

Graphical abstract
Keywords: Sars-CoV2, Next generation sequencing, Real time PCR, Nasopharyngeal swab
Abstract
Introduction
The novel coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has spread throughout the globe, causing a pandemic. In Egypt over 115,000 individuals were infected so far.
Objective
In the present study, the objective is to perform a complete genome sequence of SAR-CoV2 isolated from Egyptian coronavirus disease (COVID-19) patients.
Methods
Nasopharyngeal swabs were collected from 61 COVID-19 patients who attended at National Cancer Institute, Kasr Al-Aini Hospital and the army hospital. Viral RNA was extracted and whole genomic sequencing was conducted using Next Generation Sequencing.
Results
In all cases, the sequenced virus has at least 99% identity to the reference Wuhan 1. The sequence analysis showed 204 distinct genome variations including 114 missense mutations, 72 synonymous mutations, 1 disruptive in-frame deletion, 7 downstream gene mutations, 6 upstream gene mutations, 3 frame-shift deletions, and 1 in-frame deletion. The most dominant clades were G/GH/GR/O and the dominant type is B.
Conclusion
The whole genomic sequence of SARS-CoV2 showed 204 variations in the genomes of the Egyptian isolates, where the Asp614Gly (D614G) substitution is the most common among the samples (60/61). So far, there were no strikingly variations specific to the Egyptian population, at least for this set of samples.
Introduction
An outbreak of a viral respiratory illness (officially named by the World Health Organization coronavirus disease, COVID-19) started around mid-December 2019, in the city of Wuhan, Hubei province, China. The COVID-19 is an infectious disease of the respiratory tract triggered by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) which in severe cases can lead to acute respiratory distress syndrome and death [1]. The disease has rapidly spread to the entire world [2]. On March 11, 2020, the WHO declared COVID-19 to be pandemic. Now, > 61,654,000 confirmed cases of COVID-19, including 1,444,586 deaths have been reported worldwide according to WHO.
The SARS-CoV-2 genome size varies from 29.8 to 29.9 kb and the 5′ region represent more than two-thirds of the genome that comprises open reading frame (ORF1ab) encoding ORF1ab polyproteins, while the 3′ one third consists of genes encoding structural proteins including surface (S), envelope (E), membrane (M), and nucleocapsid (N) proteins. The SARS-CoV-2 comprises 6 accessory proteins, encoded by ORF3a, ORF6, ORF7a, ORF7b, and ORF8 [3], [4]. To date, by genetic homogeneity the closest virus phylogenetically to SARS-CoV-2 is a coronavirus isolated from the horseshoe bat (Bat CoV RaTG13) with an overall genome sequence identity of 96.2%, which is higher than that of SARS-CoV (<80%) [5], [6].
Next generation sequencing provides high-quality, full-scale genome sequences for viral isolates collected in relatively non-biased ways, irrespective of virulence or other unusual properties. Analyzes of the genome sequences gave insight into the pattern of global distribution, the genetic diversity during epidemics and the dynamics of the development of subtypes. SARS-CoV2 database, such as GISAID (www.gisaid.org), the NCBI SARS-CoV2 database (https://www.ncbi.nlm.nih.gov/sars-cov-2), and NGDC Genome Warehouse (https://bigd.big.ac.cn/gwh/), make the genomic information publicly available, together with epidemiological data for the sequenced isolates.
The development of a statistical system focused on epidemiological, antigenic and genetic knowledge may provide more insight into the rules regulating the appearance and development of antigenically novel mutations and improve the capacity for prevention and regulation of SARS-CoV2 [7], [8]. Studies strongly support an urgent necessity for additional rapid, wide-ranging investigations that incorporate genetic data, epidemiological knowledge and graph details of patients with COVID-19 with clinical characteristics [9], [10]. Therefore, we studied the molecular variation among SARS-CoV-2 genomes isolated and sequenced from COVID-19 Egyptian patients.
Materials & Methods
Ethics statement
The study was endorsed by the Ethics Committee of Ministry of Health and Populations, Training and Research Sector, with number OHRP: FWA00016183 23 March 2020, IORG0005704/ IRB0000687 31 May 2020. The Next Generation Sequencing of the SARS-CoV-2 positive samples was performed after the cases underwent standard SARS-CoV-2 diagnostic tests.
Patients and samples
The study included 61 successful whole genomic sequences from the following cases: Twenty six cases from health care workers at NCI, 13 cases from health workers at Kasr Al-Aini Hospital and 22 cases from the army hospital. All samples were collected during the period between March and April 2020. Patients in this study had symptoms and were confirmed to be SARS-CoV-2 positive by real-time PCR. The samples used for this study were nasopharyngeal swabs collected in viral transport media. Nasopharyngeal swab specimens were collected from suspected cases that met the case definition of SARS-CoV-2 infection as set out in the guidelines of the Ministry of Health and Population in Egypt.
Sample types, RNA extraction, and viral detection
A volume of 250–300 μL of each nasopharyngeal swab sample was used for viral RNA extraction using the QIAMP VIRAL RNA mini kit (Qiagen, Hilden, Germany) with internal PCR control as instructed by the manufacturer. The extracted RNA was directly used for amplification using Genesig Real-Time PCR Detection Kit for SARS-CoV2 use of two primers / probe, one for SARS-Cov2 detection, and the other for internal extraction control detection for test validation. The cycle threshold value of [C t] below 34 was considered to be positive. Compliance with the WHO-recommended research protocol confirmatory laboratory testing has been carried out.
Next Generation Sequencing of SARS-CoV-2
Volume of 250–300 μL of each nasopharyngeal swab sample (SARS-CoV-2 real-time positive at least 1.25x103 copies / μL) was used for viral RNA extraction using the QIAMP VIRAL RNA mini kit (Qiagen , Hilden , Germany) as directed by the manufacturer. The extracted genomic RNAs were quantified using a Qubit RNA High Sensitivity Kit (Invitrogen , The United States of America (USA)). The obtained genomic RNAs were retro-transcribed using the VILO-cDNA Synthesis Kit (Cat. No.11754050; Invitrogen, USA) and the custom primer COVID (Thermo Fisher Scientific) and the double-stranded DNA was subsequently obtained by the Klenow enzyme (Roche, Basel, Switzerland) as instructed by the manufacturer.
