

Direct observation of nanoparticle adsorption to the water-oil interface captures early attachment and jamming.
Abstract
Electrostatic interactions between nanoparticles (NPs) and functionalized ligands lead to the formation of NP surfactants (NPSs) that assemble at the water-oil interface and form jammed structures. To understand the interfacial behavior of NPSs, it is necessary to understand the mechanism by which the NPSs attach to the interface and how this attachment depends on the areal coverage of the interface. Through direct observation with high spatial and temporal resolution, using laser scanning confocal microscopy and in situ atomic force microscopy (AFM), we observe that early-stage attachment of NPs to the interface is diffusion limited and with increasing areal density of the NPSs, further attachment requires cooperative displacement of the previously assembled NPSs both laterally and vertically. The unprecedented detail provided by in situ AFM reveals the complex mechanism of attachment and the deeply nonequilibrium nature of the assembly.
INTRODUCTION
The assembly of solids at the liquid interface has been the subject of a substantial body of fundamental and applied research for decades. Ore purification (1), emulsions (2), and encapsulation (3–5) are all based on interfacial segregation, and as argued by Pieranski and others (6–11), it depends on the area displaced by the particle at the interface and the interfacial energies of the solid with the liquid phase(s) and between the liquids. As the particle size decreases, the binding energy of the particle to the interface decreases and, at the size scale of nanoparticles (NPs), is of order of several kT. This results in adsorption and desorption of the NPs, so the assembly is dynamic (12–14). The binding energy of NPs to the interface can be markedly increased if NPs that are soluble in one liquid interact with end-functionalized ligands in the second immiscible liquid, forming what has been termed “NP surfactants” (NPSs) (15–18). Initially, the ligands, which have a surfactant character, assemble at the interface. Then, the NPs diffuse to the interface and bind tightly with the ligands forming the NPSs. NPSs then organize at the interface, where at low areal densities they form a fluid interfacial layer, and at high areal densities the film jams forming a solid-like layer (18). The very high binding energy during adsorption drives the system into this nonequilibrium state, leading to mechanisms of attachment that may be fundamentally different from nonfunctionalized NPs. Here, we report the direct visualization of the adsorption process with exquisite spatial and temporal resolution using in situ atomic force microscopy (AFM) coupled with laser scanning confocal microscopy (LSCM). The remarkable detail of these studies reveals the attachment mechanism and provides insights into jamming phenomena.
RESULTS AND DISCUSSION
The interface between two immiscible liquids is characterized by an interfacial tension, γ (19). For the water–silicone oil interface of interest in this study, γ~40 mN m−1. If we disperse negatively charged NPs in the aqueous phase, γ is not affected because the NPs do not assemble at the interface owing to the inherent negative charge of the water-oil interface. Conversely, polymeric surfactants, for example, amine-terminated polydimethylsiloxane (PMDS-NH2), dissolved in the silicone oil will assemble into a monolayer at the interface to reduce γ. The magnitude of the reduction will depend on the concentration of PMDS-NH2 and the molecular weight of the PDMS chain (20, 21). However, with PMDS-NH2 dissolved in the oil and NPs in the aqueous solution, NPSs form with extremely high binding energy. The rate at which the NPs are attached to the interface will depend on the interfacial area available. Initially, with a completely unoccupied interface, the attachment will depend solely on the diffusion of the NPs to the interface, and therefore on the concentration of the NPs in the aqueous phase. The adsorption kinetics can be derived from Fick’s law, as discussed by Ward and Tordai (22). We assume that the adsorption is nearly irreversible, i.e., there is no back diffusion, which gives rise to a depletion layer adjacent to the interface. The areal density of adsorbed NPs is, therefore, given by
| (1) |
where M is the areal density of adsorbed NPs, D and C are the diffusion coefficient and the concentration of NPs in the aqueous phase, respectively, and t is time (23, 24).
