

Abstract
Aims
The classical Weber effect describes an increase in adverse reaction (AR) reports after medicinal product authorisation, with a peak in AR reporting at the end of the second year followed by a decline, despite increasing patient exposure. The present study aimed to evaluate the validity of the Weber effect in the context of authorised medicines in a specialty care setting.
Methods
Using 6‐monthly sales data as a proxy for exposure, the exposure–adjusted reporting rates for AR reports for 10 selected specialty care medicines were plotted against time. These data were also evaluated based on the source of report (solicited or unsolicited) and the nature of the AR contained within the reports (listed or unlisted). Unsolicited reports were analysed against sales volumes. Goodness of fit (R2) was calculated and the trend representing the highest R2 was selected.
Results
Study data comprised a total of 1 222 852 AR reports for 10 specialty care medicines. Amongst all of the products evaluated, none of the associated data represented reporting patterns entirely consistent with the classical Weber effect (see Figure 1). The results, however, showed a systemic direct correlation between AR reporting and sales volumes, especially throughout the first 5 years post‐authorisation.
Conclusion
The study not only presents evidence of the absence of the Weber effect with specialty care medicines but also provides a substantial evidence of linear AR reporting correlating with sales volumes, especially during the first 5 years after marketing.
Keywords: patient exposure, sales volume, specialty care, Weber effect
What is already known about this subject
The classical Weber effect describes a peak in adverse reaction reporting relating to new medicinal products at the end of second year after regulatory approval, followed by a continuous decline thereafter.
This effect has been observed for several decades with medicines used in primary care.
Several recent studies of the Weber effect with a wide range of medicinal products used in different healthcare settings have provided conflicting results.
What this study adds
This is the first study to refute the Weber effect based on qualitative (content of reference safety information i.e. whether the safety information is listed or unlisted in the reference safety information for the medicinal product) and quantitative safety information (number of adverse reaction reports) for a range of specialty care medicines.
This is the first study to describe a direct positive correlation between adverse reaction reporting with specialty care medicines and sales volumes.
1. INTRODUCTION
With the changing landscape of healthcare management, the majority of pharmaceutical companies are moving towards development of medications for the unmet needs of patients. 1 , 2 To assess the benefit risk profile of these medicines, pharmaceutical companies have set up organised solicited postmarketing safety data collection systems in accordance with International Conference on Harmonisation (ICH) E2D to provide a wider understanding of the safety aspects of the medicine beyond the controlled population studied within interventional clinical trials as well as unsolicited safety reporting in the form of spontaneous reports. 3 , 4
Observing adverse reaction (AR) reports from UK databases, Weber described the Weber effect in a study of 9 nonsteroidal anti‐inflammatory drugs (NSAIDs), predominantly prescribed in a primary care setting. The author observed an increase of AR reports after first marketing up to a peak at the end of the second year, followed by a plateau, then a steady decline in the number of AR reports despite a steady increase in the number of prescriptions written. 5 Hartnell and Wilson hypothesised that the increased awareness of prescribers, along with increased patient exposure during the first 2 years post‐approval of a medicine might explain the first part of the trend and familiarity with the safety profile of products could explain the plateau and subsequent decrease after 2 years, i.e. the second part of the trend in AR reporting. 6
A review of the literature indicates that this effect has been sought after on several occasions with varying results, thereby leading some authors to question the reproducibility of the Weber effect. 7 , 8 , 9 , 10 , 11 It has also been observed that most of the evaluations conducted on this topic have considered an absolute count of AR reports from the publicly available databases such as the Food and Drug Administration's Adverse Event Reporting System (FAERS), the World Health Organisation Uppsala Monitoring Centre's global safety database (VigiBase) and the European Medicines Agency's safety database EudraVigilance, without adjustment for patient exposure. It should be borne in mind that there is very limited information held in publicly available pharmacovigilance systems about the formal medical assessment of safety reports. One example of missing information is whether or not the reported AR is a listed event described in the approved prescribing information for the product.
Therefore, the current study was designed to evaluate the validity of the Weber effect in the context of medicines used in a specialty care setting on a more granular level, utilising sales volume data as a proxy for patient exposure. The study also evaluated the reporting trend differences among solicited and unsolicited safety reports, as well as the type of information reported via these systems i.e. whether the individual adverse event terms within each AR report are listed or unlisted in the reference safety information for the product.
2. MATERIALS AND METHODS
2.1. Source data
The study data comprised information pertinent to 10 Novartis medicinal products (Table 1). AR reports were retrieved from the Novartis global safety database (ARGUS Safety version 8.1.2.3). This database comprises all reports of serious adverse events from interventional clinical trials Phases I to III (published and unpublished), as well as all reports of adverse events from postmarketing sources that have been brought to the attention of the marketing authorisation holder (MAH). Novartis safety database data content is based on the format defined by the ICH guideline E2B(R3), 12 which has been adopted as the harmonised standard data model for individual case safety reports.
TABLE 1.
