

Oxidation state of iron in Earth’s magma ocean indicates it would have produced a Venus-like early atmosphere upon cooling.
Abstract
Exchange between a magma ocean and vapor produced Earth’s earliest atmosphere. Its speciation depends on the oxygen fugacity (fO2) set by the Fe3+/Fe2+ ratio of the magma ocean at its surface. Here, we establish the relationship between fO2 and Fe3+/Fe2+ in quenched liquids of silicate Earth-like composition at 2173 K and 1 bar. Mantle-derived rocks have Fe3+/(Fe3++Fe2+) = 0.037 ± 0.005, at which the magma ocean defines an fO2 0.5 log units above the iron-wüstite buffer. At this fO2, the solubilities of H-C-N-O species in the magma ocean produce a CO-rich atmosphere. Cooling and condensation of H2O would have led to a prebiotic terrestrial atmosphere composed of CO2-N2, in proportions and at pressures akin to those observed on Venus. Present-day differences between Earth’s atmosphere and those of her planetary neighbors result from Earth’s heliocentric location and mass, which allowed geologically long-lived oceans, in-turn facilitating CO2 drawdown and, eventually, the development of life.
INTRODUCTION
Earth’s present-day N2-O2 atmosphere differs markedly from those of her planetary neighbors, Venus and Mars, which both have CO2-rich atmospheres with minor N2 (1, 2). Models of their accretion suggest that the inner planets formed through an evolving hierarchy of solid-body size distributions, the terminal stages of which saw giant collisions between roughly Mars-sized planetary embryos (3). A small fraction of these embryos may have been sourced from the outer solar system and are expected to have been richer in atmophile elements such as H, C, and N. Accretion of these bodies introduces a considerable degree of stochasticity into the planetary inventories of these elements (4, 5) and, importantly, into planetary oxidation states. It is therefore ambiguous whether the differences in the contemporary atmospheres of Venus, Earth, and Mars arise because of distinct initial compositions or from their subsequent evolution governed by their differing masses and heliocentric distances (2).
The final major collision in this process occurred between the proto-Earth and a Mars-sized impactor that released energy (2 to 5 × 1031 J) sufficient to cause wholesale melting of Earth’s mantle (6). Because the impact would have removed most of the pre-existing atmosphere (7), and the mantle’s mass exceeds that of other terrestrial reservoirs, outgassing of the major volatiles (H, C, N, and O) from Earth’s molten mantle, or magma ocean, likely controlled the composition and oxidation state of its secondary (i.e., post-nebular) atmosphere (8, 9). Long-standing chemical evidence for such a secondary atmosphere comes from the marked depletion of the noble gases relative to similarly volatile yet more reactive elements, such as N (10, 11). These atmospheres are thought to have been either neutral- to oxidizing and H2O-CO2 rich (12–14) or reducing and CH4-NH3 rich (15–17) on early Earth.
The importance of discriminating between these possible atmospheric varieties lies in the potential for the presence of certain gases (NH3 and CH4) to facilitate the emergence of life (18). Models for the formation of these reducing atmospheres appeal to direct degassing of hydrogen-rich, oxygen-poor chondritic materials that accreted to Earth, together with evaporation of any pre-existing oceans (17, 19), or from stranded remnants of the nebular gas (20). On the other hand, arguments for a H2O-CO2 atmosphere are predicated on the as yet unproven assumption that early mantle-derived gases were similar to their modern-day volcanic counterparts, with oxygen fugacities (fO2) near the fayalite-magnetite-quartz buffer (21, 22). Hence, there is no consensus on which type of atmosphere existed on early Earth because of the considerable uncertainty as to the oxidation state of its mantle under the physico-chemical conditions relevant to degassing, that is, in its molten state (8, 19, 23).
Here, by establishing the relationship between fO2 and the redox state of iron in mantle-like silicate melts, we show that the atmosphere degassed from a primordial magma ocean on the early Earth, following cooling, would have resembled that of present-day Venus, implying that the present differences in their atmospheric compositions reflect their post-accretionary histories.
Thermodynamic background and approach
We examine the case in which a liquid silicate magma ocean, of bulk silicate Earth (BSE) composition, forms in equilibrium with its overlying atmosphere. To do so requires experimental constraints on how redox couples vary as a function of oxygen fugacity under compositions, temperatures, and pressures relevant to Earth’s magma ocean. At a given temperature and pressure, the composition of an H-C-N-O–bearing gas phase in chemical equilibrium with a condensed phase such as a rock or magma is specified by three intensive variables, H/C, H/N, and the oxygen fugacity, fO2. Insofar as iron is the most abundant redox-sensitive element in Earth’s mantle (24), the oxygen fugacity in an atmosphere in equilibrium with a terrestrial magma ocean is constrained by the activities of the Fe2+ and Fe3+ components in the silicate melt
| (1) |
And, at equilibrium
| (2) |
where X signifies the mole fraction, γ the activity coefficient and K(1) the equilibrium constant of Eq. 1. This reaction provides the crucial link between the Earth’s atmosphere and its mantle. Although the mole fractions of Fe2+O and Fe3+O1.5 in an isochemical system are fixed, their activity coefficients vary as a function of temperature, pressure, and the phase assemblage of the system, which is itself determined by temperature and pressure. Consequently, as the fO2 of a system with a fixed bulk composition and X(Fe3+)/X(Fe2+) ratio, such as Earth’s mantle, varies with temperature and pressure (Eq. 2), so too will the composition of an H-C-N-O gas phase in equilibrium with it.
Although the fO2 defined by spinel-bearing peridotites from Earth’s uppermost mantle is oxidizing, ~∆IW + 3.5 (25) (∆IW denotes the log10 deviation of fO2 from the iron-wüstite buffer), mantle samples that equilibrated under higher pressures, garnet peridotites, record a progressive decrease in fO2 to ~IW with depth (26, 27). This transition arises from the inability of olivine to incorporate Fe3+ into its crystal structure, prompting redistribution of ferric iron into spinel, which translates into high activity of Fe3+O1.5. At depth, the appearance of phases that can accept Fe3+, chiefly garnet, decrease the activity of Fe3+O1.5, causing ΔIW to fall (28). Melting is likewise a phase transition that induces changes in the activities of Fe3+O1.5 and Fe2+O relative to solid upper mantle peridotite. Therefore, evaluating the fO2 at the top of Earth’s magma ocean requires calibration of Eq. 1 in a silicate melt of a BSE (i.e., atmosphere + crust + mantle) composition at or above its liquidus temperature.
Experimental limitations have hitherto restricted investigation of the relationship between fO2 and Fe3+/Fe2+ in silicate melts to temperatures and compositions relevant to andesitic/basaltic magmatism (29–31). By contrast, estimates for the composition of Earth’s mantle (24) and those of the other terrestrial planets (32, 33) are broadly peridotitic, whose liquidus temperatures exceed 2000 K at 1 bar (34, 35). Thus, there exist data neither for liquids with mantle-like compositions, nor at temperatures relevant to planetary magma oceans. So when parameterizations of Fe3+/Fe2+ versus fO2, based on existing experimental data (29, 36–39), are applied to a liquid of BSE composition with a fixed Fe3+/ΣFe ratio of 0.037 (40, 41) at a temperature of 2173 K, they return values of ∆IW spanning nearly eight orders of magnitude, from −4.5 to +3.1 (Fig. 1). Over this range of oxygen fugacity, the composition of the gas phase at 1 bar changes from one dominated by reduced (CO and H2) to oxidized (CO2 and H2O) species (Fig. 1).
