

Abstract
The discovery of actionable kinase gene rearrangements has revolutionized the therapeutic landscape of thyroid carcinomas. Unsolved challenges include histopathologic recognition of targetable cases, correlation between genotypes and tumor behavior, and evolving resistance mechanisms against kinase inhibitors (KI). We present 62 kinase fusion-positive thyroid carcinomas (KFTC), including 57 papillary thyroid carcinomas (PTC), two poorly differentiated thyroid carcinomas (PDTC), two undifferentiated thyroid carcinomas (ATC), and one primary secretory carcinoma (SC), in 57 adults and 5 adolescents. Clinical records, post-operative histology, and molecular profiles were reviewed. Histologically, all KFTC showed multinodular growth with prominent intratumoral fibrosis. Lymphovascular invasion (95%), extrathyroidal extension, gross and microscopic (63%), and cervical lymph node metastasis (79%) were common. Several kinase fusions were identified: STRN-ALK, EML4-ALK, AGK-BRAF, CUL1-BRAF, MKRN1-BRAF, SND1-BRAF, TTYH3-BRAF, EML4-MET, TFG-MET, IRF2BP2-NTRK1, PPL-NTRK1, SQSTM1-NTRK1, TPR-NTRK1, TPM3-NTRK1, EML4-NTRK3, ETV6-NTRK3, RBPMS-NTRK3, SQSTM1-NTRK3, CCDC6-RET, ERC1-RET, NCOA4-RET, RASAL2-RET, TRIM24-RET, TRIM27-RET, and CCDC30-ROS1. Individual cases also showed copy number variants of EGFR and nucleotide variants and indels in pTERT, TP53, PIK3R1, AKT2, TSC2, FBXW7, JAK2, MEN1, VHL, IDH1, PTCH1, GNA11, GNAQ, SMARCA4 and CDH1. In addition to thyroidectomy and radioactive iodine, 10 patients received multi-kinase and/or selective kinase inhibitor therapy, with 6 durable, objective responses and 4 with progressive disease. Among 47 cases with >6 months of follow-up (median [range]: 41 [6 to 480] months), persistent/recurrent disease, distant metastasis and thyroid cancer-related death occurred in 57%, 38% and 6%, respectively. In summary, KFTC encompass a spectrum of molecularly diverse tumors with overlapping clinicopathologic features and a tendency for clinical aggressiveness. Characteristic histology with multinodular growth and prominent fibrosis, particularly when there is extensive lymphovascular spread, should trigger molecular testing for gene rearrangements, either in a step-wise manner by prevalence or using a combined panel. Our findings further provide information on molecularly therapy in radioiodine-refractory thyroid carcinoma.
Introduction
Protein kinases are important signaling mediators that regulate cell survival, growth and differentiation through intricate molecular pathways. Receptor tyrosine kinases (RTK), such as ALK, RET, MET, NTRK, FGFR and ROS1, upon binding ligands, transduce signals to the RAS/RAF/MAPK, PI3K/AKT/mTOR and JAK-STAT pathways, whose deregulated overactivation plays a central role in human carcinogenesis. The RAS/RAF/MAPK, PI3K/AKT/mTOR and JAK-STAT signaling cascades are composed of multiple cytoplasmic tyrosine kinases (CTK), serine/threonine kinases (S/TK) and lipid kinases (1). The activation of most RTK requires molecular dimerization. Rearrangements of RTK-encoding genes lead to ligand-independent dimerization and are well-recognized oncogenic drivers in thyroid, lung, salivary gland, breast, kidney, colorectal, pancreatic and soft tissue malignancies (2–4). Most fusion partners provide dimerization domains, such as the coiled coil domains in CCDC6, NCOA4, TRIM24, TPR, PPL, CCDC30, TPM3, ERC1, the Phox and Bem1p (PB1) interaction domains in SQSTM1 and TFG, the helix-loop-helix domain in ETV6, the zinc finger domain in IRF2BP2, the WD domains in STRN and EML4, and the RNA recognition motif of RBPMS, to be joined with the kinase domain of the RTK in forming fusion oncoproteins that are capable of autonomous activation (5–7). At a much lower frequency, transforming rearrangements of downstream signaling S/TK, such as BRAF and AKT, have also been reported. BRAF fusions are found in 60–80% of pilocytic astrocytomas and rare lung and thyroid carcinomas (8–10). The N-terminus autoinhibitory domain of BRAF is lost as the C-terminus kinase domain fuses with a partner sequence, leading to uncontrolled activation (8). Recently, rare cases of ovarian, peritoneal and breast tumors have showed AKT fusions that remove the autoregulatory pleckstrin homology domain from the kinase domain of AKT1/2/3 (11–13). In the thyroid gland, kinase fusion-related carcinomas are uncommon occurrences that have been most frequently noted in children and young adults, as well as post-Chernobyl radiation exposure victims. However, given a shift toward pre-operative molecular testing for treatment planning, these tumors are finding an established presence in sporadic thyroid malignancies in the middle age and elderly populations. Cases involving ALK, RET, MET, NTRK1/3, ROS1, FGFR1 and BRAF fusions have been increasingly discovered with a wide range of partner genes (10, 14–19).
As genomics-driven precision oncology has brought significant changes to thyroid cancer diagnosis and therapeutics over the past several years, one pivotal breakthrough has been the successful translation of protein kinase inhibitors (KI) into clinical therapeutic use. While earlier multi-kinase inhibitors (MKI), such as lenvatinib, sorafenib, vandetanib and cabozantinib, showed efficacy in thyroid cancer, MKI therapy is associated with frequent off-target side effects. Newer more selective agents, such as larotrectinib (anti-TRK), entrectinib (anti-ALK, ROS1 and TRK), selpercatinib and pralsetinib (both anti-RET), have demonstrated significant efficacy with more favorable side effect profiles. Recent trials in non-medullary thyroid carcinomas (mostly PTC) have reported an objective response rate of 62% and 79% for selpercatinib and larotrectinib, respectively; a duration of response was not reached in either study due to few progress events (20–24). Nevertheless, several challenges remain to be addressed. Firstly, immunohistochemical markers are still unavailable or yet to be fully validated for most kinase rearrangements. With costly molecular methods being the current mainstay for detecting targetable alterations, it is essential to develop a clinicopathologic case triage approach for selective genetic testing. Secondly, due to the rarity of kinase fusion-related thyroid carcinomas (KFTC), their genomic make-up and long-term clinical behavior are not well understood. Furthermore, acquired resistance to kinase inhibitors has been increasingly noted. While isolated cases have been associated with secondary mutations that cause target modifications (eg. RET G810S, NTRK G595R, ALK G1202R, ROS1 G2032R) and bypass signaling (2, 25, 26), the mechanisms of acquired resistance remain obscure in many cases. There exists an ongoing need for patient-based data focused on pre- and post-KI genotype-phenotype correlation in order to tailor individualized treatment for KFTC.
In this study, we present a large single-institutional cohort of KFTC with two main purposes: (1) to describe the cardinal clinicopathologic features of KFTC to guide diagnostic recognition and molecular testing referral, and (2) to contribute to the current literature on the genomic landscape of KFTC with correlation to clinical findings including KI therapy response.
Materials and Methods
Case Identification
With the approval by the Massachusetts General Hospital (MGH) Institutional Review Board (2012P001024), we retrospectively queried the MGH molecular pathology database for primary thyroid carcinomas that underwent the targeted gene rearrangement analysis (described below) from 2013 through the May of 2020, and reviewed the test results. Since 2013, the pathologists and other providers at MGH have been selectively submitting cases for genetic profiling based upon unusual histology, aggressive presentation, and/or negative BRAF V600E mutant-specific immunohistochemistry, or for clinical decision-making. For cases that showed kinase gene rearrangements, histologic slides were obtained from the MGH surgical pathology archive and reviewed by three pathologists (PMS, VN and YHC). Patient clinical information, surgical and molecular pathology reports were accessed through the electronic medical record interface at MGH. Histologic findings, such as clear cell, oncocytic, tall cell, hobnail and high-grade (defined as solid, trabecular or insular [STI] pattern with necrosis and/or ≥3 mitoses per 10 high-power fields [HPF]) features, were semi-quantified as focal (<30% of sampled tumor volume) versus diffuse.
Immunohistochemistry
We recently described the immunohistochemistry (IHC) setup at MGH (27). IHC markers that were employed in this study as needed to characterize morphologically unusual and genetically altered cases included TTF1 (performed in cases 1, 52, 53; clone 8G7G3/1; dilution 1:300; Dako, Carpinteria, CA), BRG1 (performed in case 9; clone EPR3912; dilution 1:50; Abcam, Cambridge, United Kingdom), GATA3 (performed in case 25; clone L50–823, dilution 1:200; Cell Marque, Rocklin, CA), mammaglobin (performed in case 25; clone 304–1A5; dilution 1:250; Dako, Carpinteria, CA), S100 protein (performed in case 25; polyclonal and prediluted; Ventana, Oro Valley, AZ), PAX8 (performed in cases 25, 52, 53; polyclonal, dilution 1:1000; Proteintech, Rosemont, IL), thyroglobulin (performed in cases 25, 52; dilution 1:800; Cell Marque, Rocklin, CA), E-cadherin (performed in case 46; clone 36B5; Leica Biosystems, Buffalo Grove, IL), p40 (performed in case 52; clone BC28, Biocare, Concord, MA) and BRAF V600E (performed in all cases; clone VE1; dilution 1:200; Abcam, Cambridge, United Kingdom).
