin polymerization to ensure our results were not specific to BMDCs. We treated a single-cell suspension from the skin dLNs of WT mice with CCL21, which led to an increase in F-actin over no chemokine in CD11c+ DCs (Figure S6H). Again, in contrast to WT, DCs from the skin dLNs of Pdl1CyMt mice did not increase actin polymerization (Figure S6H), suggesting that the results we have observed in BMDCs are consistent with changes that occur in DCs in vivo. Finally, to test whether activation downstream of the Gβγ subunit were also affected, we evaluated AKT phosphorylation following CCL21 treatment (Figure 6A). Although we did observe a slight defect in AKT phosphorylation in Pdl1CyMt BMDCs, particularly at 1 min after CCL21 treatment, the cells seemed to recover, and the differences were not significant (Figure S6I). Thus, it appears that loss of PD-L1 reverse signaling impairs DC migration to chemokines through impairment of chemokine receptor signaling via the Gα subunit of the large G protein complex and PLC activation, which in turn disrupts cell migration (Figure 5D).
DISCUSSION
We demonstrate a role for PD-L1 in the regulation of dermal DC migration to the LN during inflammation. We have identified a TSS motif within the cytoplasmic domain of PD-L1 that is required for proper chemokine-mediated DC migration. This motif is required for proper chemokine signaling, including CCR7 and CXCR4 signaling, because loss of the TSS motif reduces Gα subunit activation, ERK phosphorylation, and impedes actin polymerization. Although some of these changes could be caused by an alternative CCL21 receptor found on the X chromosome (Zhao et al., 2020), we believe this is not the case, because we found no differences in gender or specificity to CCL21. Thus, it seems plausible that upregulation of PD-L1 and CCR7/CXCR4 increases the opportunity for membrane interactions during maturation of the DC. Indeed, we found that the cDC1 population has less PD-L1 at baseline and a more dramatic increase following poly(I:C) (Figures 1A and 1B). This, in combination with our data showing a more significant defect in cDC1 migration to the dLN following poly(I:C) injection (Figure 1C), supports these conclusions. PD-L1 may then facilitate the localization of, access to, or stabilization of a specific region of CCR7 and other chemokine receptors, including CXCR4, for activation via Gα proteins, PLC, and/or RAS. A portion of the CCR7 intracellular C-terminal domain (amino acids 346–355) is required for migration via G protein activation and ERK phosphorylation (Otero et al., 2008). These findings suggest that both PD-L1 and chemokine receptor intracellular domains could interact in a way that facilitates G-protein activation (Otero et al., 2008) and downstream signaling events that lead to DC migration. This is an area of future investigation.
In many of our experiments, we use a Toll-like receptor (TLR) agonist: poly(I:C) or listeria infection. Both of these innate stimuli induce type 1 IFN and, as we show, increase PD-L1 expression (Figure 1A) (Hartley et al., 2017; Lee et al., 2005, 2006; Mühlbauer et al., 2006; Sharara et al., 1997; Staples et al., 2015; Wang et al., 2017). As mentioned earlier, this increase in expression of PD-L1 coordinated with increased expression of CCR7 could decrease the proximity between these two membrane proteins to allow productive interactions. However, there is also evidence that type 1 IFN production can inhibit migration of DCs in the skin, but not the spleen, as well as evidence that migration of DCs may rely on different signals depending on subset or location in the tissue (Pennell and Fish, 2017; Ricart et al., 2011; Tiberio et al., 2018; Yen et al., 2010). Thus, perhaps increased expression of PD-L1 during an IFN-inducing stimulus could mitigate suppressive signals initiated by type 1 IFN production. This is consistent with recent data in melanoma cells demonstrating that PD-L1 is important to protect against IFN-induced cell death (Gato-Cañas et al., 2017). In the case of DCs, this ability of PD-L1 to limit IFN signaling, or act as a rheostat, could relieve IFN-mediated inhibition of migration to ensure rapid presentation of antigens and activation of T cells. Our data examining antigen-specific T cells in response to a pathogen that requires DC migration to the LN support this idea, because we observed reduced antigen-specific CD8 T cells in both Pdl1−/− and Pdl1CyMt mice. This defect in response to dermal infection, but not systemic infection, is consistent with what others have observed with loss of dermal DCs (Tintelnot et al., 2019). Although not evaluated in this study, the migration of Langerhans’s cells, which occurs at later time points compared with migratory cDCs, could be examined to determine whether PD-L1 signaling regulates migration at later points when IFN signaling is decreased.
These studies using a Pdl1CyMt mouse identify a three-amino-acid motif that, via regulation of DC migration, manipulates the CD8 T cell response through an intracellular mechanism, rather than an extracellular mechanism. Furthermore, these findings demonstrate that although surface expression of PD-L1 is similar in WT mice and in the Pdl1CyMt mice, both during homeostasis and following poly(I:C) injection, the reduction we observed in DC migration was nearly identical to that of the Pdl1−/− mouse. This suggests that without the confounding loss of interactions between PD-L1 and CD80 or PD-1, intracellular PD-L1 signaling is required for the differences we found in DC migration. Whether PD-L1 must bind to another receptor or ligand to induce this signaling is not yet clear. Much of the published data evaluating PD-L1 intracellular signaling in cancer cells has been in vitro and in the absence of PD-1 or CD80, suggesting that at least some effects of PD-L1 reverse signaling are independent of both PD-1 and CD80, unless the tumor cells express CD80 or PD-1 (Gato-Cañas et al., 2017; Ghebeh et al., 2010; Hauser and Legler, 2016). However, CCR7 receptor, PD-L1, and CD80 are all upregulated by myeloid DCs upon recognition of pathogens, inflammatory mediators, or tissue damage, and PD-L1 interactions with CD80 have been demonstrated recently to be required in cis on the DC (Sugiura et al., 2019; Zhao et al., 2019). Whether cis interactions with CD80 contribute to PD-L1 intracellular responses that regulate CCR7 signaling, G protein coupling, RAS binding, or initiation of migration are unknown. Furthermore, studies have indicated that PD-L1 and PD-1 interact in cis, as well as in trans, to limit inhibitory signals to the T cell from either the tumor cell or the antigen-presenting cell (Zhao et al., 2018). It is possible that either CD80 or PD-1 cis interactions could influence PD-L1 reverse signaling. Interestingly, soluble PD-1 and CD80 have also been demonstrated to induce downstream signaling events such as proliferation and activation marker expression in cancer cells and macrophages expressing PD-L1 (Gato-Cañas et al., 2017; Hartley et al., 2018). In addition, a study found a role for PD-1 binding to PD-L1, which led to Th17 differentiation, in which loss of PD-L1 improved adaptive tumor immunity (Diskin et al., 2020). Thus, it appears that PD-L1 reverse signaling within the DC likely involves interactions with either CD80 or PD-1 in cis. However, which of these binding partners may induce signaling through the TSS motif of PD-L1 to regulate DC migration remains unclear and should be the subject of future studies. Finally, what effect PD-L1 antibody blockade has on intracellular PD-L1 signaling and how this may affect patients undergoing immunotherapy have yet to be evaluated. However, a study evaluating PD-L1 on macrophages found that treatment with a PD-L1 antibody leads to alterations in macrophage phenotype and function, suggesting that PD-L1 antibody therapy could potentially alter PD-L1 intracellular signaling in immune cells (Hartley et al., 2018) and/or influence DC migration.