Shown in Fig. 1A is a schematic of the attachment process where the carboxylic acid–functionalized NPs diffuse to the interface, interact with the PMDS-NH3+ assembled at the interface, and form the NPSs, anchoring the NPs to the interface. By labeling the 500-nm-diameter NPs with a fluorescent marker, LSCM can be used to image the adsorption process at low resolution. Individual NPs are high-contrast points in the image. Adsorption at two different times is shown in Fig. 1B, where the increase in the fluorescence signal and, therefore, the number of NPs attached to the interface is evident. The number of attached NPs as a function of time was determined as shown in Fig. 1C and fig. S1, where the solid line represents a 0.5 power law, i.e., Fickian diffusion control of the attachment. From the fit in Fig. 1C, the prefactor was found to be 1.2 × 102 s−0.5. Taking C = 7.6 × 1015 m−3 and interfacial area as 7.2 × 10−9 m2 (the field of view is shown in Fig. 1B), the diffusion coefficient of the 500-nm NPs in the aqueous phase was calculated to be 4.1 × 10−12 m2 s−1. For comparison, we also estimated the diffusion coefficient of 500-nm NPs in the aqueous phase as 8.8 × 10−13 m2 s−1 from the Stokes-Einstein equation, , where kB, T, η, and R are the Boltzmann constant, temperature, viscosity of the water, and diameter of NPs, respectively. The diffusion coefficient determined from dynamic light scattering (DLS) was 1.0 × 10−12 m2 s−1, in agreement with the Stokes-Einstein value. The higher diffusivity, as measured by adsorption to the interface, could possibly result from the surfactant molecules inverting the interfacial charge. The electrostatic attraction between the NPs and interface could bias the diffusion near the interface, effectively increasing the depletion (25). The 5 mM MES [2-(N-morpholino)ethanesulfonic acid] buffer solution has a Debye length of ~5 nm, which is smaller than but on the same order as the depletion region due to the lack of back diffusion. The difference in the diffusion coefficient may also result from a misestimate of the concentration of the NPs because they are reported by mass from the manufacturer, but the particle sizes vary from batch to batch. Nonetheless, the results support a diffusion-controlled adsorption to the interface, where the energy barrier to attachment is lower than the thermal energy of the system and the NPs, once in contact with the interface, remain at the interface.
Fig. 1. Attachment of NPs to sparsely populated water-oil interface.
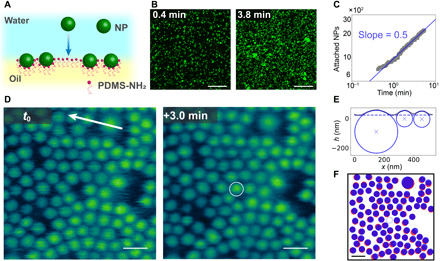
(A) Schematic diagram showing the attachment of a NP to the pristine water–oil interface. (B) Confocal microscopy images showing the attachment of 500-nm NPs to the water-oil interface. (C) Number of attached 500-nm NPs as a function of time, where the slope follows at power law of 0.5. (D) In situ AFM image of 100- and 300-nm NPs assembled at the water-oil interface at different times: t0 and t0 + 3.0 min, with a circle highlighting an attachment event. (E) Section data along arrow in (D) showing fit for NP diameter size and position in the interface. (F) Positions of the assembled NPs at different times: t0 (red) and t0 + 3.0 min (blue), where the open blue circle represents the newly attached NP. Scale bars, 20 μm (B) and 200 nm (D and F).
As more NPSs form and assemble at the water-oil interface, LSCM loses the ability to distinguish individual NPs as the separation distance between the NPs is less than the resolution of the instrument. In situ AFM, on the other hand, can directly visualize the spatial and temporal attachment of NPs to the water-oil interface. Figure 1D shows in situ AFM images of locally and almost fully packed assembly of bidisperse 100- and 300-nm NPSs. Over large length scales, Fig. 1D shows subtle variation of the out-of-plane position of the NPSs, but the change in position between nearby NPs is quite small such that the wetting of individual NPs at the interface can readily be measured. A section taken from the top of Fig. 1D across a couple 100-nm NPs and a 300-nm particle is shown in Fig. 1E. The section data are fit to a model to determine particle diameter and position relative to the interface position. The resulting diameters are 110, 110, and 290 nm, in very good agreement with DLS measurements (fig. S2), and the calculated contact angle values from the fit are 63°, 60°, and 39°, respectively. The binding energy of the NPs to the interface is a function of particle size; surface tension of the oil-water interface, which is ~23 mN m−1 for a 10% PMDS-NH2 surfactant solution (fig. S3); and the difference, ∆γ, between the NP aqueous-phase surface energy, γPW; and the NP surfactant/oil surface energy, γPO, where the larger the difference, the weaker the binding. Using the contact angle values, the difference in surface energy, ∆γ, is calculated to be 11 and 18 mJ m−2 for the 100- and 300-nm NPs, respectively. The binding energy for the two NPs is approximately the same at 5.9 × 10−17 J and 7.4 × 10−17 J for the 100- and 300-nm NPs, respectively, where the difference in surface energies is compensated for by the difference in particle size (see more details in the Supplementary Materials) (6). The binding energies are on the order of 104 kBT, which are larger than other NPS systems (6, 10, 26) but are mostly due to the relatively large NPs. The large binding energies led to no instance of the NPS system disassociation being observed.
