

Abstract
Aims
To demonstrate the bioequivalence of macitentan/tadalafil fixed‐dose combination (FDC) tablets with single‐component tablets of macitentan and tadalafil in healthy subjects.
Methods
Studies AC‐077‐101 and AC‐077‐103 were single‐centre, open‐label, single‐dose, 2‐period, randomized, crossover Phase 1 studies conducted in healthy subjects. Two FDCs were investigated: FDC‐1 and FDC‐2 in Study AC‐077‐101 and FDC‐2 in Study AC‐077‐103. Both FDCs contained 10 mg/40 mg of macitentan/tadalafil and differed in excipients and coating materials used. In both studies, pharmacokinetic sampling over 216 hours was conducted, and pharmacokinetic parameters were derived using noncompartmental methods.
Results
Bioequivalence of macitentan, its active metabolite ACT‐132577, and tadalafil was established for FDC‐2 in both studies AC‐077‐101 and AC‐077‐103 in which tadalafil as a single component was sourced from the USA and EU, respectively, to fulfil regional regulatory requirements. The area under the plasma concentration–time curve and maximum plasma concentration with 90% confidence intervals of all components were entirely within the bioequivalence limits (0.8000–1.2500). No subject died and no serious adverse events were reported in either studies.
Conclusion
The FDC‐2 tablet containing 10 mg/40 mg of macitentan/tadalafil was bioequivalent to the free combination of 10 mg macitentan and 40 mg tadalafil (both US and EU sourced). Macitentan and tadalafil were well tolerated when administered as FDC or as a free combination.
Keywords: bioequivalence, fixed‐dose combination, macitentan, pulmonary arterial hypertension, tadalafil
What is already known about this subject
Macitentan is approved for use as monotherapy or in combination with phosphodiesterase‐5 inhibitors for pulmonary arterial hypertension (PAH). No fixed‐dose combination (FDC) is available for the treatment of PAH.
An FDC would offer PAH patients a means to take concomitant macitentan and tadalafil as a single dose, facilitating compliance.
What this study adds
FDC‐2 tablet containing 10 mg/40 mg of macitentan/tadalafil was bioequivalent to the free combination of 10 mg macitentan and 40 mg tadalafil.
Patients would be able to use the FDC‐2 instead of the free combination without need for dose‐adjustments.
1. INTRODUCTION
Pulmonary arterial hypertension (PAH) is a chronic disease characterized by an increase in pulmonary vascular resistance, which leads to right ventricular failure and ultimately death if not treated. 1 , 2
Macitentan is an orally active, nonpeptide, potent dual endothelin receptor antagonist. Macitentan, brand name Opsumit (macitentan 10 mg film‐coated tablet) has been approved for the treatment of PAH at a dose of 10 mg once daily in the USA, European Economic Area, Canada, Australia, Switzerland, Japan, and additional countries in the Middle East, Asia and Latin America. Macitentan is approved for use as monotherapy or in combination with phosphodiesterase‐5 (PDE‐5) inhibitors or inhaled prostanoids.
Tadalafil is a selective PDE‐5 inhibitor approved in the USA, European Union (EU) and other countries under the brand name Adcirca (20 mg film‐coated tablet) for the treatment of PAH at a dose of 20–40 mg once daily. Endothelin receptor antagonists and PDE‐5 inhibitors are the most widely used therapies in PAH and are also the drug classes that are most frequently used in combination.
The approval of macitentan for use in monotherapy and combination therapy with PDE‐5 inhibitors was based on data generated in the long‐term, event‐driven SERAPHIN study, 3 where approximately 61% of patients received a PDE‐5 inhibitor, mainly sildenafil, as background therapy at baseline. Although sildenafil was the most frequently used PDE‐5 inhibitor in SERAPHIN, it has to be taken 3 times daily, whereas macitentan is taken only once daily. Therefore, the once daily administered tadalafil, which has the same mode of action as sildenafil and is also indicated for the treatment of PAH, was selected for use in a fixed‐dose combination (FDC) with macitentan. As sildenafil and tadalafil have similar efficacy and safety profiles, the add‐on benefit of macitentan to tadalafil was expected to be similar to that observed for sildenafil in the SERAPHIN study. 4 , 5
An FDC of macitentan and tadalafil would offer PAH patients the advantages provided by the concomitant use of macitentan and a PDE‐5 inhibitor in a single, once daily dose. This should facilitate compliance and reduce the risk of medication errors.
Both Food and Drug Administration and European Medicines Agency require that reference products used in the bioequivalence studies are marketed in the given region 6 , 7 ; hence, 2 Phase 1 clinical bioequivalence studies have been conducted, AC‐077‐101 and AC‐077‐103 using US‐ and EU‐sourced tadalafil (Adcirca), respectively. Study AC‐077‐101, conducted in the USA, compared 2 FDC tablet formulations (FDC‐1 and FDC‐2) containing 10 mg/40 mg of macitentan/tadalafil with the free combination of macitentan and tadalafil (US‐sourced) tablets in healthy subjects. Out of the tested formulations, FDC‐2 was found to be bioequivalent to the free combination and was further used for clinical development in Study AC‐077‐103 (conducted in the EU) to establish its bioequivalence with macitentan and the EU‐sourced tadalafil.
2. METHODS
2.1. Study subjects
In present studies, eligible subjects were healthy adults between the ages of 18 and 55 years with systolic blood pressure of 100–145 mmHg, diastolic blood pressure 50–90 mmHg and pulse rate of 45–90 beats/min. Key exclusion criteria included known allergic reactions or hypersensitivity to any active substance or to any excipient of the drug formulations; hepatic aminotransferases (alanine and/or aspartate) >3 × upper limit of normal at screening; history or clinical evidence of any disease and/or existence of any surgical or medical condition that could have interfered with the absorption, distribution, metabolism or excretion of the study drug(s).
Written informed consent was obtained from each subject in the study prior to any study procedure and after adequate explanation of the aims, methods, objectives and potential hazards of the study.