In summary, we have identified a signaling pathway regulating DC migration to the LN, dependent on PD-L1 reverse signaling that uses a specific TSS motif within the intracellular region of PD-L1. These studies support the growing body of evidence that PD-L1 reverse signaling plays a dynamic role in regulating the immune response. We introduce a mouse model with a three-amino-acid mutation in the cytoplasmic domain of PD-L1 that reveals the differences we observed in the total PD-L1 knockout mouse regarding DC migration are not a result of lost surface expression. Specifically, we demonstrate that the signaling mechanisms affected by the TSS motif within PD-L1 regulate Gα activation, ERK phosphorylation, and actin polymerization. This loss of actin reorganization, required for protrusions and pushing through the blood and lymphatic capillaries (Ballestrem et al., 2000; Estin et al., 2017; Jackson, 2019; Lämmermann et al., 2009; Sandig et al., 1997; Schaks et al., 2019; Thiam et al., 2016; Vargas et al., 2016; Worbs et al., 2017; Xue et al., 2010), is the likely mechanism for decreased DC migration to the LN. Finally, we confirm increased STAT3 phosphorylation in BMDCs activated with LPS, an IFN-inducing TLR agonist. However, these changes in STAT3 phosphorylation are not altered by CCL21, suggesting that STAT3 may not be involved in the differences we see in DC migration. How increased STAT3 phosphorylation affects the DCs needs further investigation. However, regarding DC migration, future studies understanding how PD-L1 can control G protein activation and if or how this is regulated by receptor binding will be of critical importance to directly understand how PD-L1 may regulate DC trafficking during inflammation. More broadly, these studies could have immediate relevance to current immunotherapy and our understanding of the mechanisms involved in the efficacy of immunotherapy.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Beth Tamburini (beth.tamburini@cuanschutz.edu)
Materials availability
The Pdl1CyMt mouse will be made available under an appropriate material transfer agreement or biological materials license. Please contact Dr. Beth Tamburini or the University of Colorado for additional information.
Data and code availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
4-6 week old mice were purchased from Charles River or Jackson laboratory, unless otherwise stated, or bred and housed in the University of Colorado Anschutz Medical Campus Animal Barrier Facility. Wild-type, Pdl1CyMt and Pdl1−/− mice were all bred on a C57BL/6 background. Male and female mice were used in the study from 5-8 weeks old and no gender differences were observed. All mice were group housed in a room with stable temperatures and humidity, with a 12 h-light cycle. Mice had free access to food and water. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Colorado.
METHOD DETAILS
FITC paint assay
Mice were anesthetized and shaved in two spots on the upper back. 4 h later, mice were again anesthetized and 25 μL of FITC (4 μg/mL) in a 1:1 ratio of acetone and dibutyl phthalate was applied via a pipette. After 24 h, the axillary and brachial LNs were harvested and processed for flow cytometry.
PolyI:C injection and Listeria infection
PolyI:C (Invivogen) (5 μg/site) was injected either subcutaneously in the hind footpad or the shoulders, in a final volume of 50 μL per injection site (Kedl et al., 2017; Tamburini et al., 2014). For some experiments, anti-CD40 (BioXcell) (5 μg/site) and ovalbumin (10 μg/site) was also injected. For Listeria monocytogenes expressing ovalbumin (LM-ova) infection LM was cultured in BHI broth with 5μg/mL erythromycin overnight and then sub-cultured in a shaking incubator for around 3hrs until an OD between 0.6 and 0.8 was reached (Tamburini et al., 2014). Mice were then infected with 1e3CFU per footpad or 2e3CFU by tail vein injection in a final volunme of 200 μL.
LN and skin harvesting and staining for flow cytometry
LNs or skin were harvested, minced with needles and then digested with collagenase D (1mg/mL) and DNase 1 (0.25mg/mL) (30min, 37°C) as described previously (Kedl et al., 2017). The cells were then stained for flow cytometry with antibodies diluted in FC block (24G2) for 30 min at 4°C (Kedl et al., 2017; Tamburini et al., 2014). Following staining, cells were washed and then run on the flow cytometer. Unless otherwise noted, all flow cytometry antibodies were purchased from Biolegend. For DCs and BMDCs cells were stained with CD11c, clone N418 (APCcy7), CD11b, clone M1/70(PEcy7), XCR1, clone ZET (PerCPcy5.5), MHC class II, clone 114.15.2 (APC), CD40, clone 3/23 (PerCPcy5.5), CD80, clone 16-10A1 (APC), CD86, clone GL-1 (PE), B220, clone RA3-6B2 (BV510), PD-L1, clone 10F.9G2 (BV421), pERK (Cell Signaling, polyclonal) and CCR7, clone 4B12 (BV421). Live/dead staining was done using Ghost Dye Red 780 (Tonbo). For T cell staining cells were stained with CD8, clone 53-6.7 (APCcy7), B220, clone RA3-6B2 (BV510), CD44, clone IM7 (PerCPcy5.5) and CD3, clone 17-A2 (PacBlue). For tetramer staining cells were first stained with an H2Kb SIINFEKL tetramer (PE) for 90 min at 37°C as described previously (Tamburini et al., 2012), followed by staining with antibodies. For F-actin staining and p-ERK staining, the F-actin Visualization Biochem Kit (fluorescence format) from Cytoskeleton Inc was used. BMDCs were fixed and permeabilized and then stained with Phalloidin (PE from kit or AF647, Invitrogen). All samples were collected on a BD LSR canto II flow cytometer using DIVA software (BD Biosciences) and analyzed with FlowJo software (Treestar).