Drugs and data range for analyses
| Selected drugs | First market authorisation date | Data cut‐off |
|---|---|---|
| Carbamazepine | 12 December 1961 a | January 2018 |
| Fluvastatin IRb | 23 August 1993 a | September 2018 |
| Basiliximab | 07 April 1998 | May 2018 |
| Fluvastatin ERc | 25 July 2000 | September 2018 |
| Zoledronic acid‐Z d | 21 August 2000 | October 2018 |
| Imatinib | 10 May 2001 | May 2018 |
| Zoledronic acid‐A d | 15 April 2005 | May 2018 |
| Deferasirox | 02 November 2005 | November 2018 |
| Vildagliptin | 14 February 2007 | March 2018 |
| Sacubitril/valsartan | 07 July 2015 | August 2018 |
sales data available from 01 January 1998; b IR: immediate release; c ER: extended release.
Formulations representing 2 separate marketed products.
AR reports were derived from both solicited (post‐authorisation safety studies, 13 post‐authorisation efficacy studies, patient support programmes, patient oriented programmes and patient assistance programmes) and unsolicited sources (spontaneous reports [SR] and literature reports).
AR reports on the Novartis database include medical and personal information about uniquely identifiable patients and individual sources of reports, hence they are subject to the European General Data Protection Regulation, 14 and these data are held securely and personally identifiable information is protected. Cumulative sales data in milligrams of the active pharmaceutical ingredient (API) were obtained from the Novartis financial reporting and accounting system. Sales data such as these are not normally made publicly available due to concerns about commercial confidentiality.
The products chosen for this study were representative of medicines developed for the use in a specialty care setting; they may be broadly defined as medicinal products intended for a specific disease or organ system. These selected medicines were considered representative of a broad array of drug classes and constituted around 5% of the innovative medicines portfolio at Novartis. These medicinal products also covered a range of indications and/or different formulations in order to further extend the coverage of planned analyses. While we included all additional therapeutic indication(s) that were approved after first launch in the first‐approved indication, for the present study we selected only the drugs that either have: (i) one indication up until data cut‐off; or (ii) subsequent approved therapeutic indication(s) similar to the first approved indication.It should be noted that not all countries across the globe had precisely the same approved indications for each of the medicines selected. In addition, given the variations in the initial approval dates in different countries across the globe for each medicine, the selection of products for the study also considered the national or regional approval dates in major markets (i.e. the USA and the EU). Considering this, only the drugs that were approved in major markets within 6 months of the international birth date were selected for this study with the only exceptions being zoledronic acid and deferasirox where the approval dates differed between the EU and the USA by approximately 12 months for zoledronic acid (for both formulations) and 7 months for deferasirox. However, given that both these products received their first approvals in a major market, and the safety data for these agents could therefore be collected in a reliable and standardised manner using existing pharmacovigilance processes, these products were included in the evaluation.
For each product, the last validated cumulative safety data set submitted to the regulatory agencies was considered as the data cut‐off. Details are provided in Table 1.
2.2. Details of analysis
Analyses were conducted in 2 tiers. All analyses were conducted using the date of the first marketing authorisation (MA) as the baseline and the data were represented as epochs at 6‐monthly intervals, each defined as a period(s) post‐approval i.e. P1, P2, P3, and so on.
2.2.1. Tier I analysis
The Tier I analysis evaluated the relationship between exposure adjusted reporting rate (EARR) and epoch post‐approval. Six‐monthly sales data were used as a proxy for exposure calculation. Exposure was represented as patient‐years (PY) and was calculated as follows.
Total drug product amount sold = defined daily dose × 365.25
The PY exposure was utilised to generate the exposure adjusted reporting rate calculated as absolute number of AR reports divided by the PY exposure.
2.2.2. Tier II analysis
It was considered important to examine potential additional factors that may contribute to changes in AR reporting rates over time, hence all reported AR terms were medically reviewed and categorised for each of the medicinal products as follows:
AR reports with listed terms (compared to the approved reference safety information).
AR reports with terms related to the approved indication(s) or with related complications or consequences.
AR reports with unlisted terms (terms not included under points 1 or 2 above).
This analysis was intended to evaluate any potential reporting bias that might occur in the post‐marketing phase based on the awareness of safety profile of the drug. A further assessment of these categories was conducted based on the source of the report (unsolicited or solicited, as defined within the Methods section above).
2.3. Additional analyses
Additional analyses were conducted to evaluate the relationship between real‐world, unsolicited reporting of ARs and patient exposure. Analyses were conducted using the precise amount of API sold as a proxy for patient exposure and, the total number of unsolicited AR reports (SR and literature reports) from first MA to the data cut‐off. To improve the presentation of these data the API quantity was converted to megagrams (i.e. metric tonnes) for fluvastatin immediate‐release (IR), fluvastatin extended‐release (ER), deferasirox, vildagliptin and sacubitril/valsartan, and to kilograms for basiliximab, zoledronic acid‐Z, zoledronic acid‐A and imatinib. These analyses were restricted to the drugs where sales data was available since the first MA for the medicinal product.