Targeted Gene Rearrangement Next-Generation Sequencing (NGS) Analysis
An Anchored Multiplex PCR (AMP) assay developed at MGH (28) was performed for detecting targeted gene fusions by next-generation sequencing (NGS) using total nucleic acid extracted from formalin-fixed paraffin embedded (FFPE) tumor tissue. The details of the assay were as we recently described (27) and covered the following genes (exons): ALK (19–22, intron 19), BRAF (7–12, 15), EGFR (2–7 exon skipping/vIII variant, 7–9, 16, 20, 24, 25), EWSR1 (4–14), FGFR2 (2, 8–10, 17), MAML2 (2,3 ), MET (exon 14 skipping), NRG1 (1–3, 6), NUTM1 (3), RET (8–13), ROS1 (31–37), AKT3 (1–3), ARHGAP26 (2, 10–12), AXL (19,20), BRAF (7–12, 15), BRD3 (9–12), BRD4 (10, 11), ERG (2–11), ESR1 (3–6), ETV1 (3–13), ETV4 (2, 4–10), ETV5 (2, 3, 7–9), ETV6 (1–7), FGFR1 (2, 8–10, 17), FGFR3 (8–10, 17, intron 17), FGR (2), INSR (12–22), JAZF1 (2–4), MAML2 (2,3 ), MAST1 (7–9, 18–21), MAST2 (2, 3, 5, 6), MET (13, 15), MSMB (2–4), MUSK (7–9, 11–14), MYB (7–9, 11–16), NOTCH1 (2, 4, 26–31, internal exon 3–27 deletion), NOTCH2 (5–7, 26–28), NRG1 (1–3, 6), NTRK1 (8,10–13), NTRK2 (11–17), NTRK3 (13–16), NUMBL (3), PDGFRA (7, exon 8 deletion, 10–14), PDGFRB (8–14), PIK3CA (2), PKN1 (10–13), PPARG (1–3), PRKCA (4–6), PRKCB (3), RAF1 (4–7, 9–12), RELA (3, 4), RSPO2 (1, 2), RSPO3 (2), TERT (2), TFE3 (2–8), TFEB (1,2), THADA (28), and TMPRSS2 (1–6). Illumina MiSeq 2x147 base paired-end reads were mapped to the hg19 human reference genome using BWA-MEM (29) and interpreted by a molecular pathologist (DDS).
SNaPshot Mutation Profiling
As previously described (30), the SNaPshot assay was AMP-based and validated for clinical application at MGH. Utilizing the ArcherDx and Illumina NextSeq platforms, the assay targeted single nucleotide variants (SNVs) and insertion/deletions (indels) in the following genes (exons): ABL1 (4–7), AKT1 (3,6), ALK (21–23,25), APC (16), ARID1A (1–20), ATM (1–63), ATRX (1–35), AURKA (2,5–8), BRAF (11,15), BRCA1 (2–23), BRCA2 (2–27), CCNB1 (2,[3-partial],5,[6-partial],7), CCND2 ([2-partial],3–4,[5-partial]), CCND3 (2–5-partial), CCNE1 (3–8,10,12), CDH1 (1–16), CDK4 (2–7), CDK6 (6), CDKN2A (1–3), CIC (1–20), CSF1R (7,22), CTNNB1 (3), DAXX (1–8), DDR2 (12–18), DDX3X (1–17), EGFR (3,7,15,18–21), ERBB2 (8,10,19–21,24), ERBB3 (2–3,7–8), ERBB4 (3–4,6–9,15,23), ESR1 (8), EZH2 (16), FBXW7 (1–11), FGFR1 (4,7–8,13,15,17), FGFR2 (7,9,12,14), FGFR3 (7–9,14–16,18), FLT3 (11,14,16,20), FOXL2 (1), GNA11 (5), GNAQ (4–5), GNAS (6–9), H3F3A (2), HNF1A (3–4), HRAS (2–3), IDH1 (3–4), IDH2 (4), JAK2 (11,13–14,16,19), JAK3 (4,13,16), KDR (6–7,11,19,21,26–27,30), KEAP1 (2–6), KIT (2,8–11,13–15,17–18), KRAS (2–5), MAP2K1 (2,3,6–7), MAP3K1 (1–20), MDM2 (2–4,6,8,10), MDM4 ([4-partial],5–6,[7,9–11-partial]), MEN1 (2–10), MET (2,11,14,16,19,21), MITF (1-partial), MLH1 (12), MPL (10), MSH6 (1–10), MSI, MYC (1–3), MYCN (3), NF1 (1–58), NF2 (1–15), NKX2–1 (1-partial), NOTCH1 (25–27,34), NPM1 (11), NRAS (2–5), PDGFRA (12,14–15,18,23), PIK3CA (2,5,7–8,10,14,19,21), PIK3R1 (1–10), POLE (9–14), PTCH1 (1–23), PTEN (1–9), PTPN11 (3,13), RB1 (1–27), RET (10–11,13–16), RHOA (2–3), RNF43 (2–10), ROS1 (36–38), SDHB (1–8), SMAD2 (7), SMAD4 (2–12), SMARCA4 (3–36), SMARCB1 (2,4,5,9), SMO (3,5–6,9,11), SRC (14), STAG2 (3–34), STK11 (1–9), SUFU (1–12), TERT (1), TP53 (1–11), TP63 (1–14), TSC1 (3–23), TSC2 (2–42), TSHR (10), and VHL (1–3). In addition, the following copy number variations (CNVs) were covered: ABL1, AKT1, ALK, APC, ARID1A, ATM, ATRX, AURKA, BRAF, BRCA1, BRCA2, CAMTA1, CCNB1, CCND1, CCND2, CCND3, CCNE1, CDK4, CDKN2A, CDK6, CIC, CDH1, CSF1R, DAXX, DDR2, DDX3X, EGFR, ERBB2 (HER-2), ERBB3, ERBB4, FBXW7, FGF19, FGFR1, FGFR2, FGFR3, FLT3, FOXL2, GLI2, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, JAK2, JAK3, KDR, KEAP1, KIT, KRAS, MAP2K1, MAP3K1, MDM2, MDM4, MEN1, MET, MITF, MLH1, MSH6, MYC, MYCN, NF1, NF2, NKX2–1, NOTCH1, NRAS, PDGFRA, PIK3CA, PIK3R1, PLAUR, POLE, PTCH1, PTEN, PTPN11, RB1, RET, RHOA, RNF43, SDHB, SMAD2, SMAD4, SMARCA4, SMARCB1, SMO, SRC, STAG2, STK11, SUFU, TERT, TP53, TP63, TSC1, TSC2, and VHL. Sequencing results were interpreted by a molecular pathologist (DDS).
Results
Clinical Presentation
A total of 395 primary thyroid carcinomas underwent the targeted gene rearrangement panel assay from 2013 to the May of 2020, including 212 (54%) papillary thyroid carcinomas (PTC), 55 (14%) medullary thyroid carcinomas, 39 (10%) undifferentiated (anaplastic) thyroid carcinomas (ATC), 36 (9%) follicular thyroid carcinomas, 24 (6%) Hürthle cell carcinomas, 23 (6%) poorly differentiated thyroid carcinomas (PDTC), one (<1%) primary thyroid secretory carcinoma (SC) and five (1%) unclassifiable carcinomas. Various kinase fusions were detected in 62 (16%) cases, including 57 (14%) PTC (including 29 classical type, 21 diffuse sclerosing variant, four solid variant, two follicular variant, and one tall cell variant), two (<1%) PDTC, two (<1%) ATC, and one SC. The most commonly rearranged kinase gene was RET, positive in 31 (8%) cases, followed by NTRK3 in 11 (3%) cases, NTRK1 in 8 (2%) cases, BRAF in 6 (2%) cases, ALK in 3 (<1%) cases, MET in two cases, and ROS1 in one case. The observed fusions and clinicopathologic characteristics of all patients are summarized in Table 1 and Figure 1, including the references for 16 previously published cases (16, 17, 27, 28, 31).