BMDC culture and in vivo transfer
GM-CSF derived BMDCs were cultured as previously described (Burchill et al., 2015; Kedl et al., 2017). Briefly, whole bone marrow was isolated from the tibia and femur of mice and RBCs were lysed. Cells were then cultured in GM-CSF (20ng/mL) for 7-10 days or FLT3L (100ng/mL) for 9 days, in MEM with 10% FBS. Cells were harvested via gentle agitation with a pipette. Cells were stained with CD11c and only cultures where greater than 85% of cells were CD11c high by flow cytometry were used for experiments. For transfer into mice, cells were released and labeled with either carboxyfluorescein succinimidyl ester (CFSE) or violet proliferation dye (VPD). Cells were combined at 1:1 ratio and approximately 1-2e6 or 5e5 cells were injected into the footpads or ears of mice that had been injected with polyI:C (5 μg/injection) 24 h previously. BMDCs were allowed to migrate for 24 h and the popliteal LNs were harvested and stained for DCs.
Transwell assays
BMDCs were treated with polyI:C (1 μg/mL, 4hrs), LPS (200ng/mL, 4hrs), or TNFα/PEG2 (30ng/mL;1 μg/mL, 48hrs). CCL21, CCL19, CXLC12 (Peprotech), and S1P (R&D) were placed in the bottom well (1 μg/mL for chemokines and 500ng/ml for S1P) of a transwell with 5 μM pores. Activated BMDCs were placed in the top of the transwell (1e4cells/well) and allowed to migrate for 4 or 12hours. Media was then removed from the top well and the membrane was wiped with a cotton swab. For transwell assays with four-hour time points, the media from the bottom well was removed and cells were counted using a hemocytometer. For 12-hour time points, media from the bottom well was saved and the bottom of the membrane was treated with trypsin for 5min to release any BMDCs from the bottom of the membrane. The trypsinized cells were then combined with the media from the bottom of the well. Cells were then counted using a hemocytometer.
PD-L1CyMt mouse generation
Guide design was done using CRISPOR and the Broad Institute sgRNA Design software. Results from each were compared, and the guide which performed best using both algorithms was chosen. The chosen guide RNA (Synthego) had the following sequence: ACAAGCTCAAAAAACCGAAA TGG. Guide activity was verified by incubating guide RNA and Cas9 protein with a PCR product containing the target sequence and comparing the ratio of cut to uncut PCR product. The ssDNA HDR template (IDT) had the following sequence:TTAAGATTGATTTCTTCTTCTTTAGTGAGAATGCTAGATGTGGAGAAATGTGGCGTTGAAGATgctgcagccAAAAACCGAA ATGGTAAGTGTGAGTAACGAGGGAGGGGCAAGCCGAGGGAATGAGTGGGACA. B6N zygotes were injected using a cytoplasmic microinjection with guide RNA (5ng/μL), Cas9 (13ng/μL, Sigma), and the ssDNA HDR template (25ng/μL). Zygotes were then transferred into outbred ICR pseudopregnant recipients. F0 pups were genotyped by PCR using primers outside the region to be modified to identify putative positive founders followed by digestion with PstI. A novel PstI restriction enzyme site was created with the introduction of the AAA motif in place of TSS in the HDR template. Five putative founder mice were then bred to wild-type B6N mice. Pups from founders that produced the largest number of males and females were sequenced to verify the desired mutation event. F1 mice from founders #7, #8, and #11 were sequenced and confirmed to have the desired modification.
Microscopy
Inguinal LNs from naive mice were fixed overnight in 10% phosphate buffered formalin and then embedded in paraffin. About 7-10 μm sections were cut using a microtome. Slides were stained with hematoxylin and eosin and then coverslipped. For F-actin staining on BMDCs the F-actin Visualization Biochem Kit (fluorescence format) from Cytoskeleton Inc was used. BMDCs were grown on glass coverslips, activated with LPS as described and fixed/permeabilized following kit directions and then stained with Phalloidin (PE from kit) Sections and slides were imaged using an IX83 Olympus fluorescent microscope.
OT1 transfer
CD8 T cells were isolated from an OT1+ mouse using the MojoSort mouse CD8 T cell isolation kit (Biolegend) and labeled with violet proliferation dye diluted 1:1000 (final concentration of 1nM) in PBS for 30 min at 37°C. Approximately 2.5e5 cells were transferred into congenically different WT, Pdl1−/− and Pdl1CyMt mice via tail vein injection. Mice were then infected in the footpad with LM-ova as described in the STAR Methods. Four days following transfer and infection the dLNs were harvested and stained for flow cytometry. OT1 division was calculated using the determination of the cells divided (percent dividing cells) as previously described (Roederer, 2011) using the equation fraction diluted where i is the generation number (0 is the undivided population), and Ni is the number of events in generation i.
DQ-OVA and DC-T cell co-culture
BMDCs were isolated and cultured as described. For antigen processing assessment with DQ-ova, BMDCs were treated with DQ-OVA (5 μg/mL) for 30min, 1hr or 2hrs at 37°C. As a control BMDCs were treated with DQ-OVA for 2hrs on ice. Following treatment cells were washed and fixed with 1%PFA and 4%sucrose for 10min at RT and then run on a flow cytometer. For DC-T cell co-culture, BMDCs were treated with OVA (25 μg/mL), polyI:C (12.5 μg/mL) and α-CD40 (12.5 μg/mL) for 24hours. Cells were washed and then co-cultured with OT1s (isolated and labeled as described) for three days and a 1:10 ratio of BMDC:OT1. Cells were cultured for three days, and then stained and run on a flow cytometer. OT1 division was calculated as described.