In addition, a subanalysis was performed to assess the relationship between the gross sales volume and the total number of unsolicited AR reports at a fixed time point 5 years post‐launch. The interval of 5 years was chosen to coincide with the initial period of additional monitoring of safety data by regulatory agencies as outlined in Guideline on good pharmacovigilance practices (GVP) Module X. 15 This coincides with the time point for first renewal of the MA and it is complemented by the requirements set down in GVP Module VII Periodic Safety Update Report. This document provides guidance MAHs on aggregate analyses in the form of the periodic benefit risk assessment, which must be provided to the regulatory agencies by the MAH every 6 months for first 2 years of marketing and annually for another 2 years. 16 One additional year was conservatively added to the GVP recommendations, making the data‐lock point 5 years, which is the targeted time for submission of the first renewal of the MA in Europe.
The additional analyses were restricted to unsolicited reporting i.e. only SR and literature reports to exclude any external influence that may affect safety reporting, such as healthcare provider influence on AR information (e.g. in patient support programmes, patient oriented programmes and patient assistance programmes) and structured safety data collection in post‐authorisation studies.
2.3.1. Analysis tools
All analyses were conducted using Microsoft Excel v16.0. The investigation of trending was performed using regression analysis. A graphical trend line depicting the goodness of fit (R 2) value i.e. explained variation/total variation was used, with the trend line depicting the highest R 2 value being selected to represent the model with best fit. The respective analysis method was presented alongside the trend analysis. Regression parameters inbuilt within Microsoft Excel v16.0 as detailed immediately below were employed in the analyses:
Linear trend: y = m * x + b; where, m = SLOPE(y,x) and, b = INTERCEPT(y,x)
Logarithmic trend: y = (c * LN(x)) + b; where, c = INDEX (LINEST(y,LN(x)),1) and, b = INDEX (LINEST(y,LN(x)),1,2)
Power trend: y = c*x ˆb; where, c = EXP (INDEX (LINEST (LN(y),LN(x),),1,2)) and, b = INDEX (LINEST (LN(y),LN(x),),1)
Exponential trend: y = c *e ˆ(b * x); where c = EXP (INDEX (LINEST (LN(y),x),1,2)), b = INDEX (LINEST (LN(y),x),1) and, e = EXP(1)
3. RESULTS
The study data comprised a total of 1 222 852 AR reports for 10 medicinal products used in a specialty care setting. The analysis against baseline could not be performed for carbamazepine and the fluvastatin IR formulation, as the sales data for these products were only available from January 1998, despite the fact that these medicinal products were marketed before that date (see Table 1). Therefore, the analyses for these drugs represented the reporting rate trends in the later part of the products' life cycle. An overview of the study AR data is displayed in Table 2.
TABLE 2.
Study data overview
| Drugs | Unsolicited reports | Solicited reports | ||||
|---|---|---|---|---|---|---|
| ID reports | Listed reports | Unlisted reports | ID reports | Listed reports | Unlisted reports | |
| Carbamazepine | 5334 | 58 734 | 85 291 | 386 | 1704 | 9031 |
| Fluvastatin IRb | 45 | 13 501 | 13 820 | 3 | 658 | 4626 |
| Basiliximab | 1316 | 2866 | 5207 | 665 | 2169 | 4988 |
| Fluvastatin ERc | 27 | 5309 | 5897 | 5 | 404 | 1607 |
| Zoledronic acid‐Z d | 8305 | 66 800 | 109 979 | 1162 | 10 166 | 18 641 |
| Imatinib | 21 383 | 134 806 | 37 901 | 16 396 | 71 083 | 23 759 |
| Zoledronic acid‐A d | 1688 | 90 810 | 49 985 | 1697 | 25 921 | 32 736 |
| Deferasirox | 2932 | 30 877 | 35 352 | 2920 | 18 014 | 32 850 |
| Vildagliptin | 2468 | 13 991 | 10 673 | 7502 | 17 831 | 20 658 |
| Sacubitril/valsartan | 4252 | 12 982 | 10 747 | 10 175 | 14 344 | 27 473 |
sales data available from 01 January 1998; b IR: immediate release; c ER: extended release.
Formulations representing 2 separate marketed products.
3.1. Tier I analysis
Our analyses did not reveal many observations similar to the classical Weber effect for products within the study. None of the trends depicted a peak in AR reports after around 2 years of marketing. For vildagliptin there was a gradual decrease of the reporting rate (in Tiers I and Tier II of the study). The effect occurred during the first year of product launch, and not at 2 years. There was very limited evidence suggestive that reporting waned over time. Based on the results presented, our assertion is that, in general for specialist medicines reporting of ARs increases in proportion to the prescribing rate. Among the various products evaluated in the study, deferasirox, fluvastatin ER, zoledronic acid‐A and imatinib, to some extent, yielded a higher EARR immediately after launch, but the trends did not align with the classical Weber effect (Figure 1). It was notable that for the medicines that are usually used in specialty care but are also prescribed in an outpatient setting, or at the primary care (general practitioner) level (fluvastatin ER, vildagliptin and sacubitril/valsartan), there was a gradual decrease in EARR as compared to the early postmarketing phase of the medicinal product (Figure 1).
FIGURE 1.