Table 1:
Clinical characteristics of kinase fusion-related thyroid carcinomas
| Case # | Rearrangement | Fusion Exons | Age (years) | Sex | Stage | Distant Metastasis | Treatment | Followup (months) | Outcome | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | STRN-ALK | S3; A20 | 61 | M | T3 N1b | Skin, mediastinum (both R) | Sorafenib, crizotinib, lenvatinib# | 22 | R; on hospice | |
| 2 | STRN-ALK | S3; A20 | 66 | F | T1 N0 | - | - | 19 | NED | |
| 3 | EML4-ALK | E6; A20 | 32 | M | T1 N0 | - | - | 96 | NED | (17) |
| 4 | AGK-BRAF | A2; B8 | 15 | F | T3 N1b | Lung (R) | RAI | 33 | AWD | |
| 5 | CUL1-BRAF | C7; B9 | 67 | M | T4a N1b | - | RAI | 27 | AWD | |
| 6 | MKRN1-BRAF | M3; B10 | 55 | M | T1 N1a | - | RAI | 5 | NED | |
| 7 | SND1-BRAF | S14; B11 | 75 | M | T3 N0 | - | RAI | 15 | NED | |
| 8 | SND1-BRAF | S14; B9 | 76 | F | T2 N0 | - | RAI | 30 | NED | |
| 9 | TTYH3-BRAF | T12; B8 | 60 | M | T4a N1b | - | XRT, RAI | 24 | NED | |
| 10 | EML4-MET | E6; M15 | 61 | F | T2 N0 | - | RAI | 46 | NED | |
| 11 | TFG-MET | T5; M15 | 44 | F | T1 N0 | - | - | 2 | NED | (17) |
| 12 | IRF2BP2-NTRK1 | I1; N10 | 36 | F | T1 N0 | - | - | 1 | NED | |
| 13 | PPL-NTRK1 | P21; N11 | 62 | M | T3 N1b | Mediastinum (D), lung (R) | RAI, trametinib, larotrectinib# | 80 | R; AWD | (28) |
| 14 | SQSTM1-NTRK1 | S5; N10 | 37 | F | T2 N1a | - | RAI | 44 | AWD | (27) |
| 15 | SQSTM1-NTRK1 | S5; N10 | 74 | F | T1 N0 | - | - | 1 | NED | |
| 16 | TPR-NTRK1 | T20; N11 | 54 | M | T3 N1b | Lung (R) | RAI, larotrectinib# | 144 | R x2; AWD | (27) |
| 17 | TPR-NTRK1 | T21; N12 | 24 | M | T3 N1b | Lung (D) | RAI | 34 | AWD | (27) |
| 18 | TPM3-NTRK1 | T7; N10 | 40 | M | T3 N1a | - | - | 1 | AWD | |
| 19 | TPM3-NTRK1 | T7; N12 | 27 | M | T3 N1b | - | - | 0.2 | AWD | |
| 20 | EML4-NTRK3 | E2; N13 | 42 | M | T3 N1b | Lung, brain (both R) | RAI, XRT, entrectinib# | 480 | R x2; AWD | (27) |
| 21 | ETV6-NTRK3 | E3; N13 | 74 | F | T3 N1b | - | RAI; XRT | 13 | R; AWD | (27) |
| 22 | ETV6-NTRK3 | E4; N13 | 32 | F | T3 N1b | - | RAI | 39 | NED | (27) |
| 23 | ETV6-NTRK3 | E4; N13 | 14 | F | T3 N1b | Lung (D) | RAI | 80 | R; AWD | (27) |
| 24 | ETV6-NTRK3 | E5; N14 | 71 | F | T3 N1a | - | RAI, XRT, larotrectinib# | 71 | R x2; NED | (27) |
| 25 | ETV6-NTRK3 | E4; N13 | 23 | F | T1 N0 | - | - | 1 | NED | |
| 26 | RBPMS-NTRK3 | R5; N13 | 22 | F | T3 N1a | - | RAI | 44 | NED | (27) |
| 27 | RBPMS-NTRK3 | R5; N13 | 60 | F | T3 N1a | Lung (R) | RAI, proton beam radiation | 121 | R x2; AWD | (27) |
| 28 | SQSTM1-NTRK3 | S5; N13 | 43 | F | T3 N1b | Lung (R) | RAI | 16 | AWD | (27) |
| 29 | SQSTM1-NTRK3 | S5; N13 | 29 | F | T1 N0 | - | - | 0.5 | AWD | |
| 30 | SQSTM1-NTRK3 | S5; N13 | 26 | F | T2 N1a | - | - | 1 | AWD | |
| 31 | CCDC6-RET | C1; R12 | 72 | M | T3 N1b | Brain, lung (both R) | RAI, SRS, doxorubicin, docetaxel, selpercatinib# | 35 | R; AWD | (31) |
| 32 | CCDC6-RET | C1; R12 | 40 | F | T3 N1b | - | RAI | 41 | R x2; AWD | |
| 33 | CCDC6-RET | C1; R12 | 19 | F | T3 N1b | - | RAI | 69 | R; AWD | |
| 34 | CCDC6-RET | C1; R12 | 19 | F | T3 N1b | - | RAI | 27 | AWD | |
| 35 | CCDC6-RET | C1; R12 | 37 | F | T1 N1b | - | RAI | 19 | R; AWD | |
| 36 | CCDC6-RET | C1; R12 | 17 | F | T3 N1b | - | RAI | 49 | AWD | |
| 37 | CCDC6-RET | C1; R12 | 23 | F | T1 N1b | - | RAI | 20 | R; AWD | |
| 38 | CCDC6-RET | C1; R12 | 23 | F | T3 N1b | - | RAI | 63 | R; AWD | |
| 39 | CCDC6-RET | C1; R12 | 26 | M | T3 N1b | - | RAI | 6 | AWD | |
| 40 | CCDC6-RET | C1; R12 | 16 | M | T2 N1b | - | RAI | 2 | AWD | |
| 41 | CCDC6-RET | C1; R12 | 44 | M | T4a N1b | Spine, axilla (both R) | RAI, XRT, SBRT, selpercatinib# | 136 | R x2; AWD | |
| 42 | CCDC6-RET | C1; R12 | 51 | M | T3 N1b | Lung (R) | RAI | 47 | AWD | |
| 43 | CCDC6-RET | C1; R12 | 39 | F | T3 N1b | Lung (R) | RAI | 41 | AWD | |
| 44 | CCDC6-RET | C1; R12 | 53 | M | T2 N1a | - | RAI | 3 | AWD | |
| 45 | CCDC6-RET | C1; R12 | 67 | F | T1 N1a | - | RAI | 54 | NED | |
| 46 | CCDC6-RET | C1; R12 | 33 | F | T1 N1a | - | RAI | 6 | NED | |
| 47 | CCDC6-RET | C1; R12 | 25 | F | T1 N0 | - | RAI | 27 | NED | |
| 48 | CCDC6-RET | C1; R12 | 19 | F | T3 N0 | - | RAI | 32 | NED | |
| 49 | CCDC6-RET | C1; R12 | 51 | F | T2 N0 | - | - | 0.03 | NED | |
| 50 | CCDC6-RET | C1; R12 | 21 | M | T3 N1b | - | - | 1 | AWD | |
| 51 | CCDC6-RET | C1; R12 | 25 | F | T3 N1b | - | - | 1 | AWD | |
| 52 | ERC1-RET | E7; R12 | 13 | F | Tx N1* | Lung, brain, bone, breast (all R) | RAI, XRT, SRS, kinase inhibitors# | 441 | R x2; DOD | |
| 53 | NCOA4-RET | N8; R12 | 61 | M | T4a N1b | Lung (D), bone (D) | Inoperable presentation; XRT, kinase inhibitors# | 35 | DOD | |
| 54 | NCOA4-RET | N8; R12 | 58 | F | T2 N1b | - | RAI | 3 | AWD | |
| 55 | NCOA4-RET | N8; R12 | 31 | M | T3 N1b | - | RAI | 21 | R; AWD | |
| 56 | NCOA4-RET | N8; R12 | 26 | F | T3 N1b | - | RAI | 63 | NED | |
| 57 | NCOA4-RET | N7; R12 | 55 | F | T3 N1b | - | RAI | 7 | NED | |
| 58 | NCOA4-RET | N8; R12 | 36 | F | T1 N1b | Liver, lung, pleura, bone, abdominal wall (all R) | RAI, XRT, lenvatinib# | 250 | R x2; DOD | |
| 59 | RASAL2-RET | R17; R12 | 33 | F | T1 N1a | - | RAI | 61 | NED | |
| 60 | TRIM24-RET | T10; R12 | 20 | F | T3 N1* | Lung (R) | RAI | 372 | AWD | |
| 61 | TRIM27-RET | T3; R12 | 26 | M | T3 N1b | Lung (R) | RAI | 54 | AWD | |
| 62 | CCDC30-ROS1 | C10; R36 | 24 | F | T4a N1b | - | RAI | 20 | NED | (16) |
Abbreviations: D, at diagnosis; na, not available; R, recurrence; RAI, radioactive iodine; XRT, external radiation therapy; SRS, stereotactic radiosurgery; SBRT, stereotactic body radiation therapy; AWD, alive with disease; NED, alive with no evidence of disease; DOD, died of disease.
Thyroidectomy was performed prior to 2000 with limited staging information in the archived pathology reports; histologic slides were no longer in storage.
See Table 2 for treatment details.
Figure 1:

Summary of the clinicopathologic and molecular findings in kinase fusion-related thyroid carcinomas.
The median (range) age for patients with kinase fusion-related thyroid carcinoma (KFTC) was 36 (13 to 76) years, including 57 adults and five adolescents. The female-to-male ratio was 1.8 (40:22). Except for case 16, who received low-dose radiation therapy for acne >30 years prior to developing thyroid cancer, all other cases were radiation-naïve. Tumor size ranged from 0.2 to 10.5 cm (median 3.0 cm), staged as pT1 in 15 (24%, including five microcarcinomas), pT2 in 8 (13%), pT3 in 33 (53%) and pT4 in 5 (8%) at the time of diagnosis. T stage was unavailable for case 52, whose thyroidectomy was in 1984, and neither the original histopathologic report nor the slides were available for review. Cervical lymph node metastases were noted in 49 (79%) patients, and the lateral compartment was involved (N1b) in 36 (58%). Four patients had distant metastases to lung (cases 17, 23, 53), mediastinum (case 13) and bone (case 53) at the initial presentation.
Histologic and Molecular Features
As listed in Table 1, 25 different kinase fusions were found, each with a preserved kinase domain. Interestingly, all cases showed remarkably similar histologic features at low magnification. Multinodular growth was consistently observed, with dense arborizing bands of fibrosis entrapping and encircling irregular-shaped tumor nodules in a pattern somewhat reminiscent of hepatic cirrhosis (Figures 2 to 6). At higher magnification, the cellular architecture varied with the underlying molecular alterations, as described below. Extrathyroidal extension was present in 39 (63%) cases, involving the strap muscle in seven (11%) cases and beyond (T4 disease) in five (8%) (Figure 1). Lymphovascular invasion was seen in most patients (95%) except for cases 3, 10 and 49. The histopathologic and genetic findings of all the cases are summarized in Figure 1. A list of gene transcripts based on which we annotated the observed fusions is provided in Supplementary Table 1. All the kinase fusion-related cases underwent the BRAF V600E mutant-specific immunohistochemical staining and were all negative. All the cases profiled using SNaPshot lacked the BRAF V600E mutation (Figure 1).
Figure 2:

ALK-rearranged thyroid carcinomas. Case 2, an ALK-STRN fusion PTC, showed multinodular growth at low magnification with traversing fibrosis (A). Follicular architecture was seen at high magnification (B). Case 1, an ALK-STRN fusion PDTC with a concurrent TP53 mutation c.772G>C, consisted of numerous solid tumor nodules with comedo-type necrosis and extensive fibrosis (C). One 0.1 cm focus of follicular-patterned PTC was found within the tumor with abrupt transition to predominantly high-grade morphology characterized by admixed clear, oncocytic and squamoid cells and necrosis (D).