Western blots
BMDCs were treated with LPS (200ng/ml, 4hrs) in serum free MEM. Treated cells were then washed 2x with serum free media. Cells were then resuspended at a concentration of 10e7 cells/ml. 100ul of cells were added to 1.5ml microcentrifuge tube. 100ul of pre-warmed CCL21 (2 μg/ml) was then added to cells and cells were immediately incubated in 37C water bath for 1 or 5 min. Then 200ul of 4x Laemmli buffer with 10% 2-mercaptoethanol, and moved to 90C heat block for 10 min. Samples were run on 10% acrylamide gels and transferred to 0.45μm PVDF membrane. Membranes were blocked with 5%BSA/TBST for 30–60 min at room temp or 4C overnight while rocking. Antibodies: total ERK1/2 (Biolegend W15133B), phospho-ERK1/2 (Cell Signaling 9109), total P38 (Cell Signaling D13E1), phoshpo-P38 (Cell Signaling D3F9), total STAT3 (Cell Signaling 79D7), phospho-STAT3 (Cell Signaling 9145), phospho-AKT (Cell Signaling, Cat#9271S), phospho-AKT (Cell Signaling, Cat#4060S), and GAPDH-HRP (Invitrogen MA5-15738-HRP). All primary antibodies were diluted 1:1,000 in 3%BSA/TBST and incubated with blocked membranes for 2 h at room temperature or 4C overnight, while rocking. Membranes were washed 3 × 5 min in TBST Secondary antibodies used were anti-Rabbit-HRP (abcam AB7090) and anti-Rat-HRP (Abcam ab97057). All secondary antibodies were diluted 1:10,000 in 3% BSA/TBST and incubated at room temperature for 1 h while rocking. Membranes were then washed 3 × 5 min in TBST and imaged using Pierce ECL western blotting substrate and the ChemoDoc MP imaging system.
IP-1 measuremen
BMDCs were treated with LPS (200ng/ml, 4hrs). Treated cells were then washed 3x with media and resuspend in MEM and plated in a 96well or 384well white, opaque bottomed plate. CCL21 was diluted using the buffer from the IP-One Gq Kit (CisBio) to a 2X stock (2 μg/ml), and then added to the BMDCs. Cells were treated with CCL21 for 30min at 37°C, and then lysed and stained for IP-1 using the reagents and protocol in the IP-One Gq Kit. Following staining, the plate was imaged on an EnVision HTRF compatible plate reader at 630nm and 665nM. The concentration of IP-1 was calculated using the standard curve as outlined in the protocol.
qRT-PCR
RNA was isolated from BMDCs with the RNeasy micro kit (QIAGEN-74004). Complimentary DNA (cDNA) was made using the QIAGEN QuantiTect Reverse Transcription kit (QIAGEN-205314). Primers to CCR7 and control gene 18 s were purchased from QIAGEN and run on Thermo Fisher Step 1 plus real-time PCR machine. Quantification was performed using the delta-delta CT method (Livak and Schmittgen, 2001).
Bone marrow chimeras
WT or Pdl1−/− were irradiated with 1000 Rads using a Cesium-137 gamma irradiator and rested for 4 h before reconstitution with WT or Pdl1−/− bone marrow. Bone marrow was isolated and red blood cells were lysed prior to intravenous transfer and allowed to reconstitute for 12 weeks. Mice were bled and checked for appropriate reconstitution by staining for T cells and B cells before used in experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS
To calculate statistical significance, unpaired Student’s t tests, paired Student’s t tests and 1-way Anova were used. Software Graph-Pad Prism 8.0 was used for statistical analysis. All error bars displayed are the standard error of the mean (SEM). Please see figure legends for details.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD11c clone N148 APC-Cy7 | Biolegend | Cat#117324; RRID: AB_830649 |
| Anti-mouse CD11b clone M1/70 PE-Cy7 | Biolegend | Cat#101216; RRID: AB_312799 |
| Anti-mouse XCR1 clone ZET PerCP-Cy5.5 | Biolegend | Cat#148208; RRID: AB_2564364 |
| Anti-mouse I-A/I-E clone M5/114.15.2 APC | Biolegend | Cat#107614; RRID: AB_313329 |
| Anti-mouse CD40 clone 3/32 PerCP-Cy5.5 | Biolegend | Cat#124624; RRID: AB_2561474 |
| Anti-mouse CD80 clone 16-10A1 APC | Biolegend | Cat#104714; RRID: AB_313135 |
| Anti-mouse CD86 clone GL-1 PE | Biolegend | Cat#105008; RRID: AB_313151 |
| Anti-mouse B220 clone RA3-6B2 BV510 | Biolegend | Cat#103248; RRID: AB_2650679 |
| Anti-mouse PD-L1 clone 10F.9G2 BV421 | Biolegend | Cat#124315; RRID: AB_10897097 |
| Anti-mouse CCR7 clone 4B12 BV421 | Biolegend | Cat#120120; RRID: AB_2561446 |
| Anti-mouse CD8 clone 53-6.7 APCcy7 | Biolegend | Cat#100714; RRID: AB_312753 |
| Anti-mouse CD44 clone IM7 PerCPcy5.5 | Biolegend | Cat#103032; RRID: AB_2076204 |
| Anti-mouse CD3 clone 17-A2 Pacific Blue | Biolegend | Cat#100214; RRID: AB_493645 |
| H2Kb SIINFEKL tetramer PE | NIH Tetramer Core Facility | https://tetramer.yerkes.emory.edu/reagents/class-I-mhc |
| Anti-mouse CD40 | BioXcell | Cat#BE0016-2; RRID: AB_1107647 |
| Anti-phosphoAKT | Cell Signaling | Cat#9271S; RRID: AB_329825 |
| Anti-phosphoAKT | Cell Signaling | Cat#4060S; RRID: AB_2315049 |
| Anti-ERK1/2-Total | Biolegend | Cat# 686902; RRID: AB_2629535 |
| Anti-phosphoERK1/2 | Cell Signaling | Cat#9101S; RRID: AB_331646 |
| Anti-GAPDH-HRP | Invitrogen | Cat#MA5-15738-HRP; RRID: AB_2537659 |
| Anti-p38 MAPK | Cell Signaling | Cat#8690S; RRID: AB_10999090 |
| Anti-phosphop38 MAPK | Cell Signaling | Cat#4511S; RRID: AB_2139682 |
| Anti-STAT3 | Cell Signaling | Cat#4904S; RRID: AB_331269 |
| Anti-phosphoStat3 (Tyr705) (D3A7) | Cell Signaling | Cat#9145S; RRID: AB_2491009 |
| Anti-Rabbit IgG H&L (HRP) | Abcam | ab7090; RRID: AB_955417 |
| Anti-Rat IgG H&L (HRP) | Abcam | ab97057; RRID: AB_10680316 |
| Bacterial and Virus Strains | ||
| Listeria monocytogenes-expressing ovalbumin | Ross Kedl | N/A |
| Biological Samples | ||
| Murine lymph nodes | This paper | N/A |
| Murine skin | This paper | N/A |
| Murine bone marrow | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fluorescein isothiocyanate isomer 1 | Sigma-Aldrich | Cat#F7250 |