Although both types of NPs have nominally the same carboxyl surface chemistry, a difference in the ∆γ values led to different wetting conditions. This is most likely due to different surface charge densities between the batches of NPs. Using titration to neutralize the streaming potential, surface charge densities of −3.2 × 10−6 nC μm−2 and −1.9 × 10−5 nC μm−2 were measured for the 100- and 300-nm NPs, respectively (reported by Micromod Partikeltechnologie GmbH). The increase in surface charge density will more strongly influence the attachment of the surfactant to the NPs and increase the NPS surface energy, γPO, driving the particle further into the oil phase.
In situ AFM also probes the dynamics of the NPs in the interface. The white circled particle in Fig. 1D is a 100-nm NP that attached to the interface in the 3-min time interval. Figure 1F shows the positions of NPs at t0 and t0 + 3.0 min in red and blue, respectively. From the comparison image, small displacements in random directions of all the assembled NPs indicate that the free attachment of NPs to the water-oil interface does not require a structural rearrangement of assembled NPs. When there is sufficient space to accommodate the addition of a new NP, the motion is simply due to diffusion at the interface. The NPs in the interface may also have dynamics in the normal direction, such as the previously observed slow relaxation into the interface of micrometer-sized particles (27–30). Similar relaxation is not evident in the results in Fig. 1 because most of the NPs have comparable contact angles, and the settling likely happens faster than the time resolution of the measurement. However, the dynamics slow down when the NPs are more densely packed at the interface.
When the local areal density of the NPSs assembled at the interface increases and there is insufficient free space to accommodate the entry of a new NP, the assembled NPs do require rearrangement. Time-dependent in situ AFM images of the interface with coassembled 100- and 300-nm NPSs are shown in Fig. 2B at time t0 and t0 + 5 min. At t0, there is a very dense layer of NPSs assembled on the interface (both 100 and 300 nm), and fluctuations in the areal density are evident along with regions of closest packed structure. While these fluctuations are quite interesting, they were not quantified in this study. However, there is no bare interface with sufficient area to accommodate the entry of a new 300-nm NP to the assembly. After a 5-min time period, a 300-nm NP (circled in white) does attach to the interface, accompanied by several 100-nm NPs that disappear from view (labeled by faded red circles in Fig. 2C) and two 100-nm NPs that markedly change their positions (indicated by the arrows in Fig. 2C). These results show that a cooperative structural change of the NPSs assembled at the interface is required to accommodate the attachment of additional NPs from the bulk phase at these high packing densities as has been previously modeled using Monte Carlo simulations (31). In addition to the 100-nm NPs that laterally shifted their positions, several are no longer detectable. NPSs dissociating into their constituent parts have not been observed by AFM, nor has complete transfer of the NPs into the oil phase away from the interface been measured by fluorescence. The missing NPs are likely entrapped beneath the 300-nm NP, while lack of access to the oil side of the assemblies prevents ruling out the possibility of NPs completely transferring into the oil phase.
Fig. 2. Attachment of 300-nm NPs to thoroughly populated water-oil interface.
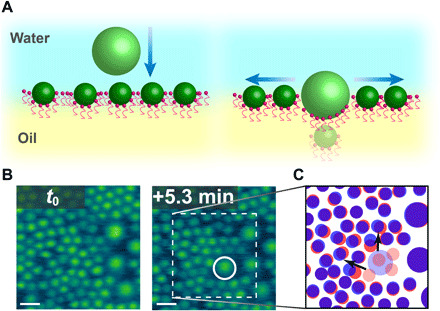
(A) Schematic diagram of the attachment of a 300-nm NP where previous NPs are displaced laterally and vertically. (B) Time-dependent in situ AFM showing the attachment of 300-nm NP to the water-oil interface, where the white circle indicates the newly adsorbed NP. (C) NPS positions at t0 (red) and t0 + 5.3 min (blue), where the faded blue dot represents the newly attached NP and the faded red dots represent NPSs that cannot be located by AFM after attachment of the newly attached NP. Scale bars, 200 nm.