2.2. Study design
The present studies were single‐center, open‐label, single‐dose, 2‐period, randomized, crossover Phase 1 studies. The primary objective was to demonstrate bioequivalence of the maximum plasma concentration (Cmax), the area under the plasma concentration–time curve (AUC) from time 0 to time t of the last measured concentration above the lower limit of quantification (AUC0‐t), and AUC from time 0 to infinity (AUC0‐∞) of macitentan and tadalafil given as an FDC (test) of macitentan/tadalafil (10 mg/40 mg) and as a free combination (reference) of 10 mg macitentan (Opsumit) and 40 mg tadalafil (Adcirca). The secondary objectives were to evaluate the safety and tolerability of concomitant macitentan and tadalafil administered as an FDC product or as a free combination and to investigate other pharmacokinetic (PK) parameters of concomitant macitentan and tadalafil administered as an FDC product or as a free combination.
With the crossover study design, the influence of sex on the results was lowered due to the intrasubject comparison.
Both studies were approved by the institutional review board/independent ethics committee (Study AC‐077‐101, IRB Services, Ontario, Canada; Study AC‐077‐103, Ethik‐Kommission bei der Landesärztekammer Baden‐Württemberg, Stuttgart, Germany) and were performed in accordance with the Declaration of Helsinki and with the laws and regulations of the USA (Study AC‐077‐101) and Germany (Study AC‐077‐103).
Study AC‐077‐101: Two FDC formulations were investigated in Study AC‐077‐101, FDC‐1 and FDC‐2, each containing 10 mg/40 mg of macitentan/tadalafil and with different composition. Subjects were allocated to either Group 1 (FDC‐1) or Group 2 (FDC‐2) and within each group subjects were randomized to 1 of the 2 possible treatment sequences, A/B or B/A. Treatment A was a single FDC (test treatment: FDC‐1 or FDC‐2) tablet of macitentan/tadalafil (10 mg/40 mg). Treatment B was a reference treatment of the free combination, single macitentan 10 mg tablet (Opsumit) taken together with 2 × tadalafil 20 mg tablets (US‐sourced). Both FDCs were oval film‐coated tablets and differed with respect to the excipients and coating materials used.
In both groups, study drug (Treatment A or Treatment B) was administered to the subjects on Day 1 in the fasted state. Fluids were restricted 1 hour before and lasting until 1 hour after dosing, except for water taken with the study drug. Following study drug administration, there was an observation period of 216–288 hours. At the end of the observation period, there was an 11–14 days washout period, prior to the administration of the second treatment in the sequence. The maximum duration on study for each subject including follow‐up was 12 weeks, which included 3 weeks of screening period, about 2 weeks of Treatment A sequence, about 2 weeks of washout period, about 2 weeks of Treatment B sequence and about 3 weeks of follow‐up period.
Study AC‐077‐103: In Study AC‐077‐103, subjects were randomized to 2 possible treatment sequences (A/B or B/A). Treatment A (test treatment) was a single oral dose of FDC (FDC‐2 from Study AC‐077‐101 containing 10 mg/40 mg macitentan/tadalafil). Treatment B (reference treatment) was a single oral dose of macitentan 10 mg taken together with a single oral dose of tadalafil 40 mg (given as 2 × tadalafil 20 mg tablets; EU‐sourced). Study drugs were administered to the subjects on Day 1 in the fasted state. The subjects remained fasted from at least 10 hours prior to each study drug administration until 4 hours after. Fluids were restricted 1 hour before and lasting until 1 hour after dosing, except for water taken with the study drug.
End‐of‐period (EOP) assessments were performed 216 hours after study drug administration in Period 1. The 2 treatment and observation periods were to be separated by a 7–10 day washout period.
In both studies, the FDCs and the reference treatments macitentan (Opsumit) and tadalafil (Adcirca) were provided by Actelion Pharmaceuticals Limited.
2.3. PK evaluations
2.3.1. Sample collection and analytical methods
Study AC‐077‐101: Blood samples (7.0 mL) were collected at predose, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 24, 48, 72, 120, 168 and 216 hours postdose into Monovette or equivalent tubes containing ethylene diamine tetra‐acetic acid as anticoagulant. Within 30 minutes of collection, the tubes were centrifuged at approximately 1500 g for 10 minutes at 4 ± 2°C. All samples were stored in an upright position below −20°C.
Study AC‐077‐103: Blood samples (7.0 mL) were collected at predose, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 7, 7.5, 8, 8.5, 9, 10, 12, 24, 48, 72, 120, 168 and 216 hours postdose into Monovette or equivalent tubes containing tri‐potassium ethylene diamine tetra‐acetic acid as anticoagulant. Within 30 minutes of collection, the tubes were centrifuged at approximately 1,500 g for 10 minutes at 4 ± 2°C. All samples were stored in an upright position at −75°C or below, protected from light.
For both studies, the plasma concentrations of macitentan, its active metabolite ACT‐132577, and tadalafil were measured with 2 independent validated methods using liquid chromatography with tandem mass spectrometry. 8 , 9 , 10
The concentrations of macitentan and ACT‐132577 in human plasma were determined using liquid chromatography coupled to tandem mass spectrometric detection after precipitation of human plasma proteins with acetonitrile/ethanol (1:1, v/v) containing the stable isotope‐labelled internal standards. The method was successfully validated with respect to specificity/selectivity, response, precision and accuracy, recovery, matrix effect, interference testing with tadalafil, effect of sample dilution, and carry over. Stability in plasma samples was demonstrated for at least 3 days at room‐temperature, 36 months storage at −20°C and for 3 freeze/thaw cycles.
The concentration of tadalafil in human plasma were determined using liquid chromatography coupled to tandem mass spectrometric detection after liquid–liquid extraction with tert‐butyl methyl ether using a stable isotope‐labelled internal standard. The method was successfully validated with respect to specificity/selectivity, response, precision and accuracy, recovery, matrix effect, interference testing with macitentan/ACT‐1325777, effect of sample dilution, and carry over. Stability in plasma samples were demonstrated for at least 6 hours at room‐temperature, 408 days storage at −20°C and for 3 freeze/thaw cycles.