The Ion AmpliSeq Library Kit Plus (Thermo Fisher Scientific) was used for the preparation of the library. Two pre-mixed pools of 275 amplicons were used to generate sequencing libraries. Clonal amplification of the libraries was performed using the Ion-PI-Hi-Q Sequencing 200 Kit (Thermo Fisher Scientific) PCR emulsion. The detailed protocol was as instructed by the manufacturer(http://tools.thermofisher.com/content/sfs/manuals/MAN0010947_Ion_PI_HiQ_Seq_200_Kit_UG.pdf). The whole genome sequencing was performed by using Ion PI Hi-Q Sequencing 200 Kit –Chef Kit (all Thermo Fisher Scientific) on the Ion proton Sequencer.
Data analysis
Viral Sequence Assembly
The Ion Torrent package (v.5.12) was used to perform the base calling of the raw data. Two strategies were utilized for genome assembly and these were executed in two independent tracks: The first is de-novo assembly, where the reads were assembled using the IRMA (v0.9.3) workflow. The second is based on reference-based assembly, where the reads were assembled by mapping (aligning) them to a reference genome. The tmap program (v.512) was used to align the reads to the reference corona virus genome Wuhan 1 (RefSeq; NC_045512.2). The consensus sequence was then computed from the aligned reads. After finishing the assembly, the de-novo assembly was compared against the reference-based assembly to assure consistency of the results. In fact, for this target amplicon based panel, we see that the reference-based assembly is enough to reconstruct the viral sequence. Samples with <99% coverage or with gaps >30 bps were excluded. The final successful set included 61 complete genome sequences and these were uploaded to NCBI/GISAID repositories (Supplementary File S1).
Preparing world datasets
All the complete SARS-CoV2 genome sequences were first retrieved from the GISAID website on 30 June 2020 and all sequences with long internal gaps or ambiguities (>30bps) were excluded. The final dataset included 46,612 sequences. On October 2020, we updated this dataset and the number of sequences increased to 89632. On these sequences, the following processing was conducted to create different collections of samples:
Collecting Subsampled Nextstrain sequences: To facilitate visualization of the phylogenetic tree, the Nextstrain team (www.nextstrain.org) subsamples the huge virus dataset into smaller collection of sequences representing different geographical areas. The latest version (October 2020) has about 5000 sequences. We sub-sampled this list further and collected about 250 sequences.
Clustering of Genome Sequences: All sequences were formatted into a blast database by the 'makeblastdb' command to allow all vs. all blast comparison. Then all sequences were queried against this reference set. The sequences that matched other sequences with 100% identity without indels over 99% of genome length are considered identical and grouped into clusters. There were 1729 clusters comprising>2 sequences. The total number of sequences in clusters with size more than or equal 2 is 9253 (Supplementary File S3). For each cluster, one sequence is selected to represent the respective cluster in further analysis. That is, the set of unique sequences/clusters is 37359. We used this clustering information to compute the Computing Extended Neighbor Set as follows: Each of the 61 Egyptian sequences was used as a query against the BLAST coronavirus database we constructed. (-qcov_hsp_perc 95% -perc_identity 95%). We also used the virus data set of 157 sequences compiled by Forster et al [11] as queries against BLAST corona virus database. The hits from the Egyptian and Forster et al. collections were considered as neighboring sequences. The IDs of these hits were used to collect the respective neighboring sequences. Sequences belonging to the same cluster were tagged with the respective cluster ID. There were 786 neighboring sequences (some of them represent bigger clusters). The sequences in this set cover the landscape of the virus phylogeny, around the Egyptian set. This way we avoided computing phylogeny from all the GISAID sequences, as we are not interested in other parts of the tree.
Phylogenetic analysis
For different groups of sequences (Egyptian plus Public ones), we first ran the multiple sequence alignment step using MAFFT (v7.450) [12]. The terminal sequences were trimmed to assure gapless terminals of the alignment. To deal with large number of sequences, we used the nextstrain pipeline deployed via the mini-conda environment: First, the sequences were re-aligned using MAFFT. Then the iqtree packages is used to compute the phylogeny. The iqtree package selects the best nucleotide substitution model and runs bootstrapping to assure high confidence of tree topology.
Clade Assignment
For clade assignment, we used two approaches: The first is to pick the nearest neighbor in the phylogeny tree. The second is by running BLAST against the well-annotated GISAID dataset. The clade classification of the best hit with minimum error and longest match is used to label our viral sequence.
Variation analysis
World dataset
All the GISAID sequences we collected and revised were aligned to the reference viral sequence using the nucmer program [13]. The output delta file of nucmer is parsed to extract the variations and this file is then transformed to VCF format using in-house script. Each VCF is then annotated using the snpEff package [14] dedicated to the corona virus (snpEff_v4_5covid19_core.zip). All the VCFs were then processed to compute the frequency of each variation in the world population.
Egyptian dataset
The variations (mutations) in the Egyptian genomes were examined for quality and depth. A variation is filtered out if its depth is<50 reads. It is also filtered out if its depth is<100, appears only once in our dataset, and did not appear in world population. We also checked if the variations occur in a homopolymer region or not, especially if it appears once in our dataset and not present in the world population. (Homo-polymer errors are frequent and well known sequencing errors for the Ion Torrent technology.) The remaining variations were then annotated with snpEff to add information about the gene and mutation effect. They were also annotated with their frequencies in the Egyptian dataset. Moreover, the variations were annotated with their frequencies in the world population and in different regions, including USA, Europe, China, Middle East. Also, we annotated them with their frequencies in the Saudi dataset published in GISAID (Saudi Arabia is the closest country to Egypt in terms of Geography and people movement). The variation frequencies in the world and other regions were computed at different time points: June 2020, when first version of the paper was prepared, and October 2020, at the time of preparing a revised version of the paper.
