

Abstract
Aim
Regorafenib is an oral multikinase inhibitor with clinical efficacy in a range of advanced solid tumours. A population pharmacokinetic (PK) model was developed to evaluate the variability of the PK of regorafenib and its pharmacologically active metabolites M‐2 and M‐5 in solid tumours.
Methods
The model was initially developed using densely sampled phase 1 data and information on food intake to incorporate enterohepatic circulation (EHC) that was identified to considerably contribute to the PK of regorafenib. This was then applied to sparsely sampled data from four phase 3 studies in patients with advanced solid tumours. The need for exact food intake data to estimate individual drug exposure was evaluated.
Results
By incorporating EHC, the model adequately described the PK profiles of regorafenib, M‐2 and M‐5 after single and multiple doses in patients from phase 1 studies. Individual exposure in phase 3 studies was adequately described based on assumptions on the time and frequency of food intake, although exact food intake data are recommended to improve the estimation. Covariate analysis identified sex and body mass index (BMI) as impacting exposure to regorafenib, and sex as strongly impacting exposure to M‐2 and M‐5 (also influenced by the BMI effect on parent regorafenib in the joint model developed); however, these factors accounted for a small portion of the overall variability in exposure.
Conclusions
The adequate description of regorafenib PK after multiple dosing requires the incorporation of EHC. Neither single nor combined covariates predicted exposures that would warrant a priori regorafenib dose adjustment.
Keywords: clinical pharmacology, drug metabolism, population pharmacokinetics, regorafenib
1. What is already known about this subject
Regorafenib exposure in clinical practice may be variable, and may be influenced by new dosing strategies, where fixed dosing may no longer be standard practice.
Enterohepatic circulation has been identified as a significant absorption and clearance pathway of regorafenib that should be taken into account in PK studies.
What this study adds
The model adequately estimates exposure of regorafenib and its metabolites, based on sparse clinical study/practice data without information on food intake, although recording mealtimes or PK sampling after prolonged post‐prandial periods are recommended for more reliable exposure estimates.
Identified patient characteristic covariates impart only minor contributions to observed PK variability.
1. INTRODUCTION
Regorafenib (4‐[4‐[[4‐chloro‐3‐(trifluoromethyl)phenyl]carbamoylamino]‐3‐fluorophenoxy]‐N‐methylpyridine‐2‐carboxamide) is an oral multikinase inhibitor with anti‐angiogenic, anti‐metastatic and immunomodulatory effects. 1 , 2 Early‐phase clinical studies demonstrated the antitumour activity of regorafenib in various solid tumours, 3 , 4 , 5 , 6 , 7 , 8 which translated into clinical benefit in phase 3 studies in patients with treatment‐refractory metastatic colorectal cancer (mCRC), hepatocellular carcinoma and advanced gastrointestinal stromal tumours. 9 , 10 , 11 , 12
Regorafenib is metabolised primarily in the liver by oxidative metabolism, mediated by cytochrome P450, family 3, subfamily A, polypeptide 4 (CYP3A4), and glucuronidation, mediated by uridine diphosphate glucuronosyltransferase family 1 member A9 (UGT1A9). 13 , 14 , 15 Two major and six minor metabolites have been identified in human plasma. 13 , 16 The two major metabolites, M‐2 (N‐oxide; BAY 75‐7495) and M‐5 (N‐oxide and N‐desmethyl; BAY 81‐8752), are pharmacologically active and reach nearly similar concentrations to regorafenib at steady state. 13 , 16 Moreover, the estimated free plasma concentrations of regorafenib and M‐2, but not M‐5, exceeded the half maximal inhibitory concentration (IC50) for human kinase insert domain receptor (vascular endothelial growth factor receptor 2), suggesting that regorafenib and M‐2 are the primary contributors to the pharmacologic activity of the drug in humans. 16
Although there are clear differences in the fraction of unbound regorafenib and metabolites M‐2 and M‐5 in human plasma (0.5%, 0.2% and 0.05%, respectively 16 ), these metabolites should be taken into account when considering active drug exposure in clinical practice. Here we describe the development of a comprehensive population pharmacokinetic (PK) model for regorafenib, M‐2 and M‐5 after a single 160 mg dose and after multiple doses. Furthermore, during the clinical development of regorafenib, enterohepatic circulation (EHC) was identified as a significant disposition pathway of regorafenib, with the gallbladder acting as a collection and storage repository for regorafenib and its metabolites. When bile is secreted into the gut through gallbladder emptying after food intake, 17 regorafenib and its metabolites may be reabsorbed. 18 Therefore, the comprehensive population PK model for regorafenib and its metabolites described here incorporates EHC.