Precision and bias—study AC‐077‐101: The descriptive statistics of the quality control (QC) samples of macitentan showed that the interbatch precision was ≤4.3%, whereas the interbatch bias was in the range from 2.1 to 7.4%. The descriptive statistics of the QC samples of ACT‐132577 showed that the interbatch precision was ≤4.0%, whereas the interbatch bias was in the range from 1.4 to 7.5%. The descriptive statistics of the QC samples for tadalafil showed that the interbatch precision was <7.9%, whereas the interbatch bias was in the range from 0.4 to 5.0%. Incurred sample analysis was performed for both methods. For macitentan/ACT‐132577, 10% of the number of study samples were reanalysed and 99.2% met the criteria (±20% of the mean of the original and reanalysed results) for macitentan and ACT‐132577. For tadalafil 6.4% of number of samples were reanalysed, and 97.4% were in criteria.
Precision and bias—study AC‐077‐103: The descriptive statistics of the QC samples of macitentan showed that the interbatch precision was ≤7.0%, whereas the interbatch bias was in the range from −2.1 to 2.6%. The descriptive statistics of the QC samples for ACT‐132577 showed that the interbatch precision was ≤6.6%, whereas the interbatch bias was in the range from −1.9 to 1.9%. The descriptive statistics of the QC samples for tadalafil showed that the interbatch precision was ≤4.9%, whereas the interbatch bias was in the range from 1.1% to 7.1%. Incurred sample analysis was performed for both methods. For macitentan/ACT‐132577, 9% of the number of study samples were reanalysed and 98.1% met the criteria for macitentan and 94.3% for ACT‐132577. For tadalafil 8.1% of number of samples were reanalysed, and 100% were within criteria.
2.3.2. PK analysis
PK parameters (AUC0‐∞, AUC0‐t, Cmax, time to reach Cmax [tmax] and terminal half‐life [t½]) were derived by noncompartmental analysis using Phoenix WinNonlin version 6.4 (Certara, Princeton, NJ, USA). PK parameters were summarized by study group and treatment in Study AC‐077‐101 and by study treatment in Study AC‐077‐103 with arithmetic mean, geometric mean, minimum, median, maximum, standard deviation (SD), standard error, intersubject coefficient of variation (in %), intrasubject coefficient of variation (in %), and 95% confidence interval (CI) of the arithmetic and geometric means. Arithmetic mean, minimum, median, maximum, SD, standard error and 95% CI of the arithmetic mean were calculated for tmax. Median and range for tmax and geometric mean with 95% CI are presented for all other PK parameters in Tables 2 and 4.
TABLE 2.
Study AC‐077‐101: Summary of pharmacokinetic parameters of macitentan, ACT‐132577 and tadalafil by treatment in group 1 (FDC‐1) and group 2 (FDC‐2), pharmacokinetic analysis set
| Geometric means (95% CI); tmax: Median (range) | ||||||
|---|---|---|---|---|---|---|
| Macitentan | Tadalafil | ACT‐132577 | ||||
| Parameter | Treatment A FDC | Treatment B free combination | Treatment A FDC | Treatment B free combination | Treatment A FDC | Treatment B free combination |
| Group 1 (FDC‐1) | ||||||
| N | 57 | 57 | 57 | 57 | 56 | 56 |
| Cmax, ng/mL | 168.73 (154.87, 183.83) | 167.93 (153.73, 183.44) | 384.56 (353.66, 418.17) | 506.45 (471.51, 543.98) | 157.67 (144.85, 171.63) | 158.77 (145.26, 173.54) |
| AUC0‐t, ng h/mL | 4324.13 (4032.28, 4637.11) | 4404.26 (4081.93, 4752.05) | 12 733.26 (11 440.52, 14 172.08) | 13 908.13 (12 522.48, 15 447.11) | 16 534.42 (15 297.50, 17 871.36) | 16 821.24 (15 485.60, 18 272.09) |
| AUC0‐∞, ng h/mL | 4429.18 (4134.83, 4744.48) | 4509.81 (4187.36, 4857.09) | 12 825.31 (11 523.07, 14 274.71) | 13 975.28 (12 583.17, 15 521.40) | 17 799.76 (16 458.30, 19 250.56) | 18 090.62 (16 670.17, 19 632.11) |
| tmax, h | 9.00 (5.0–10.1) | 9.00 (4.0–10.1) | 4.00 (1.0–24.0) | 3.00 (1.0–5.0) | 48.00 (24.0–72.0) | 48.00 (24.0–72.0) |
| t1/2, h | 13.821 (13.037, 14.652) | 13.957 (13.209, 14.747) | 18.372 (16.637, 20.287) | 18.066 (16.472, 19.815) | 49.112 46.979, 51.343) | 48.526 (46.023, 51.166) |
| Group 2 (FDC‐2) | ||||||
| N | 58 | 58 | 58 | 58 | 56 | 56 |
| Cmax, ng/mL | 193.66 (181.52, 206.60) | 163.02 (151.29, 175.66) | 468.72 (438.22, 501.33) | 489.94 (460.06, 521.76) | 164.53 (154.17, 175.58) | 149.71 (139.70, 160.45) |
| AUC0‐t, ng h/mL | 4740.06 (4415.13, 5088.91) | 4296.41 (3989.60, 4626.81) | 13 995.62 (12 750.92, 15 361.83) | 14 316.68 (13 118.08, 15 624.80) | 17 924.06 (16 799.59, 19 123.80) | 16 415.81 (15 281.60, 17 634.19) |
| AUC0‐∞, ng h/mL | 4833.20 (4506.52, 5183.55) | 4394.02 (4086.54, 4724.64) | 14 056.64 (12 807.84, 15 427.20) | 14 378.46 (13 175.79, 15 690.89) | 19 406.69 (18 127.98, 20 775.59) | 17 833.72 (16 556.31, 19 209.70) |
| tmax, h | 9.00 (3.0–10.1) | 9.00 (4.0–12.0) | 3.00 (1.0–7.0) | 3.00 (1.0–9.0) | 48.00 (24.0–72.0) | 48.00 (24.0–72.0) |
| t1/2, h | 13.794 (12.808, 14.857) | 14.410 (13.419, 15.474) | 18.141 (16.709, 19.695) | 18.503 (16.878, 20.283) | 50.478 (47.992, 53.093) | 50.332 (48.000, 52.778) |
AUC = area under the plasma concentration–time curve; AUC0‐t = AUC from time 0 to time t of the last measured concentration above the lower limit of quantification; AUC0‐∞ = AUC from time 0 to infinity; CI = confidence interval; Cmax = maximum plasma concentration; FDC = fixed‐dose combination; N = maximum number of subjects with data; PK = pharmacokinetic; t1/2 = terminal half‐life; tmax = time to reach maximum plasma concentration.