Statistical analysis
Fisher exact test and Chi square test were used (the default one and the G-test version for large numbers) to check whether the frequency of a variation is different between a pair of sub-populations. For each variation in question, we created a contingency table including the count of wild type cases (i.e., number of cases with no mutations) and the count of cases with variations in the two sub-populations under examination. Supplementary Table 5, shows the contingency table for the variations in Table 3 and the sub-populations listed above. For comparing the distribution of variations in the set of virus genes (Table 2), we also used Chi square test. The P-value threshold 0.05 is used to confirm or reject differences between the variables in the test. Specifically, P-value > 0.05 means that there is no difference between the two sub-populations (two groups) with respect to the variations (categories) at hand.
Table 3.
High frequency mutations in SARS-CoV-2 sequences of Egypt and the world. The table shows the most frequent variations in the Egyptian population. The table also includes the frequency of these variations in different populations. The numbers in bold indicate that this frequency is significantly different from the Egyptian frequency using Chi-Square/Fisher Exact test (P < 0.05). Supplementary Table S5 includes comparisons among other populations.
| Genome Change | Position | Gene | Protein Change | Mutation type | Egypt count (n = 61) | EgyFreq |
|---|---|---|---|---|---|---|
| c.1841A > G | 23,403 | S | p.Asp614Gly | Missense | 60 | 0.98361 |
| c.-25C > T | 241 | ORF1ab | Non-coding | Upstream | 59 | 0.96721 |
| c.2772C > T | 3037 | ORF1ab | p.Phe924Phe | Synonymous | 57 | 0.93443 |
| c.14144C > T | 14,408 | ORF1ab | p.Pro4715Leu | Missense | 56 | 0.91803 |
| c.171G > T | 25,563 | ORF3a | p.Gln57His | Missense | 30 | 0.4918 |
| c.18613C > T | 18,877 | ORF1ab | p.Leu6205Leu | Synonymous | 17 | 0.27869 |
| c.3108C > A | 3373 | ORF1ab | p.Asp1036Glu | Missense | 13 | 0.21311 |
| c.12269C > T | 12,534 | ORF1ab | p.Thr4090Ile | Missense | 10 | 0.16393 |
| c.2169C > T | 23,731 | S | p.Thr723Thr | Synonymous | 10 | 0.16393 |
| c.3737C > T | 4002 | ORF1ab | p.Thr1246Ile | Missense | 10 | 0.16393 |
| Genome Change | EgyFreq | WorldFreq June 2020 (n=46,612) | WorldFreq Oct. 2020 (n=89632) | AFR (n=1820) Oct. 2020 | EU (n=48716) Oct. 2020 | USA (n=20758) Oct. 2020 | China (n=743) Oct. 2020 | MENA (n=1133) Oct. 2020 | Saudia (n=560) Oct. 2020 | Compare To EgyPop |
|---|---|---|---|---|---|---|---|---|---|---|
| c.1841A>G | 0.9836 | 0.76056 | 0.8050 | 0.9412 | 0.8187 | 0.8459 | 0.0767 | 0.7485 | 0.7250 | 7/8; P<10E-3 |
| c.-25C>T | 0.9672 | 0.74560 | 0.7921 | 0.9335 | 0.8121 | 0.8304 | 0.0781 | 0.8402 | 0.9357 | 6/8; P<0.05 |
| c.2772C>T | 0.9344 | 0.75654 | 0.8041 | 0.9368 | 0.8158 | 0.8443 | 0.0740 | 0.8270 | 0.8786 | 5/8; P<0.05 |
| c.14144C>T | 0.9180 | 0.75755 | 0.8028 | 0.9390 | 0.8152 | 0.8423 | 0.0579 | 0.8570 | 0.9393 | 2/8; P<0.01 |
| c.171G>T | 0.4918 | 0.22243 | 0.2365 | 0.0462 | 0.1089 | 0.6006 | 0.0135 | 0.3928 | 0.7107 | 6/8; P<10E-3 |
| c.18613C>T | 0.2787 | 0.02581 | 0.0312 | 0.0077 | 0.0126 | 0.0479 | 0.0000 | 0.2913 | 0.5554 | 7/8; P<1-E-5 |
| c.3108C>A | 0.2131 | 0.00002 | 0.0044 | 0.0126 | 0.0065 | 0.0002 | 0.0000 | 0.0079 | 0.0000 | 8/8; P<10E-9 |
| c.12269C>T | 0.1639 | 0.00024 | 0.0007 | 0.0005 | 0.0007 | 0.0008 | 0.0013 | 0.0009 | 0.0000 | 8/8; P<10E-9 |
| c.2169C>T | 0.1639 | 0.04304 | 0.0395 | 0.1016 | 0.0668 | 0.0004 | 0.0000 | 0.0000 | 0.0000 | 7/8; P<0.01 |
| c.3737C>T | 0.1639 | 0.01425 | 0.0139 | 0.1027 | 0.0197 | 0.0006 | 0.0000 | 0.0000 | 0.0000 | 7/8; P<10E-5 |
Table 2.
Distribution of variations in different genes. Total world Samples in June and October are 46,612 and 89,632, respectively. The VarFreq is the number of variations in the gene divided by the total number of samples. The relative frequency is the number of variations divided. Freq norm is the frequency divided by the total number of variations in each group. N/S is the ration between the number of non-synonymous and synonymous variations in the same gene.