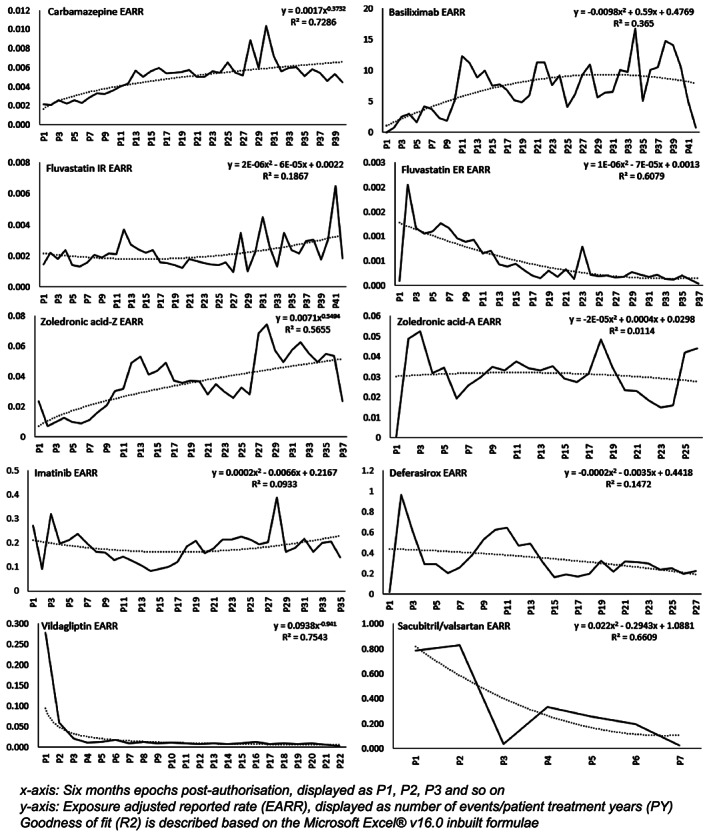
Time relationship of exposure adjusted reporting rate (EARR)
3.2. Tier II analysis
The EARR analysis for the indicated disease‐related terms could not be performed for fluvastatin ER and fluvastatin IR due to a relatively small number of AR reports on file. Similarly, due to the low number of reports, the AR analyses for unlisted events in solicited reports for carbamazepine could not be performed.
The EARR analyses based on the type of safety information in the AR report i.e. listed, unlisted or indicated disease‐related terms did not show any pattern similar to the Weber effect (Figure 2, Supplementary Figure S1). Instead, the reporting trends displayed a discordance, with an increasing reporting trend for events observed in years 3 –5 post‐launch, for majority of the products under study.
FIGURE 2.
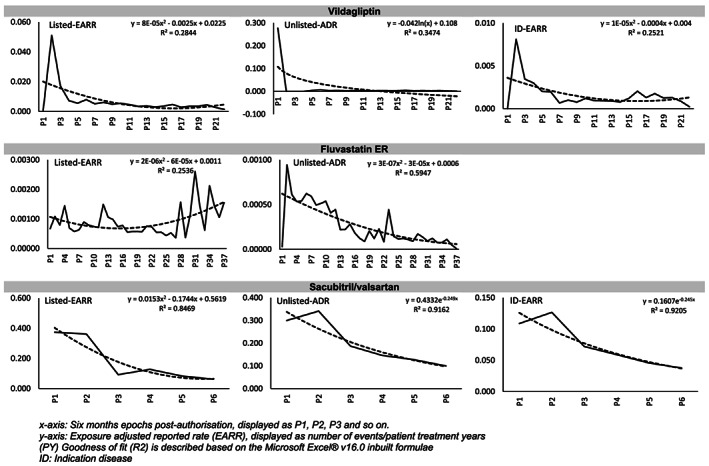
Exposure adjusted reporting rate (EARR) based on the listedness of the safety information content
Exceptionally, fluvastatin ER yielded an early pattern of AR reporting, which appeared consistent with the Weber effect; however, the latter trend was not representative of the Weber effect (Figure 2). In this case we observed a declining trend in the EARR for unlisted events, and rising trend for listed events. This is in contrast to the classical Weber effect.
Vildagliptin displayed a pattern that superficially resembled the Weber effect across all of the categories (Figure 2). However, the peak reporting rate for the product occurred during the first year post‐launch and not at the 2‐year time point described in the original observation of the Weber effect.
All of the patterns described above were also replicated across the various sources of information i.e. solicited vs unsolicited (Figure 3, Supplementary Figure S2). Overall, no pattern similar to the classical Weber effect was observed under any of the variables tested with each of the 10 specialty care medicines.
FIGURE 3.