The objective of this study was to investigate the ability of a model based on sparse PK sampling used in phase 3 studies to predict drug exposure. This model may provide a tool that can be used to estimate individual patient's drug exposure based on sparse PK sampling, particularly as exposure may be highly variable and flexible dosing strategies are becoming standard clinical practice. 19
Since daily food intake has a significant effect on bile secretion, it is important to consider the timing of food intake. Therefore, since the timing of food intake was not reported in the phase 3 studies, the effect of imputing mealtimes on the estimated exposure of regorafenib and its metabolites was also investigated. The population PK model was used to evaluate the effects of covariates (patient [including age, sex, race, body mass index (BMI) and alcohol aetiology] and clinical [including indication and hepatic and renal function] parameters). Exposure estimates derived with population PK models either with or without EHC incorporated were compared.
2. METHODS
The population PK and covariate data used in this study originate from a number of clinical studies (Appendix Table A1 in the Supporting information). All protocols were approved by the ethics committees or institutional review boards of participating institutions and complied with the Declaration of Helsinki, current Good Clinical Practice guidelines and applicable local laws/regulations. All participants provided written informed consent.
Population PK modelling was performed using NONMEM version 7.4 (ICON plc, Gaithersburg, MD, USA). GFortran (gcc version 5.4.0) was used as compiler. Diagnostic graphics, statistical tests, exploratory analyses and post‐processing of NONMEM output were performed using R version 3.3.2 (The R Foundation for Statistical Computing, Vienna, Austria) and RStudio Version 1.0.44 (RStudio Inc., Boston, MA, USA). The first‐order conditional estimation approximation (FOCE with η‐ε interaction) was used as the estimation method with a convergence criterion of at least three significant digits in the parameter estimation, using NONMEM mixed‐effects modelling to describe the relationship between independent variables (such as dose and time) and the dependent variable (concentration). 20
2.1. PK sampling and analysis
The population PK model incorporating EHC was initially developed based on densely sampled PK data from phase 1 studies, and the model was qualified and subsequently applied to sparsely sampled PK data from phase 3 studies; sampling designs and schedules are provided in Appendix Table A1 in the Supporting information.
2.1.1. Data
The phase 1 dataset for EHC model development was constructed from densely sampled PK data obtained from the two phase 1 studies, comprising data from 62 patients (1,340 observations for parent regorafenib). For model qualification and the covariate analysis, sparsely sampled PK data from four phase 3 clinical trials were added involving another 906 patients (Appendix Table A1 in the Supporting information). Population PK models for parent regorafenib and its M‐2 and M‐5 metabolites were developed and qualified with the data from all 968 patients treated with a regorafenib starting dose of 160 mg daily.
2.1.2. PK analysis
After single and multiple dosing, PK samples for the first 8 hours post‐dose and 7‐day post‐dose washout were included. To determine absorption in the population PK model, a dataset based only on the phase 1 study 15823 was constructed. A full dataset was obtained by adding data from the other phase 1 study 14814 to this dataset. For the phase 3 model, a dataset was constructed using PK data from four phase 3 clinical studies (Appendix Table A1 in the Supporting information). PK samples were analysed using previously published methodology. 21 Briefly, samples were analysed using a triple‐quadrupole tandem mass spectrometer with a validated analyte concentration range of 2.0 μg/L (lower limit of quantification) to 2.0 mg/L (see Appendix Section 1 in the Supporting information for further details).
2.2. Handling missing mealtimes
Mealtimes in studies 14814 and 15823 were not reported on all days; therefore, for the preparation of the phase 1 dataset, mealtimes were related to regorafenib dose intake per protocols (0.5 h before dosing). For days where no meal/dosing times were available, three generally accepted mealtimes (based on Leech et al. 22 ) were assumed: 08:00, 12:00 and 18:00. At days where at least one mealtime was reported, others were calculated assuming a period of 4 h between breakfast and lunch, and 6 h between lunch and dinner. In the four phase 3 studies, mealtimes were not available and were set to be relative to dose with time between breakfast and dose increased from 0.5 h to 1 h.