Fig. 1. Effect of oxygen fugacity on the relative abundances of major gas species.
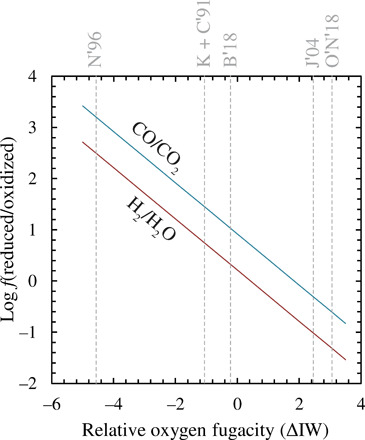
Calculated H2/H2O (red) and CO/CO2 (blue) ratios of an ideal gas at 2173 K as a function of oxygen fugacity expressed relative to the ΔIW. These ratios are independent of molar H/C of the gas phase, as well as its total pressure (provided it remains ideal), and are controlled only by temperature (which determines the intercept, corresponding to the logarithm of the equilibrium constant of reaction) and (fO2)0.5 (the exponent being the slope of the line). Dashed gray lines denote the fO2 of a peridotite liquid with a BSE composition (24) and an Fe3+/ΣFe ratio 0.037 (fig. S8) (40, 41) calculated according to different model parameterizations of the relationship between fO2 and Fe3+/Fe2+ in silicate melts; K + C′91 = Kress and Carmichael (29); N′96 = Nikolaev et al. (36); B′18 = Borisov (37); J′04 = Jayasuriya et al. (38); O′N′18 = O’Neill et al. (39).
RESULTS
Accordingly, we have measured the dependence of Fe3+/Fe2+ on fO2 in a silicate melt of BSE-like composition at a temperature (1900 ± 50°C) relevant to the surface of a terrestrial magma ocean. The experiments were conducted by aerodynamic levitation with laser heating over a range of fO2 from ∆IW −1.5 to +6.4, imposed by CO2-H2-Ar gas mixtures and O2 at a total pressure of 1 bar (see table S1 and Materials and Methods). The method enables extremely rapid quenching, which produces glass with no evidence of quench crystallization, suitable for the precise determination of Fe3+/Fe2+ by Fe K-edge x-ray absorption near-edge structure (XANES) spectroscopy (30, 31). The XANES spectra were collected at the 13-IDE (GSECARS) beamline at Advanced Photon Source (APS), Chicago, Illinois (see Materials and Methods). Quantitative Fe3+/Fe2+ ratios in the glasses were determined by calibration of the energies of the edge and centroid features of the XANES spectra against those of mid-ocean ridge basalt (MORB) glasses (31) whose iron redox state was independently measured by Mössbauer spectroscopy (Supplementary Materials). The major element compositions of the glasses were determined with a JEOL 8530F Plus electron microprobe (Materials and Methods).
The levitation method allowed the silicate melts to be quenched into homogeneous glasses in all cases (table S2). Two glasses equilibrated under the most reducing conditions (logfO2−6.74 and −7.86) experienced evaporative loss of Fe (7 and 9%, respectively, compared to the initial composition) consistent with increasing volatility according to the vaporization reaction FeO(l) = Fe(g) + 1/2O2(g) (42). The measured Fe3+/ΣFe ratios vary from 0.014 ± 0.009 at ∆IW −1.5 to 0.440 ± 0.008 at ∆IW +6.4. The values of log(Fe3+/Fe2+) (n = 11) against logfO2 (expressed as ΔIW) define a trend whose functional form is given by a straight line (Fig. 2), adhering to the stoichiometry of Eq. 1;
| (3) |
Fig. 2. Relationship between iron oxidation state and oxygen fugacity in quenched peridotite liquid.
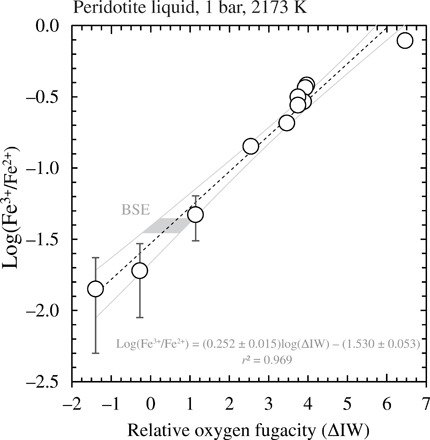
The log(XFe3+/XFe2+) ratios determined by Fe K-edge XANES in a peridotite liquid (1 bar, 2173 K) quenched to glass as a function of the imposed oxygen fugacity, expressed as the log unit deviation from the IW buffer (ΔIW). Error bars on points are the SD. The dashed black line is a linear regression through the data, and the gray curves represent the 95% confidence envelope on the regression. The shaded gray area pertains to the range of log(XFe3+/XFe2+) defined by the BSE, as inferred from Fe3+/Fe2+ ratios of whole-rock peridotites (table S6) (40, 41).
The data define a slope of 0.252 ± 0.015 and a logK* (=logK(1) + log[γFeO1.5/γFeO]) of 0.096 ± 0.053, which may be written with respect to the IW buffer at 2173 K
| (4) |
This slope falls within uncertainty of that expected from Eq. 1, 0.25, indicating that the activity coefficients of both iron melt oxide components (γFeO and γFeO1.5) are constant as a function of fO2 (31). The logK* is much lower than that for MORB at 1400°C [0.58 ± 0.02; (31)], indicating higher stability of FeO relative to FeO1.5 at the same absolute fO2 in peridotite liquid. Comparing the equilibrium constant determined for reaction (1) in peridotite liquid at 2173 K and 1 bar, K*(1), with that of the reaction of pure phases at the same temperature and pressure, K(1) [(43); Supplementary Materials], yields γFeO1.5/γFeO within uncertainty of unity, 1.00 ± 0.13, whereas it is 0.64 ± 0.05 (31) to 0.79 ± 0.12 (38) for basaltic melts at 1400°C. The ratio of the activity coefficients thus tends to unity at higher temperatures, as expected theoretically.
DISCUSSION
A Venus-like atmosphere on the early earth
The fraction of the magma ocean that is able to equilibrate with the overlying atmosphere depends upon the rate of energy delivery to the base of the atmosphere relative to that of heat loss to space. The latter is proportional to T4r2 (T = effective temperature, r = atmospheric radius), the radiative black- or gray-body cooling rate, which in turn depends on composition. Because of the high Rayleigh number of a fully molten magma ocean, ~1030, mixing time scales are rapid; of the order of days to weeks, depending on its viscosity (44, 45). As a result, heat transfer is initially very efficient such that the energy balance (as quantified in the Nusselt number) between radiative cooling for fictive CO2-H2O atmospheres on Earth and convective heat flux to the surface maintains a surface melt over 103 to 104 years (45, 46). Thus, equilibrium over the entire mass of the mantle is readily achieved with the atmosphere.