| Gene | Len | Var Count World June | Var Count World June | Var Count | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| M | 669 | 464 | 515 | 2 | ||||||
| N | 908 | 1107 | 1371 | 23 | ||||||
| E | 228 | 266 | 320 | 1 | ||||||
| ORF1ab | 21,290 | 14,779 | 16,221 | 131 | ||||||
| ORF3a | 828 | 809 | 1003 | 6 | ||||||
| ORF6 | 186 | 191 | 230 | 1 | ||||||
| ORF7a | 498 | 427 | 524 | 6 | ||||||
| ORF8 | 193 | 366 | 439 | 4 | ||||||
| S | 3822 | 3405 | 3146 | 30 | ||||||
| Gene | len | World June | World Oct. | |||||||
| Gene | len | RelativeFreq | RelFreqNorm | N/S | RelativeFreq | RelFreqNorm | N/S | RelativeFreq | RelFreqNorm | N/S |
| M | 669 | 0.022 | 0.032 | 1.03 | 0.022 | 0.033 | 1.14 | 0.010 | 0.015 | 1 |
| N | 908 | 0.051 | 0.057 | 1.88 | 0.058 | 0.064 | 2.14 | 0.113 | 0.125 | 1.75 |
| E | 228 | 0.012 | 0.054 | 2.02 | 0.014 | 0.060 | 3.2 | 0.005 | 0.021 | 0 |
| ORF1ab | 21,290 | 0.686 | 0.032 | 1.24 | 0.692 | 0.032 | 1.25 | 0.645 | 0.030 | 1.48 |
| ORF3a | 828 | 0.038 | 0.045 | 2.38 | 0.043 | 0.052 | 2.5 | 0.030 | 0.036 | 4 |
| ORF6 | 186 | 0.009 | 0.048 | 1.91 | 0.010 | 0.053 | 2.17 | 0.005 | 0.026 | 0 |
| ORF7a | 498 | 0.020 | 0.040 | 1.92 | 0.022 | 0.045 | 2.08 | 0.030 | 0.059 | 2 |
| ORF8 | 193 | 0.017 | 0.088 | 1.9 | 0.019 | 0.097 | 1.88 | 0.020 | 0.102 | 3 |
| S | 3822 | 0.158 | 0.041 | 1.31 | 0.134 | 0.035 | 1.16 | 0.148 | 0.039 | 2 |
Results
Clinical data
The main clinical symptoms of patients with COVID-19 were fever 43/61 (70%), cough 14/61 (23%), asthma 9/61 (14.7%), myalgia or fatigue 30/61 (49%), nasal congestion 16/61 (26%), sputum production 4/61 (5.7%) and dyspnea 14/61 (23%). Minor symptoms include headache or dizziness 3/61 (5%), diarrhea 3/61 (5%), nausea and vomiting 4/61 (5.7%). All patients recovered without complications.
Data availability
Analyzing all of the Egyptian SARS-CoV-2 genome sequences in positive cases has shown that the nucleotide and amino acid percentage variations between SARS-CoV-2 cases are 0.4% and 0.25% respectively. The Egyptian samples have been given the name “hCoV-19/Egypt/CUNCI-HGC” and the sample numbers were added to that prefix. They were submitted to GenBank and GISAID (Supplementary Table 1) https://github.com/mabouelhoda/nCovEgypt. Additionally, all sequences have also been stored in GISAID (https://www.gisaid.org/). Supplementary S1 includes list of all sequences, and their IDs in all databases.
Genes and mutations in SARS-CoV2 genomes
All retrieved sequences were aligned and trimmed based on the reference Wuhan 1 sequence NC_045512.2. All sequences were trimmed to 29698 bp and a total number of 204 mutations were detected in the Egyptian strains (Fig. 1, Supplementary File S2) https://github.com/mabouelhoda/nCovEgypt. We found that more than half of the variations were in the ORF1ab polyprotein (64%). The least number of variations were related to the ORF6 and E protein sequences (0.5%) (Table 1). Specifically, of the 204 mutations, there were 131 ones in ORF1ab, followed by 30 in S, 23 in N, 6 in ORF3a, 6 in ORF7a, 4 in ORF8, 2 in M, 1 in E, and 1 in ORF6. ORF1ab is transcribed into a multi-protein and subsequently divided into 16 non-structural proteins (NSPs). Of these proteins, NSP3 has 34 variations (20 missense, 13 synonymous and one frameshift) of ORF1ab proteins. Of the NSP3, c.2772C > T (p. Phe924Phe) was detected in 57 of the 61 samples followed by c.5019C > T (p. Asn1673Asn) detected in 6/61 samples, followed by c.2772delC (p. Tyr925fs) in 3/61 samples. Three variations were found in the RNA dependent RNA polymerase area: c.14144C > T (p.Pro4715Leu) in 56/61 samples followed by c.16193C > T in 3/61 samples (p. Ser5398Leu) and c.13794A > G (p.Thr4598Thr) in 2/61 samples.
Fig. 1.

Variant frequencies in SARS-Cov2 isolate in Egypt in comparison to world population. Part (a) frequency of all variants. Part (b), frequency of variants in ORF genes.
Table 1.
Number of gene variations in SARS-CoV2 genomes. E: envelope protein; M: membrane glycoprotein; N: nucleocapsid phosphoprotein; ORF: open reading frame; S: spike glycoprotein; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2. Note: We compared 61 whole genomes to the NC_045512.2 genome sequence.
| Genome segment | Missense mutation | Synonymous mutation | Non-coding region |
Other mutation |
Frameshift deletion/in frame del | Stop-gained | Total | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | Deletion | Insertion | Upstream | downstream | ||||||
| ORF1ab | 74 | 50 | 0 | 0 | 0 | 5 | 0 | 2(1,1) | 0 | 131 |
| S | 14 | 7 | 0 | 0 | 0 | 0 | 7 | 2 (1,1) | 0 | 30 |
| ORF3a | 4 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 6 |
| E | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| M | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| ORF6 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| ORF7 | 4 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 6 |
| ORF8 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 |
| N | 14 | 8 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 23 |
| Total | 114 | 72 | 0 | 0 | 0 | 6 | 7 | 5 (3,2) | 0 | 204 |
Of the 114 missense variations, 4, 4, and 3 (3.5%, 3.5%, and 2.63%) are found in ORF3a ORF7a, and ORF8, respectively. The most frequent ones within these 11 variations are c.171G > T (p.Gln57His) in 30/61, c.512C > T (p. p.Ser171Leu) in 2/61, and c.251 T > C (p.Leu84Ser) in 2/61. Of the 100 missense variations, 14 (12.28%) variations are found in each of S and N genes. The most common variation of the S gene is the missense mutation c.1841A > G (p. Asp614Gly). In this study, one (0.8%) missense mutation was detected in the M gene c.374A > G (p. His125Arg) and one in ORF8 c.251 T > C (p.Leu84Ser). There are 72 synonymous mutations, two of them are in E and M regions {c.222G > C (p. Leu74Leu) and c.213C > T (p.Tyr71Tyr) respectively (supplementary S2) https://github.com/mabouelhoda/nCovEgypt.
In this study, three frameshift mutations were detected, one of which was detected in ORF1ab, c.10818delG (p. Leu3606fs) in 3c like proteinase, and one frameshift mutation was detected in S gene c.13delC (p.Val6fs). Synonym mutations were detected in 72 positions, 50 of which 50 were detected in ORF1ab, 7 in S, 8 in N, 2 in ORF7, and one in remaining genes (Table 1).