2.3. Population PK models
2.3.1. Phase 1 single‐dose and multiple‐dose regorafenib model
As a starting point for model development, the PK of parent regorafenib was described by a two‐compartmental linear model with transit compartments to describe the absorption profile (Figure 1). To incorporate EHC into the model, a gallbladder compartment was included, integrating transfer to and from the central compartment. It was assumed that regorafenib is transported from the central compartment to the gallbladder compartment following first‐order kinetics, independent of other processes, such as food effects. However, the kinetics of the reverse transfer of regorafenib from the gallbladder to the central compartment was assumed to depend on food intake, since this is the major trigger for bile release via gallbladder emptying. 23 , 24 A switch function was included in the model to mimic gallbladder‐mediated drug release. 25 For the purposes of this model, it was assumed that three releases, complete and (almost) instantaneous emptying (assumed first‐order rate constant 100 h−1) occurred per day, and that the duration of transfer of regorafenib from the gallbladder compartment to the central compartment was approximately 30 min. 17 , 24 PK data from study 15823 up to 8 h post‐dose after a single dose of regorafenib were used to determine the absorption process. The absorption rate constant k a (Figure 1) was fitted and the number of transit compartments required to get an adequate description of the absorption was determined. After an appropriate description of the fixed‐effects part of the model, interindividual variability (IIV) of k a was included. k a, the variance of random variability of k a, and the number of transit compartments, were all fixed in subsequent steps of the model development. Subsequently, the EHC model was fitted to all regorafenib data (single and multiple doses) from studies 15823 and 14814, resulting in the phase 1 regorafenib model.
FIGURE 1.
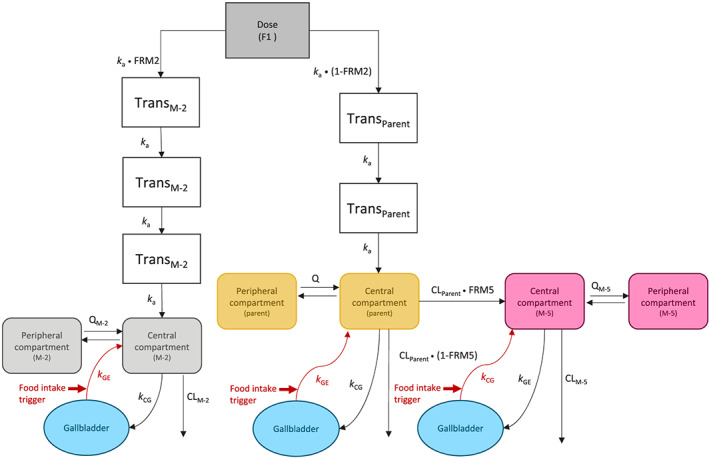
Final population PK model for regorafenib, M‐2 and M‐5, incorporating enterohepatic circulation. Volume and flow parameters are relative, and apparent clearances and volume of distribution are shown. Gallbladder emptying for parent regorafenib is described by the following equations (this also occurs in the model for M‐2 and M‐5, but these similar equations are not presented here):
Not during meal:
dA3/dt = k a A2 – k CG A3 – (CLP/VCP) A3 – (QP/VCP) A3 + (QP/VPP) A5
dA4/dt = k CG A3
During meal:
dA3/dt = k a A2 – k CG A3 – (CLP/VCP) A3 – (QP/VCP) A3 + (QP/VPP) A5 + A4 k GE
dA4/dt = k CG A3 – A4 k GE
A3, parent amount in central compartment; A4, parent amount in gallbladder compartment; A5, parent amount in peripheral compartment; CG, central compartment to gallbladder; CLP/CLM‐2/CLM‐5, apparent parent/M‐2/M‐5 clearance; FRM2/5, fraction of formation of metabolite M‐2/M‐5; F1, oral bioavailability; GE, gallbladder emptying; k, rate constant; k a, absorption rate constant; PK, pharmacokinetic; Q, intercompartmental clearance; Trans, transit compartment; VC, central compartment volume; VP, peripheral compartment volume
2.3.2. Phase 1 regorafenib–metabolite model
As a next step, the M‐2 and M‐5 metabolite data were included in the analysis to develop a combined parent–metabolite model, in which the structural model parameters for parent regorafenib were fixed to their values obtained when fitted to data for regorafenib only. It was assumed that M‐2 is formed presystemically from regorafenib, and M‐5 is formed from regorafenib in the central compartment. The PK of M‐2 and M‐5 could also be described by a two‐compartmental model (Figure 1), and both metabolites are also subject to EHC. The apparent fractions of formation of M‐2 and M‐5 (FRM2 and FRM5; Figure 1), and apparent oral clearance (referred to as clearance [CL] throughout) of M‐2 and M‐5 (CLM‐2 and CLM‐5) and IIV were then estimated by fitting the model to the complete dataset. Since no PK data are available after direct administration of M‐2 and M‐5, the volume of distribution of M‐2 and M‐5 cannot be estimated, and it is assumed that their volume is the same as that of parent regorafenib. Modelling methodology and evaluation are described in detail in Appendix Section 2 in the Supporting information.