In situ LSCM imaging provides insight into the process of addition of NPs to already dense assemblies. Here, the process of adding large 500-nm NPs to a dense film of small 70-nm NPs was investigated. To prepare a self-assembled layer of NPSs, a dispersion of 70-nm NPs was placed over a droplet of PMDS-NH2 in silicone oil on a glass-bottom Petri dish. After 1 hour of assembly, a dispersion of 500-nm NPs was introduced into the water phase. After a set period of time, all excess NPs were removed by flushing with pure water, and the sample was imaged immediately. Because the washing process freezes the assembly at a specific time point, new samples were required for each exposure time of the 500-nm NP dispersion in contact with the already assembled layer of 70-nm NPSs at the interface. Figure 3A shows the LSCM images of two samples with different exposure times. Only the 500-nm NPs are distinctly observable at this resolution, so the fluorescence from the 70-nm NPs is not shown. It is seen that the number of 500-nm NPs assembled at the interface increases with time. The attachment kinetics of the 500-nm NPs is shown in Fig. 3B. Compared with Fig. 1C, a substantially longer time is required to attach the same number of 500-nm NPs to the already densely assembled interface than onto a bare interface. Because the 500-nm NPs must first displace the 70-nm NPs before or during attachment, the attachment can be viewed as a reaction-controlled process (32). In movies S1 and S2, it is evident that the 500-nm NPs (green) diffuse to the assembled interfacial layer, but many do not attach, indicating that attachment requires multiple attachment attempts, further supporting the conclusion that this is a reaction-controlled process, i.e., attachment with an energy barrier. When NPs approach the interface, there is initially a charge repulsion, because the surface of the NPSs assembled at the interface exposed to the water phase will be negatively charged. Yet, interstitial sites, local fluctuations in the packing density of the NPSs assembled at the interface, and intermittent excursions of the PDMS-NH3+ into the aqueous phase provide sufficient opportunities for the arriving NPs to make contact with the PDMS-NH3+ at the interface and hold the newly arrived NPs at the interface. Subsequently, more PDMS-NH3+ attach to the NPs as it is drawn to its steady-state position. The time scale in which this settling occurs depends strongly on the local spatial distribution of other NPs at the interface and the coordinated displacements required to accommodate the new NP. Some NPs may be pushed into the oil phase and be lost from the field of view. Alternatively, a graded anchoring of the ligands could occur, causing a physical lateral displacement of the NPs already assembled at the interface. There is evidence for both mechanisms by in situ AFM.
Fig. 3. Attachment of NPs to the water-oil interface with assembled NPs.
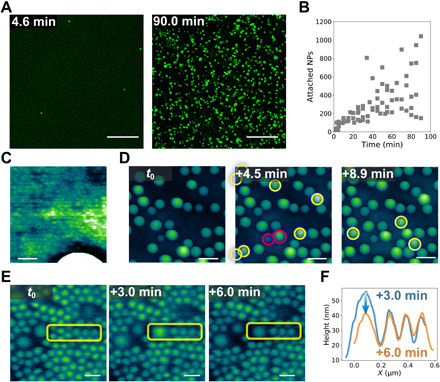
(A) LSCM images showing the attachment of 500-nm NPs to the water-oil interface with assembled 70-nm NPSs. (B) Number of 500-nm NPSs in the field of view as a function of time. (C) In situ AFM images show the coassembly of 30- and 300-nm NPSs at the water-oil interface. (D) In situ AFM images showing the attachment of 300-nm NPs to the water-oil interface with assembled 30- and 300-nm NPSs. (E) Time-dependent in situ AFM images showing the attachment of a 300-nm NP to the water-oil interface covered by 100- and 300-nm NPSs, where the yellow rectangles indicate the affected areas. (F) Line profiles of the region shown in (E) indicate the relaxation of the newly attached 300-nm NP. Scale bars, 20 μm (A), 100 nm (C), 500 nm (D), and 200 nm (E).