Distribution of variations
Fig. 1 shows the distribution of the Egyptian variations throughout the genome. No wonder that most of the variegations (148 ones) are in the ORF part due to its size (Fig. 1 b). We compared this pattern of distribution in Egyptian world datasets. Table 2 includes this comparison. The table also includes the relative frequencies among the genes in the same dataset. The results in the table show that the number of variations in the whole world increased by maximum 24% from June to October 2020, which is not proportional to the doubling of deposited sequences. Also, the relative frequencies in the world in June did not change from that of October (P- value > 0.05; Chi-square test). This indicates there is no increasing pressure on certain genes than the others. Comparing the RelativeFreq columns between the Egyptian and world population in June and October did not show significant difference (P-value > 0.05; Chi square).
The table also shows the ratio between the synonymous and non-synonymous mutations in each gene. The ratio is larger than one for most of the genes. Also the distribution of the ratio of N/S among the different genes in world in June and October is very similar (P-value > 0.05; Chi square). The minor differences between the Egyptian N/S values and the corresponding world ones cannot be confirmed statistically (P-value > 0.05; Chi square).
Highly frequent mutations
The frequencies of the Egyptian 204 variations were also computed in other sequences from different regions including USA, Europe, China, Middle East, and Saudi Arabia. Supplementary Table S2 includes these frequencies at two time points: June and October 2020. Only one mutation was novel and specific to the Egyptian dataset. The other 203 ones exist in the world population. Eleven out of the 204 variable sites in the Egyptian virus genomes were the most prevalent. Table 3 lists these variations and their frequencies in world and regional datasets. It is interesting to note that these variations are also abundant in the world and regional sequences, except for the Chinese sequences (P-value < 0.00001 based on Chi Square tests for almost all variants in Chinese and other populations as in Supplementary Table 5). The bold values in Table 3 highlights that there is statistically confirmed increase in variant counts between Egyptian and other populations (P-value < 0.05 for Chi Square and Fisher Exact test as in Supplementary Table S5). However, this should be taken with caution due to the small size of Egyptian samples.
The observation that the difference in variation frequencies between the Egyptian and Chinese populations is much higher than the difference between the corresponding ones for the Egyptian and non-Chinese (up to 5 folds for top frequent variants; P-value < 0.05) points to the non-Asian origin of Egyptian isolates. Phylogenetic analysis introduced below confirms this and indicates that the source of infection of the Egyptian population is most likely Europe and United States of America.
Phylogenetic analysis:
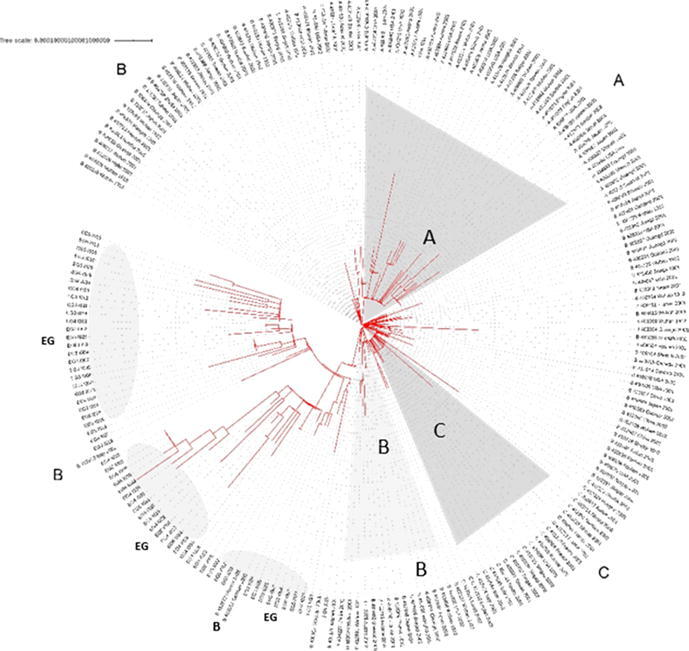
Level 1: Phylogenetic tree was constructed using the Egyptian sequences plus the 250 sequences sub-sampled from the Nextstrain (Fig. 2). The tree shows that most of the Egyptian cohorts of samples can be assigned clades G/GR/GH/O (as per GISAID system).
Fig. 2.

Phylogenetic analysis of the Egyptian sequences plus 250 sequences sub-sampled from the Nextstrain dataset. The tree is annotated with GISAID clade information. (High resolution plot is in Supplementary File S6). Each sequence is named as follows: “Type:GISAID_ID:Country:Year:Month”.
Level 2: Phylogenetic analysis including the 61 Egyptian sequences and the extended neighbor set composed of 786 sequences (Figure 3). The tree shows more in-depth clades over large landscape of the virus phylogeny. We could confirm that most of the Egyptian cohort of samples can be assigned the clades G/GR/GH/O (as per GISAID system), which is in accordance with the clade assignment conducted by the GISAID team (Supplementary S4) https://github.com/mabouelhoda/nCovEgypt.
Fig. 3.

Phylogenetic analysis of Egyptian sequences and extended neighbor set composed of 786 genome sequences. The tree is annotated with GISAID clade information. The black dots show location of the Egyptian sequences. (High resolution plot is in Supplementary File S6). Each sequence is named as follows: “Type:GISAID_ID:Country:Year:Month”.
High resolution images of the phylogenetic trees are in Supplementary File S6 where one can zoom in to see the sequence information https://github.com/mabouelhoda/nCovEgypt.
In detail, the phylogenetic study of the full sequences (Figure 3) showed that the Egyptian cohorts of samples EG5_I003, EG3_I021, EG3_I025, EG3_I005, and EG3_I004 are within the G clade close to the German sample with ID G425139 and Cluster 286 including four samples from The United States of America, The samples EG5_I033 and EG3_I023 are also from the G clade and close to Cluster 7136 (including four samples from Austria) and the English sample 461486; respectively. The sample EG6_I007 is close to 461,505 from India. EG3_I013 close to 470,427 from India. EG3_I014 close to 471,854 from The United States of America.