Figure 6:
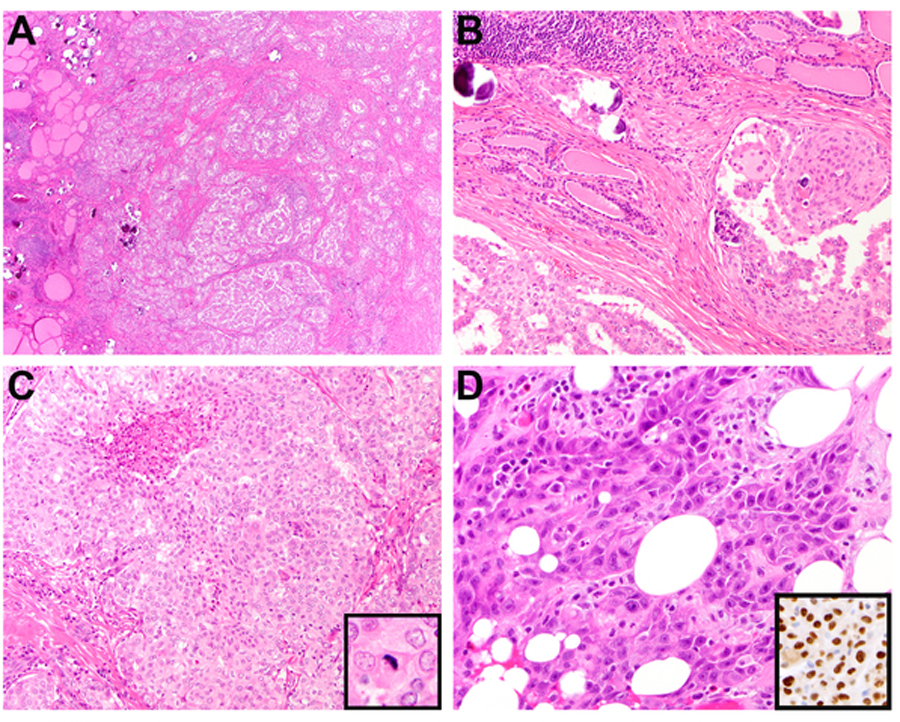
RET fusion-related thyroid carcinomas. Case 51, a CCDC6-RET fusion PTC, showed numerous cellular nodules and stromal sclerosis (A). Frequent, variably-sized psammoma bodies and tumor nests with squamous morular metaplasia were seen scattered within background thyroid (A-B). Case 46, a CCDC6-RET fusion PTC with concurrent CDH1 mutation, showed a solid architecture with up to three mitoses per 10 high-power fields and focal necrosis (C). Case 52, after a long-standing history of PTC since childhood, showed anaplastic transformation in the scapular metastatic tumor. The cells displayed moderate pleomorphism, frequent mitoses and squamoid cytology (D). PAX8 was diffusely positive (D, inset).
ALK-rearranged thyroid carcinomas
ALK rearrangements were found in one PDTC (case 1) and two PTC (cases 2 and 3). The two PTC, despite having different fusion partners (STRN and EML4), were morphologically similar with multinodular growth and predominantly follicular architecture (Figure 2A–2B). Scattered papillary architecture was present in case 3 (5% of tumor volume) whereas case 2 was entirely follicular-patterned. Calcifications were seen in case 2 (mostly dystrophic with rare psammoma bodies) but not in cases 3. There was no additional genetic alteration in either PTC by SNaPshot. Case 1, a PDTC harboring both a STRN-ALK fusion and a pathogenic TP53 mutation c.772G>C (p.Glu258Gln), consisted of solid nests of polygonal malignant cells with admixed clear, oncocytic and squamoid changes, along with frequent mitoses and necrosis (Figure 2C–2D). One 0.1 cm focus of follicular-patterned PTC was found within the tumor with abrupt transition into the predominantly poorly differentiated morphology (Figure 2D). IHC showed diffuse TTF1 expression, supporting a PDTC diagnosis.
BRAF-rearranged thyroid carcinomas
BRAF fusions were found in six PTC (cases 4 to 9) including two with high-grade features (cases 7 and 8). A packeted, papillary appearance was seen in cases 4, 5, 6 and 9, characterized by nodules of papillae encircled by dense fibrosis (Figure 3A–3B). The two BRAF-SND1 fusion PTC (cases 7 and 8) were histologically unique for having admixed classical (accounting for 40% and 20% of tumor volume, respectively), tall cell (40% and 10%), solid (10% and 30%) and follicular (10% and 40%) components and focal high-grade features (up to 4 mitoses/10 HPF in case 7; focal necrosis in both cases; Figure 3C–3D). Psammomatous calcifications were numerous in case 4, rare in case 5, and absent in cases 6 to 9.
Figure 3:
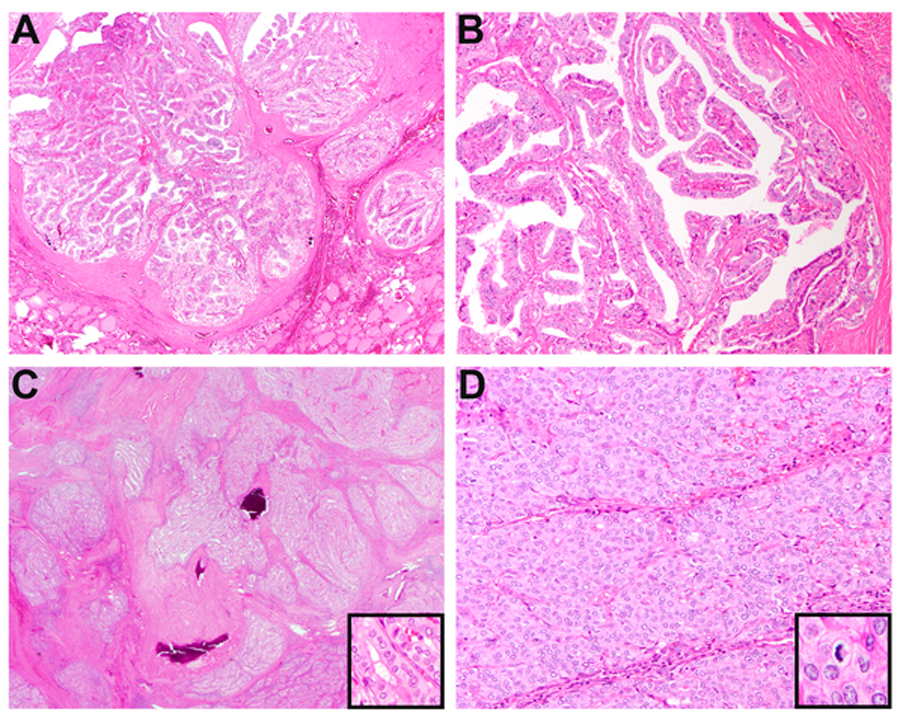
BRAF fusion-related thyroid carcinomas. Case 5, despite having additional TP53 and TERT promoter mutations, was histologically similar to cases 4, 6 and 9, consisting of multiple nodules of well-formed papillae encircled by dense fibrosis (A-B). Case 7, an SND1-BRAF fusion tall cell variant of PTC, showed extensive fibrosis that divided neoplastic cells into irregularly shaped islands (C, inset showing tall cell features). In approximately 10% of the tumor volume, a solid to trabecular architecture was noted along with increased mitotic activity (up to 4 mitoses per 10 high-power fields, inset) and focal necrosis (D).
SNaPshot mutational profiling revealed TERT pathogenic mutation c.−146C>T (C250T) in 3 out of the 5 tested (60%) patients (Figure 1). A TP53 in-frame deletion (p.Val218_Pro219delinsAla) was found in case 5. A SMARCA4 mutation (p.Gly1030Ser) of unknown significance was detected in case 9, which showed retained nuclear BRG1 expression by IHC.
MET-rearranged thyroid carcinomas
EML4-MET and TFG-MET fusions were detected in cases 10 and 11, respectively. The two PTC were similar in having a lobulated appearance, traversing fibrosis, and predominantly follicular architecture (Figure 4A). A papillary component was present in case 10 (20% of tumor volume; Figure 4A–4B) whereas case 11 was entirely follicular-patterned. In addition, case 11 showed diffuse clear cell change, as previously described (17).
Figure 4:

Case 10, an EML4-MET fusion PTC, was characterized by a lobulated appearance, traversing fibrosis, and predominantly follicular architecture with a minor papillary component (around 20% of tumor volume) (A-B). Case 62, a CCDC30-ROS1 fusion PTC, was composed of numerous solid nodules of pale to clear tumor cells with arborizing fibrosis (C-D).
NTRK-rearranged thyroid carcinomas (NRTC)
We recently described 11 NRTC (27), and since then seven new NRTC have been identified (cases 12, 15, 18, 19, 25, 29, 30) using our proposed morphologic approach. The new cases were all PTC and harbored fusions that included IRF2BP2-NTRK1, SQSTM1-NTRK1, TPM3-NTRK1, ETV6-NTRK3 and SQSTM1-NTRK3 (Table 1). In addition, although case 13 has been previously included in a validation study for the gene rearrangement NGS panel (28), its histopathologic features are first reported here. Overall, cases 12 (harboring IRF2BP2-NTRK1), 13 (PPL-NTRK1) and 25 (ETV6-NTRK3) were morphologically similar, composed of lobules of partially fused papillae with scattered glomeruloid structures and scant colloid production (Figure 5A–5B). IHC was performed in case 25 to exclude the possibility of SC phenotype and were positive for PAX8 (diffuse) and thyroglobulin (focal); S100, mammaglobin and GATA3 were all negative. The two TPM3-NTRK1 fusion PTC (cases 18 and 19) were both predominantly follicular-patterned with abundant colloid production and isolated foci of papillary architecture (Figure 5C). Case 15 (SQSTM1-NTRK1) had a mixture of 60% follicular, 20% solid and 20% papillary growth. The two SQSTM1-NTRK3 fusion PTC (cases 29 and 30) were architecturally different. Case 29 was entirely solid with scattered microfollicles (Figure 5D), whereas case 30 was entirely papillary. Psammomatous calcifications were seen in all the cases except for case 29.