The in situ AFM images in Fig. 3D show time-dependent imaging of 300-nm NPs attaching to an interface already occupied by 30-nm NPSs. Several 300-nm NPs are seen to attach to the interface (the newly attached NPs are indicated). Images at time t0 and t0 + 4.5 min show seven new 300-nm NPs attached to the interface in this 4.5-min time period. The 300-nm NPs attaching to the interface already populated with 30-nm NPs show a large variation in position normal to the interface. Two extreme examples are highlighted by red circles in Fig. 3D. Further analysis by single-particle fitting (fig. S4) yields the distribution based on the center of mass (Z0) and size of the assembled NPs (fig. S5) at t0. The far left value of the center-of-mass distribution is comparable to the position in the interface of the 300-nm particle that had likely equilibrated to a contact angle of 39° in Fig. 1. Thus, the vast majority of NPs measured at the interface have not yet reached an equilibrium position. Figure S6 unambiguously shows the local height fluctuations of 30-nm NPSs assembled at the water-oil interface and the local jamming of these NPs. The bending of the 30-nm NPs near the adsorbed 300-nm NP, i.e., the dark ring, provides additional evidence of local jamming (Fig. 3C). Furthermore, the interface is pinned to the 30-nm NPs and only wets a small area on newly attached 300-nm particles further slowing its equilibration. The existence of a dense layer of 30-nm NPSs already assembled at the interface slows the equilibration of the larger 300-nm NPs into the interface, resulting in a large distribution depending on their relative arrival time to the interface and the local reorganization kinetics of the existing NPS assemblies.
The time-dependent adsorption process of a 300-nm NP is shown in Fig. 3E, where both the adsorption and relaxation of the NP can be observed. In Fig. 3F, the line profiles of the region of interest in Fig. 3E show that at t0 + 3.0 min, the 300-nm NP attaches to the water-oil interface; then, at t0 + 6.0 min, the out-of-plane displacement of the NP, Z0, decreases, as the NP settles into the interface. This is similar to the slow relaxation of larger NPs into the interface that are dependent on the contact line hopping along the rough particle surface (27–30). While the relaxation shown here is dependent on a different mechanism, namely, the smaller NPs rearranging at the interface, both processes have complex energy landscapes with local energetic minima instead of a single energetic well associated with simple adsorption of smooth particles. When large NPs adsorb to an interface already populated by very small NPs, such as the data shown in Fig. 3 (D and E), the system is so far from equilibrium that no contact angle information can be measured because the large NP sit atop the small NP, but the interface is pinned at the small NP.
LCSM experiments were used to investigate a 50:50 (by weight) mixed dispersion of 70- and 500-nm NPs to probe the dynamic coassembly. Figure 4A shows the time-dependent LSCM images of the interfacial attachment of 70-nm (yellow) and 500-nm (green) NPs to the oil-water interface. Both the small and large NPSs coassemble at the interface, although only the 500-nm NPSs are clearly resolved. An interesting feature to note is the existence of many dark areas, i.e., cracks, at the interface. These cracks, more than likely, arise from a retraction of the three-phase contact line (water-oil-substrate) after the contact between the water and oil phases is made and the oil droplet volume rising up at the center creating more area as the interfacial film is pinned at the edges. The formation of cracks instead of a homogeneous expansion of the NP density suggests attractive pair interactions between the assembled NPSs due to the partial screening of electrostatic repulsions between NPSs at the interface (33) or an entangling of the ligands anchored to the NPs. It also shows the nonequilibrium nature of the assembly of NPSs in the in-plane direction, as discussed previously. After crack formation, new interfacial areas are exposed. As a result, NPs in the bulk water phase can further attach to the interface, as shown in Fig. 4B. Continuous imaging of the same spot shows that cracks eventually self-heal (movie S3), an important trademark of structured liquids that maintains the integrity of the overall structured liquids. Similar to the results shown in Figs. 2 and 3, a 500-nm NP can also attach to the water-oil interface that has been covered by 70-nm NPSs, as shown in Fig. 4C (yellow-colored area labeled by white circle). Both free attachment and reaction-controlled attachment are observed simultaneously.
Fig. 4. Attachment of 70- and 500-nm NPs to the water-oil interface.

(A) LSCM images showing the attachment of both 70-nm (yellow) and 500-nm (green) NPs. (B) Zoomed-in view of (A) showing the crack of the assemblies and the attachment of 500-nm NPs to the open area. (C) Zoomed-in view of (A) showing the attachment of a 500-nm NP to the water-oil interface already assembled with 70-nm NPSs. Scale bars, 20 μm (A) and 2 μm (B and C).
To summarize, we investigated NPS assembly at the water-oil interface and the factors that control the adsorption process. We found that the early stage of attachment of NPs to the interface followed a diffusion-controlled process and demonstrated, by in situ AFM, that structural changes could occur, depending on the local areal density of NPs. In addition to the free attachment of NPs to the bare interface, we showed that NPs could also attach to the water-oil interface even with a dense assembly of NPSs already present at the interface. The attachment process is a reaction-controlled process with the existing assembly providing an electrostatic barrier to the approach of the NPs to the interface and a coordinated rearrangement of the NPSs to accommodate the attachment of the NP. In situ AFM showed both a slow relaxation of the assemblies in the plane of the interface and laterally inhomogeneous areal densities of the assemblies, with lateral fluctuations in the packing density. Details on the attachment process under different conditions are uncovered with unprecedented spatial and temporal resolution, providing invaluable insight into adsorption and jamming, which aids in the design and fabrication of responsive assemblies.