The samples EG3_I007, EG3_I003, EG3_I006, EG3_I008, EG3_I016, EG4_I026 are from clade GH and close to the, The United States of America sample 424857, the Saudi sample 437699. The samples EG3_I009 and EG5_I016 are also from clade G close to the French sample 447,689 and Cluster 1079 including 13 samples from France, Australia, Sweden, The United States of America, Russia, Israel, Belgium, and England. The samples EG4_I025, EG5_I029, EG4_I030, EG5_I014, EG5_I015 belong to the same clade and close to sample 435,524 from USA. Samples EG3_I012 close to the GH sample 435,498 from USA. The EG3_I010 sample is close to the GH samples 437,743 from Saudi Arabia and to Cluster 10,508 including 2 samples from India. The sample EG6_I016 is close to the GH Saudi samples 437,748 and 437739.
The Egyptian cohorts of samples EG5_I001, EG5_I011, EG4_I004, EG4_I032, EG5_I010 belong to the GR clade and close to the sample 437,990 from Austria and the Cluster 1018 including 6 samples from Portugal, Morocco, and Russia. The samples EG4_I015, EG4_I003, EG6_I024, EG4_I023, EG5_I004, EG5_I012, EG5_I022 belong to the same clade and close to Cluster 9310 (including 2 samples from England) and Cluster 1220 (including 2 samples from Sweden and England). The samples EG6_I015, EG4_I022 are close to the GR sample 429,143 from Sweden and the samples EG4_I029, EG6_I026, EG4_I031, EG6_I031, EG6_I033, EG6_I002, EG6_I009 are close to the GR samples 468,024 from Australia and 420,723 from England.
Samples EG3_I002, EG4_I033, EG3_I015, EG5_I029, EG6_I010, EG5_I025, EG5_I026, EG5_I034, EG6_I003, EG5_I030, are from clade O and close to the samples 437,300 from Austira, 447,659 from France, Cluster 34, Cluster 9717, and Cluster 3443.
Evolutionary selection
We used the program HyPhy (http://hyphy.org/) on the Egyptian sequences to compute the site specific dN/dS ratio to determine the genome sites under selection pressure. However, this analysis, as expected, did not yield any significant result due to the small size of the dataset (almost no polymorphism at each site.). To overcome this limitation, we used the list of sites under evolutionary selection computed by the HyPhy team (http://hyphy.org/) using the world sequences up to September 2020. The list included 977 sites in the corona virus genome under selection pressure: 267 positive and 710 negative. We annotated our variations with this list and found that only 15 of our variations (15/204 = 7.35%) are in regions under positive pressure and there is no site in this dataset under negative pressure The variations under positive pressure are N:c.623C > T; ORF1ab:c.10818delG; N:c.605G > A; N:c.15560C > T; N:c.974C > G; ORF1ab:c.17765C > T; ORF1ab:c.17414C > T; ORF1ab:c.16193C > T; ORF1ab:c.926C > T; S:c.293C > T; ORF1ab:c.2675C > T; ORF1ab:c.10058A > G; ORF1ab:c.3737C > T; ORF1ab:c.14144C > T; S:c.1841A > G. The last three ones are of high frequency in the population. Four of them are in the N gene, two in the S gene, and the remaining are in the ORF1ab gene.
Discussion
The quick increase in people infected with SARS-CoV-2 will provide the opportunity to conduct more genome studies. Over the last five months the number of individuals infected with COVID-19 reported increased steadily, with no sign of any decrease. Four structural proteins are encoded in the SARS-CoV-2 genome. Structural proteins are much more immunogenic than non-structural protein to T cell responses [15]. In various viral processes, including virus particle formation, the structural proteins are involved. Spike (S), envelope (E), protein membrane (M) and nucleoprotein (N), specific to all coronaviruses, are found in structural proteins [16], [17]. In the current study, we detect 204 variations in the Egyptian strains. We did not observe relevant novel variations.
In the current study, synonymous variations were detected in one position of E and one position of M genes: c.222G > C (p. Leu74Leu) and c.213C > T (p. Tyr71Tyr), respectively. The missense variations of S and N genes were found in the present study. The spike S protein is an infection-initiating glycoprotein [18], [19], the virion binds to the cell membrane by communicating with the host receptors angiotensin-converting enzyme 2 (ACE2). The efficacy of sub-genomic viral RNA transcription and viral replication is improved by nucleoprotein. Nucleoprotein (ORF9a), during viral assembly via its interactions with the virus genome and membrane protein M, packs the positive-strand RNA genomic into a ribonucleocapsid (RNP) helical. In the budding compartment of the host cell, the protein envelope (E) interacts with membrane protein M. The M protein has overriding cell immunogenicity [20]. A total of 204 mutations of which 30 were found in the region S, 23 in region N, 1 in region E and 2 in region M were identified in a current genomic region report.
In viral glycoprotein-mediated binding to host cells, the only major difference in SARS-CoV-2 viral surface spikes, and subsequent fusion of virus and host cell membranes, is aspartate (D) mutation at position 614 found in most a subset of the sequences from China to glycine (G) enriched in another one from Western Europe [21]. While in this epitope the amino acids are well preserved, 14 other variations besides D614 G have been identified. Virus replication speeds can also be affected by nearly all strains that have D614 G mutation in their protein responsible for Replication (Orf1ab P4715L; RdRp P323L) [22].
This protein is the target of antiviral, remdesivir, and favipiravir and is susceptible to mutations that suggest the rapid production of treatment resistive strains. Likewise the most common variation in the current study in the S gene is the missense mutation c.1841A > G (p. Asp614Gly), which also has a replication mutation in ORF1ab P4715L. Recent study of the SARS-CoV-2 isolate fine-scale sequence variation found many areas with an increased genetic variation [23], [24]. One of these variations is the S-protein mutation, D614 G, in the carboxy(C)-terminal region of the S1 domain [24], [25]. This mutation with residual glycine 614 (G614) was previously detected to increase at an alarming rate and were observed at low frequency in March (26%), but increased rapidly by April (65%) and May (70%), indicating a transmission advantage over D614 viruses [26]. This shift was also related to an increased viral charge among patients with COVID-19, but the role in these observations of the S-protein remained unclear because this shift is also associated with mutations in viral nsp3 and in RdRp proteins [26].