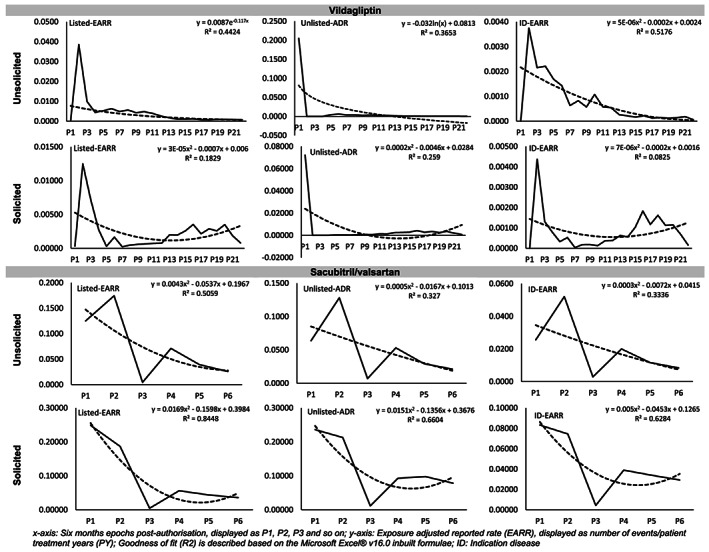
Reporting trends based on safety information content (listedness) and the source of the reports
3.3. Additional analyses
The regression analyses to evaluate the correlation between the total API amount and the total number of unsolicited AR reports since the first marketing of the drug indicated a linear correlation for majority of the products. Although a linear trend was not apparent for imatinib and deferasirox, the numerical data represented a steadily increasing number of AR reports that was systematically related to increasing sales volumes. This trend was not observed for vildagliptin (Supplementary Figure S3).
For the subanalysis using the first 5 years' marketing data, there was a clear linear trend between the total amount of API sold and the number of AR reports, with the sole exception of fluvastatin ER (Figure 4).
FIGURE 4.
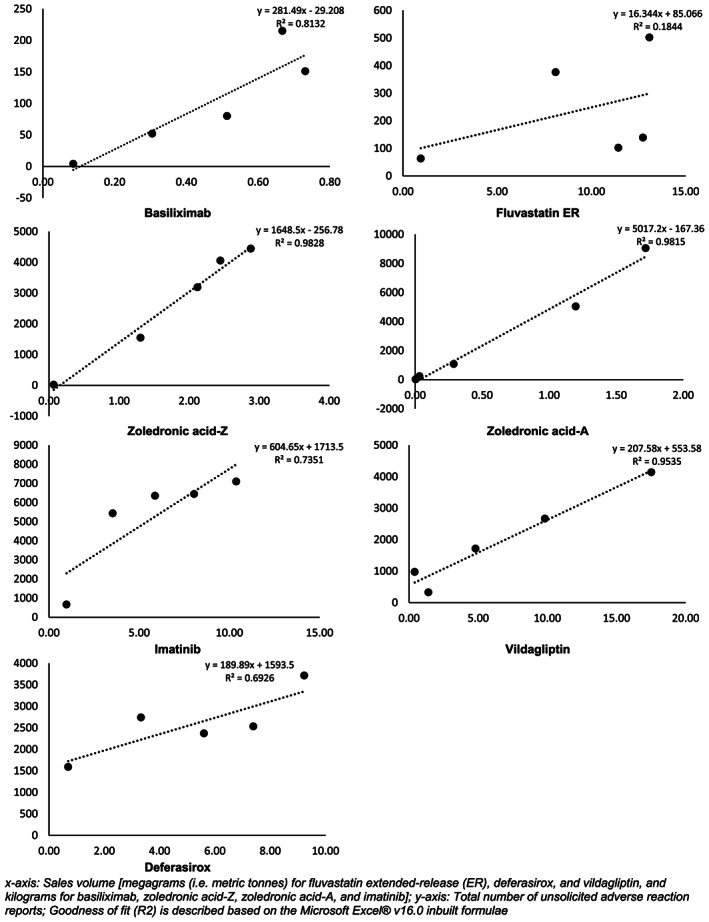
Total sales volume vs the number of unsolicited reports (first 5 years postlaunch)
4. DISCUSSION
Since the first postulation of the Weber effect in 1984, the pharmaceutical paradigm has substantially changed. 17 , 18 There have been significant improvements to pharmacovigilance systems across the globe, with growing numbers of healthcare professionals, patients and consumers becoming aware of, and making positive contributions to safety information within pharmacovigilance systems. 19
The Weber effect has often been used to rationalise the frequency of AR reporting and thereby associating these observations with signal evaluation. However, it certainly appears to be valid to question the applicability of the Weber effect based on the evidence presented above. The authors believe that exploring different determinants of AR reporting may not only help in supporting better signal detection, but may also contribute to the comprehension of the actual incidence and prevalence of safety risks. The authors believe that such explorations can aid in more robust research, shaping and defining public policies, financial and pharmacoeconomic studies.
In the present study, including data from over 1 million AR reports, the most noteworthy finding was that for majority of the 10 medicinal products evaluated, the numbers of AR reports systematically correlated with the sales volume. Utilising linear regression, the authors tried to comprehend the relationship between number of AR reports and sales, independent of the product or the disease under treatment. In all these analyses, a consistent high R 2 value was observed for a linear relationship. This linearity was even more pronounced during the first 5 years of marketing during which time there is intensive scrutiny of the safety profile of the medicinal product. It should be noted that data suggested a lack of linearity for fluvastatin ER. However, this observation strengthens the hypothesis that for a compound where safety profile is relatively known (in this case knowledge was gleaned from the IR formulation which was introduced earlier), a change in formulation is less likely to trigger a higher reported rate of ARs, despite increasing sales volumes.