2.3.3. Phase 3 regorafenib–metabolite covariate model
The regorafenib phase 1 model was fitted to sparsely sampled regorafenib PK data from the phase 3 studies to generate the regorafenib phase 3 model. The shift (time between meal and dose) was allowed to change based on phase 3 data, driven by an improvement of fit (based on objective function value [OFV] and goodness of fit [GOF]). As the sparsely sampled phase 3 studies do not contain sufficient information on all model parameters, we assessed which parameters could be estimated based on the phase 3 data, and which parameters had to be fixed to the values of the phase 1 model. The phase 3 parent regorafenib covariate model was developed first, followed by an optimisation of the model for the parent and the two metabolites, developed by combining the phase 3 regorafenib covariate and phase 1 parent–metabolite models. This model was used as a starting point for the analysis of covariates in the phase 3 parent–metabolite covariate model (Appendix Section 2 in the Supporting information). Empirical Bayes estimates of individual model parameters were used to derive the individual exposure to regorafenib (Cav,p; mean concentration of regorafenib over 24 h at steady state of 160 mg), M‐2 and M‐5. To confirm detected covariate effects and visualise the extent of the effect on a clinically relevant measure of exposure to parent regorafenib, M‐2 and M‐5, forest plots were created (Appendix Section 2 in the Supporting information).
2.4. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, 26 and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18. 27
3. RESULTS
3.1. Phase 1 regorafenib model
A description of the regorafenib absorption process after a single dose in study 15823 required two transition compartments. Fitting the EHC population PK model, with fixed k a and IIV of k a from the single‐dose step, to the full phase 1 dataset from 62 patients provided a successful minimisation and covariance step. Inclusion of EHC resulted in a significantly improved fit versus an equivalent model without EHC and was able to describe the observed higher steady state concentration caused by gallbladder emptying and rapid reabsorption, followed by a rapid decrease after dosing through distribution and elimination, and subsequent slow absorption step 3–4 h later (Appendix Figure A1 in the Supporting information). Visual predictive check (VPC) plots for the phase 1 regorafenib model are shown in Figure 2. The model adequately predicted the central tendency and the variability of the observed data, both after a single dose and at steady state, and the higher pre‐dose concentrations observed after multiple dosing were well described by inclusion of EHC. Parameter values and their uncertainties for the parent regorafenib model with EHC, based on both phase 1 studies, with fixed absorption parameters based on single‐dose data of study 15823 only, are shown in Table 1.
FIGURE 2.