Knowledge of Fe3+/ΣFe in the terrestrial magma ocean is required to specify the fO2 of the atmosphere with which it is in equilibrium (Eq. 4). Determination of fO2 in mantle-derived magmas suggests little [≤1 log unit (47, 48)] to no systematic secular change (49, 50) since 4.4 Ga. On this basis, modern peridotites and partial melts thereof, basalts, whose Fe3+/ΣFe ratios are precisely known, are used to define that of the magma ocean. Inversion of the Fe3+/ΣFe ratios measured in MORB glasses, 0.10 ± 0.02 (31) and 0.12 ± 0.02 (51) yield Fe3+/ΣFemantle between 0.023 ± 0.006 and 0.028 ± 0.007 (39). These values are minima because MORB is produced from depleted mantle that contains only a fraction of its original (i.e., BSE-like) Fe3+ inventory. A complementary estimate is obtained via reconstruction of whole-rock ferric-ferrous ratios from measurement of iron oxidation states in minerals of a global compilation of xenolith and massif peridotites, which gives Fe3+/ΣFemantle = 0.037 ± 0.005 (fig. S8) (40, 41), a value that is adopted hereafter.
Assuming that the magma ocean had the composition of the BSE, its oxygen fugacity would have been ∆IW = +0.5 ± 0.7 at its surface at a temperature of 1900°C (Eq. 4). This is ~1.9 log units lower than predicted using Mössbauer spectroscopy data for andesitic glasses at low pressure extrapolated to peridotitic compositions and temperatures (52) using the calibration of (38) with a dependence of log(Fe3+/Fe2+) on 0.25logfO2. Because both studies derive Fe3+/Fe2+ from Mössbauer calibrations, this result underlines the importance of the effects of melt composition and temperature on fO2 for a given Fe3+/Fe2+ratio, and the inaccuracy of existing parameterizations in accounting for them (see Fig. 1).
An fO2 of ∆IW + 0.5 fixes the ratios of the fugacities of gas species that depend solely on oxygen fugacity, namely, fH2O/fH2 = 1.05 and fCO2/fCO = 0.21 at 2173 K. However, the fugacities of H-, C-, and N-bearing species also reflect their (i) abundances in the BSE (24, 53) and (ii) solubilities in the magma ocean (8). Their solubilities in molten silicate depend on the speciation of these elements in both the gas and liquid phase (54–57). At oxygen fugacities of ∆IW + 0.5, H, C, and N dissolve as O-bearing; CO32−, H2O, and OH (55, 58) or molecular; N2 (56) components in mafic silicate melts. Nevertheless, their solubilities are known only over a limited temperature (≤1400°C) and/or compositional range (59, 60), largely restricted to basaltic melts at high pressures (>0.2 GPa). As such, the identity and solubility of the major volatile species for magma ocean compositions, temperatures, and pressures remain poorly constrained.
Given estimates of H, C, and N contents in the BSE (table S7) (24, 53), existing solubility laws predict that ~99% of the H2O (54) and ~35% of the CO2 (55) are initially dissolved in the magma ocean at 2173 K, while N2 (56) remains almost entirely in the atmosphere (see Supplementary Materials). These solubilities confer molar ratios of between 0.07 and 0.23 (H/C) and 2.25 to 6.79 (H/N) in the atmosphere complementary to the magma ocean, depending on the model compositions used for the BSE (24, 53).
At a logfO2 fixed by the magma ocean of ΔIW + 0.5 and at 2173 K, the speciation of the model atmosphere with H/C = 0.22 and H/N = 5.8 is calculated by Gibbs free energy minimization (61) and is found to comprise ~100 bar of CO and ~16 bar of CO2, with minor quantities of H2 and H2O (~7 bar each) and N2 (2.5 bar), yielding a Ptotal of 140 bar (Fig. 3A). The value of ΔIW is insensitive to the initial temperature (2173 K) because the temperature dependence of fO2 defined by most mineral buffers (such as IW) parallels that fixed by Fe3+/Fe2+ in silicate melts (40). Moreover, upon closed-system cooling, the partial pressures of the major volatile species vary little over the peridotite liquidus-solidus interval (down to ~1600 K). Below 1000 K, graphite precipitates, as suggested by (62), because the reaction H2(g) + 3CO(g) ↔ CO2(g) + H2O(g) + 2C(s) proceeds strongly to the right, leading to an increase in fO2 in the atmosphere relative to the IW buffer and the formation of a “neutral” CO2-N2-H2O atmosphere between 400 and 600 K. Condensation of H2O engenders further increases in ΔIW and leads to the generation of a CO2-N2 atmosphere at low temperatures.
Fig. 3. Speciation during closed-system cooling of a magma ocean–generated atmosphere.
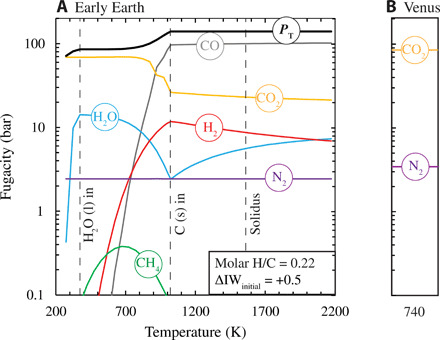
(A) A model atmosphere on the early Earth initially in equilibrium with a magma ocean of BSE composition with H = 0.01 weight % (wt %); C = 0.01 wt %, N = 0.0002 wt % (24, 53) before cooling to 300 K. Molar abundances of H, C, and N in the atmosphere are calculated with existing solubility laws (54–56) yielding H/C = 0.22 and H/N = 5.8, while O is fixed to give an fO2 of ΔIW + 0.5 at 2173 K. The resultant atmosphere of 140 bar is allowed to cool in a closed system. Vertical lines denote the temperatures of condensation of H2O, graphite, and the 1-bar peridotite solidus. Speciation of the real gas is calculated at 50 K intervals using FactSage 7.3 (61) and is reported as fugacity (bar). (B) The present-day partial pressures of CO2 and N2 on Venus are shown for comparison at its surface temperature of 740 K.
Chemical modification of cooling atmospheres
The degree to which cooling atmospheres remain closed to chemical exchange with the underlying mantle, as modeled above, depends on the thermal and dynamic evolution of the coupled magma ocean-atmosphere system. A rheological transition from a suspension to an interlocking matrix in partially molten systems occurs when the melt fraction decreases below ~0.4 (63), causing a decrease in convective vigor and therefore heat delivery to the base of the atmosphere (46). This threshold is crossed when mantle potential temperatures decline to ~1700 K (45), with atmospheric cooling occurring over short time scales of ~103 years thereafter (45, 46). At this point, the mantle and atmosphere are no longer in thermochemical equilibrium and can be considered as discrete thermodynamic entities.
Although the solubility of H2O in silicate melts is sufficiently high that 99% of the BSE’s water budget is initially dissolved in the magma ocean, as it cools and crystallizes, H2O may be partially expelled (64, 65) because of its lower solubility in nominally anhydrous mantle minerals (66) relative to melt. To place limits on the potential influence of H degassing on the speciation of the atmosphere, Gibbs free energy minimizations are performed at 2173 K and at 300 K as a function of the atmospheric H/C ratio and ΔIW (Fig. 4). These are combined with models for H degassing from a magma ocean initially at ΔIW + 0.5 and log(H/C) = −0.66, for different H2/H2O ratios in the gas.
Fig. 4. Effects of oxygen fugacity and H/C on atmospheric speciation.