The replicase enzyme is shown as two polyproteins (ORF1a and ORF1ab) [27], [28]. The ORF1ab is the most important factor among coronaviruses [28]. In this study, 204 mutations were identified (including 131 ORF1ab, 6 ORF3a, 6 ORF7, 4 ORF8 and one ORF6) according to the genomic regions. The 131 high-frequency variation was observed in ORF1ab relative to the global population frequency.
The relationship between ORFs and COVID-19, e.g. 8782C > T(ORF1ab) and 28144 T > C (ORF8), has been identified among researchers in several genome databases [28], [29]. The biological role of a specific protein ORF1ab in SARS-CoV-2 will therefore be clinically significant. The ORF1ab is two-thirds of the genome and is transcribed into a multiprotein and then split into several nonstructural proteins (NSP1-NSP16). Among the analyzed samples of NSPs, NSP3 has more variation [28]. The most widely defined clade was a vaccine-based variation D614G, which is located in a B cell epitope with a highly immuno-dominant region [30]. In this study, NSP3 has 34 variation (20 missense, 13 interchangeable and 1 framework-shift), out of the 121 variation of ORF1ab. Wang and coworkers have recently identified 13 variation sites in SARS-CoV-2 ORF1ab, S, ORF3a, ORF8 and N regions, of which 28,144 in ORF8 and 8782 in ORF1a showed mutation rates of 30.53% and 29.47% respectively [31].
The Nsp3 is an integral component of the replication and transcription complex, and the Nsp4 encrypted into ORF1ab forms the dual membrane vesicle (DMV) [32]. In the current analysis, c.2772C > T (p. Phe924Phe) (detected by 57/61 samples) was the highest mutation observed in NSP3, followed by c.5019C > T (p. Asn1673 Asn) in 6 samples and finally, c.2772delC (p. Tyr925fs) in 3 of these samples. C.2772delC (p. Tyr925fs) was observed in the current studies in ORF1ab as frameshift mutations in NSP3, as was in 3C LIKE PROTEIASE as in the 3C region. Synonyms mutation in 69 positions were observed, 50 of 69 in ORF1ab, ORF6, and ORF7 genes were identified in the present study. Therefore, the NSP3 mutation may affect the virus replication or transcription as nsp3, nsp4 and nsp6 together induce DMV.
The RNA replication of SARS-coronavirus is unique with two RNA-based RNA (RdRp) polymerases involved. A non-structural protein 12 (nsp12) is the first polymerase is RNA, while the second RNA is nsp8. Nsp8 has primase ability for RNA replication without primers for novo initiation [33], [34]. SARS-CoV-2 isolates are the most common SNP mutation in nsp8 proteins, where leucine (L) amino acid is mutated to serine(S) (28,144 T > C). The previous research was carried out on 103 genomes of SARS-CoV-2 for both co-mutations (8782C > T and 28,144 T > C) which classified the virus as S / L types (Yin, 2020). A main component of the replication and transcription machinery is the SARS-CoV-2 RdRp (also called nsp12). RdRps against a wide range of viruses are known as key targets for antiviral medicinal products. The target of SARS-CoV-2 has been taken into account for a number of RdRp inhibitors such as favipiravir [35]. In contrast to SARS-CoV-2, SARS-CoV-2 shares a high homology for nsp 12, which indicates that its function and mechanism of action can be properly preserved [36]. The analysis currently investigates the 10 variation in 56/61 samples, followed by c.16193C > T (p. Ser5398Leu), and c.137994A > G (p. Ther4598Thr) for 3/61 samples in 10 RNA-dependent RNA Polymerase (RdRp) region (one missense variation c.14144C > T (p. Pro4715Leu)). In a recent study that reveals SARS-CoV-2 RdRp / nsp7 / nsp8 complex structure, Kirchdoerfer and colleagues showed that nsp7 and nsp8 are involved in RdRp supercomplex formation in SARS CoV [29], as was also reported for SARS-CoV-2. This complex guarantees RdRp processivity, which is important in the fidelity of transcription [36].
ORF10 is a 38-deposit small protein or peptide. Koyama et al . identified COVID-19 as ORF10 which has no comparative NCBI proteins. [37]. In the current study, the missense variation was found in ORF3a c.171G > T (p.Gln57His), ORF7a c.21C > A (p.Phe7Leu), ORF8, and ORF10 genes. Many non-recurring frameshift variation have been observed which can sequence apps. ORF10′s Y3 frameshift may be inaccessible to the survival of the new coronavirus but is identified only in one sample because ORF10 is not homologous to another NCBI protein, which is a small 38 residue peptide. The phylogenetic analysis provides an independent test of the major clades that have been identified. In late January, in China, D614 G was first observed and in three months became the largest clade. The mutation rate for 1.12 × 10−3 mutations per site-year is similar to 0.80 × 10−3 to 2.38 × 10−3 mutations per site-year reported for SARS [38].
In the current study, the Egyptian SARS-CoV-2 viruses have been placed in different clusters. The phylogenetic tree shows a main clade containing several clusters. The large number of patients' viral genome sequences thirteen cases was identical to those from The United States of America, Austria, Sweden, Saudi Arabia and France. The 8 viral genome sequences followed were identical to those from England, the United State of America, Wales, Chile, Austria, Russia, Vietnam and Belgium. Five viral genome sequences were identical to those from Germany , Sweden, India, England and the United States of America. Another Five viral genome sequences were identical to those from the United States of America, Latvia, Sweden, Belgium and England. Four samples were like to those taken from Austria. Another four viral genome sequences were identical to those recovered from India and Latvia. Four other samples were identical to those collected from Canada, the United State of America and Brazil. Three samples were identical to those taken from Bangla and England. Two samples were identical to those collected from the United States of America and Taiwan. The other two samples were identical to those collected from the United States of America. One sample was identical to the one from Sweden, England and Israel. One sample was identical to that from the United States of America, Saudi Arabia, India and Colombia. One sample was the same as that collected from Saudi Arabia.