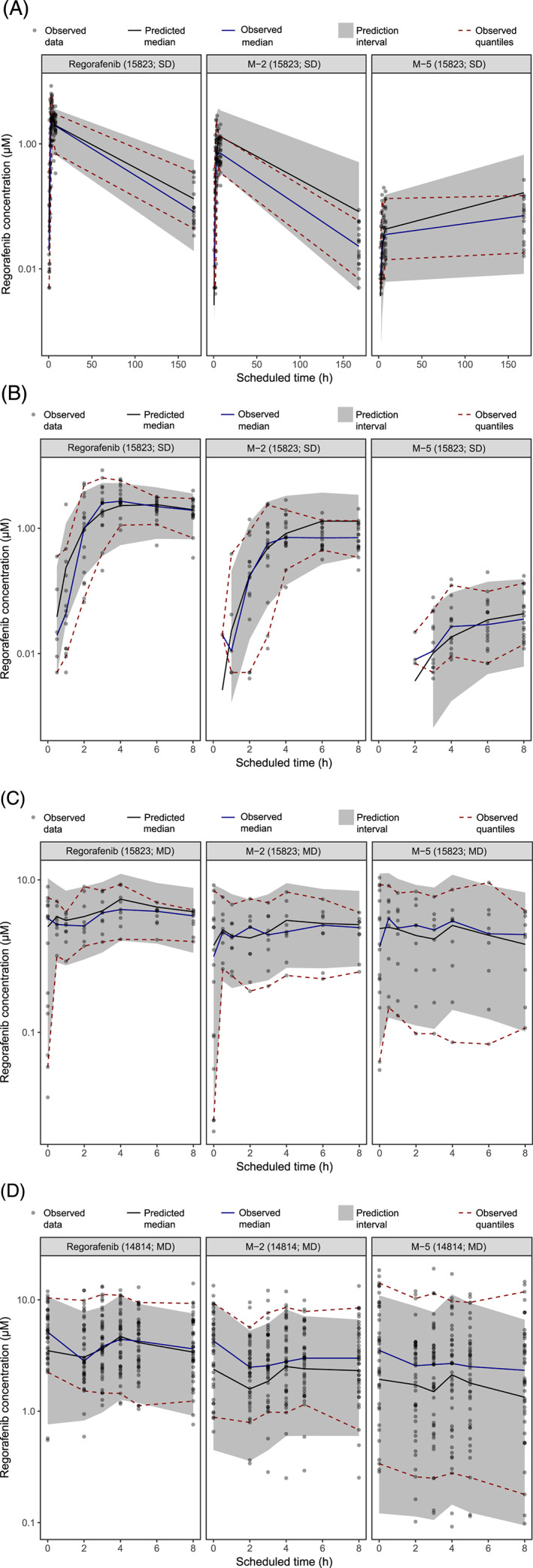
VPCs for the phase 1 regorafenib–metabolite enterohepatic circulation population PK model. Predicted concentrations of regorafenib and its metabolites, M‐2 and M‐5, after a single dose (A and B) and multiple doses (C and D) in two phase 1 studies [14814 (n = 44) and 15823 (n = 18)]. Black dots represent observed concentrations in the two phase 1 studies. The grey area represents the 90% prediction interval; the red dashed lines represent the 5% and 95% percentiles of the observed concentrations. The black lines represent the median of the model predictions, and the blue lines represent the median of the observations.
MD, multiple dose; PK, pharmacokinetic; SD, single dose; VPC, visual predictive check
TABLE 1.
Model parameter estimates of the phase 1 regorafenib enterohepatic circulation model, showing parameter estimates and precision (see Figure 3 plots)
| Parameter | Value | SE | RSE % | L95% CI | U95% CI |
|---|---|---|---|---|---|
| Fixed effects | |||||
| k a, h−1 | 0.482 | Fixed | ‐ | ‐ | ‐ |
| CL/Foral, L h−1 | 4.02 | 0.266 | 6.60 | 3.50 | 4.54 |
| VC/Foral, L | 10.7 | 1.92 | 18.1 | 6.88 | 14.4 |
| k GE, h−1 | 100 | Fixed | ‐ | ‐ | ‐ |
| Breakfast, h | 08:00 | Fixed | ‐ | ‐ | ‐ |
| Lunch, h | 12:00 | Fixed | ‐ | ‐ | ‐ |
| Dinner, h | 18:00 | Fixed | ‐ | ‐ | ‐ |
| Q/Foral, L h−1 | 11.0 | 1.37 | 12.5 | 8.30 | 13.7 |
| VP/Foral, L | 162 | 26.2 | 16.1 | 111 | 214 |
| k CG, h−1 | 0.141 | 0.0190 | 13.5 | 0.103 | 0.178 |
| DGE, h | 0.500 | Fixed | ‐ | ‐ | ‐ |
| Individual random effects | |||||
| ω2 k a | 0.127 | Fixed | ‐ | ‐ | ‐ |
| ω2CL | 0.123 | 0.0250 | 20.3 | 0.0741 | 0.172 |
| Residual error | |||||
| SD of proportional error | 0.413 | 0.0214 | 5.18 | 0.371 | 0.455 |
CG, central compartment to gallbladder; CI, confidence interval; CL, apparent total body clearance; DGE, duration of gallbladder emptying; Foral, oral bioavailability; GE, gallbladder emptying; k, rate constant; k a , absorption rate constant; L, lower; Q, intercompartmental clearance; RSE, relative standard error; SD, standard deviation; SE, standard error; U, upper; VC, central compartment volume; VP, peripheral compartment volume.