| polyI:C | Invivogen | Cat#Vac-PIC |
| Recombinant Murine Exodus-2 (CCL21) | Peprotech | Cat#250-13 |
| Recombinant Murine MIP-3β (CCL19) | Peprotech | Cat#250-27B |
| Recombinant Murine SDF-1β (CXCL12) | Peprotech | Cat#250-20B |
| Sphingosine-1-phosphate | R&D Systems | Cat#1370/1 |
| Ghost Dye Red 780 | Tonbo Biosciences | Cat#13-0865 |
| Violet proliferation dye | BD Biosciences | Cat#562158 |
| Critical Commercial Assays | ||
| IP-One Gq kit | Cis-Bio | Cat#62IPAPEB |
| MojoSort Mouse CD8 T Cell Isolation Kit | Biolegend | Cat#480008 |
| F-actin Visualization Biochem Kit (fluorescence format) | Cytoskeleton Inc. | Cat#BK005 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Pdl1−/−: CD274 KO (C57BL/6) | Mayo Clinic | N/A |
| Mouse: OT1: C57BL/6-Tg(TcraTcrb) 1100Mjb/J | Jackson Labs | JAX: 003831 |
| Mouse: WT: C57BL/6 | Charles River Labs | C57BL/6 (B6) Mouse Inbred 027 |
| Mouse: Pdl1CyMt: CD274(TSS277-279AAA) | Regional Mouse Genomics Core at National Jewish Health, This paper | N/A |
| Oligonucleotides | ||
| Mm_Rn18s_3_SG QuantiTect Primer Assay | QIAGEN | Cat#QT02448075 |
| Mm_Ccr7_1_SG QuantiTect Primer Assay | QIAGEN | Cat#QT00240975 |
| Guide RNA: ACAAGCTCAAAAAACCGAAA TGG | Synthego | N/A |
| Recombinant DNA | ||
| ssDNA HDR template: TTAAGATTGATTTCTTCTTCTTTAGTGAGAATGCTAGATGTGGAGAAATGTGGCGTTGAAGATgctgcagccAAAAACCGAAATGGTAAGTGTGAGTAACGAGGGAGGGGCAAGCCGAGGGAATGAGTGGGACA. | IDT | N/A |
| Software and Algorithms | ||
| Prism Version 8 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| CRISPOR | Jean-Paul Concordet and Macimilian Haeussler | http://crispor.tefor.net/ |
| GPP sgRNA Designer: CRISPRko | Broad Institute | https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
| FlowJo | Becton, Dickinson and Company | https://www.flowjo.com/ |
Highlights.
Loss of PD-L1 impairs dendritic cell (DC) migration from the skin to the lymph node
Mutation of 3 amino acids in the PD-L1 cytoplasmic tail impairs DC migration
PD-L1-dependent DC migration is required for cytotoxic T cell priming
Loss of PD-L1 reverse signaling impedes chemokine receptor signaling
ACKNOWLEDGMENTS
The Kb SIINFEKL PE tetramer was provided by the NIH Tetramer Core Facility. The Pdl1−/− mice were a gift from Haidong Dong (Mayo Clinic, Rochester, MN). Pdl1CyMt was created by the Regional Mouse Genomics Core housed at National Jewish Health (Denver, CO). We thank Dr. David Orlicky, a pathologist at the University of Colorado, for evaluation of the LN and spleen of the Pdl1CyMt mice. We thank Thu Doan, Jeffrey Finlon, and Alyssa Goldberg for critical reading of the manuscript. B.A.J.T. was funded by NIH R01 AI121209 and NIH R01 AI155474 and the Department of Medicine Outstanding Early Career Scholar, RNA Biosciences Initiative Clinical Scholar Award, and University of Colorado Anschutz Medical Campus GI and Liver Innate Immune Programs. E.D.L. was funded by NIH T32 AI007405. J.B.S. was funded by NIH T32 GM008730-20S1.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108258.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aliberti J, Schulz O, Pennington DJ, Tsujimura H, Reis e Sousa C, Ozato K, and Sher A (2003). Essential role for ICSBP in the in vivo development of murine CD8alpha + dendritic cells. Blood 101, 305–310 [DOI] [PubMed] [Google Scholar]
- Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, and Chen L (2008). B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 111, 3635–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachem A, Hartung E, Güttler S, Mora A, Zhou X, Hegemann A, Plantinga M, Mazzini E, Stoitzner P, Gurka S, et al. (2012). Expression of XCR1 Characterizes the Batf3-Dependent Lineage of Dendritic Cells Capable of Antigen Cross-Presentation. Front. Immunol. 3, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballestrem C, Wehrle-Haller B, Hinz B, and Imhof BA (2000). Actin-dependent lamellipodia formation and microtubule-dependent tail retraction control-directed cell migration. Mol. Biol. Cell 11, 2999–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazhin AV, von Ahn K, Fritz J, Werner J, and Karakhanova S (2018). Interferon-α Up-Regulates the Expression of PD-L1 Molecules on Immune Cells Through STAT3 and p38 Signaling. Front. Immunol 9, 2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, and Springer TA (1996). The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 382, 829–833. [DOI] [PubMed] [Google Scholar]
- Brown CC, Gudjonson H, Pritykin Y, Deep D, Lavallée V-P, Mendoza A, Fromme R, Mazutis L, Ariyan C, Leslie C, et al. (2019). Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 179, 846–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchill MA, Tamburini BA, and Kedl RM (2015). T cells compete by cleaving cell surface CD27 and blocking access to CD70-bearing APCs. Eur. J. Immunol 45, 3140–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursch LS, Wang L, Igyarto B, Kissenpfennig A, Malissen B, Kaplan DH, and Hogquist KA (2007). Identification of a novel population of Langerin+ dendritic cells. J. Exp. Med 204, 3147–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CC, Di Meglio P, and Nestle FO (2011). Harnessing dendritic cells in inflammatory skin diseases. Semin. Immunol 23, 28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeloth N, Bernhardt G, Hofmann F, Genth H, and Förster R (2005). Sphingosine-1-phosphate mediates migration of mature dendritic cells. J. Immunol 175, 2960–2967. [DOI] [PubMed] [Google Scholar]
- Dai S, Jia R, Zhang X, Fang Q, and Huang L (2014). The PD-1/PD-Ls pathway and autoimmune diseases. Cell. Immunol 290, 72–79. [DOI] [PubMed] [Google Scholar]