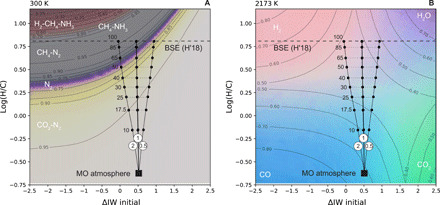
Approximately 21,000 Gibbs free energy minimizations using FactSage 7.3 (61) were performed each at 100 bar and (A) 300 K and (B) 2173 K. Mole fractions of the gas species were linearly interpolated over a grid with variables of log(H/C) and logfO2 expressed relative to the IW buffer. Colors shown are mixtures between end-members in (A) light green = CO2; purple = N2; gray = CH4; white = NH3; red = H2, where contour lines correspond to the mole fractions of CO2 below the N2 band and CH4 above; and in (B) red = H2; purple = H2O; green = CO2; blue = CO. Contour lines show the mole fractions of each species listed in each corner. Overlain on these calculations are lines showing the atmospheric log(H/C) ratio (−0.66) and ΔIW (+0.5) calculated in equilibrium with the Earth’s magma ocean (MO) at 2173 K with an Fe3+/ΣFe = 0.037. Hydrogen degassing models (black curves) are shown for three different H2/H2O ratios whose value is listed in the white circles. Numbers next to dashed horizontal lines refer to the percentage of H degassed from the BSE composition of Hirschmann (2018) (H′18) (53). The BSE is a strict upper limit because it neglects the small solubility of H in nominally anhydrous minerals and/or inefficient degassing.
At ΔIW + 0.5, H2/H2O ratios in the vapor are close to unity over the liquidus-solidus interval of peridotite (see Fig. 3), and relative oxygen fugacity varies little with respect to the IW buffer during degassing (Fig. 4). The eventual atmospheric speciation after cooling at 300 K remains in the CO2-N2 field (i.e., with a composition similar to that modeled in Fig. 3) until up to ~80% of the H inventory of the BSE has degassed (Fig. 4A). This figure decreases to ~30% should the initial oxygen fugacity of the magma ocean have been set by core formation (≈ΔIW − 2) as would be expected for smaller planetary bodies or planetesimal precursors to Earth. An fO2 > ΔIW + 1 precludes the formation of a reduced atmosphere for bulk log(H/C) ratios equal to or less than that of the BSE. Recent numerical models of magma ocean crystallization indicate only 23 to 81% of the H2O budget is released (65), suggesting that a CO2-N2 atmosphere is likely to have resulted on early Earth.
The extensive stability field of CO2-N2 atmospheres predicted thermodynamically at an initial fO2 of ΔIW + 0.5 is enhanced when photochemical reactions and the kinetics of homogeneous gas reactions occurring in planetary atmospheres are considered. Although methane occurs as a minor gas at low H/C (XCH4 ~ 10−3; Fig. 3A), it is expected to predominate for log(H/C) > 0.7 (Fig. 4A). Methane is chiefly formed by hydrogenation; CO + 3H2 = CH4 + H2O, which proceeds to the right down-temperature such that graphite saturation is no longer reached in high H/C atmospheres (Supplementary Materials). However, this reaction is kinetically inhibited below ~900 to 1000 K (14, 67), meaning the high temperature state of the atmosphere with low CH4 mole fractions becomes “frozen-in.” Moreover, CH4 is readily photolysed (68) and is also susceptible to decomposition by oxidative radicals produced from photolysis of CO2 and H2O. Ammonia, a minor gas only at very high log(H/C) (>1; Fig. 4A) has even higher quench temperatures owing to the sluggish kinetics of N2 + 3H2 = 2NH3 (67) and is even more susceptible to photolytic destruction than is CH4. By contrast, CO2 and particularly CO triple bonds are more resistant to photolysis (69), resulting in increased stability of these species relative to that predicted from Fig. 4, at the expense of CH4 and NH3.
Should the Earth’s secondary atmosphere have been modified by vapor-producing impacts post-magma ocean crystallization, particularly associated with the late veneer, more reducing NH3-CH4 atmospheres may have developed (17, 19). Nevertheless, the lifetimes of these reducing atmospheres are fleeting (several million years) in the absence of protection from photolysis by organic hazes (17) and revert to CO- or CO2-N2 atmospheres. Moreover, the prevalence of NH3 and CH4 is sensitive not only to the nature of the pre-existing atmosphere but also to the redox state of the incoming material (19) whose identity in a late veneer remains uncertain (70). Contrastingly, atmospheres derived from a partially crystallized magma ocean, because of the incompatibility of Fe3+ relative to Fe2+ in major silicates (71), are likely to yield more oxidizing atmospheres than shown in Fig. 4, as predicted on the basis of models for the vaporization of the continental crust (14, 69).
Atmospheric evolution of the terrestrial planets
We have demonstrated that the earliest prebiotic atmosphere on Earth was likely saturated in graphite and water and dominated by CO2 and N2 gases in proportions and at pressures akin to those presently observed on Venus. The same CO2-N2 ratio, albeit at much lower total pressures (~0.0061 bar), also occurs on Mars, meaning the Earth is bracketed heliocentrically by planets with 97:3 CO2-N2 atmospheres. This observation therefore points to a distinct evolutionary path of the terrestrial atmosphere compared with those of Venus and Mars following magma ocean crystallization.
The likelihood that the three planets had similar bulk initial volatile contents (72, 73), together with their inferred common early CO2-N2 atmospheres permit first-order simplifications to be made in assessing their respective atmospheric evolution. The two principal differences lie in (i) their masses and (ii) their heliocentric distances (and hence equilibrium radiating temperatures). Both factors are important in quantifying the tendency for atmospheric loss to occur, and can be expressed in the escape parameter, λ, describing the competition between thermal motion of the gas relative to the gravitational pull of the body
| (5) |
Where mi is the mean molecular mass of the atmosphere, vesc is the escape velocity and is given by , kB is Boltzmann’s constant, and Tesc is the temperature at the escaping surface. Here, lower values of λesc favor escape. Because the atmospheric composition is taken to be identical for all three bodies, mi cancels, while vesc = 11.18 km/s for Earth, 10.37 km/s for Venus, and 5.02 km/s for Mars. The remaining variable, the effective radiating temperature, Tesc, is proportional to the amount of incoming solar radiation, which is itself proportional to 1/d2, where d = heliocentric distance. The resulting escape parameter ratios are = 2.37 and = 2.10 at any temperature, indicating that the ease of atmospheric escapes is in the order Venus > Mars > > Earth.
This first-order hierarchy is borne out in the isotopic evidence for extreme hydrogen loss from the Venusian and, to a lesser extent, Martian atmospheres relative to the terrestrial atmosphere. The Venusian and Martian atmospheres are enriched by a factor of 150 and 6, respectively, in D/H relative to that of Earth (1), suggesting water on Venus was almost entirely lost early in its evolution (74). Geological observations on Mars imply that any surface water either also escaped or was sequestered into ice caps before 3.8 Ga (75).
The prolonged presence of surface water on Earth is key to the subsequent development of its atmosphere, because the combined action of dissolution of CO2 into seawater through the Urey Reaction (16, 76) and its burial in the crust and mantle via a global crustal recycling process such as plate tectonics (77, 78) may have instigated its drawdown from Earth’s early atmosphere. That neither of these ingredients existed on Venus or Mars for any prolonged period (79, 80) allowed them to retain much of their dry, inhospitable CO2-N2 atmospheres. Earth’s mass and heliocentric distance favored the preservation of liquid H2O, thereby enabling the extraction CO2 and providing an environment germane to the development of life (81).