It is noteworthy that the Weber effect was initially described for drugs predominantly used in a primary care setting (i.e. NSAIDs and selective serotonin‐reuptake inhibitors). 6 , 8 The original findings made by Weber5 differ markedly from later observations by Wallenstein and Fife. 11 In that study, while the authors studied NSAIDs, and thus this was akin to the original study that postulated the Weber effect, the results did not show the classical temporal rise and fall of number of AR reports after launch. The authors attributed these observations to the change in the processes for reporting safety data in the USA (i.e. reports of all ARs not just suspected ARs from all sources not just physician reports), as well as to the public communications that might have stimulated the reporting of ARs during peak reporting rate periods. 11
The review of literature for drugs intended to be used in specialty care settings showed observations different from Weber effect. McAdams et al. 9 studied the rate of AR reports of 4 angiotensin receptor blockers and observed patterns different from the Weber effect. The authors evaluated AR reports from the FAERS database, and adjusted the number of reports to the number of dispensed prescriptions from US outpatient retail pharmacies, as a proxy for drug exposure. 9 Similarly, analyses conducted by Arora et al. 7 and Hoffman et al. 20 studied recently approved drugs and utilised data from FAERS. There was no observation of a second‐year peak nor of a subsequent fall in the volume of AR reports. One important observation evident from the review of studies conducted with drugs used in a specialty care setting is that whilst Weber5 attributed the changing trends in AR reporting to drug exposure and familiarity of the drug, the studies thus far have primarily relied upon the absolute number of ARs from the source system (usually the Adverse Event Reporting System for the USA, which was the predecessor of FAERS). Additionally, studies evaluating the reported number of ARs adjusted for exposure have attempted to evaluate data utilising information from 2 unrelated sources such as number of AR reports from FAERS and exposure estimates from dispensed prescriptions. We believe that this approach is flawed in that it confers an undesirable heterogeneity because the sources of ARs in source database may be different from the sources of the prescription data that is to say that the 2 key variables are to an extent independent of each other. Additionally, it should be noted that although the FAERS database is a comprehensive repository of safety data, there are several limitations of this data set. Examples of these limitations include the duplication of reports for individual patients, which occurs when a consumer or patient reports an AR to both Food and Drug Administration and to the MAH and the exclusion of non‐expedited reports for example reports of non‐serious adverse events. If the MAH is granted a waiver, there is no requirement to automatically submit AE reports of listed non‐serious events. 21 These drawbacks tend to prevent a comprehensive evaluation of AR reporting from FAERS and EudraVigilance and from other regulatory‐sourced databases including VigiBase. Also, the difference between regulatory obligations for the reporting of serious vs non‐serious reports may also provide for a misleading assessment, if seriousness is taken as the parameter for important (as opposed to non‐serious equating to trivial or not clinically important) safety information as postulated by Weber. 5 In summary, it is our view that although the methodologies reported in the available literature provide a high level understanding of the AR reporting patterns, the more granular approach taken in the present study, employing patient exposure from direct sales data and qualitative safety information (listed vs unlisted AR), provides a more comprehensive and detailed evaluation of AR reporting patterns. In addition, the potential limitation for this study (in that we used Novartis' global safety database, which is not as heterogeneous and diversified as reference databases such as the FAERS database, VigiBase or the EudraVigilance database) could be considered a strength because of the legal obligation of the MAH to record all reports of ARs. It should be noted that a detailed comparison of all safety data content in these reference databases is not possible due to data privacy restrictions, 14 which restrict access and, to some extent, publication.
The evidence presented in this study does not appear to substantiate the hypotheses linking the Weber effect to the physician's knowledge and confidence in prescribing medicines after around 2 years of availability. In particular, we provide evidence that contradicts this hypothesis. We found no evidence to affirm that the collective increase in physicians' awareness during the first 2 years after MA is followed by familiarity with the safety profile of the product thereby leading to a subsequent decrease in AR reporting. The evidence that we have presented shows that the absolute number of AR reports correlates directly with patient exposure over an extended period of time (at least 5 years, and for several years beyond). Based on these comprehensive data, it is our view that the vast majority of the data from the study were discordant with the classical Weber effect. There was 1 exception, fluvastatin ER, where a partial trend consistent with the classical Weber effect was observed. This could be mitigated by the fact that whilst fluvastatin ER was a new approved formulation, the API is identical to the previously approved product fluvastatin IR, which was marketed for 7 years prior to the launch of the ER formulation. Therefore, the early peak of reporting rate for fluvastatin ER, which was observed at 2 years post‐launch should be viewed with caution. It is plausible that this is a true reflection of the first phase Weber effect in that physician awareness of the safety profile first‐approved IR formulation could have been established prior to the approval of the ER formulation. Additionally, when this trend is evaluated utilising the listedness assessment and source of safety information, the trend was apparent only for unlisted events, and was limited to the setting of solicited data collection. By contrast, the peak in unsolicited, unlisted AR reports occurred much earlier than would be the case if this were a classical Weber effect. Hence, in our view, even this isolated finding is not consistent with the postulates of the Weber effect.