Regorafenib exposure was calculated at steady state (AUC0−8h,ss) for the phase 1 dataset patients using the regorafenib EHC model with different mealtime imputations and a standard two‐compartment model without EHC fitted to the same dataset. When reported mealtimes were included, the EHC model exposure estimates were found to be closer to those calculated using the trapezoidal non‐compartmental method (Figure 3A). When all available mealtimes were disregarded, the model performed poorly, and calculated exposures were similar to the standard two‐compartmental model. These results indicate that including meal information is valuable and provides similar exposures to the model‐independent non‐compartmental method. This was also seen when reduced datasets that mimic sparse sampling were used in an EHC model, suggesting the model's suitability for application in a sparse sampling setting.
FIGURE 3.

(A) Estimated AUC0‐8h,ss for patients in the phase 1 dataset, using a two‐compartmental model (2 Comp model; yellow), the phase 1 regorafenib enterohepatic circulation (EHC) model with fixed standard mealtimes (orange) or with reported mealtime if available and fixed standard times if not (brown), and by using the trapezium method (NCA; grey). Model‐based estimates were derived for reduced datasets with only observations included as indicated in the figure. (B) Estimated AUC0‐24h,ss based on the EHC model for patients in the phase 1 dataset based on a PK profile at steady state using different assumptions on the mealtimes. In brown are the results using the reported mealtime at the study day, and in orange are the results for fixed mealtimes, with results for the two‐compartment model shown in yellow. In blue are the results when available mealtime was disregarded and the mealtimes were related to the dosing time; the periods between food intake were 0.25 h (dark blue), 0.5 h (blue), or 1 h (light blue). The effect of imputing mealtimes was assessed by using all available data, as well as in sparse sampling settings, as indicated in the figure.
AUC, area under curve; EHC, enterohepatic circulation; NCA, non‐compartmental analysis; PK, pharmacokinetic; ss, steady state
Calculated AUC0−24h,ss based on the EHC model with fixed mealtimes provided higher exposures compared with a model using available actual mealtimes (Figure 3B). In contrast, by disregarding mealtime data and imputing mealtimes related to dosing schedule, similar and consistent exposures were observed compared with the model using all available data. Results were similar in sparse sampling settings, particularly when assuming a period of 0.5 h between mealtime and regorafenib dosing (Figure 3B). These data indicate that the EHC model still leads to reliable exposure estimates in a phase 3 sparse sampling setting where mealtimes are not reported but are assumed to be relative to the dosing schedule, and that the methodology may thus be applied in phase 3 studies.
3.2. Phase 1 regorafenib–metabolite model
The PK of parent regorafenib and its metabolites were well described by the phase 1 model, as indicated by the VPC plots in Figure 2. The PK profile of M‐2 was similar to that of regorafenib (Figure 2), with a similar structural model incorporating pre‐systemic formation from parent, best describing the observed profile. The PK profile for M‐5 (Figure 2) showed a slower formation of this metabolite, best described by systemic formation from regorafenib in the central compartment and incorporating EHC. CLM‐2 and CLM‐5 were estimated by fitting the model to the complete dataset, with parameters for volume of distribution (central and peripheral), inter‐compartmental flow, rate constant from the central compartment to the gallbladder compartment and the rate constant of drug release from the gallbladder compartment to the central compartment, fixed to their respective parent regorafenib values. Model parameter estimates for this model are provided in Table 2. All structural parameters and all variances/covariances of the included random effects could be estimated with sufficient precision (relative standard errors [RSE] <50%), apart from the covariance of IIV between CL of parent regorafenib (CLP) and CLM‐5 (Table 2).
TABLE 2.