Figure 5:
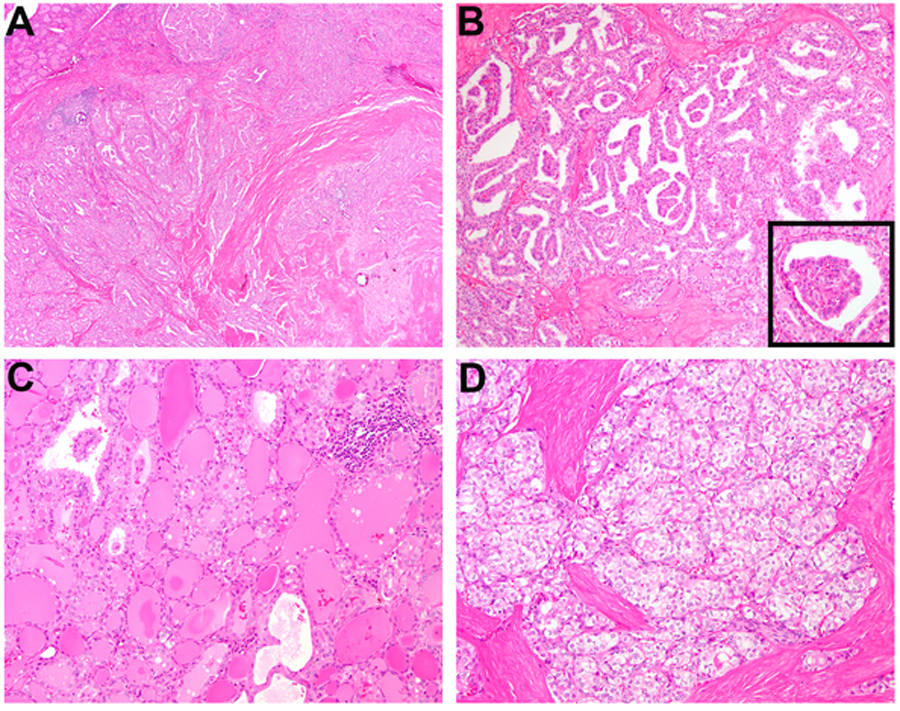
NTRK-rearranged thyroid carcinomas. Case 12, an IRF2BP2-NTRK1 fusion PTC, was composed of lobules of partially fused papillae with scant colloid production (A) and scattered glomeruloid structures and (B). Case 18, a TPM3-NTRK1 fusion PTC, was predominantly follicular-patterned with abundant colloid production and isolated foci of papillary formation (C). Case 29, an SQSTM1-NTRK3 fusion PTC demonstrated solid architecture with scattered microfollicles and prominent streaming fibrosis (D).
RET-rearranged thyroid carcinomas
RET fusions were detected in 28 PTC, one PDTC and two ATC. As summarized in Figure 1, most RET-rearranged PTC were morphologically similar, composed of nodules of crowded papillae with a minor follicular component and arborizing sclerosis (Figure 6A). The majority also showed squamous metaplasia, lymphocytic inflammation, and extensive lymphovascular spread of tumor cells and numerous psammoma bodies throughout the thyroid, thus fulfilling the defining features of the so-called diffuse sclerosing variant (Figure 6A–6B). Nine other cases had fewer psammoma bodies and lacked squamous metaplasia and were hence categorized as classical PTC. Case 46 was unique as a solid variant PTC with focal high-grade features (3 mitoses/10 HPF and focal necrosis; Figure 6C). Several mutations of uncertain significance were found in RET-rearranged PTC, including an in-frame duplication within VHL (p.Ser43_Glu47dup; case 32), a PIK3R1 splice site variant (c.1020G>A; case 33), and missense mutations in GNAQ (p.Arg166His; case 34), AKT2 (p.Asp151Glu; case 42), CDH1 (p.Asp334Glu in case 46, with retained membranous E-cadherin expression by IHC), JAK2 (p.Met88Thr; case 54) and TP53 (p.Val31Ile; case 61).
The only RET-rearranged PDTC was case 53, who presented with a mostly intra-mediastinal mass extending from the left inferior thyroid. The mass remained subclinical until extensive intrathoracic metastatic disease caused hoarseness due to recurrent laryngeal nerve involvement. Given the unresectable presentation, the patient underwent a core biopsy that showed monotonous malignant cells diffusely expressing TTF1 and PAX8. SNaPshot did not reveal additional genetic changes.
The two RET-rearranged ATC were cases 31 and 52. The histology and genetic abnormalities of case 31 were previously reported (31) with recent clinical progress updated in Table 2. Case 52 initially presented as a childhood PTC in 1984, which, after two recurrences in 1998 and 2012, exhibited accelerated progression in 2013 with widespread metastases to the sternum, scapula, brain, lung and breast. Resections of sternal and scapular metastases showed high-grade carcinoma with squamous differentiation (Figure 6D). There was diffuse expression of PAX8 (Figure 6D, inset) and p40 but TTF1 and thyroglobulin were lacking. The initial molecular characterization performed in a sample of post-radiation sternal metastatic tumor revealed a ERC1-RET fusion, a PTCH1 mutation of uncertain pathogenicity (p.His739Phe), a TERT promoter mutation c.−124C>T (C228T) and an NF2 deletion/insertions (delins) (p.GluLeu-GluArgArgLeuLeuGln355TerLeuGluArgArgLeuLeuHis). After progressing on lenvatinib and selpercatinib therapy, re-profiling of her intrapulmonary tumor showed the same RET fusion as well as pTERT and PTCH1 mutations but the NF2 deletion was no longer detected. In addition, a copy number gain of EGFR was newly identified.
Table 2:
Treatment history of patients receiving kinase inhibitor (KI) therapy
| Case #, Diagnosis | Pre-KI Management | Kinase Inhibitor Therapy and Responsea | Toxicity (gradeb) | ||
|---|---|---|---|---|---|
| First Regimen | Second Regimen | Third Regimen | |||
| 1, PDTC | T, M (STRN-ALK fusion, TP53 c.772G>C) | Sorafenib 400 mg BID, stopped after 19 days due to ischemic stroke. | Crizotinib 250 mg BID, stopped after 11 months due to PD. The BOR was SD. | Lenvatinib 14mg daily while on hospice, stopped after 1 month for lethargy. | Crizotinib: fatigue (1). Lenvatinib: small oral ulcers. |
| 13, PTC | T, RAI, M (PPL-NTRK1 fusion) | Trametinib 2 mg daily (redifferentiation therapy) for 30 days, followed by RAI; restaging imaging showed PD. | Larotrectinib* 100 mg BID; CR after 10 cycles; now 45 cycles into therapy with ongoing CR. | - | Trametinib: fatigue (1), dysphagia (1), acneiform eruption (3). Larotrectinib: fatigue (1), muscle cramping (1), dysgeusia (1). |
| 16, PTC | T, RAI, revision neck dissection, M (TPR-NTRK1 fusion) | Larotrectinib* 100 mg BID; now on cycle 21 with ongoing PR (33% decrease). | - | - | Fatigue (1), joint pain (1); transaminitis (1). |
| 20, PTC | T, RAI, resection and irradiation of brain metastasis, M (EML4-NTRK1 fusion; pTERT c.−124C >T; MEN1 frameshift deletion) | Entrectinib** 400 mg daily; now on cycle 38 with ongoing PR (69% decrease). | - | - | Orthostatic hypotension (3), gait disturbance (2), exertional dyspnea (1), fatigue (1), anemia (2), leukopenia (1), serum creatinine elevation (1). |
| 24, SC | T, RAI, irradiation of cervical recurrence; M (ETV6-NTRK3 fusion) | Larotrectinib* 100 mg BID; now on cycle 29 with ongoing PR (100% decrease of measurable tumor and undetectable thyro-globulin, but deemed PR due to small persistent lung lesions likely granulomata). | - | - | Instability (1), hypophosphatemia (1). |
| 31, ATC | T, RAI, radiosurgery of brain metastasis, one cycle of Adriamycin/Taxotere; M (CCDC6-RET, pTERT, TP53, TSC2, PTCH1 mutations) | Selpercatinib*** 160 mg BID; now on cycle 28 with ongoing PR (64% decrease). | - | - | Bowel edema (1). |
| 41, PTC | T, RAI, resection and irradiation of paraspinal metastasis, M (CCDC6-RET fusion) | Selpercatinib*** 160 mg BID; now on cycle 18 with ongoing PR (61% decrease). | - | - | Hypertension (1). |
| 52, ATC | T, RAI, resection and irradiation of metastases, M (ERC1-RET fusion; pTERT, PTCH1 and NF2 mutations) | Lenvatinib 14mg daily, with a BOR of SD, stopped after 20 months of treatment due to PD. |
Selpercatinib*** 160 mg BID, stopped due to PD after 22 cycles (21 months). The BOR was PR (63% decrease). Posttreatment M: new EGFR copy number gain. |
Cabozantinib 40 mg daily for 3 months followed by 60 mg daily for one month, then stopped due to wound infection; restaging imaging showed PD. Pembrolizumab was subsequently started one month prior to death. | Lenvatinib: hand-foot syndrome (1); transaminitis (1), hyperbilirubinemia (2). Selpercatinib: fatigue (1), transaminitis (1), hypertension (1), thrombocytopenia (1). Cabozantinib: transaminitis (2). |
| 53, PDTC | Radiotherapy; M (NCOA4-RET fusion). | Lenvatinib 24 mg daily, stopped after five months due to PD. | Selpercatinib*** 60–80 mg BID for 8 cycles, followed by 160 mg BID for 10 cycles and then stopped due to PD. The BOR was PR (72% decrease). | Cabozantinib 40 mg daily for 6 months, then combined with pembrolizumab for 2 months before patient died of advanced disease. | Lenvatinib: fatigue (1). Selpercatinib: diarrhea (1), transaminitis (3). Cabozantinib: diarrhea (2), mucositis (1). |
| 58, PTC | T, RAI, radiotherapy, M (NCOA4-RET fusion) | Lenvatinib 24 mg daily; PD leading to death 8 months after treatment initiation. | - | - | None documented. |
Abbreviations: T, thyroidectomy; M, molecular profiling of tumor (see text for alteration details); BID, twice daily; RAI, radioactive iodine; BOR, best overall response; SD, stable disease; PD, progressive disease; CR, complete response; PR, partial response; PTC, papillary thyroid carcinoma; PDTC, poorly differentiated thyroid carcinoma; SC, secretory carcinoma; ATC, anaplastic thyroid carcinoma.