- Dejani NN, Brandt SL, Piñeros A, Glosson-Byers NL, Wang S, Son YM, Medeiros AI, and Serezani CH (2016). Topical Prostaglandin E Analog Restores Defective Dendritic Cell-Mediated Th17 Host Defense Against Methicillin-Resistant Staphylococcus aureus in the Skin of Diabetic Mice. Diabetes 65, 3718–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, Buttar C, Li E, Sundberg B, Salas RD, et al. (2020). PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat. Immunol 21, 442–454. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LE, Haluszczak C, and Kedl RM (2013). Phenotype and function of protective, CD4-independent CD8 T cell memory. Immunol. Res 55, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbarth SC (2019). Dendritic cell subsets in T cell programming: location dictates function. Nat. Rev. Immunol 19, 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estin ML, Thompson SB, Traxinger B, Fisher MH, Friedman RS, and Jacobelli J (2017). Ena/VASP proteins regulate activated T-cell trafficking by promoting diapedesis during transendothelial migration. Proc. Natl. Acad. Sci. USA 114, E2901–E2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco LM, Sage PT, and Sharpe AH (2010). The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev 238, 219–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. (2017). Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 19, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gato-Cañas M, Zuazo M,Arasanz H, Ibañez-Vea M, Lorenzo L, Fernandez-Hinojal G, Vera R, Smerdou C, Martisova E, Arozarena I, et al. (2017). PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 20, 1818–1829. [DOI] [PubMed] [Google Scholar]
- Gerner MY, Casey KA, Kastenmuller W, and Germain RN (2017). Dendritic cell and antigen dispersal landscapes regulate T cell immunity. J. Exp. Med 214,3105–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghebeh H, Lehe C, Barhoush E, Al-Romaih K, Tulbah A, Al-Alwan M, Hendrayani SF, Manogaran P, Alaiya A, Al-Tweigeri T, et al. (2010). Doxorubicin downregulates cell surface B7-H1 expression and upregulates its nuclear expression in breastcancer cells: role of B7-H1 as an anti-apoptotic molecule. Breast Cancer Res. 12, R48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E,Tussiwand R, and Yona S (2014). Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat. Rev. Immunol 14, 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. (2016). Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley G, Regan D, Guth A, and Dow S (2017). Regulation of PD-L1 expression on murine tumor-associated monocytes and macrophages by locally produced TNF-α. Cancer Immunol. Immunother 66, 523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley GP, Chow L, Ammons DT, Wheat WH, and Dow SW (2018). Programmed Cell Death Ligand 1 (PD-L1) Signaling Regulates Macrophage Proliferation and Activation. Cancer Immunol. Res 6, 1260–1273. [DOI] [PubMed] [Google Scholar]
- Hauser MA, and Legler DF (2016). Common and biased signaling pathways of the chemokine receptor CCR7 elicited by its ligands CCL19 and CCL21 in leukocytes. J. Leukoc. Biol 99, 869–882. [DOI] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Sakaguchi S, Nakajima C, Watanabe A, Yanai H, Matsumoto M, Ohteki T, Kaisho T, Takaoka A, Akira S, et al. (2003). Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc. Natl. Acad. Sci. USA 100, 10872–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Jacobson K, and Schaller MD (2004). MAP kinases and cell migration. J. Cell Sci. 117, 4619–4628. [DOI] [PubMed] [Google Scholar]
- Jackson DG (2019). Leucocyte Trafficking via the Lymphatic Vasculature–Mechanisms and Consequences. Front. Immunol 10, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen P, Storni T, Rettig L, Qiu Z, Der-Sarkissian A, Smith KA, Manolova V, Lang KS, Senti G, Müllhaupt B, et al. (2008). Antigen kinetics determines immune reactivity. Proc. Natl. Acad. Sci. USA 105, 5189–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedl RM, Lindsay RS, Finlon JM, Lucas ED, Friedman RS, and Tamburini BAJ (2017). Migratory dendritic cells acquire and present lymphatic endothelial cell-archived antigens during lymph node contraction. Nat. Commun 8, 2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore AM, Pennock ND, and Kedl RM (2020). cDC1 IL-27p28 Production Predicts Vaccine-Elicited CD8+ T Cell Memory and Protective Immunity. J. Immunol 204, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissenpfennig A, Henri S, Dubois B, Laplace-Builhé C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, et al. (2005). Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity 22, 643–654. [DOI] [PubMed] [Google Scholar]
- Kobayashi D, Endo M, Ochi H, Hojo H, Miyasaka M, and Hayasaka H (2017). Regulation of CCR7-dependent cell migration through CCR7 homo-dimer formation. Sci. Rep 7, 8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, Keller M, Förster R, Critchley DR, Fässler R, and Sixt M (2008). Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 453, 51–55. [DOI] [PubMed] [Google Scholar]
- Lämmermann T, Renkawitz J, Wu X, Hirsch K, Brakebusch C, and Sixt M (2009). Cdc42-dependent leading edge coordination is essential for interstitial dendritic cell migration. Blood 113, 5703–5710. [DOI] [PubMed] [Google Scholar]
- Lee SK, Seo SH, Kim BS, Kim CD, Lee JH, Kang JS, Maeng PJ, and Lim JS (2005). IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J. Dermatol. Sci 40, 95–103. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, Oh S, Shin JG, Yao S, Chen L, and Choi IH (2006). Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett. 580, 755–762. [DOI] [PubMed] [Google Scholar]
- Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, Klinakis A, Charo IF, Jung S, Gommerman JL, et al. (2011). Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 35, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Titus S, Southall N, Zhu P, Inglese J, Austin CP, and Zheng W (2008). Comparison on functional assays for Gq-coupled GPCRs by measuring inositol monophospate-1 and intracellular calcium in 1536-well plate format. Curr. Chem. Genomics 1, 70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Loo CP, Nelson NA, Lane RS, Booth JL, Loprinzi Hardin SC, Thomas A, Slifka MK, Nolz JC, and Lund AW (2017). Lymphatic Vessels Balance Viral Dissemination and Immune Activation following Cutaneous Viral Infection. Cell Rep. 20, 3176–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas ED, Finlon JM, Burchill MA, McCarthy MK, Morrison TE, Colpitts TM, and Tamburini BAJ (2018). Type 1 IFN and PD-L1 Coordinate Lymphatic Endothelial Cell Expansion and Contraction during an Inflammatory Immune Response. J. Immunol 201, 1735–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macatonia SE, Edwards AJ, and Knight SC (1986). Dendritic cells and the initiation of contact sensitivity to fluorescein isothiocyanate. Immunology 59, 509–514. [PMC free article] [PubMed] [Google Scholar]
- Macatonia SE, Knight SC, Edwards AJ, Griffiths S, and Fryer P (1987). Localization of antigen on lymph node dendritic cells after exposure to the contact sensitizer fluorescein isothiocyanate. Functional and morphological studies. J. Exp. Med 166, 1654–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, and Bachmann MF (2008). Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol 38, 1404–1413. [DOI] [PubMed] [Google Scholar]
- Mendoza MC, Vilela M, Juarez JE, Blenis J, and Danuser G (2015). ERK reinforces actin polymerization to power persistent edge protrusion during motility. Sci. Signal 8, ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M, Sathe P, Helft J, Miller J, and Mortha A (2013). The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol 31, 563–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalska A, Blaszczyk K, Wesoly J, and Bluyssen HAR (2018). A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol 9, 1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlbauer M, Fleck M, Schütz C, Weiss T, Froh M, Blank C, Schölmerich J, and Hellerbrand C (2006). PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J. Hepatol 45, 520–528. [DOI] [PubMed] [Google Scholar]
- Muto J, Morioka Y, Yamasaki K, Kim M, Garcia A, Carlin AF, Varki A, and Gallo RL (2014). Hyaluronan digestion controls DC migration from the skin. J. Clin. Invest 124, 1309–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschké M, Aebischer D, Abadier M, Haener S, Lucic M, Vigl B, Luche H, Fehling HJ, Biehlmaier O, Lyck R, and Halin C (2012). Differential requirement for ROCK in dendritic cell migration within lymphatic capillaries in steady-state and inflammation. Blood 120, 2249–2258. [DOI] [PubMed] [Google Scholar]
- Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, Blankenstein T, Henning G, and Förster R (2004). CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 21, 279–288. [DOI] [PubMed] [Google Scholar]
- Otero C, Eisele PS, Schaeuble K, Groettrup M, and Legler DF (2008). Distinct motifs in the chemokine receptor CCR7 regulate signal transduction, receptor trafficking and chemotaxis. J. Cell Sci 121, 2759–2767. [DOI] [PubMed] [Google Scholar]
- Pennell LM, and Fish EN (2017). Interferon-β regulates dendritic cell activation and migration in experimental autoimmune encephalomyelitis. Immunology 152, 439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu XY, Hu DX, Chen WQ, Chen RQ, Qian SR, Li CY, Li YJ, Xiong XX, Liu D, Pan F, et al. (2018). PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochim. Biophys. Acta Mol. Basis Dis 1864 (5 Pt A), 1754–1769. [DOI] [PubMed] [Google Scholar]
- Ricart BG, John B, Lee D, Hunter CA, and Hammer DA (2011). Dendritic cells distinguish individual chemokine signals through CCR7 and CXCR4. J. Immunol. 186, 53–61. [DOI] [PubMed] [Google Scholar]
- Riol-Blanco L, Sánchez-Sánchez N, Torres A, Tejedor A, Narumiya S, Corbí AL, Sánchez-Mateos P, and Rodríguez-Fernández JL (2005). The chemokine receptor CCR7 activates in dendritic cells two signaling modules that independently regulate chemotaxis and migratory speed. J. Immunol 174, 4070–4080. [DOI] [PubMed] [Google Scholar]
- Roederer M (2011). Interpretation of cellular proliferation data: avoid the panglossian. Cytometry A 79, 95–101. [DOI] [PubMed] [Google Scholar]
- Sandig M, Negrou E, and Rogers KA (1997). Changes in the distribution of LFA-1, catenins, and F-actin during transendothelial migration of monocytes in culture. J. Cell Sci 110, 2807–2818. [DOI] [PubMed] [Google Scholar]
- Schaks M, Giannone G, and Rottner K (2019). Actin dynamics in cell migration. Essays Biochem. 63, 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, Ho AW, See P, Shin A, Wasan PS, et al. (2013). IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity 38, 970–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann K, Lämmermann T, Bruckner M, Legler DF, Polleux J, Spatz JP, Schuler G, Förster R, Lutz MB, Sorokin L, and Sixt M (2010). Immobilized chemokine fields and soluble chemokine gradients cooperatively shape migration patterns of dendritic cells. Immunity 32, 703–713. [DOI] [PubMed] [Google Scholar]