MATERIALS AND METHODS
Experimental
A peridotite composition, modeled after that of KLB-1 (34), was synthesized by mixing and grinding a combination of oxide powders (>99.95% pure), MgO, SiO2, Fe2O3, Al2O3, and CaCO3 three times under ethanol in an agate mortar. The mixture was then decarbonated for 24 hours in air at ~1000°C, and the resultant powder was pressed into pellets. Roughly 20-mg pieces of the pellet were placed into the conical nozzle of the furnace, from which was fluxed a gas-mixture providing sufficient buoyancy to cause the molten silicate bead to levitate (82). Heating was achieved by a continuous wave 125-W CO2 laser focused by mirrors to a spot diameter of ~5 mm. All samples were melted once at 1900° ± 50°C for 30 s in pure O2 to achieve complete fusion of the synthetic KLB-1 peridotite and quenched to glass. Subsequently, the glass beads, with diameters between 1.45 and 2.05 mm, were equilibrated under different gas mixtures (from pure CO2 to CO2:H2 10:90, diluted in each case with a 92% Ar carrier gas metered by Bronkhorst mass flow controllers with a precision of ±0.1 standard cubic centimeter per minute) to impose the desired oxygen fugacity to a precision of ±0.05 log units, which was calculated by Gibbs free energy minimization. In all cases, temperatures were kept constant to 1900° ± 50°C, as continuously recorded by an optical pyrometer. These temperatures were held for ~30 s before quenching, except in the case of two time-series experiments of 10 and 120 s. Quenching below the glass transition was achieved by switching off the laser, resulting in cooling rates of ~800°C per second.
Electron-probe microanalysis
Electron probe microanalysis of the glasses was performed using a JEOL JXA-8530F Plus at the Australian National University, operating in wavelength-dispersive mode at 20 kV and 20 nA. The calibration standards used were SiO2 (Si), albite (Na, Al), hematite (Fe), MgO (Mg), diopside (Ca), and sanidine (K). Ten or eleven analyses were obtained per sample in a core-to-rim traverse. Each bead was internally homogeneous. The two most reduced samples had lost ~10% of their Fe by volatility, with a concomitant decrease in Si/Mg by ~4% relative. The average composition for the peridotite glass, by weight, was 46.53(26) % SiO2, 4.37(4) % Al2O3, 8.44(29) % FeO (all Fe as FeO), 38.05(42) % MgO, and 2.06(5) % CaO, with Na2O and K2O both <0.01%. See “Compositions of experimental glasses” section in the Supplementary Materials for a full set of analyses of each glass.
X-ray absorption near-edge structure
Fe K-edge XANES spectra were collected at beamline 13IDE of the Advanced Photon Source, Argonne National Laboratories (IL, USA). The x-ray energy was selected using a Si(311) double-crystal monochromator and calibrated to place the first peak in the derivative of a transmission spectrum of Fe foil at 7110.7 eV. A focused beam of 10-μm diameter was used for all analyses. Fluorescence spectra of the peridotite glasses and basaltic glass standards (31) were obtained using a four-element Vortex silicon drift detector. Data were collected in ~2-eV steps from 7061 to 7107 eV, ~0.1-eV steps from 7107.2 to 7137.2 eV, and ~2-eV steps from 7139 to 7309 eV with a dwell time of 1 s per energy. Beam damage was evaluated by holding the monochromator at 7112.7 eV (Fe2+ peak) and 7114.2 eV (Fe3+ peak) and monitoring absorption intensity as a function of time. The time-resolved data indicated no change in relative peak intensity over the 9-min analysis duration. XANES spectra were normalized to the average intensity above 7270 eV. The background beneath the pre-edge peaks was fitted up to a normalized intensity of 0.3 (excluding the range 7108.3 to 7115.5 eV) as the sum of one Gaussian and one Lorentzian peak, the former centered at ~7520 eV and the latter at ~7130 eV. This background was subtracted from the spectra. The centroid of the extracted peaks was calculated by integrating from 7107.4 to 7116.4 eV using the trapezium rule. The relationship between the centroid energy and Fe3+/ΣFe, together with Fe3+/ΣFe and 0.8 × the edge energy for the standards (31), was used in tandem to determine Fe3+/ΣFe for the peridotite glasses (see “Treatment and quantification of XANES spectra” section in the Supplementary Materials for details).
Thermodynamic models
All equilibrium thermodynamic calculations of the composition of the H-C-N-O atmosphere were performed using the Gibbs free energy minimization Equilib module of the FactSage Thermochemical Software and Database (61). Input and outputs of these calculations are reproduced in full in the “Thermodynamic models of atmospheric composition” subsection of the Supplementary Materials.
Supplementary Material
Acknowledgments
We appreciate the editorial handling of C.T. Lee, and acknowledge constructive comments from four anonymous reviewers, particularly as regards the evolution of mantle fO2 with time, the boundary conditions of the models, and the effects of H/C on atmospheric composition. P.A.S. thanks D. J. Bower for helpful discussions. Funding: P.A.S. acknowledges support from SNSF Ambizione Fellowship grant no. 180025 and H.S.O. on IPGP visiting professorship in September, 2017. A.D.B. and H.S.O. acknowledge support from ARC Laureate Fellowship no. FL130100066. Parts of this work were supported by the UnivEarthS Labex program (ANR-10-LABX-0023 and ANR-11-IDEX-0005-02) and IPGP multidisciplinary program PARI (Paris–IdF region SESAME Grant no. 12015908). The authors acknowledge the facilities and the scientific and technical assistance of Microscopy Australia at the Centre for Advanced Microscopy, Australian National University, a facility that is funded by the University and the Federal Government. Portions of this work were performed at GeoSoilEnviroCARS (The University of Chicago, Sector 13), Advanced Photon Source (APS), Argonne National Laboratory. GeoSoilEnviroCARS is supported by the National Science Foundation–Earth Sciences (EAR: 1634415) and Department of Energy- GeoSciences (DE-FG02-94ER14466). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. Author contributions: P.A.S. conceived the study, fit XANES spectra, performed the laser levitation experiments, and thermodynamic calculations. A.D.B. performed the microprobe analyses and collected and fit XANES spectra. J.B. conceived the study and performed the laser levitation experiments. A.L. and M.N. assisted with XANES spectra collection on the beamline. H.S.O. conceived the study, performed the laser levitation experiments, and fit XANES spectra. P.A.S. wrote the paper with contributions from H.S.O., A.D.B., and J.B. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data are available in the main text of in the Supplementary Materials. Additional information related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eabd1387/DC1