Treatment A = FDC of macitentan and tadalafil (test).
Treatment B = free combination of macitentan and tadalafil (reference).
Only subjects who had a full set of PK parameters for both treatments were included in the analysis.
TABLE 4.
Study AC‐077‐103: summary of pharmacokinetic parameters of macitentan, ACT‐132577 and tadalafil by treatment (FDC‐2), pharmacokinetic analysis set
| Parameter | Geometric means (95% CI); tmax: median (range) | |||||
|---|---|---|---|---|---|---|
| Macitentan | Tadalafil | ACT‐132577 | ||||
| Treatment A FDC (N = 34) | Treatment B free combination (N = 34) | Treatment A FDC (N = 34) | Treatment B free combination (N = 34) | Treatment A FDC (N = 34) | Treatment B free combination (N = 34) | |
| Cmax, ng/mL | 174.64 (161.08, 189.35) | 164.63 (152.48, 177.74) | 538.91 (492.00, 590.29) | 597.78 (542.32, 658.91) | 162.72 (145.78, 181.62) | 159.57 (144.49, 176.21) |
| AUC0‐t, ng h/mL | 5809.26 (5274.06, 6398.77) | 5787.14 (5270.38, 6354.57) | 17 159.15 (15 516.18, 18 976.09) | 17 091.33 (15 210.43, 19 204.81) | 18 656.33 (16 888.63, 20 609.04) | 18 711.87 (17 144.29, 20 422.79) |
| AUC0‐∞, ng h/mL | 5890.08 (5354.81, 6478.85) | 5881.08 (5365.79, 6445.84) | 17 268.40 (15 589.27, 19 128.39) | 17 187.34 (15 278.78, 19 334.30) | 20 373.98 (18 416.42, 22 539.63) | 20 504.95 (18 767.02, 22 403.83) |
| tmax, h | 9.00 (4.0–12.0) | 9.00 (4.2–24.0) | 2.00 (0.5–5.0) | 1.75 (1.0–5.0) | 48.00 (24.0–72.0) | 48.00 (24.0–72.1) |
| t1/2, h | 16.146 (14.778, 17.642) | 16.106 (14.841, 17.478) | 23.319 (21.092, 25.781) | 22.592 (20.460, 24.945) | 49.405 (46.240, 52.787) | 50.401 (47.640, 53.322) |
AUC = area under the plasma concentration–time curve; AUC0‐t = AUC from time 0 to time t of the last measured concentration above the lower limit of quantification; AUC0‐∞ = AUC from time 0 to infinity; Cmax = maximum plasma concentration; CI = confidence interval; FDC = fixed‐dose combination; N = maximum number of subjects with data; t½ = terminal half‐life; tmax = time to reach maximum plasma concentration.
Treatment A = FDC of macitentan and tadalafil (test).
Treatment B = free combination of macitentan and tadalafil (reference).
In Study AC‐077‐101, bioequivalence was evaluated within each study group by comparing test treatment (Treatment A [FDC‐1 in Group 1 and FDC‐2 in Group 2]) to reference treatment (Treatment B [free combination]). In Study AC‐077‐103, bioequivalence was evaluated by comparing test treatment (Treatment A [FDC]) to reference treatment (Treatment B [free combination]). In both studies, determination of bioequivalence was based upon 90% CI for the ratios of the geometric means (test/reference) for macitentan and tadalafil AUC0‐∞, AUC0‐t and Cmax. For acceptance of bioequivalence, the 90% CIs had to be within the range of ≥0.8000 and ≤1.2500, when rounded to 4 decimal places. 7
The treatments were compared with a mixed‐effects model using log‐transformed values of the primary endpoint as dependent variables, treatment, treatment sequence and period as fixed effects, and subject as a random effect. Geometric mean ratios (test/reference) and 90% CIs were calculated from the corresponding back‐transformed contrasts for treatment of the mixed‐effects models. In addition, the inter‐ and intrasubject variability were estimated from the mixed‐effects model.
The PK analysis were performed in the PK set, which comprised all subjects included in the All‐treated set (all randomized subjects who received at least 1 dose of study drug) who did not deviate from the protocol in a way that could have affected the outcome of the study (i.e. did not have major protocol deviations).
2.3.3. Safety evaluation
The safety evaluation included change from baseline to each scheduled time point of measurement and to EOP/end‐of‐study (EOS) in vital signs, body weight, electrocardiogram (ECG) variables, clinical laboratory tests, treatment‐emergent ECG abnormality, treatment‐emergent adverse events (AEs) and treatment‐emergent serious AEs (SAEs) up to 240 hours after drug intake, and AEs leading to premature discontinuation of study drug.
Safety and tolerability data were listed by study group, subject number and sex, and summarized descriptively by study group and treatment.
2.3.4. Sample size calculation
A formal sample size calculation was performed for the primary PK endpoints. The number of subjects required for a bioequivalence study depended on the expected deviation of the test drug from the reference drug, and the error variance associated with the PK parameters (Cmax, AUC0‐t and AUC0‐∞) of the study drug.