Response was evaluated based on the Response Evaluation Criteria In Solid Tumors (RECIST) criteria version 1.1.
Based on the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. ClinicalTrials.gov identifiers:
ROS1-rearranged thyroid carcinomas
A CCDC30-ROS1 fusion was observed in a young adult with a solid variant PTC (case 62), which had a prior case report at the time of diagnosis in 2016 (16). Briefly, the 3.5 cm tumor was composed of numerous oval solid nodules of pale to clear cells encircled by branching collagenous bundles (Figure 4C–4D). SNaPshot revealed an FBXW7 splice acceptor variant (c.585–5T>A). The patient was subsequently followed for 20 months as presented below.
Treatment and Outcome
All patients except for case 53 had total thyroidectomy, and most received adjuvant radioactive iodine (RAI)(Table 1). We evaluated patients for response to RAI; 32 out of 62 did not have adequate follow-up information or received RAI in the adjuvant setting, and their response was thus not evaluable. Among the 30 patients with evaluable data, there were 26 showing evidence of refractoriness based on the 2015 American Thyroid Association guidelines (32), whereas four patients were responsive (Supplementary Table 2). Overall, among the 47 KFTC cases with at least six months of follow-up (median [range]: 41 [6 to 480] months), 20 (43%) patients had no evidence of recurrent disease, despite 10 of them having presented with nodal metastasis. The other 27 (57%) patients had persistent or recurrent disease and 18 (38%) developed distant metastases. The distant metastatic rate appeared to be the highest in NTRK-rearranged thyroid carcinomas (7 out of 12 cases [58%] with >6 months follow-up), followed by RET-rearranged thyroid carcinomas (9/25, 36%), ALK-rearranged thyroid carcinomas (1/3, 33%) and BRAF-rearranged thyroid carcinomas (1/5, 20%). All MET- or ROS1-rearranged cases remained free of locoregional and distant metastatic disease at last follow-up. Three (6%) patients eventually died of advanced RET-rearranged thyroid carcinomas (cases 52, 53 and 58), while mortality was not noted in patients with tumors driven by other kinases. When only PTC cases were considered, a total of 42 PTC cases had >6 months of follow-up (median [range]: 41 [6 to 480] months); persistent/recurrent disease, distant metastasis and thyroid cancer-related death was noted in 59% (25/42), 33% (14/42) and 2% (1/42) of PTC, respectively, at the last follow-up (Table 1, Figure 1). Multi-kinase and/or selective kinase inhibitors were prescribed to ten patients, and their treatment history, response and toxicity profiles are summarized in Table 2.
Discussion
Targeted kinase inhibitors (KI) play a prominent role in the modern advancement of oncologic care. Their successful application has proven highly efficacious in patients with advanced cancers who had previously been deemed to be terminal. KI also represent a substantial cause of healthcare expenditure, and the importance of laboratory guidance, both histologic and molecular, in identifying targetable cases cannot be overstated. In this study, we pointed out a characteristic and reproducible histologic pattern of KFTC. The triad of multinodularity, prominent fibrosis, and lymphovascular invasion were seen in 95% of the cases, and the former two features were present in 100% of our cohort, which encompassed PTC, SC, PDTC and ATC driven by a wide range of kinase fusions involving ALK, BRAF, MET, NTRK1/3, RET and ROS1. In addition, we also observed high percentages of extrathyroidal extension (ETE), micro and macroscopic (63%), lymph node involvement, central and lateral neck (79%) and distant metastasis (38%). Of note, our cohort was mostly radiation-naïve adults, whereas most of the existing KFTC literature had been conducted in pediatric and radiation-associated patients. Recently, Prasad et al. reported similar histologic findings of multinodularity, scar-like fibrosis and frequent lymphovascular spread in pediatric NTRK-rearranged thyroid carcinomas (33). In another study, Pekova et al. examined 52 pediatric KFTC carrying RET, NTRK1/3, ALK, BRAF and MET fusions and noted frequent multifocality (53.8%), extrathyroidal extension (40.9%) and lymph node metastasis (62.4%). Elevated metastatic rates have also been reported by other researchers in post-radiation and sporadic thyroid carcinomas driven by RET (34–36), BRAF (37), NTRK (38) and ALK (39, 40) fusions. Although our MET fusion cases were non-metastatic, one of the only two previously published MET-rearranged PTC (15, 41) had lymph node and pulmonary metastases. Both case 62 and the only other known case of ROS1-rearranged thyroid carcinoma were metastatic (42). Overall, KFTC appeared to form a group of clinically aggressive tumors with overlapping clinicopathologic characteristics, which can serve as helpful clues for pathologists to initiate molecular testing. An algorithmic approach for identifying actionable kinase fusions in thyroid carcinomas is proposed in Figure 7.
Figure 7:
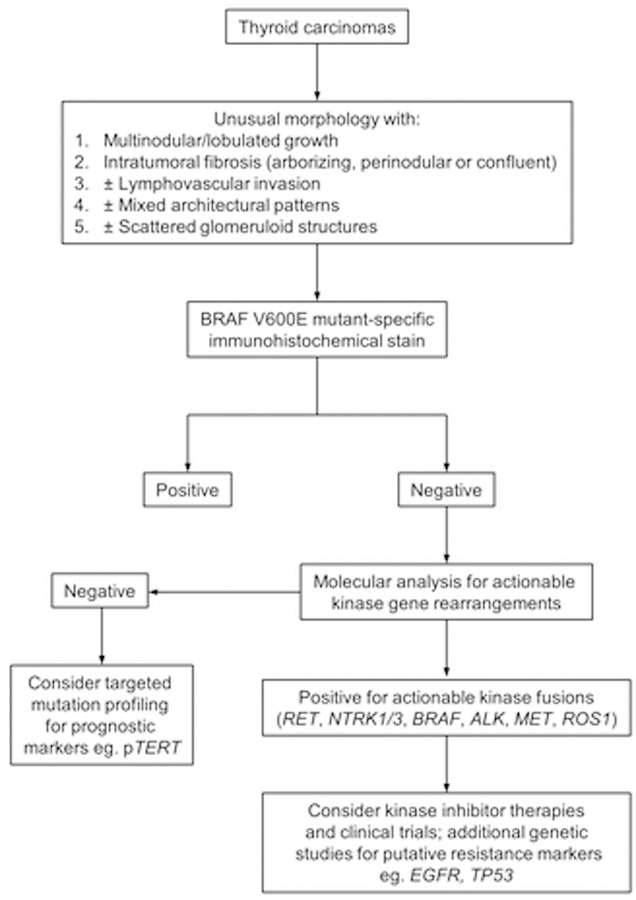
Proposed algorithmic approach for identifying actionable kinase gene fusions in thyroid carcinomas
The histologic concept of “diffuse sclerosing variant (DSV)” of PTC has been commonly associated with RET rearrangements in the literature (43, 44). Rendering a DSV diagnosis can be somewhat subjective, but the main defining features include extensive lymphovascular involvement, stromal sclerosis, prominent lymphocytosis, squamous metaplasia, and numerous psammoma bodies. Although the former two features were present in the vast majority of our KFTC cohort regardless of fusion type, we saw squamous morule formation exclusively in the RET fusion cases. Moreover, while most of our RET fusion tumors showed countless psammoma bodies, cases driven by other kinases had few or none with rare exceptions (cases 4, 13, 16, 17). As a result, among a total of 21 DSV PTC in our cohort, 18 (86%) were associated with RET fusions, while NTRK1 and BRAF fusions were implicated in 2 (10%) and one (5%) case, respectively. Previously, Joung et al, in examining 37 DSV PTC, reported BRAF V600E mutation in 24% (44). Taken together, one plausible approach for identifying therapeutic targets in DSV cases may include immunohistochemical staining for BRAF V600E mutation, followed by molecular testing for RET, NTRK and BRAF rearrangements if negative. Another histologic type that is historically associated with RET fusions is the solid variant PTC, which was also seen in NTRK- and ROS1-rearranged PTC in our cohort (Figure 1).
Although previous studies have suggested that CCDC6-RET fusion PTC were generally indolent with a low propensity for high grade transformation (45), cases 31 (a CCDC6-RET fusion ATC) and 46 (a CCDC6-RET fusion PTC with high-grade features) exemplified the dedifferentiating potential of these tumors. It was noteworthy that both cases had genetic co-alterations. In addition to the CCDC6-RET fusion, case 46 also carried a CDH1 missense variant of uncertain pathogenicity. We performed immunohistochemistry and noticed retained membranous E-cadherin staining. Nevertheless, it has been documented in the literature that aberrant E-cadherin expression can occasionally occur with carcinogenic CDH1 mutations (46). Case 46 had four co-occurring mutations involving TP53, TERT promoter, TSC2 and PTCH1 (31). We also noted various degree of high-grade features in other RET fusion partners (cases 52 and 53) as well as ALK, BRAF, and NTRK-rearranged tumors (cases 1, 7, 8 and 28; Figure 1). The average number of co-altered genes were 3.50, 0.50 and 0.50 and 0.45 per case for ATC, PDTC, PTC with high-grade features, and PTC without high-grade features, respectively (Figure 1). The increasing number of co-alterations accompanied by higher histologic grade supports a significant role for mutation accumulation in escalating the biological potential of KFTC. Previous genomic studies in RET, ALK, BRAF and NTRK fusion-related ATC and PDTC using larger sequencing panels have reported similar mutation burdens involving a similar set of genes, such as TP53, pTERT and SMARCA4 (47–49). It should be noted that we performed molecular analysis only in tumor tissue samples without paired normal tissue testing. Therefore, we cannot exclude the possibility that some of the detected variants, particularly those that lack clear pathogenicity data in the literature, may represent rare germline variants.