In conclusion, we detect 204 unique sequence variations in genomes isolated from Egyptian patients. Most Egyptian genomic strains sequenced so far are similar to isolates from United States of America, Austria, Sweden, Saudi Arabia and France.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
The authors are thankful to Prof. Dr. Mohamed Othman Elkhosht the President of Cairo University for his support of this work. Also the authors’ thank go to Science and Technology Development Fund (STDF) Egypt, Grant ID grg(ACSE41907).
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2020.11.012.
Contributor Information
Abdel-Rahman N. Zekri, Email: ncizekri@yahoo.com.
Mohamed Abouelhoda, Email: mabouelhoda@yahoo.com.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Pal M. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus. 2020;12(3) doi: 10.7759/cureus.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ojha V. CT in coronavirus disease 2019 (COVID-19): a systematic review of chest CT findings in 4410 adult patients. Eur Radiol. 2020 doi: 10.1007/s00330-020-06975-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yadav P.D. Full-genome sequences of the first two SARS-CoV-2 viruses from India. Indian J Med Res. 2020;151(2 & 3):200–209. doi: 10.4103/ijmr.IJMR_663_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mousavizadeh L., Ghasemi S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J Microbiol Immunol Infect. 2020 doi: 10.1016/j.jmii.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Letko M., Marzi A., Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5(4):562–569. doi: 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rihtaric D. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch Virol. 2010;155(4):507–514. doi: 10.1007/s00705-010-0612-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ge X.Y. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503(7477):535–538. doi: 10.1038/nature12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang X.L. Isolation and Characterization of a Novel Bat Coronavirus Closely Related to the Direct Progenitor of Severe Acute Respiratory Syndrome Coronavirus. J Virol. 2015;90(6):3253–3256. doi: 10.1128/JVI.02582-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang Y.C., Leung T.K. Establishment of a basic medical science system for Traditional Chinese medicine education: A suggestion based on the experience of BIOCERAMIC technology. J Tradit Complement Med. 2020;10(2):95–103. doi: 10.1016/j.jtcme.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madec F.X., Dariane C., Cornu J.N. Evaluation and comparison of basic gestures in ex vivo laparoscopic surgery using a robotic instrument and traditional laparoscopic instruments. Prog Urol. 2020;30(1):58–63. doi: 10.1016/j.purol.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Forster P. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Natl Acad Sci U S A. 2020;117(17):9241–9243. doi: 10.1073/pnas.2004999117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katoh K., Standley D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics. 2016;32(13):1933–1942. doi: 10.1093/bioinformatics/btw108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurtz S. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cingolani P. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li C.K. T cell responses to whole SARS coronavirus in humans. J Immunol. 2008;181(8):5490–5500. doi: 10.4049/jimmunol.181.8.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruan Y.J. Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet. 2003;361(9371):1779–1785. doi: 10.1016/S0140-6736(03)13414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marra M.A. The Genome sequence of the SARS-associated coronavirus. Science. 2003;300(5624):1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 18.Wong S.K. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J Biol Chem. 2004;279(5):3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan Y. Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J Virol. 2020;94(7) doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J. The membrane protein of severe acute respiratory syndrome coronavirus acts as a dominant immunogen revealed by a clustering region of novel functionally and structurally defined cytotoxic T-lymphocyte epitopes. J Infect Dis. 2010;202(8):1171–1180. doi: 10.1086/656315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Becerra-Flores M., Cardozo T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int J Clin Pract. 2020 doi: 10.1111/ijcp.13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koyama T., Platt D., Parida L. Variant analysis of SARS-CoV-2 genomes. Bull World Health Organ. 2020;98(7):495–504. doi: 10.2471/BLT.20.253591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lokman S.M. Exploring the genomic and proteomic variations of SARS-CoV-2 spike glycoprotein: A computational biology approach. Infect Genet Evol. 2020;84 doi: 10.1016/j.meegid.2020.104389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laha S. Characterizations of SARS-CoV-2 mutational profile, spike protein stability and viral transmission. Infect Genet Evol. 2020;85 doi: 10.1016/j.meegid.2020.104445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bal A. Molecular characterization of SARS-CoV-2 in the first COVID-19 cluster in France reveals an amino acid deletion in nsp2 (Asp268del) Clin Microbiol Infect. 2020;26(7):960–962. doi: 10.1016/j.cmi.2020.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv. 2020 [Google Scholar]
- 27.van der Meer Y. ORF1a-encoded replicase subunits are involved in the membrane association of the arterivirus replication complex. J Virol. 1998;72(8):6689–6698. doi: 10.1128/jvi.72.8.6689-6698.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khailany R.A., Safdar M., Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020 doi: 10.1016/j.genrep.2020.100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirchdoerfer R.N., Ward A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun. 2019;10(1):2342. doi: 10.1038/s41467-019-10280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korber B. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020 doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang C. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. 2020;92(6):667–674. doi: 10.1002/jmv.25762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angelini M.M. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio. 2013;4(4) doi: 10.1128/mBio.00524-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Q. Structural Basis for RNA Replication by the SARS-CoV-2 Polymerase. Cell. 2020;182(2):417–428 e13. doi: 10.1016/j.cell.2020.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yin C. Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics. 2020;112(5):3588–3596. doi: 10.1016/j.ygeno.2020.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furuta Y. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res. 2013;100(2):446–454. doi: 10.1016/j.antiviral.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pachetti M. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. 2020;18(1):179. doi: 10.1186/s12967-020-02344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koyama T. Emergence of Drift Variants That May Affect COVID-19 Vaccine Development and Antibody Treatment. Pathogens. 2020;9(5) doi: 10.3390/pathogens9050324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao Z. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol Biol. 2004;4:21. doi: 10.1186/1471-2148-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Analyzing all of the Egyptian SARS-CoV-2 genome sequences in positive cases has shown that the nucleotide and amino acid percentage variations between SARS-CoV-2 cases are 0.4% and 0.25% respectively. The Egyptian samples have been given the name “hCoV-19/Egypt/CUNCI-HGC” and the sample numbers were added to that prefix. They were submitted to GenBank and GISAID (Supplementary Table 1) https://github.com/mabouelhoda/nCovEgypt. Additionally, all sequences have also been stored in GISAID (https://www.gisaid.org/). Supplementary S1 includes list of all sequences, and their IDs in all databases.