MATERIALS AND METHODS
MES (low moisture content, ≥99%) and silicone oil (60,000 cSt, PDMS) were bought from Sigma-Aldrich. Polydimethylsiloxane (monoaminopropyl terminated, asymmetric, 18 to 25 cSt, PMDS-NH2) was bought from Gelest. Carboxylated silica NPs with fluorescent dye were bought from Micromod Partikeltechnologie GmbH. All chemicals were used without any further treatment.
Sample preparation
NP solutions were made by diluting the stock NP solutions with 5 mM MES (pH 6.5) buffer solution. Depending on the experiments, the final NP solutions were 70 nm (1 mg ml−1; WS-1), 500 nm (1 mg ml−1; WS-2), 50:50 mixture of 30 and 300 nm (1 mg ml−1; WS-3), 50:50 mixture of 100 and 300 nm (1 mg ml−1; WS-4), and 50:50 mixture of 70 and 500 nm (1 mg ml−1; WS-5). Oil solutions were made by dissolving PMDS-NH2 in silicon oil (60,000 cSt), where the final solutions had two concentrations: 1% (w/w; OS-1) and 10% (w/w; OS-2).
LSCM imaging: The attachment of single-sized NPs and two-sized NPs
A tiny oil droplet (OS-1, less than 1 μl) was dipped on a glass-bottom Petri dish, followed by adding 50 μl of WS-2 or WS-5 to completely cover the oil droplet immediately followed by the LSCM imaging, where the delay time between the water-oil first in contact and the first frame of LSCM images was recorded. The number of NPs in each frame was counted by a homemade Python Script involving the package of Trackpy (34). NPs (500 nm) were used for LSCM measurements to better resolve individual particle locations.
LSCM imaging: The attachment of 500-nm NPs to the interface with assembled NPs
A tiny oil droplet (OS-1, less than 1 μl) was dipped on a glass-bottom Petri dish, followed by adding 50 μl of WS-1 to completely cover the oil droplet. One hour later, 50 μl of WS-2 was added, and the time was recorded as T1. After a predetermined adsorption time, the sample was washed by pure water to remove excess NPs in the water phase, and the time was recorded as T2. Last, all samples were measured in situ by LSCM, where the adsorption time was defined as T = T2 − T1.
In situ AFM
Using in situ AFM to observe the assembly of NPs at the liquid interface is a relatively new technique. It was first introduced by Costa et al. (35, 36) in 2016 using a thin layer of water solution (less than 100 μm) placed on a piece of mica, confined by another mica washer, and covered by organic solvent. We later developed another sample geometry, where the oil phase was below the water phase (18). Several advantages result from this new geometry. First, it gives more freedom to choose the oil phase, including organic solvent (37, 38) and silicone oil (39, 40). Second, it minimizes evaporation as the organic/oil phase is covered by the water phase. It allows surfactants and NPs to be dispersed in different phases; therefore, the different species only interact at the interface, greatly increasing the range of application for in situ AFM in NP assembly. A tiny oil droplet (OS-2) was dipped on a glass-bottom Petri dish, followed by adding 60 μl of WS-4 to completely cover the oil droplet. All in situ AFM imaging was conducted on a Cypher ES AFM (Asylum Research, Oxford Instruments) with BioLever mini (Olympus) probes in tapping mode. Imaging large topographical features is challenging by AFM, so the largest NP used for AFM experiments was 300 nm. A detailed description of the sample preparation and imaging conditions for in situ AFM experiments can be found in the Supplementary Materials.