We were particularly interested in correlating genetic abnormalities with patient response to kinase inhibitor therapy. As summarized in Table 2, four cases exhibiting kinase inhibitor resistance were encountered (cases 1, 52, 53 and 58). Case 1 was found harboring both a STRN-ALK fusion and TP53 c.772G>C mutation before starting crizotinib treatment. His tumor burden initially appeared to stabilize but progressed within a year. STRN-ALK fusion thyroid and lung carcinomas, although rare, have been putatively sensitive to crizotinib based on a total of three case reports and one in vitro study (39, 40, 50, 51). Although no prior study has examined the drug sensitivity of TP53-mutant STRN-ALK fusion tumors, Kron et al. have recently demonstrated that concurrent TP53 mutations represent a significant unfavorable outcome predictor in ALK-rearranged lung cancer patients in all the analyzed treatment subgroups (crizotinib, next-generation ALK inhibitors, chemotherapy) (52). Several other studies have also associated TP53 mutations with poor crizotinib response (53–55). Cases 52, 53 and 58 initially received lenvatinib, a repurposed VEGFR inhibitor with suboptimal efficacy against RET-driven tumors (2), and experienced disease progression. Cases 52 and 53 were subsequently treated with selpercatinib. Interestingly, both patients had been showing clear response (63% and 72% reduction of tumor burden) for longer than a year before developing resistance (Table 2). Post-treatment tumor genetic profiling was performed in case 52 and revealed a new copy number gain of EGFR, which had been absent in the pre-treatment sample. Activation of bypass signaling is a well-recognized mechanism for kinase inhibitor resistance (2). Although secondary EGFR gain has not been previously reported in selpercatinib-treated patients, EGFR is a well-established mediator of acquired resistance to kinase inhibitor therapy in ALK, RET and ROS1 fusion-driven lung cancers (56–59). The mechanism of acquired resistance in case 53 was unclear since no post-treatment molecular profiling was performed. It was noteworthy that several co-alterations were found in patients with durable response to kinase inhibitors, such as pTERT c.−124C >T (cases 20 and 31), MEN1 p.Pro55LeufsTer64 (case 20) (27), TP53 p.Pro152TrpfsTer10 (case 31), PTCH1 (p.Ala4Thr, case 31) and TSC2 (p.Gly1356Ser, case 31) (31).
BRAF fusions occur in around 1–2% of thyroid carcinomas, mainly PTC, with a largely uncharacterized prognosis (60, 61). The frequency is higher in pilocytic astrocytoma (60–80%), cutaneous Spitzoid melanomas (75%), and pancreatic acinar cell carcinomas (20%) (61–63). The clinical management of BRAF fusion-driven tumors represents an active area of research that currently has very limited patient data. Isolated case studies in astrocytoma and melanoma have showed highly variable patient response to RAF and MEK inhibitors (64, 65). Most interestingly, one recent study using patient-derived melanoma cells expressing BRAF fusion transcripts have found the sensitivity to RAF inhibitors dependent on whether the 5’ end fusion partner contributes additional dimerization domain(s). Partner-derived dimerization domains cause conformation change-mediated paradoxical activation of MAPK pathway and accelerated tumor growth when treated with first and second generation RAF inhibitors (66). Although BRAF fusions with (eg. AKAP9-BRAF) and without (eg. AGK-BRAF, CUL1-BRAF) partner-derived dimerization domains have both been discovered in thyroid carcinomas (14, 15, 67), none of the fusion partners in our cases have a known dimerization domain. Since our cases did not receive kinase inhibitor therapy, their sensitivity cannot be determined. Nevertheless, compared to the NTRK and RET fusion cases, BRAF fusion tumors appeared to have a better prognosis in our cohort, with the majority achieving remission after thyroidectomy and RAI treatment (Figure 1). Additional clinical research is needed to elucidate the behavior of BRAF fusion-driven thyroid carcinomas and the therapeutic utility of MAPK pathway inhibitors.
In summary, our findings highlight a unique histologic pattern shared by a wide morphologic (PTC, SC, PDTC and ATC) and molecular (RET, NTRK, BRAF, ALK, MET, and ROS1 driven) spectrum of kinase fusion-driven thyroid carcinomas (KFTC). The triad of multinodularity, intratumoral fibrosis and lymphovascular involvement can serve as a helpful diagnostic aid for pathologists’ recognition of these clinically aggressive but highly treatable tumors. In addition, we uncovered multiple genetic co-alterations in KFTC, which demonstrated significant consequence to patient response to targeted kinase inhibitor therapy. The study had several limitations, including a lack of paired normal tissue genetic testing and possible selection bias resulting from preferential sequencing of clinically aggressive cases. Nevertheless, we expect this data to contribute to the current understanding of KFTC with anticipated benefit for patients.
Supplementary Material
Footnotes
Disclosure
Dr. Wirth received support from Loxo Oncology, Inc. and Genentech USA, Inc. for costs of the clinical trials listed in the footnotes of Table 2. All the other authors declare no conflict of interest.
References
- 1.Bhullar KS, Lagarón NO, McGowan EM, Parmar I, Jha A, Hubbard BP, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 2018;17:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Subbiah V, Cote GJ. Advances in Targeting RET-Dependent Cancers. Cancer Discov 2020;10:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuroda N, Trpkov K, Gao Y, Tretiakova M, Liu YJ, Ulamec M, et al. ALK rearranged renal cell carcinoma (ALK-RCC): a multi-institutional study of twelve cases with identification of novel partner genes CLIP1, KIF5B and KIAA1217. Mod Pathol 2020. [DOI] [PubMed]
- 4.Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep 2018;23:227–38.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wai DH, Knezevich SR, Lucas T, Jansen B, Kay RJ, Sorensen PH. The ETV6-NTRK3 gene fusion encodes a chimeric protein tyrosine kinase that transforms NIH3T3 cells. Oncogene 2000;19:906–15. [DOI] [PubMed] [Google Scholar]
- 6.Farago AF, Taylor MS, Doebele RC, Zhu VW, Kummar S, Spira AI, et al. Clinicopathologic Features of Non-Small-Cell Lung Cancer Harboring an NTRK Gene Fusion. JCO Precis Oncol 2018;2018. [DOI] [PMC free article] [PubMed]
- 7.Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 2018;15:731–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shin CH, Grossmann AH, Holmen SL, Robinson JP. The BRAF kinase domain promotes the development of gliomas in vivo. Genes Cancer 2015;6:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vojnic M, Kubota D, Kurzatkowski C, Offin M, Suzawa K, Benayed R, et al. Acquired BRAF Rearrangements Induce Secondary Resistance to EGFR therapy in EGFR-Mutated Lung Cancers. J Thorac Oncol 2019;14:802–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ricarte-Filho JC, Li S, Garcia-Rendueles ME, Montero-Conde C, Voza F, Knauf JA, et al. Identification of kinase fusion oncogenes in post-Chernobyl radiation-induced thyroid cancers. J Clin Invest 2013;123:4935–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slotkin EK, Diolaiti D, Shukla NN, Dela Cruz FS, Clark JJ, Gundem G, et al. Patient-Driven Discovery, Therapeutic Targeting, and Post-Clinical Validation of a Novel AKT1 Fusion-Driven Cancer. Cancer Discov 2019;9:605–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kannan K, Coarfa C, Chao PW, Luo L, Wang Y, Brinegar AE, et al. Recurrent BCAM-AKT2 fusion gene leads to a constitutively activated AKT2 fusion kinase in high-grade serous ovarian carcinoma. Proc Natl Acad Sci U S A 2015;112:E1272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matissek KJ, Onozato ML, Sun S, Zheng Z, Schultz A, Lee J, et al. Expressed Gene Fusions as Frequent Drivers of Poor Outcomes in Hormone Receptor-Positive Breast Cancer. Cancer Discov 2018;8:336–53. [DOI] [PubMed] [Google Scholar]
- 14.Cordioli MI, Moraes L, Bastos AU, Besson P, Alves MT, Delcelo R, et al. Fusion Oncogenes Are the Main Genetic Events Found in Sporadic Papillary Thyroid Carcinomas from Children. Thyroid 2017;27:182–8. [DOI] [PubMed] [Google Scholar]
- 15.Pekova B, Sykorova V, Dvorakova S, Vaclavikova E, Moravcova J, Katra R, et al. RET, NTRK, ALK, BRAF and MET fusions in a large cohort of pediatric papillary thyroid carcinomas. Thyroid 2020. [DOI] [PubMed]
- 16.Ritterhouse LL, Wirth LJ, Randolph GW, Sadow PM, Ross DS, Liddy W, et al. ROS1 Rearrangement in Thyroid Cancer. Thyroid 2016;26:794–7. [DOI] [PubMed] [Google Scholar]
- 17.Cipriani NA, Agarwal S, Dias-Santagata D, Faquin WC, Sadow PM. Clear Cell Change in Thyroid Carcinoma: A Clinicopathologic and Molecular Study with Identification of Variable Genetic Anomalies. Thyroid 2017;27:819–24. [DOI] [PubMed] [Google Scholar]
- 18.Okamura R, Boichard A, Kato S, Sicklick JK, Bazhenova L, Kurzrock R. Analysis of NTRK Alterations in Pan-Cancer Adult and Pediatric Malignancies: Implications for NTRK-Targeted Therapeutics. JCO Precis Oncol 2018;2018. [DOI] [PMC free article] [PubMed]
- 19.Liang J, Cai W, Feng D, Teng H, Mao F, Jiang Y, et al. Genetic landscape of papillary thyroid carcinoma in the Chinese population. J Pathol 2018;244:215–26. [DOI] [PubMed] [Google Scholar]
- 20.Hong DS, DuBois SG, Kummar S, Farago AF, Albert CM, Rohrberg KS, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 2020;21:531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wirth LJ, Sherman E, Drilon A, Solomon B, Robinson B, Lorch J, et al. Registrational results of LOXO-292 in patients with RET-altered thyroid cancers. In: European Society for Medical Oncology 2019. Congress.