AC‐077‐101: The sample size calculated for the study was based on a conservative intersubject coefficient of variation value of 30%. With an adjusted by Bonferroni adjustment Type II error set to 0.10, it was estimated that a total of 52 evaluable subjects per group was required for the study to have at least 90% overall power to reject the null hypothesis, given the 0.80 and 1.25 no‐effect boundaries and assuming a true mean ratio of 1.
AC‐077‐103: For sample size calculations, a conservative intrasubject coefficient of variation of 20% was chosen. With an adjusted by Bonferroni adjustment Type II error set to 0.10, it was estimated that a total of 32 evaluable subjects were required in order for the study to achieve a power of at least 90% for the analysis of the primary endpoints, given the 0.80 and 1.25 no‐effect boundaries and assuming a true mean ratio of 1.
2.3.5. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
3. RESULTS
3.1. Study subjects
In Study AC‐077‐101, a total of 124 subjects were planned and enrolled, 62 subjects to each group. All subjects received study drug. In Group 1 (FDC‐1 vs free combination), 5 subjects discontinued study drug (4 subjects withdrew consent and 1 subject discontinued the study due to an AE of increased blood calcium). All 5 subjects completed their EOS visit and were therefore considered to have completed the study. In Group 2 (FDC‐2 vs free combination), 4 subjects discontinued study drug. Of these 4 subjects, 2 completed their EOS, whereas the other 2 were lost to follow‐up. Therefore, 60 out of 62 subjects in Group 2 completed the study. A total of 57 out of 62 subjects in Group 1 and 58 out of 62 subjects in Group 2 were evaluable for PK.
In Study AC‐077‐103, a total of 38 subjects were planned and enrolled, and all subjects received study drug. Two subjects discontinued study drug as well as the study. Therefore, 36 subjects completed the study. A total of 34 out of 38 subjects were evaluable for PK.
3.2. Demographics and baseline characteristics
The demographics and baseline characteristics were generally similar for all treated subjects in both studies (Table 1).
TABLE 1.
All studies: demographics characteristics, all‐treated set
| Arithmetic mean (SD); range | |||
|---|---|---|---|
| AC‐077‐101 | AC‐077‐103 | ||
| Variable | Group 1 FDC‐1 vs free combination (N = 62) | Group 2 FDC‐2 vs free combination (N = 62) | FDC‐2 vs free combination (N = 38) |
| Females, n (%) | 17 (27.4) | 20 (32.3) | 16 (42.1) |
| Males, n (%) | 45 (72.6) | 42 (67.7) | 22 (57.9) |
| Age, y | 31.3 (9.97); 18–55 | 29.1 (8.73); 19–54 | 44.6 (10.29); 19–55 |
| Weight, kg | 79.2 (14.36); 48.8–107.0 | 76.8 (14.09); 41.6–111.8 | 74.7 (13.32); 53.0–100.6 |
| Height, cm | 175.0 (9.03); 149.8–189.1 | 172.7 (9.66); 149.0–189.8 | 172.6 (9.48); 151.0–188.0 |
| BMI, kg/m2 | 25.8 (3.54); 18.2–31.9 | 25.6 (3.52); 18.7–31.8 | 24.9 (2.65); 19.9–29.4 |
BMI = body mass index; FDC = fixed‐dose combination; N = maximum number of subjects with data; SD = standard deviation; % = percentage of subjects calculated on the basis of N specified in the header.
In Study AC‐077‐101, there were 72.6% males and 27.4% females in Group 1 and 67.7% males and 32.3% females in Group 2. The mean (SD) age was 31.3 (9.97) years in Group 1 and 29.1 (8.73) years in Group 2. The respective body mass index values were 25.8 and 25.6 kg/m2.
In Study AC‐077‐103, there were 57.9% males and 42.1% females. The mean (SD) age was 44.6 (10.29) years. The mean body mass index was 24.91 kg/m2.
3.3. PK
3.3.1. Study AC‐077‐101
Macitentan and ACT‐132577
The arithmetic mean plasma concentrations and exposure PK parameters AUC0‐∞, AUC0‐t and Cmax were similar following administration of FDC‐1 or macitentan (Opsumit) in the free combination in Group 1 (Table 2, Figures 1 and 2). The plasma concentrations and PK parameters of ACT‐132577 were also similar after FDC‐1 and free combination administration in Group 1.
FIGURE 1.
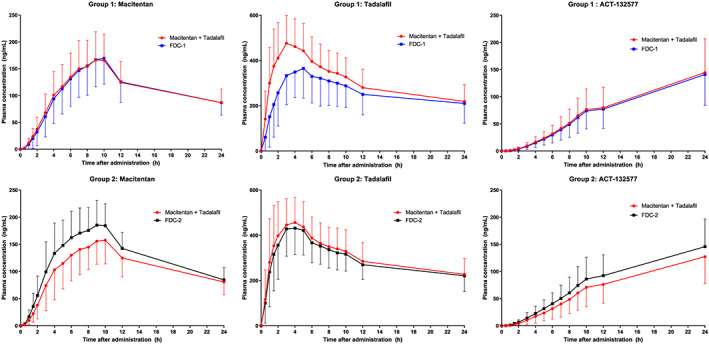
Study AC‐077‐101: Arithmetic mean plasma concentration vs time profiles for macitentan, ACT‐132577 and tadalafil during the first 24 hours after administration of fixed‐dose combination (FDC) or free combination of macitentan and US‐sourced tadalafil in group 1 and group 2, pharmacokinetics analysis set. Data are plotted on a linear scale
FIGURE 2.