It is noteworthy that for the majority of the medicinal products studied, the increase in EARR for listed events preceded the increase in reporting rates for unlisted events. This is in contrast to the postulate proposed to explain the Weber effect, where it was hypothesised that with increasing awareness of the safety profile for a medicinal product the reporting rate should decrease over time. However, for relatively newer products this observation could potentially be related to the rapid dissemination of safety data in the digital age. Healthcare providers and consumers have greater access to the safety information online, with both authorised and anecdotal reports available from multiple sources including social media. 22
Lastly, although it was not intent of the study, it was notable that for the drugs that are usually used in specialty care but are also prescribed in the outpatient setting at a general practitioner level (fluvastatin ER, vildagliptin and sacubitril/valsartan), there was a declining trend of the EARR. This declining rate may imply that although the prescribers of these drugs may be becoming more confident in the safety profile of the drug in the context of the potential benefits, there is a need to reinforce understanding that pharmacovigilance is important for the identification of new potential ARs as it is valuable for characterising known adverse effects and tolerability issues in real world use. In other words, the reporting, collection and collation of AR reports throughout the life‐cycle of medicinal products informs the ongoing benefit–risk assessment.
4.1. Study limitations
Although this study has some strengths, we recognise that additional exploration of our observations concerning reporter data (i.e. healthcare provider vs consumer reports) and the important of AR seriousness would strengthen our hypothesis of a direct correlation between AR reports and sales volume. We recognise that our study is limited by the closed nature of the Novartis global safety database, in the sense that this dataset is not in the public domain thus cannot be accessed by external researchers. Nevertheless it is our assertion that this is a reliable source system, as it is a good practice quality guidelines and regulations‐validated relational patient safety database based on the ICH E2B harmonised data model, and the data content is controlled according to a wide range of data quality standards. Also, the study utilised the MA approval date as the data‐lock point. We recognise that the approval of reimbursement, and the formulation of national or regional treatment guidelines and the actual launch date for the product, may be delayed. Practically, this potential delay of market access when compared to the MA approval could have influenced the study results. It is our belief that because the actual product launch date was invariably within 6 months of MA approval in at least 1 of the major markets identified, and utilisation of EARR instead of the absolute AR report count, has reduced any impact of these variables. Finally, the study referred to the reference safety information available at the data‐lock point. It is inevitable that medically important AR terms would have been incorporated into the reference safety information after the first approval of the drug, based on the evaluation of postmarketing safety data. Whilst such terms may have been considered unlisted at first approval, in the current study, based on the latest data cut‐off, these terms were considered as listed. It is our view that the ARs identified during the early postmarketing phase represent uncommon events (with a frequency of <1/100); hence, the numerical impact of such a change was minimal.
The authors acknowledge that there are several additional variables that may influence AR reporting including, but not limited to:
Stimulated reporting after identification of important new safety information;
Changes in safety reporting obligations enforced by new or amended regulations;
Awareness of safety reporting platforms; and
Local treatment practices (e.g. prevalence of polypharmacy, prescribing guidelines).
We believe that composite analyses including controls for these variables would potentially yield a more robust assessment of AR reporting trends.
COMPETING INTERESTS
Dr Vikas Modgill is a full‐time employee of Novartis Pharmaceuticals AG, Basel Switzerland and Dr David J. Lewis is a full‐time employee of Novartis Pharma GmbH, Wehr, Germany. Dr Modgill holds options on Novartis stock. Dr Lewis holds shares in Novartis and GlaxoSmithKline, and has options on Novartis stock. Dr Lea Dormegny has no conflicts of interest that are relevant to the content of this study. Dr Dormegny was an associate of Novartis Pharmaceuticals AG, Basel Switzerland. The authors have no conflicts of interest that are directly relevant to the content of this study.
The views and opinions expressed in this article are those of the authors, and they do not reflect in those of the institutions to which they are affiliated.
CONTRIBUTORS
DJL conceived the idea and, with LD, developed the early manuscript. VM and DJL extended the first draft and developed the theory. Calculations were performed by VM. All authors contributed to the final manuscript, with DJL providing critical feedback.
Supporting information
FIGURE S1 Exposure adjusted reporting rate (EARR) based on the listedness of the safety information content
FIGURE S2 Reporting trends based on safety information content (listedness) and the source of the reports
FIGURE S3 Total sales volume vs number of unsolicited reports
ACKNOWLEDGEMENTS
The authors would like to thank Vikas Vaishnavi, Srilatha R.K., Hemanth Naraparaju and Hemang Soni from Novartis Healthcare Pvt. Ltd., India for their logistical support of this study.
Modgill V, Dormegny L, Lewis DJ. Reporting rates of adverse reactions to specialty care medicines exhibit a direct positive correlation with patient exposure: A lack of evidence for the Weber effect. Br J Clin Pharmacol. 2020;86:2393–2403. 10.1111/bcp.14342
The authors confirm that the Principal Investigator for this paper is Vikas Modgill.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Khanna I. Drug discovery in pharmaceutical industry: productivity challenges and trends. Drug Discov Today. 2012;17(19–20):1088‐1102. [DOI] [PubMed] [Google Scholar]
- 2. Scavone C, di Mauro G, Mascolo A, Berrino L, Rossi F, Capuano A. The new paradigms in clinical research: from early access programs to the novel therapeutic approaches for unmet medical needs. Front Pharmacol. 2019;10 10.3389/fphar.2019.00111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Council for International Organisations of Medical Sciences . Current challenges in pharmacovigilance: pragmatic approaches. Report of CIOMS Working Group V. Geneva: CIOMS; 2001.