REFERENCES AND NOTES
- 1.Hunten D. M., Atmospheric evolution of the terrestrial planets. Science 259, 915–920 (1993). [Google Scholar]
- 2.Lammer H., Zerkle A. L., Gebauer S., Tosi N., Noack L., Scherf M., Pilat-Lohinger E., Güdel M., Grenfell J. L., Godolt M., Nikolaou A., Origin and evolution of the atmospheres of early Venus, Earth and Mars. Astron. Astrophys. Rev. 26, 2 (2018). [Google Scholar]
- 3.Morbidelli A., Lunine J. I., O’Brien D. P., Raymond S. N., Walsh K. J., Building terrestrial planets. Annu. Rev. Earth Planet. Sci. 40, 251–275 (2012). [Google Scholar]
- 4.Marty B., The origins and concentrations of water, carbon, nitrogen and noble gases on Earth. Earth Planet. Sci. Lett. 313–314, 56–66 (2012). [Google Scholar]
- 5.Albarède F., Volatile accretion history of the terrestrial planets and dynamic implications. Nature 461, 1227–1233 (2009). [DOI] [PubMed] [Google Scholar]
- 6.H. J. Melosh, Giant impacts and the thermal state of the early Earth, in Origin of the Earth, H. E. Newsom, J. H. Jones, Eds. (Oxford Univ., New York, 1990), pp. 69–83. [Google Scholar]
- 7.Genda H., Abe Y., Modification of a proto-lunar disk by hydrodynamic escape of silicate vapor. Earth Planets Sp. 53, 53–57 (2003). [Google Scholar]
- 8.Hirschmann M. M., Magma ocean influence on early atmosphere mass and composition. Earth Planet. Sci. Lett. 341–344, 48–57 (2012). [Google Scholar]
- 9.Zahnle K., Schaefer L., Fegley B., Earth’s earliest atmospheres. Cold Spring Harb. Perspect. Biol. 2, a004895 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aston F. W., The rarity of the inert gases on the Earth. Nature 114, 786 (1924). [Google Scholar]
- 11.H. Brown, Rare gases and the formation of Earth’s atmosphere, in The Atmosphere of the Earth and Planets, G. Kuiper, Ed. (University of Chicago Press, Chicago, 1949), pp. 258–266. [Google Scholar]
- 12.Poole J. H. J., The evolution of the earth’s atmosphere. Sci. Proc. R. Dublin Soc. 25, 201–224 (1951). [Google Scholar]
- 13.H. D. Holland, Model for the evolution of the Earth’s atmosphere, in Petrologic Studies: A Volume in Honor of A.F. Buddington, E. J. Engle, H. L. James, B. F. Leonard, Eds. (Geological Society of America, 1962), pp. 447–477. [Google Scholar]
- 14.Lupu R. E., Zahnle K., Marley M. S., Schaefer L., Fegley B., Morley C., Cahoy K., Freedman R., Fortney J. J., The atmospheres of Earth-like planets after giant impact events. Astrophys. J. 784, 1–19 (2014). [Google Scholar]
- 15.Tian F., Toon O. B., Pavlov A. A., De Sterck H., A hydrogen-rich early Earth atmosphere. Science 308, 1014–1017 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Urey H. C., On the early chemical history of the Earth and the origin of life. Proc. Natl. Acad. Sci. U.S.A. 38, 351–363 (1952). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zahnle K. J., Lupu R., Catling D. C., Wogan N., Creation and evolution of impact-generated reduced atmospheres of early Earth. Planet. Sci. J. 1, 1–21 (2020). [Google Scholar]
- 18.Miller S. L., A production of amino acids under possible primitive earth conditions. Science 117, 528–529 (1953). [DOI] [PubMed] [Google Scholar]
- 19.Schaefer L., Fegley B. Jr., Redox states of initial atmospheres outgassed on rocky planets and planetesimals. Astrophys. J. 843, 120 (2017). [Google Scholar]
- 20.Sharp Z. D., Nebular ingassing as a source of volatiles to the terrestrial planets. Chem. Geol. 448, 137–150 (2017). [Google Scholar]
- 21.Kasting J. F., Earth’s early atmosphere. Science 259, 920–926 (1993). [DOI] [PubMed] [Google Scholar]
- 22.Stagno V., Carbon, carbides, carbonates and carbonatitic melts in the Earth’s interior. J. Geol. Soc. Lond. 176, 375–387 (2019). [Google Scholar]
- 23.Frost D. J., Mann U., Asahara Y., Rubie D. C., The redox state of the mantle during and just after core formation. Philos. Trans. A. Math. Phys. Eng. Sci. 366, 4315–4337 (2008). [DOI] [PubMed] [Google Scholar]
- 24.H. Palme, H. St. C. O’Neill, Cosmochemical estimates of mantle composition, in Treatise on Geochemistry, Vol. 3: The Mantle and Core, R. W. Carlson, Ed. (Elsevier B.V., ed. 2, 2014), pp. 1–39. [Google Scholar]
- 25.Woodland A. B., Kornprobst J., Wood B. J., Oxygen thermobarometry of orogenic lherzolite massifs. J. Petrol. 33, 203–230 (1992). [Google Scholar]
- 26.Woodland A. B., Koch M., Variation in oxygen fugacity with depth in the upper mantle beneath the Kaapvaal craton, Southern Africa. Earth Planet. Sci. Lett. 214, 295–310 (2003). [Google Scholar]
- 27.Stagno V., Ojwang D. O., McCammon C. A., Frost D. J., The oxidation state of the mantle and the extraction of carbon from Earth’s interior. Nature 493, 84–88 (2013). [DOI] [PubMed] [Google Scholar]
- 28.H. St. C. O’Neill, D. C. Rubie, D. Canil, C. A. Geiger, C. R. Ross II, F. Seifert, A. B. Woodland, Ferric iron in the upper mantle and in transition zone assemblages: Implications for relative oxygen fugacities in the mantle, in Evolution of the Earth and Planets, E. Takahashi, R. Jeanloz, D. C. Rubie, Eds. (American Geophysical Union, 1993), pp. 73–88. [Google Scholar]
- 29.Kress V. C., Carmichael I. S. E., The compressibility of silicate liquids containing Fe2O3 and the effect of composition, temperature, oxygen fugacity and pressure on their redox states. Contrib. Mineral. Petrol. 108, 82–92 (1991). [Google Scholar]
- 30.Cottrell E., Kelley K. A., The oxidation state of Fe in MORB glasses and the oxygen fugacity of the upper mantle. Earth Planet. Sci. Lett. 305, 270–282 (2011). [Google Scholar]
- 31.Berry A. J., Stewart G. A., O’Neill H. S. C., Mallmann G., Mosselmans J. F. W., A re-assessment of the oxidation state of iron in MORB glasses. Earth Planet. Sci. Lett. 483, 114–123 (2018). [Google Scholar]
- 32.Wänke H., Dreibus G., Chemical composition and accretion history of terrestrial planets. Philos. Trans. R. Soc. Lond. 325, 545–557 (1988). [Google Scholar]
- 33.Ringwood A. E., Chemical evolution of the terrestrial planets. Geochim. Cosmochim. Acta 30, 41–104 (1966). [Google Scholar]
- 34.Takahashi E., Melting of a dry peridotite KLB-1 up to 14 GPa: Implications on the origin of peridotitic upper mantle. J. Geophys. Res. 91, 9367–9382 (1986). [Google Scholar]