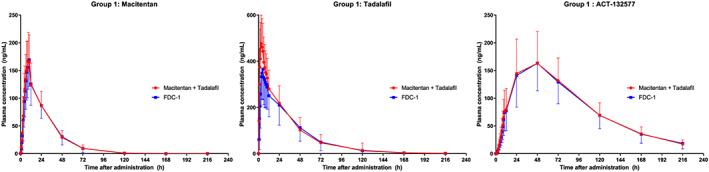
Study AC‐077‐101: Arithmetic mean plasma concentration vs time profiles for macitentan, ACT‐132577 and tadalafil over 216 hours after administration of fixed‐dose combination (FDC)‐1 or free combination of macitentan and US‐sourced tadalafil in group 1, pharmacokinetics analysis set. Data are plotted on a linear scale
In Group 2, after administration of macitentan (Opsumit) in the free combination, PK parameters for macitentan and ACT‐132577 were similar to those observed in Group 1 (Table 2). Compared to administration as the free combination, the PK profiles of macitentan and ACT‐132577 after administration of FDC‐2 showed slight differences (Table 2, Figures 1 and 3). While tmax and t½ values were comparable to those observed after administration of the free combination, exposure to macitentan (in terms of Cmax and AUC) was higher (approximately 19 and 10% higher, respectively) after administration of FDC‐2 (Table 2). These differences were less noticeable for ACT‐132577, where ACT‐132577 exposure (in terms of Cmax and AUC) was slightly higher (approximately 10 and 9% higher, respectively) when given as the FDC‐2. The PK parameters tmax and t½ for ACT‐132577 were similar for FDC‐2 and the free combination.
FIGURE 3.
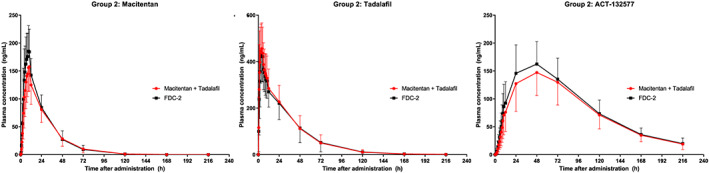
Study AC‐077‐101: Arithmetic mean plasma concentration vs time profiles for macitentan, ACT‐132577 and tadalafil over 216 hours after administration of fixed‐dose combination (FDC)‐2 or free combination of macitentan and US‐sourced tadalafil in group 2, pharmacokinetic analysis set. Data are plotted on a linear scale
In Group 1, the 90% CIs for the geometric mean ratios (FDC‐1 vs free combination) of AUC0‐∞, AUC0‐t and Cmax for macitentan were within the bioequivalence limits (0.8000–1.2500; Table 3). In Group 2, macitentan, geometric mean ratios (FDC‐2 vs free combination) of AUC0‐∞, AUC0‐t and Cmax were slightly above 1, indicating that exposure to macitentan when administered in the FDC‐2 was slightly higher compared to the free combination. However, all 90% CIs were within the bioequivalence limits of 0.8000–1.2500, fulfilling bioequivalence criteria (Table 3).
TABLE 3.
Study AC‐077‐101: Results of bioequivalence determination, pharmacokinetic analysis set
| Parameter | Geometric means ratio (treatment A/B; 90% CI) CVw % | |||||
|---|---|---|---|---|---|---|
| Group 1 | Group 2 | |||||
| FDC‐1 vs free combination | FDC‐2 vs free combination | |||||
| Macitentan (N = 57) | Tadalafil (N = 57) | ACT‐132577 (N = 56) | Macitentan (N = 58) | Tadalafil (N = 58) | ACT‐132577 (N = 56) | |
| Cmax, ng/mL | 1.0045 (0.9631, 1.0478) 13.50 | 0.7588 (0.7167, 0.8034) 18.38 | 0.9935 (0.9616, 1.0264) 10.32 | 1.1901 (1.1374, 1.2451) 14.64 | 0.9576 (0.9063, 1.0118) 17.86 | 1.0983 (1.0470, 1.1520) 15.15 |
| AUC0‐t, ng h/mL | 0.9815 (0.9527, 1.0113) 9.54 | 0.9151 (0.8687, 0.9639) 16.71 | 0.9826(0.9550, 1.0110) 9.03 | 1.1014 (1.0652, 1.1389) 10.80 | 0.9764 (0.9293, 1.0259) 16.01 | 1.0925 (1.0545, 1.1319) 11.20 |
| AUC0‐∞, ng h/mL | 0.9818 (0.9537, 1.0109) 9.31 | 0.9172 (0.8707, 0.9662) 16.70 | 0.9836(0.9572, 1.0107) 8.61 | 1.0981 (1.0641, 1.1332) 10.16 | 0.9765 (0.9293, 1.0260) 16.02 | 1.0884 (1.0502, 1.1281) 11.31 |
AUC = area under the plasma concentration–time curve; AUC0‐t = AUC from time 0 to time t of the last measured concentration above the lower limit of quantification; AUC0‐∞ = AUC from time 0 to infinity; CI = confidence interval; Cmax = maximum plasma concentration; CVw = intrasubject (within‐subject) coefficient of variation; FDC = fixed‐dose combination; N = maximum number of subjects with data.
Treatment A = FDC‐1 in Group 1 and FDC‐2 in Group 2 (test).
Treatment B = free combination of macitentan and tadalafil (reference).
Tadalafil
After administration of tadalafil (Adcirca) in the free combination, tadalafil plasma concentrations reached a peak at about 3 hours and decreased with a t½ of approximately 18 hours (Table 2). When administered as part of the FDC‐1, the Cmax of tadalafil was reached later at 4 hours, and exposures in terms of Cmax and AUC were lower (approximately 24 and 8% lower, respectively) when compared to administration as a free combination. The t½ was comparable after administration of the free combination.
The PK parameters of tadalafil in Group 2 after administration of the free combination were similar to those observed when the free combination was administered in Group 1 (Table 2). Comparison of the tadalafil plasma concentration–time profiles and exposures revealed no relevant PK differences between the 2 treatments (Figure 1 and 3). The tmax values and t½ were similar when tadalafil was administered as either the FDC or free combination.
For tadalafil in FDC‐1, the 90% CIs for the geometric mean ratios of AUC0‐∞ and AUC0‐t were both within the bioequivalence limits (Table 3). For the Cmax of tadalafil, bioequivalence was not achieved as the lower limit of the 90% CI of the geometric mean ratio was below 0.8000. This indicated that, at peak, subjects would be exposed less to tadalafil after administration of the FDC‐1, than after the free combination. Therefore, bioequivalence of FDC‐1 with the free combination of macitentan and tadalafil was not achieved.