- Sharara AI, Perkins DJ, Misukonis MA, Chan SU, Dominitz JA, and Weinberg JB (1997). Interferon (IFN)-alpha activation of human blood mononuclear cells in vitro and in vivo for nitric oxide synthase (NOS) type 2 mRNA and protein expression: possible relationship of induced NOS2 to the anti-hepatitis C effects of IFN-alpha in vivo. J. Exp. Med 186, 1495–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solodova E, Jablonska J, Weiss S, and Lienenklaus S (2011). Production of IFN-β during Listeria monocytogenes infection is restricted to monocyte/macrophage lineage. PLoS ONE 6, e18543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples KJ, Nicholas B, McKendry RT, Spalluto CM, Wallington JC, Bragg CW, Robinson EC, Martin K, Djukanović R, and Wilkinson TM (2015). Viral infection of human lung macrophages increases PDL1 expression via IFNβ. PLoS ONE 10, e0121527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura D, Maruhashi T, Okazaki IM, Shimizu K, Maeda TK, Takemoto T, and Okazaki T (2019). Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 364, 558–566. [DOI] [PubMed] [Google Scholar]
- Talay O, Shen CH, Chen L, and Chen J (2009). B7-H1 (PD-L1) on T cells is required for T-cell-mediated conditioning of dendritic cell maturation. Proc. Natl. Acad. Sci. USA 106, 2741–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamburini BA, Kedl RM, and Bellgrau D (2012). IL-6-inducing whole yeast-based immunotherapy directly controls IL-12-dependent CD8 T-cell responses. J. Immunother 35, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamburini BA, Burchill MA, and Kedl RM (2014). Antigen capture and archiving by lymphatic endothelial cells following vaccination or viral infection. Nat. Commun 5, 3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelen M (2001). Dancing to the tune of chemokines. Nat. Immunol 2, 129–134. [DOI] [PubMed] [Google Scholar]
- Thiam HR, Vargas P, Carpi N, Crespo CL, Raab M, Terriac E, King MC, Jacobelli J, Alberts AS, Stradal T, et al. (2016). Perinuclear Arp2/3-driven actin polymerization enables nuclear deformation to facilitate cell migration through complex environments. Nat. Commun 7, 10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiberio L, Del Prete A, Schioppa T, Sozio F, Bosisio D, and Sozzani S (2018). Chemokine and chemotactic signals in dendritic cell migration. Cell. Mol. Immunol 15, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintelnot J, Ufer F, Engler JB, Winkler H, Lücke K, Mittrücker HW, and Friese MA (2019). Arc/Arg3.1 defines dendritic cells and Langerhans cells with superior migratory ability independent of phenotype and ontogeny in mice. Eur. J. Immunol 49, 724–736. [DOI] [PubMed] [Google Scholar]
- Tomura M, Hata A, Matsuoka S, Shand FH, Nakanishi Y, Ikebuchi R, Ueha S, Tsutsui H, Inaba K, Matsushima K, et al. (2014). Tracking and quantification of dendritic cell migration and antigen trafficking between the skin and lymph nodes. Sci. Rep 4, 6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas P, Maiuri P, Bretou M, Sáez PJ, Pierobon P, Maurin M, Chabaud M, Lankar D, Obino D, Terriac E, et al. (2016). Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat. Cell Biol 18, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, and You Z (2017). Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol. Lett 184, 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilflingseder D, Müllauer B, Schramek H, Banki Z, Pruenster M, Dierich MP, and Stoiber H (2004). HIV-1-induced migration of monocyte-derived dendritic cells is associated with differential activation of MAPK pathways. J. Immunol 173, 7497–7505. [DOI] [PubMed] [Google Scholar]
- Worbs T, Hammerschmidt SI, and Förster R (2017). Dendritic cell migration in health and disease. Nat. Rev. Immunol 17, 30–48. [DOI] [PubMed] [Google Scholar]
- Wymann MP, Zvelebil M, and Laffargue M (2003). Phosphoinositide 3-kinase signalling–which way to target? Trends Pharmacol. Sci 24, 366–376. [DOI] [PubMed] [Google Scholar]
- Xue F, Janzen DM, and Knecht DA (2010). Contribution of Filopodia to Cell Migration: A Mechanical Link between Protrusion and Contraction. Int. J. Cell Biol 2010, 507821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa Y, and Onoe K (2002). CCL19 induces rapid dendritic extension of murine dendritic cells. Blood 100, 1948–1956. [DOI] [PubMed] [Google Scholar]
- Yen JH, Kong W, and Ganea D (2010). IFN-beta inhibits dendritic cell migration through STAT-1-mediated transcriptional suppression of CCR7 and matrix metalloproteinase 9. J. Immunol 184, 3478–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen JH, Kocieda VP, Jing H, and Ganea D (2011). Prostaglandin E2 induces matrix metalloproteinase 9 expression in dendritic cells through two independent signaling pathways leading to activator protein 1 (AP-1) activation. J. Biol. Chem 286, 38913–38923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida R, Imai T, Hieshima K, Kusuda J, Baba M, Kitaura M, Nishimura M, Kakizaki M, Nomiyama H, and Yoshie O (1997). Molecular cloning of a novel human CC chemokine EBI1-ligand chemokine that is a specific functional ligand for EBI1, CCR7. J. Biol. Chem 272, 13803–13809. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Harrison DL, Song Y, Ji J, Huang J, and Hui E (2018). Antigen-Presenting Cell-Intrinsic PD-1 Neutralizes PD-L1 in cis to Attenuate PD-1 Signaling in T Cells. Cell Rep. 24, 379–390.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Lee CK, Lin CH, Gassen RB, Xu X, Huang Z, Xiao C, Bonorino C, Lu LF, Bui JD, et al. (2019). PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity 51, 1059–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Chen X, Ma W, Zhang J, Guo J, Zhong X, Yao J, Sun J, Rubinfien J, Zhou X, et al. (2020). A GPR174-CCL21 module imparts sexual dimorphism to humoral immunity. Nature 577, 416–420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.