- 35.Walter M. J., Melting of garnet peridotite and the origin of komatiite and depleted lithosphere. J. Petrol. 39, 29–60 (1998). [Google Scholar]
- 36.Nikolaev G. S., Borisov A. A., Ariskin A. A., Calculation of the ferric–ferrous ratio in magmatic melts: Testing and additional calibration of empirical equations for various magmatic series. Geochem. Int. 34, 641–649 (1996). [Google Scholar]
- 37.Borisov A., Behrens H., Holtz F., Ferric/ferrous ratio in silicate melts: A new model for 1 atm data with special emphasis on the effects of melt composition. Contrib. Mineral. Petrol. 173, 98 (2018). [Google Scholar]
- 38.Jayasuriya K. D., O’Neill H. S. C., Berry A. J., Campbell S. J., A Mössbauer study of the oxidation state of Fe in silicate melts. Am. Mineral. 89, 1597–1609 (2004). [Google Scholar]
- 39.O’Neill H. S. C., Berry A. J., Mallmann G., The oxidation state of iron in mid-ocean ridge basaltic (MORB) glasses: Implications for their petrogenesis and oxygen fugacities. Earth Planet. Sci. Lett. 504, 152–162 (2018). [Google Scholar]
- 40.Canil D., O’Neill H. S. C., Pearson D. G., Rudnick R. L., McDonough W. F., Carswell D. A., Ferric iron in peridotites and mantle oxidation states. Earth Planet. Sci. Lett. 123, 205–220 (1994). [Google Scholar]
- 41.Woodland A. B., Kornprobst J., Tabit A., Ferric iron in orogenic lherzolite massifs and controls of oxygen fugacity in the upper mantle. Lithos 89, 222–241 (2006). [Google Scholar]
- 42.Sossi P. A., Klemme S., O’Neill H. S. C., Berndt J., Moynier F., Evaporation of moderately volatile elements from silicate melts: Experiments and theory. Geochim. Cosmochim. Acta 260, 204–231 (2019). [Google Scholar]
- 43.M. W. Chase, NIST-JANAF Thermochemical Tables, 4th Ed., in Monograph 9 (Part I and Part II) (Journal of Physical and Chememical Reference Data Monographs, ed. 4, 1998). [Google Scholar]
- 44.V. Solomatov, Magma oceans and primordial mantle differentiation, in Treatise of Geophysics Second Edition, G. Schubert, Ed. (Elsevier B.V., ed. 2, 2015), pp. 81–104. [Google Scholar]
- 45.Nikolaou A., Katyal N., Tosi N., Godolt M., Greenfell J. L., Rauer H., What factors affect the duration and outgassing of the terrestrial magma ocean? Astrophys. J. 875, 11 (2019). [Google Scholar]
- 46.Bower D. J., Kitzmann D., Wolf A. S., Sanan P., Dorn C., Oza A. V., Linking the evolution of terrestrial interiors and an early outgassed atmosphere to astrophysical observations. Astron. Astrophys. 631, A103 (2019). [Google Scholar]
- 47.Aulbach S., Stagno V., Evidence for a reducing Archean ambient mantle and its effects on the carbon cycle. Geology 44, 751–754 (2016). [Google Scholar]
- 48.Nicklas R. W., Puchtel I. S., Ash R. D., Piccoli P. M., Hanski E., Nisbet E., Waterton P., Pearson D. G., Anbar A. D., Secular mantle oxidation across the Archean-Proterozoic boundary: Evidence from V partitioning in komatiites and picrites. Geochim. Cosmochim. Acta 250, 49–75 (2019). [Google Scholar]
- 49.Delano J. W., Redox history of the Earth’s interior since ~3900 Ma: Implications for prebiotic molecules. Orig. Life Evol. Biosph. 31, 311–341 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Trail D., Watson E. B., Tailby N. D., The oxidation state of Hadean magmas and implications for early Earth’s atmosphere. Nature 480, 79–82 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Bézos A., Humler E., The Fe3+/ΣFe ratios of MORB glasses and their implications for mantle melting. Geochim. Cosmochim. Acta 69, 711–725 (2005). [Google Scholar]
- 52.Armstrong K., Frost D. J., McCammon C. A., Rubie D. C., Boffa Ballaran T., Deep magma ocean formation set the oxidation state of Earth’s mantle. Science 365, 903–906 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Hirschmann M. M., Comparative deep Earth volatile cycles: The case for C recycling from exosphere/mantle fractionation of major (H2O, C, N) volatiles and from H2O/Ce, CO2/Ba, and CO2/Nb exosphere ratios. Earth Planet. Sci. Lett. 502, 262–273 (2018). [Google Scholar]
- 54.Moore G., Vennemann T., Carmichael I. S. E., An empirical model for the solubility of H2O in magmas to 3 kilobars. Am. Mineral. 83, 36–42 (1998). [Google Scholar]
- 55.Dixon J. E., Stolper E. M., Holloway J. R., An experimental study of water and carbon dioxide solubilities in mid-ocean ridge basaltic liquids. Part I: Calibrations and solubility models. J. Petrol. 36, 1607–1631 (1995). [Google Scholar]
- 56.Libourel G., Marty B., Humbert F., Nitrogen solubility in basaltic melt. Part I. Effect of oxygen fugacity. Geochim. Cosmochim. Acta 67, 4123–4135 (2003). [Google Scholar]
- 57.Wetzel D. T., Rutherford M. J., Jacobsen S. D., Hauri E. H., Saal A. E., Degassing of reduced carbon from planetary basalts. Proc. Natl. Acad. Sci. U.S.A. 110, 8010–8013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stolper E., Water in silicate glasses: An infrared spectroscopic study. Contrib. Mineral. Petrol. 81, 1–17 (1982). [Google Scholar]
- 59.Grewal D. S., Dasgupta R., Sun C., Tsuno K., Costin G., Delivery of carbon, nitrogen, and sulfur to the silicate Earth by a giant impact. Sci. Adv. 5, eaau3669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshioka T., Nakashima D., Nakamura T., Shcheka S., Keppler H., Carbon solubility in silicate melts in equilibrium with a CO-CO2 gas phase and graphite. Geochim. Cosmochim. Acta 259, 129–143 (2019). [Google Scholar]
- 61.Bale C. W., Bélisle E., Chartrand P., Decterov S. A., Eriksson G., Gheribi A. E., Hack K., Jung I.-H., Kang Y.-B., Melançon J., Pelton A. D., Petersen S., Robelin C., Sangster J., Spencer P., Van Ende M.-A., FactSage thermochemical software and databases, 2010-2016. Calphad 55, 1–19 (2016). [Google Scholar]
- 62.Keppler H., Golabek G., Graphite floatation on a magma ocean and the fate of carbon during core formation. Geochem. Perspect. Lett. 11, 12–17 (2019). [Google Scholar]
- 63.Toramaru A., Fujii N., Connectivity of melt phase in a partially molten peridotite. J. Geophys. Res. 91, 9239–9252 (1986). [Google Scholar]
- 64.Elkins-Tanton L. T., Linked magma ocean solidification and atmospheric growth for Earth and Mars. Earth Planet. Sci. Lett. 271, 181–191 (2008). [Google Scholar]
- 65.Hier-Majumder S., Hirschmann M. M., The origin of volatiles in the Earth’s mantle. Geochem. Geophys. Geosyst. 18, 3078–3092 (2017). [Google Scholar]