For tadalafil in FDC‐2, the 90% CIs for the geometric mean ratios of all 3 parameters (AUC0‐∞, AUC0‐t and Cmax) were within the bioequivalence limits similar to macitentan (Table 3). Therefore, bioequivalence of FDC‐2 with the free combination of macitentan and tadalafil sourced from the USA was demonstrated.
3.3.2. Study AC‐077‐103
Macitentan and ACT‐132577
The arithmetic mean plasma concentrations of macitentan were similar following administration of FDC‐2 or the free combination (Figure 4). Exposure to macitentan and ACT‐132577 in terms of AUC0‐∞, AUC0‐t and Cmax was similar following administration of FDC‐2 or the free combination (Table 4). The median tmax of macitentan and ACT‐132577 (9 and 48 hours, respectively) was the same when macitentan was administered as either the FDC or free combination (Table 4). Geometric mean t½ of macitentan and ACT‐132577 was comparable when macitentan was administered as either the FDC or free combination.
FIGURE 4.
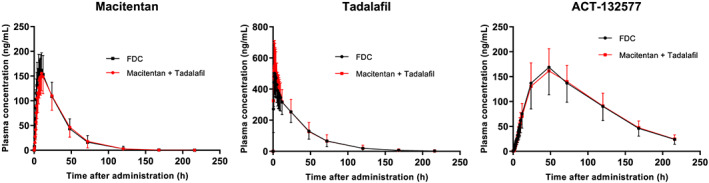
Study AC‐077‐103: Arithmetic mean plasma concentration vs time profiles of fixed‐dose combination (FDC)‐2 for macitentan, ACT‐132577 and tadalafil over 216 hours after administration of FDC‐2 or free combination of macitentan and EU‐sourced tadalafil, pharmacokinetic analysis set. Data are plotted on a linear scale
Tadalafil
The arithmetic mean plasma concentrations of tadalafil were similar following administration of FDC‐2 or the free combination (Figure 4). Exposure to tadalafil in terms of Cmax, AUC0‐t and AUC0‐∞ was similar following administration of FDC‐2 or the free combination (Table 4). The tmax and t½ values were comparable when tadalafil was administered as either the FDC (2.0 and 23.3 h, respectively) or free combination (1.8 and 22.6 h, respectively).
All 90% CIs for the geometric mean ratios (FDC‐2 vs free combination) of Cmax, AUC0‐t and AUC0‐∞ for both macitentan and tadalafil were within the bioequivalence limits (0.8000 to 1.2500; Table 5). Therefore, bioequivalence of FDC‐2 with the free combination of macitentan and tadalafil sourced from the EU was demonstrated.
TABLE 5.
Study AC‐077‐103: Results of bioequivalence determination, pharmacokinetic analysis set
| Ratio of geometric means (treatment A/B; 90% CI) CVw % | |||
|---|---|---|---|
| FDC‐2 vs free combination | |||
| Parameter | Macitentan (N = 34) | Tadalafil (N = 34) | ACT‐132577 (N = 34) |
| Cmax, ng/mL | 1.0608 (1.0014, 1.1238) 14.10 | 0.9015 (0.8413, 0.9661) 16.96 | 1.0198 (0.9819, 1.0591) 9.24 |
| AUC0‐t, ng h/mL | 1.0038 (0.9677, 1.0413) 8.94 | 1.0040 (0.9584, 1.0517) 11.35 | 0.9970 (0.9651, 1.0300) 7.93 |
| AUC0‐∞, ng h/mL | 1.0015 (0.9675, 1.0367) 8.42 | 1.0047 (0.9591, 1.0525) 11.34 | 0.9936 (0.9619, 1.0264) 7.91 |
AUC = area under the plasma concentration–time curve; AUC0‐t = AUC from time 0 to time t of the last measured concentration above the lower limit of quantification; AUC0‐∞ = AUC from time 0 to infinity; CI = confidence interval; Cmax = maximum plasma concentration; CVw = intrasubject (within‐subject) coefficient of variation; FDC = fixed‐dose combination; N = maximum number of subjects with data.
Treatment A = FDC of macitentan and tadalafil (test).
Treatment B = free combination of macitentan and tadalafil (reference).
3.4. Safety
In the present studies, the AE profiles for the FDCs and the free combination were consistent with the known safety profiles of macitentan and tadalafil.
3.4.1. Study AC‐077‐101
No subject died and no SAEs were reported. Three subjects prematurely discontinued treatment due to AEs: 1 subject discontinued 8 days after administration of FDC‐1 due to increased blood calcium; 1 subject discontinued due to increased blood triglycerides 11 days after administration of FDC‐2; and 1 subject discontinued due to pain (general soreness) on the day of administration of FDC‐2. The majority of the subjects in the study had at least 1 AE. The proportion of subjects who had at least 1 AE was similar for the FDCs and the free combination for both groups and varied between 70.0 and 78.7%. Headache was the most frequently reported AE for the FDCs and the free combination, with an incidence of 45.8–67.2%. Other frequently reported AEs included back pain, pain in extremity, nausea and dizziness.
3.4.2. Study AC‐077‐103
No deaths, other SAEs, or other significant AEs occurred during the study. The proportion of subjects who had at least 1 AE was similar for the FDC and the free combination (92.1% and 88.9%, respectively). The most frequently reported AE in both treatments was headache (60.5% and 66.7%, respectively). Other frequently reported AEs included back pain, pain in extremity, nasal congestion, nausea, dizziness, myalgia and head discomfort. All reported AEs were mild or moderate in intensity, except 1 severe intensity AE of upper respiratory tract infection reported after administration of free combination.
For both studies, the mean changes from baseline (predose) to each time point of measurement up to EOP/EOS in vital signs and ECG variables were small and similar for the FDCs and the free combination.
4. DISCUSSION
The present studies evaluated the bioequivalence of FDCs of macitentan/tadalafil (10 mg/40 mg) with a free combination of 10 mg macitentan and 40 mg tadalafil in healthy subjects.