Supplementary Material
Acknowledgments
Yu Chai would like to acknowledge Behzad Rad for training and support at the LSCM. Funding: This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division under contract no. DE-AC02-05CH11231 within the Adaptive Interfacial Assemblies Towards Structuring Liquids program (KCTR16). Portions of the work—including in situ AFM imaging—were carried out at the Molecular Foundry, which is supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. Author contributions: Y.C., T.P.R., and P.D.A. designed and directed the experiments. Y.C., K.B., M.W., and P.D.A. performed the in situ AFM experiments. Y.C., K.B., and M.W. performed LSCM. Y.C., J.H., and P.D.A. performed the data analysis and modeling. All authors were involved in the data interpretation. Y.C., T.P.R., and P.D.A. wrote the manuscript with contributions from all coauthors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eabb8675/DC1
REFERENCES AND NOTES
- 1.Crossley S., Faria J., Shen M., Resasco D. E., Solid nanoparticles that catalyze biofuel upgrade reactions at the water/oil interface. Science 327, 68–72 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Deshmukh O. S., van den Ende D., Stuart M. C., Mugele F., Duits M. H. G., Hard and soft colloids at fluid interfaces: Adsorption, interactions, assembly & rheology. Adv. Colloid Interface Sci. 222, 215–227 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Zhu Y.-J., Chen F., pH-responsive drug-delivery systems. Chem. Asian J. 10, 284–305 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Bikram M., West J. L., Thermo-responsive systems for controlled drug delivery. Expert Opin. Drug Deliv. 5, 1077–1091 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Huang X., Li M., Green D. C., Williams D. S., Patil A. J., Mann S., Interfacial assembly of protein–polymer nano-conjugates into stimulus-responsive biomimetic protocells. Nat. Commun. 4, 2239 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Pieranski P., Two-dimensional interfacial colloidal crystals. Phys. Rev. Lett. 45, 569–572 (1980). [Google Scholar]
- 7.Binks B. P., Whitby C. P., Nanoparticle silica-stabilised oil-in-water emulsions: Improving emulsion stability. Colloids Surf. A Physicochem. Eng. Asp. 253, 105–115 (2005). [Google Scholar]
- 8.Binks B. P., Lumsdon S. O., Influence of particle wettability on the type and stability of surfactant-free emulsions. Langmuir 16, 8622–8631 (2000). [Google Scholar]
- 9.Binks B. P., Rodrigues J. A., Inversion of emulsions stabilized solely by ionizable nanoparticles. Angew. Chem. Int. Ed. 44, 441–444 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Binks B. P., Particles as surfactants—Similarities and differences. Curr. Opin. Colloid Interface Sci. 7, 21–41 (2002). [Google Scholar]
- 11.Razavi S., Hernandez L. M., Read A., Vargas W. L., Kretzschmar I., Surface tension anomaly observed for chemically-modified Janus particles at the air/water interface. J. Colloid Interface Sci. 558, 95–99 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Hua X., Bevan M. A., Frechette J., Reversible partitioning of nanoparticles at an oil–water interface. Langmuir 32, 11341–11352 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Hua X., Bevan M. A., Frechette J., Competitive adsorption between nanoparticles and surface active ions for the oil–water interface. Langmuir 34, 4830–4842 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Hua X., Frechette J., Bevan M. A., Nanoparticle adsorption dynamics at fluid interfaces. Soft Matter 14, 3818–3828 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Huang C., Sun Z., Cui M., Liu F., Helms B. A., Russell T. P., Structured liquids with pH-triggered reconfigurability. Adv. Mater. 28, 6612–6618 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Cui M., Emrick T., Russell T. P., Stabilizing liquid drops in nonequilibrium shapes by the interfacial jamming of nanoparticles. Science 342, 460–463 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Liu X., Kent N., Ceballos A., Streubel R., Jiang Y., Chai Y., Kim P. Y., Forth J., Hellman F., Shi S., Wang D., Helms B. A., Ashby P. D., Fischer P., Russell T. P., Reconfigurable ferromagnetic liquid droplets. Science 365, 264–267 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Chai Y., Lukito A., Jiang Y., Ashby P. D., Russell T. P., Fine-tuning nanoparticle packing at water–oil interfaces using ionic strength. Nano Lett. 17, 6453–6457 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Ross S., Chen E. S., Adsorption and thermodynamics at the liquid-liquid interface. Ind. Eng. Chem. 57, 40–52 (1965). [Google Scholar]