- 4. International Conference on Harmonisation . Post‐approval safety data management: Definitions and standards for expedited reporting E2D. May 2004. https://database.ich.org/sites/default/files/E2D_Guideline.pdf
- 5. Weber J.C.P. (1987) Epidemiology in the United Kingdom of adverse drug reactions from non‐steroidal anti‐inflammatory drugs In: Rainsford K.D., Velo G.P. (eds) Side‐Effects of Anti‐Inflammatory Drugs. Inflammation and Drug Therapy Series, vol 1 Springer, Dordrecht: 10.1007/978-94-010-9772-7_2 [DOI] [Google Scholar]
- 6. Hartnell NR, Wilson JP. Replication of the Weber effect using postmarketing adverse event reports voluntarily submitted to the United States Food and Drug Administration. Pharmacotherapy. 2004;24(6):743‐749. [DOI] [PubMed] [Google Scholar]
- 7. Arora A, Jalali RK, Vohora D. Relevance of the Weber effect in contemporary pharmacovigilance of oncology drugs. Ther Clin Risk Manag. 2017;13:1195‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hartnell NR, Wilson JP, Patel NC, Crismon ML. Adverse event reporting with selective serotonin‐reuptake inhibitors. Ann Pharmacother. 2003;37(10):1387‐1391. [DOI] [PubMed] [Google Scholar]
- 9. McAdams MA, Governale LA, Swartz L, Hammad TA, Dal Pan GJ. Identifying patterns of adverse event reporting for four members of the angiotensin II receptor blockers class of drugs: revisiting the Weber effect. Pharmacoepidemiol Drug Saf. 2008;17(9):882‐889. [DOI] [PubMed] [Google Scholar]
- 10. de Graaf L, Fabius MA, Diemont WL, van Puijenbroek EP. The Weber‐curve pitfall: effects of a forced introduction on reporting rates and reported adverse reaction profiles. Pharm World Sci. 2003;25(6):260‐263. [DOI] [PubMed] [Google Scholar]
- 11. Wallenstein EJ, Fife D. Temporal patterns of NSAID spontaneous adverse event reports: the Weber effect revisited. Drug Saf. 2001;24(3):233‐237. [DOI] [PubMed] [Google Scholar]
- 12. ICH guideline E2B (R3) on electronic transmission of individual case safety reports (ICSRs) ‐ data elements and message specification ‐ implementation guide . [PubMed]
- 13. European Medicines Agency (EMA). Guideline on good pharmacovigilance practices (GVP) . Module VIII – Post‐authorisation safety studies (Revision 3). October 2017. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-module-viii-post-authorisation-safety-studies-rev-3_en.pdf
- 14. European Commission . General Data Protection Regulation (GDPR) 2016/679. 2016. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32016R0679
- 15. Guideline on good pharmacovigilance practices (GVP) Module X Additional Monitoring . April 2013. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-module-x-additional-monitoring_en.pdf
- 16. Guideline on good pharmacovigilance practices (GVP) Module VII—periodic safety update report (Rev 1) . https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-module-vii-periodic-safety-update-report_en.pdf
- 17. Finkelstein SN and Sinskey AJ. The coming paradigm shift in pharmaceuticals http://web.mit.edu/popi/PharmaGenomics/PharmaGenomics.Sept.Oct.2002.pdf
- 18. Kunst M Natanek R Plantevin L et al. A new pharma launch paradigm: From one size fits all to a tailored product approach. June 2013. https://www.bain.com/insights/a-new-pharma-launch-paradigm/
- 19. Fornasier G, Francescon S, Leone R, Baldo P. An historical overview over pharmacovigilance. Int J Clin Pharmacol. 2018;40(4):744‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoffman KB, Dimbil M, Erdman CB, Tatonetti NP, Overstreet BM. The Weber effect and the United States Food and Drug Administration's adverse event reporting system (FAERS): analysis of sixty‐two drugs approved from 2006 to 2010. Drug Saf. 2014;37(4):283‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. FDA . Providing Postmarketing Periodic Safety Reports in the ICH E2C(R2) Format (Periodic Benefit‐Risk Evaluation Report). Guidance for Industry. November 2016. https://www.fda.gov/media/85520/download
- 22. Sloan R, Osanlou O, Lewis D, Bollegala D, Maskell S, Pirmohamed M. Social media and pharmacovigilance: a review of the opportunities and challenges. Br J Clin Pharmacol. 2015;80(4):910‐920. 10.1111/bcp.12717 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Exposure adjusted reporting rate (EARR) based on the listedness of the safety information content
FIGURE S2 Reporting trends based on safety information content (listedness) and the source of the reports
FIGURE S3 Total sales volume vs number of unsolicited reports
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.