Two formulations of the FDC (FDC‐1 and FDC‐2) were developed and their bioequivalence was compared in Study AC‐077‐101 to the free combination of macitentan and tadalafil (US‐sourced). When administered in the free combination, the PK profiles of macitentan and tadalafil, respectively, were similar to those observed in previous studies. 11 , 12 , 13
FDC‐1 was not bioequivalent to the free combination. The 90% CIs for the geometric mean ratios for AUC0‐∞, AUC0‐t and Cmax for macitentan (and ACT‐132577) were within the bioequivalence limits (0.8000–1.2500). However, the Cmax for tadalafil was lower following administration of FDC‐1 (lower bound of the 90% CI = 0.7167). FDC‐2 was bioequivalent to the free combination, with the 90% CIs for AUC0‐∞, AUC0‐t and Cmax for macitentan, ACT‐132577 and tadalafil falling entirely within the range of 0.8000–1.2500. The difference in PK between FDC‐1 and FDC‐2 seems to be caused by the difference in composition of the 2 FDC formulations.
To ensure that the FDC‐2 could also be used in patients in the EU, bioequivalence had to be demonstrated using EU‐sourced tadalafil. FDC‐2 was further investigated in Study AC‐077‐103 and its bioequivalence was compared with the EU‐sourced tadalafil. The FDC‐2 was bioequivalent to the free combination, with the 90% CIs for the geometric mean ratios for AUC0‐∞, AUC0‐t and Cmax for macitentan, ACT‐132577 and tadalafil falling entirely within the bioequivalence range of 0.8000–1.2500.
In both studies, the safety profiles of both the FDCs and the free combination were comparable, suggesting that FDC can be used instead of the free combination, without any dose alterations.
5. CONCLUSION
The FDC‐2 tablet containing 10 mg/40 mg of macitentan/tadalafil was bioequivalent to the free combination of 10 mg macitentan and 40 mg tadalafil (both US‐ and EU‐sourced). Macitentan and tadalafil were well tolerated when administered as FDC product or as a free combination. There were no apparent differences in the safety profile between the FDC formulation and the free combination.
COMPETING INTERESTS
Si.G., S.B. and D.C. are employees of Actelion Pharmaceuticals Ltd. Su.G. and P.N.S. are employees of Idorsia Pharmaceuticals Ltd. M.A. is an employee of Aixial s.r.o. J.C. is an employee of Reven, LLC.
CONTRIBUTORS
J.C. and A.S. were responsible for the study implementation and had direct responsibility for the subjects. P.N.S., S.B. and Si.G. were responsible for the PK analysis and for the interpretation of the data. Su.G. was responsible for the bioanalytical activities. M.A. was responsible for the statistical analysis. D.C. was responsible for the interpretation of the data. All authors reviewed, contributed to and approved the manuscript.
DATA ACCESSIBILITY STATEMENT
The data sharing policy of the Sponsor is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
ACKNOWLEDGEMENTS
This study was sponsored by Actelion Pharmaceuticals Ltd (Allschwil, Switzerland), a Janssen pharmaceutical company of Johnson & Johnson. Medical writing and editorial support were provided by Barbara Boggetti of Trilogy Writing and Consulting GmbH. This support was funded by Actelion Pharmaceuticals Ltd (Allschwil, Switzerland), a Janssen pharmaceutical company of Johnson & Johnson.
Grill S, Bruderer S, Sidharta PN, et al. Bioequivalence of macitentan and tadalafil given as fixed‐dose combination or single‐component tablets in healthy subjects. Br J Clin Pharmacol. 2020;86:2424–2434. 10.1111/bcp.14347
The authors confirm the Principal Investigators for this paper are James Carlson and Armin Schultz and they had direct clinical responsibility for subjects.
REFERENCES
- 1. D'Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343‐349. [DOI] [PubMed] [Google Scholar]
- 2. Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J am Coll Cardiol. 2013;62(25 Suppl):D60‐D72. [DOI] [PubMed] [Google Scholar]
- 3. Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809‐818. [DOI] [PubMed] [Google Scholar]
- 4. Archer SL, Michelakis ED. Phosphodiesterase type 5 inhibitors for pulmonary arterial hypertension. N Engl J Med. 2009;361(19):1864‐1871. [DOI] [PubMed] [Google Scholar]
- 5. Falk JA, Philip KJ, Schwarz ER. The emergence of oral tadalafil as a once‐daily treatment for pulmonary arterial hypertension. Vasc Health Risk Mana. 2010;6:273‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Federal Food , Drug and Cosmetic Act (FD&C Act). Chapter V, Part A, Sec. 355 (b; 2). 1938.
- 7. European Medicines Agency , Evaluation of Medicines for Human Use, CPMP. Guideline on the investigation of bioequivalence. CPMP/QWP/EWP/1401/98 Rev. 1. London: 2010.
- 8. Zimmermann T. Validation of an analytical method for the determination of ACT‐064992 and its metabolite ACT‐132577 in human plasma samples by LC MS/MS, Actelion study no. BA 13.225. 2014.
- 9. Steurer A. Supplementary validation of an analytical method for the determination of ACT‐064992 and its metabolite ACT‐132577 in human plasma samples by LC MS/MS, Actelion study no. BA 14.033. 2016.
- 10. Steigerwald K. Validation of determination of tadalafil in plasma samples of subjects. ACC (Analytical Clinical Concept) 316B14‐Val. 2014.
- 11. Forgue ST, Patterson BE, Bedding AW, et al. Tadalafil pharmacokinetics in healthy subjects. Br J Clin Pharmacol. 2006;61(3):280‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sidharta PN, van Giersbergen PL, Halabi A, Dingemanse J. Macitentan: entry‐into humans study with a new endothelin receptor antagonist. Eur J Clin Pharmacol. 2011;67(10):977‐984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sidharta PN, Treiber A, Dingemanse J. Clinical pharmacokinetics and pharmacodynamics of the endothelin receptor antagonist macitentan. Clin Pharmacokinet. 2015;54(5):457‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sharing policy of the Sponsor is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.