- 22.Shah MH, Sherman EJ, Robinson B, Solomon BJ, Kang H, Lorch JH, et al. Selpercatinib (LOXO-292) in patients with RET-mutant medullary thyroid cancer. Journal of Clinical Oncology 2020;38:3594. [Google Scholar]
- 23.Subbiah V, Hu MI-N, Gainor JF, Mansfield AS, Alonso G, Taylor MH, et al. Clinical activity of the RET inhibitor pralsetinib (BLU-667) in patients with RET fusion+ solid tumors. Journal of Clinical Oncology 2020;38:109. [Google Scholar]
- 24.Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, Lee J, et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372–001 and STARTRK-1). Cancer Discov 2017;7:400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer Discov 2017;7:963–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cocco E, Schram AM, Kulick A, Misale S, Won HH, Yaeger R, et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat Med 2019;25:1422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chu YH, Dias-Santagata D, Farahani AA, Boyraz B, Faquin WC, Nosé V, et al. Clinicopathologic and molecular characterization of NTRK-rearranged thyroid carcinoma (NRTC). Mod Pathol 2020. [DOI] [PMC free article] [PubMed]
- 28.Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med 2014;20:1479–84. [DOI] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med 2010;2:146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dias-Santagata D, Lennerz JK, Sadow PM, Frazier RP, Raju SG, Henry D, et al. Response to RET-Specific Therapy in RET Fusion-Positive Anaplastic Thyroid Carcinoma. Thyroid 2020. [DOI] [PMC free article] [PubMed]
- 32.Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016;26:1–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prasad ML, Vyas M, Horne MJ, Virk RK, Morotti R, Liu Z, et al. NTRK fusion oncogenes in pediatric papillary thyroid carcinoma in northeast United States. Cancer 2016;122:1097–107. [DOI] [PubMed] [Google Scholar]
- 34.Tallini G, Santoro M, Helie M, Carlomagno F, Salvatore G, Chiappetta G, et al. RET/PTC oncogene activation defines a subset of papillary thyroid carcinomas lacking evidence of progression to poorly differentiated or undifferentiated tumor phenotypes. Clin Cancer Res 1998;4:287–94. [PubMed] [Google Scholar]
- 35.Adeniran AJ, Zhu Z, Gandhi M, Steward DL, Fidler JP, Giordano TJ, et al. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am J Surg Pathol 2006;30:216–22. [DOI] [PubMed] [Google Scholar]
- 36.Pisarchik AV, Ermak G, Fomicheva V, Kartel NA, Figge J. The ret/PTC1 rearrangement is a common feature of Chernobyl-associated papillary thyroid carcinomas from Belarus. Thyroid 1998;8:133–9. [DOI] [PubMed] [Google Scholar]
- 37.Sisdelli L, Cordioli M, Vaisman F, Moraes L, Colozza-Gama GA, Alves PAG Jr., et al. AGK-BRAF is associated with distant metastasis and younger age in pediatric papillary thyroid carcinoma. Pediatr Blood Cancer 2019;66:e27707. [DOI] [PubMed] [Google Scholar]
- 38.Musholt TJ, Musholt PB, Khaladj N, Schulz D, Scheumann GF, Klempnauer J. Prognostic significance of RET and NTRK1 rearrangements in sporadic papillary thyroid carcinoma. Surgery 2000;128:984–93. [DOI] [PubMed] [Google Scholar]
- 39.Kelly LM, Barila G, Liu P, Evdokimova VN, Trivedi S, Panebianco F, et al. Identification of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci U S A 2014;111:4233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pérot G, Soubeyran I, Ribeiro A, Bonhomme B, Savagner F, Boutet-Bouzamondo N, et al. Identification of a recurrent STRN/ALK fusion in thyroid carcinomas. PLoS One 2014;9:e87170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agrawal N, Akbani R, Aksoy B, Ally A, Arachchi H, Asa S, et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014;159:676–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu SV, Macke LA, Colton BS, Imran SS, Christiansen J, Chow-Maneval E, et al. Response to Entrectinib in Differentiated Thyroid Cancer With a ROS1 Fusion. JCO Precision Oncology 2017:1–5. [DOI] [PMC free article] [PubMed]
- 43.Sheu SY, Schwertheim S, Worm K, Grabellus F, Schmid KW. Diffuse sclerosing variant of papillary thyroid carcinoma: lack of BRAF mutation but occurrence of RET/PTC rearrangements. Mod Pathol 2007;20:779–87. [DOI] [PubMed] [Google Scholar]
- 44.Joung JY, Kim TH, Jeong DJ, Park SM, Cho YY, Jang HW, et al. Diffuse sclerosing variant of papillary thyroid carcinoma: major genetic alterations and prognostic implications. Histopathology 2016;69:45–53. [DOI] [PubMed] [Google Scholar]
- 45.Acquaviva G, Visani M, Repaci A, Rhoden KJ, de Biase D, Pession A, et al. Molecular pathology of thyroid tumours of follicular cells: a review of genetic alterations and their clinicopathological relevance. Histopathology 2018;72:6–31. [DOI] [PubMed] [Google Scholar]
- 46.Grabenstetter A, Mohanty A, DeLair D, Tan L, Ross D. Abstract PD7–05: Correlation of CDH1 alterations and aberrant E-cadherin expression in lobular carcinomas. Cancer Research 2019;79:PD7-05-PD7-. [Google Scholar]
- 47.Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest 2016;126:1052–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, et al. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clinical Cancer Research 2018;24:3059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu B, Fuchs T, Dogan S, Landa I, Katabi N, Fagin JA, et al. Dissecting Anaplastic Thyroid Carcinoma: A Comprehensive Clinical, Histologic, Immunophenotypic, and Molecular Study of 360 Cases. Thyroid 2020. [DOI] [PMC free article] [PubMed]
- 50.Zhou C, Zeng L, Zhang Y, Yang N. Responder of Gefitinib Plus Crizotinib in Osimertinib Failure EGFR-mutant NSCLC-Resistant With Newly Identified STRN-ALK by Next-Generation Sequencing. J Thorac Oncol 2019;14:e143–e4. [DOI] [PubMed] [Google Scholar]
- 51.Yang Y, Qin SK, Zhu J, Wang R, Li YM, Xie ZY, et al. A Rare STRN-ALK Fusion in Lung Adenocarcinoma Identified Using Next-Generation Sequencing-Based Circulating Tumor DNA Profiling Exhibits Excellent Response to Crizotinib. Mayo Clin Proc Innov Qual Outcomes 2017;1:111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kron A, Alidousty C, Scheffler M, Merkelbach-Bruse S, Seidel D, Riedel R, et al. Impact of TP53 mutation status on systemic treatment outcome in ALK-rearranged non-small-cell lung cancer. Ann Oncol 2018;29:2068–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Couëtoux du Tertre M, Marques M, Tremblay L, Bouchard N, Diaconescu R, Blais N, et al. Analysis of the Genomic Landscape in ALK+ NSCLC Patients Identifies Novel Aberrations Associated with Clinical Outcomes. Mol Cancer Ther 2019;18:1628–36. [DOI] [PubMed] [Google Scholar]
- 54.Lin C, Shi X, Yang S, Zhao J, He Q, Jin Y, et al. Comparison of ALK detection by FISH, IHC and NGS to predict benefit from crizotinib in advanced non-small-cell lung cancer. Lung Cancer 2019;131:62–8. [DOI] [PubMed] [Google Scholar]
- 55.Pailler E, Faugeroux V, Oulhen M, Mezquita L, Laporte M, Honoré A, et al. Acquired Resistance Mutations to ALK Inhibitors Identified by Single Circulating Tumor Cell Sequencing in ALK-Rearranged Non-Small-Cell Lung Cancer. Clin Cancer Res 2019;25:6671–82. [DOI] [PubMed] [Google Scholar]
- 56.Chang H, Sung JH, Moon SU, Kim HS, Kim JW, Lee JS. EGF Induced RET Inhibitor Resistance in CCDC6-RET Lung Cancer Cells. Yonsei Med J 2017;58:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nelson-Taylor SK, Le AT, Yoo M, Schubert L, Mishall KM, Doak A, et al. Resistance to RET-Inhibition in RET-Rearranged NSCLC Is Mediated By Reactivation of RAS/MAPK Signaling. Mol Cancer Ther 2017;16:1623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A Novel ALK Secondary Mutation and EGFR Signaling Cause Resistance to ALK Kinase Inhibitors. Cancer Research 2011;71:6051–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song A, Kim TM, Kim D-W, Kim S, Keam B, Lee S-H, et al. Molecular Changes Associated with Acquired Resistance to Crizotinib in ROS1-Rearranged Non–Small Cell Lung Cancer. Clinical Cancer Research 2015;21:2379–87. [DOI] [PubMed] [Google Scholar]
- 60.Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun 2014;5:4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ross JS, Wang K, Chmielecki J, Gay L, Johnson A, Chudnovsky J, et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 2016;138:881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nikiforova MN, Hamilton RL. Molecular diagnostics of gliomas. Arch Pathol Lab Med 2011;135:558–68. [DOI] [PubMed] [Google Scholar]
- 63.Chmielecki J, Hutchinson KE, Frampton GM, Chalmers ZR, Johnson A, Shi C, et al. Comprehensive genomic profiling of pancreatic acinar cell carcinomas identifies recurrent RAF fusions and frequent inactivation of DNA repair genes. Cancer Discov 2014;4:1398–405. [DOI] [PubMed] [Google Scholar]
- 64.Johnson DB, Zhao F, Noel M, Riely GJ, Mitchell EP, Wright JJ, et al. Trametinib Activity in Patients with Solid Tumors and Lymphomas Harboring BRAF Non-V600 Mutations or Fusions: Results from NCI-MATCH (EAY131). Clin Cancer Res 2020;26:1812–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Botton T, Yeh I, Nelson T, Vemula SS, Sparatta A, Garrido MC, et al. Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment Cell Melanoma Res 2013;26:845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Botton T, Talevich E, Mishra VK, Zhang T, Shain AH, Berquet C, et al. Genetic Heterogeneity of BRAF Fusion Kinases in Melanoma Affects Drug Responses. Cell Rep 2019;29:573–88.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ciampi R, Knauf JA, Kerler R, Gandhi M, Zhu Z, Nikiforova MN, et al. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest 2005;115:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.