- 20.Mucha M., Frigato T., Levering L. M., Allen H. C., Tobias D. J., Dang L. X., Jungwirth P., Unified molecular picture of the surfaces of aqueous acid, base, and salt solutions. J. Phys. Chem. B 109, 7617–7623 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Roger K., Cabane B., Why are hydrophobic/water interfaces negatively charged? Angew. Chem. Int. Ed. 51, 5625–5628 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Ward A. F. H., Tordai L., Time-dependence of boundary tensions of solutions I. The role of diffusion in time-effects. J. Chem. Phys. 14, 453–461 (1946). [Google Scholar]
- 23.Riechers B., Maes F., Akoury E., Semin B., Gruner P., Baret J.-C., Surfactant adsorption kinetics in microfluidics. Proc. Natl. Acad. Sci. U.S.A. 113, 11465–11470 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mysels K. J., Diffusion-controlled adsorption kinetics. General solution and some applications. J. Phys. Chem. 86, 4648–4651 (1982). [Google Scholar]
- 25.Jalilvand Z., Haider H., Cui J., Kretzschmar I., Pt-SiO2 Janus particles and the water/oil interface: A competition between motility and thermodynamics. Langmuir 36, 6880–6887 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Zell Z. A., Isa L., Ilg P., Leal L. G., Squires T. M., Adsorption energies of poly(ethylene oxide)-based surfactants and nanoparticles on an air–water surface. Langmuir 30, 110–119 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Kaz D. M., McGorty R., Mani M., Brenner M. P., Manoharan V. N., Physical ageing of the contact line on colloidal particles at liquid interfaces. Nat. Mater. 11, 138–142 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Colosqui C. E., Morris J. F., Koplik J., Colloidal adsorption at fluid interfaces: Regime crossover from fast relaxation to physical aging. Phys. Rev. Lett. 111, 028302 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Wang A., McGorty R., Kaz D. M., Manoharan V. N., Contact-line pinning controls how quickly colloidal particles equilibrate with liquid interfaces. Soft Matter 12, 8958–8967 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Wang A., Zwanikken J. W., Kaz D. M., McGorty R., Goldfain A. M., Rogers W. B., Manoharan V. N., Before the breach: Interactions between colloidal particles and liquid interfaces at nanoscale separations. Phys. Rev. E 100, 042605 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Schwenke K., Isa L., Del Gado E., Assembly of nanoparticles at liquid interfaces: Crowding and ordering. Langmuir 30, 3069–3074 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Carroll K. M., Knoll A. W., Wolf H., Duerig U., Explaining the transition from diffusion limited to reaction limited surface assembly of molecular species through spatial variations. Langmuir 34, 73–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim P. Y., Gao Y., Chai Y., Ashby P. D., Ribbe A. E., Hoagland D. A., Russell T. P., Assessing pair interaction potentials of nanoparticles on liquid interfaces. ACS Nano 13, 3075–3082 (2019). [DOI] [PubMed] [Google Scholar]
- 34.D. Allan, C. van der Wel, N. Keim, T. A. Caswell, D. Wieker, R. Verweij, C. Reid, Thierry, L. Grueter, K. Ramos, Apiszcz, Zoeith, R. W. Perry, F. Boulogne, P. Sinha, Pfigliozzi, N. Bruot, L. Uieda, J. Katins, H. Mary, A. Ahmadia, soft-matter/trackpy: Trackpy v0.4.2 (2019); 10.5281/ZENODO.3492186. [DOI]
- 35.Costa L., Li-Destri G., Pontoni D., Konovalov O., Thomson N. H., Liquid-liquid interfacial imaging using atomic force microscopy. Adv. Mater. Interfaces 4, 1700203 (2017). [Google Scholar]
- 36.Costa L., Li-Destri G., Thomson N. H., Konovalov O., Pontoni D., Real space imaging of nanoparticle assembly at liquid–liquid interfaces with nanoscale resolution. Nano Lett. 16, 5463–5468 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Wu X., Yuan Q., Liu S., Shi S., Russell T. P., Wang D., Nanorod–surfactant assemblies and their interfacial behavior at liquid–liquid interfaces. ACS Macro Lett. 8, 512–518 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Zhang Z., Jiang Y., Huang C., Chai Y., Goldfine E., Liu F., Feng W., Forth J., Williams T. E., Ashby P. D., Russell T. P., Helms B. A., Guiding kinetic trajectories between jammed and unjammed states in 2D colloidal nanocrystal-polymer assemblies with zwitterionic ligands. Sci. Adv. 4, eaap8045 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng W., Chai Y., Forth J., Ashby P. D., Russell T. P., Helms B. A., Harnessing liquid-in-liquid printing and micropatterned substrates to fabricate 3-dimensional all-liquid fluidic devices. Nat. Commun. 10, 1095 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu P.-Y., Chai Y., Hou H., Xie G., Jiang Y., Xu Q.-F., Liu F., Ashby P. D., Lu J.-M., Russell T. P., Stabilizing liquids using interfacial supramolecular polymerization. Angew. Chem. 131, 12240–12244 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/48/eabb8675/DC1