- 66.Bai Q., Kohlstedt D. L., Substantial hydrogen solubility in olivine and implications for water storage in the mantle. Nature 357, 672–674 (1992). [Google Scholar]
- 67.Zahnle K. J., Marley M. S., Methane, carbon monoxide, and ammonia in brown dwarfs and self-luminous giant planets. Astrophys. J. 797, 41 (2014). [Google Scholar]
- 68.Lasaga A. C., Holland H. D., Dwyer M. J., Primordial oil slick. Science 174, 53–55 (1971). [DOI] [PubMed] [Google Scholar]
- 69.Schaefer L., Lodders K., Fegley B., Vaporization of the earth: Application to exoplanet atmospheres. Astrophys. J. 755, 41 (2012). [Google Scholar]
- 70.Fischer-Gödde M., Elfers B.-M., Münker C., Szilas K., Maier W. D., Messling N., Morishita T., van Kranendonk M., Smithies H., Ruthenium isotope vestige of Earth’s pre-late-veneer mantle preserved in Archaean rocks. Nature 579, 240–244 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mallmann G., O’Neill H. S. C., The crystal/melt partitioning of V during mantle melting as a function of oxygen fugacity compared with some other elements (Al, P, Ca, Sc, Ti, Cr, Fe, Ga, Y, Zr and Nb). J. Petrol. 50, 1765–1794 (2009). [Google Scholar]
- 72.Dreibus G., Wanke H., Volatiles on Earth and Mars: A comparison. Icarus 71, 225–240 (1987). [Google Scholar]
- 73.O’Brien D. P., Izidoro A., Jacobson S. A., Raymond S. N., Rubie D. C., The delivery of water during terrestrial planet formation. Space Sci. Rev. 214, 47 (2018). [Google Scholar]
- 74.Kasting J. F., Runaway and moist greenhouse atmospheres and the evolution of Earth and Venus. Icarus 74, 472–494 (1988). [DOI] [PubMed] [Google Scholar]
- 75.Carr M. H., Mars: A water-rich planet? Icarus 68, 187–216 (1986). [Google Scholar]
- 76.Kellogg L. H., Lokavarapu H., Turcotte D. L., Carbonation and the Urey reaction. Am. Mineral. 104, 1365–1368 (2019). [Google Scholar]
- 77.Liu L.-g., The inception of the oceans and CO2-atmosphere in the early history of the Earth. Earth and Planet. Sci. Lett. 227, 179–184 (2004). [Google Scholar]
- 78.Sleep N. H., Zahnle K., Neuhoff P. S., Initiation of clement surface conditions on the earliest Earth. Proc. Natl. Acad. Sci. U.S.A. 98, 3666–3672 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nimmo F., McKenzie D., Volcanism and tectonics on Venus. Annu. Rev. Earth Planet. Sci. 26, 23–51 (1998). [Google Scholar]
- 80.Van Thienen P., Vlaar N. J., Van Den Berg A. P., Plate tectonics on the terrestrial planets. Phys. Earth Planet. Inter. 142, 61–74 (2004). [Google Scholar]
- 81.Cleaves H. J., Chalmers J. H., Lazcano A., Miller S. L., Bada J. L., A reassessment of prebiotic organic synthesis in neutral planetary atmospheres. Orig. Life Evol. Biosph. 38, 105–115 (2008). [DOI] [PubMed] [Google Scholar]
- 82.Auzende A.-L., Gillot J., Coquet A., Hennet L., Ona-Nguema G., Bonnin D., Esteve I., Roskosz M., Fiquet G., Synthesis of amorphous MgO-rich peridotitic starting material for laser-heated diamond anvil cell experiments – application to iron partitioning in the mantle. High Press. Res. 31, 199–213 (2011). [Google Scholar]
- 83.Jackson W. E., Farges F., Yeager M., Mabrouk P. A., Rossano S., Waychunas G. A., Solomon E. I., Brown G. E. Jr., Multi-spectroscopic study of Fe(II) in silicate glasses: Implications for the coordination environment of Fe(II) in silicate melts. Geochim. Cosmochim. Acta 69, 4315–4332 (2005). [Google Scholar]
- 84.Bajt S., Sutton S. R., Delaney J. S., X-ray microprobe analysis of iron oxidation states in silicates and oxides using x-ray absorption near edge structure (XANES). Geochim. Cosmochim. Acta 58, 5209–5214 (1994). [Google Scholar]
- 85.Mungall J. E., Empirical models relating viscosity and tracer diffusion in magmatic silicate melts. Geochim. Cosmochim. Acta 66, 125–143 (2002). [Google Scholar]
- 86.Zhang Y., Ni H., Chen Y., Diffusion data in silicate melts. Rev. Mineral. Geochem. 72, 311–408 (2010). [Google Scholar]
- 87.O’Neill H. S. C., Eggins S. M., The effect of melt composition on trace element partitioning: An experimental investigation of the activity coefficients of FeO, NiO, CoO, MoO2 and MoO3 in silicate melts. Chem. Geol. 186, 151–181 (2002). [Google Scholar]
- 88.V. P. Glushko, V. A. Medvedev, L. V. Gurvich, V. S. Yungman, Thermal Constants of Substances (John Wiley & Sons, ed. 1, 1999). [Google Scholar]
- 89.McDonough W. F., Sun S.-s., The composition of the Earth. Chem. Geol. 120, 223–253 (1995). [Google Scholar]
- 90.Canil D., O’Neill H. S. C., Distribution of ferric iron in some upper-mantle assemblages. J. Petrol. 37, 609–635 (1996). [Google Scholar]
- 91.Boyd F. R., Pearson D. G., Nixon P. H., Mertzman S. A., Low-calcium garnet harzburgites from southern Africa: Their relations to craton structure and diamond crystallization. Contrib. Mineral. Petrol. 113, 352–366 (1993). [Google Scholar]
- 92.Frey F. A., Green D. H., The mineralogy, geochemistry and origin of Iherzolite inclusions in Victorian basanites. Geochem. Cosmochim. Acta 38, 1023–1059 (1974). [Google Scholar]
- 93.Wasylenki L. E., Baker M. B., Kent A. J. R., Stolper E. M., Near-solidus melting of the shallow upper mantle: Partial melting experiments on depleted peridotite. J. Petrol. 44, 1163–1191 (2003). [Google Scholar]
- 94.M. J. Walter, Melt extraction and compositional variability in mantle lithosphere, in Treatise on Geochemistry, R. W. Carlson, Ed. (Elsevier, 2003), vol. 2, pp. 363–394. [Google Scholar]
- 95.Workman R. K., Hart S. R., Major and trace element composition of the depleted MORB mantle (DMM). Earth Planet. Sci. Lett. 231, 53–72 (2005). [Google Scholar]
- 96.Wang Z., Becker H., Ratios of S, Se and Te in the silicate Earth require a volatile-rich late veneer. Nature 499, 328–331 (2013). [DOI] [PubMed] [Google Scholar]
- 97.Hirschmann M. M., Withers A. C., Ardia P., Foley N. T., Solubility of molecular hydrogen in silicate melts and consequences for volatile evolution of terrestrial planets. Earth Planet. Sci. Lett. 345–348, 38–48 (2012). [Google Scholar]
- 98.Stolper E., Holloway J. R., Experimental determination of the solubility of carbon dioxide in molten basalt at low pressure. Earth Planet. Sci. Lett. 87, 397–408 (1988). [Google Scholar]
- 99.Hamilton D. L., Oxtoby S., Solubility of water in albite-melt determined by the weight-loss method. J. Geol. 94, 626–630 (1986). [Google Scholar]
- 100.Pan V., Holloway J. R., Hervig R. L., The pressure and temperature dependence of carbon dioxide solubility in tholeiitic basalt melts. Geochim. Cosmochim. Acta 55, 1587–1595 (1991). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eabd1387/DC1