Model parameter estimates of the phase 1 parent–metabolite enterohepatic circulation model showing parameter estimates and precision (see Figure 2 VPCs)
| Parameter | Value | SE | RSE % | L95% CI | U95% CI |
|---|---|---|---|---|---|
| Fixed effects | |||||
| k a, h−1 | 0.482 | Fixed | ‐ | ‐ | ‐ |
| FRM2 | −0.355 | 0.0946 | 26.7 | −0.540 | −0.169 |
| CLP/(1‐FRM2), L h−1 | 4.02 | Fixed | ‐ | ‐ | ‐ |
| VCP/(1‐FRM2), L | 10.7 | Fixed | ‐ | ‐ | ‐ |
| k GE, h−1 | 100 | Fixed | ‐ | ‐ | ‐ |
| Breakfast, h | 08:00 | Fixed | ‐ | ‐ | ‐ |
| Lunch, h | 12:00 | Fixed | ‐ | ‐ | ‐ |
| Dinner, h | 18:00 | Fixed | ‐ | ‐ | ‐ |
| QP/(1‐FRM2), L h−1 | 11.0 | Fixed | ‐ | ‐ | ‐ |
| VPP/(1‐FRM2), L | 162 | Fixed | ‐ | ‐ | ‐ |
| k CG, h−1 | 0.141 | Fixed | ‐ | ‐ | ‐ |
| DGE, h | 0.5 | Fixed | ‐ | ‐ | ‐ |
| CLM‐2, L h−1 | 2.45 | 0.122 | 4.98 | 2.21 | 2.69 |
| CLM‐5, L h−1 | 0.746 | 0.215 | 28.8 | 0.325 | 1.17 |
| FRM5 | −1.09 | 0.212 | 19.4 | −1.51 | −0.679 |
| Individual random effects | |||||
| ω2 k a | 0.127 | Fixed | ‐ | ‐ | ‐ |
| ω2CLP | 0.117 | 0.0231 | 19.7 | 0.0718 | 0.162 |
| ω2CLM‐2 | 0.267 | 0.0505 | 18.9 | 0.168 | 0.367 |
| ω2CLM‐5 | 1.95 | 0.469 | 24.0 | 1.03 | 2.87 |
| ω2FRM2 | 0.156 | 0.0759 | 48.7 | 0.00725 | 0.305 |
| ω2FRM5 | 0.841 | 0.258 | 30.7 | 0.334 | 1.35 |
| ω CLP, CLM‐2 | 0.122 | 0.0261 | 21.4 | 0.0708 | 0.173 |
| ω CLP, CLM‐5 | 0.157 | 0.0824 | 52.4 | −0.00412 | 0.319 |
| ω CLM‐2, CLM‐5 | 0.656 | 0.121 | 18.4 | 0.419 | 0.893 |
| Residual error | |||||
| Parent Prop. error, SD | 0.406 | 0.0208 | 5.11 | 0.366 | 0.447 |
| M‐2 Add. error, SD | 0.001 | Fixed | ‐ | ‐ | ‐ |
| M‐2 Prop. error, SD | 0.380 | 0.0231 | 6.07 | 0.335 | 0.425 |
| M‐5 Add. error, SD | 0.001 | Fixed | ‐ | ‐ | ‐ |
| M‐5 Prop. error, SD | 0.455 | 0.0256 | 5.63 | 0.405 | 0.506 |
FRM2 and FRM5 on logit scale corresponds with 41% and 25%, respectively.
Volumes, clearances and intercompartmental flow are relative to oral bioavailability (Foral), which is not listed for clarity.
Add., additive; CG, central compartment to gallbladder; CI, confidence interval; CL, apparent total body clearance; DGE, duration of gallbladder emptying; FRM2/5, fraction of formation of metabolite M‐2/M‐5; GE, gallbladder emptying; k, rate constant; k a, absorption rate constant; L, lower; P, parent (e.g. in CLP); Prop., proportional; Q, intercompartmental clearance; RSE, relative standard error; SD, standard deviation; SE, standard error; U, upper; VC, central compartment volume; VP, peripheral compartment volume, VPC, visual predictive check.
3.3. Phase 3 model development and covariate analysis
3.3.1. Phase 3 regorafenib model
The regorafenib phase 1 model was applied to data from four phase 3 studies. The CORRECT, GRID and CONCUR protocols indicate medication should be taken after light breakfasts, whereas the RESORCE protocol mentions a 2‐h window after food. Using the developed model based on phase 1 data with all parameters fixed, several values for the time between mealtime and reported dosing were tested (per study) separately. A 1‐h period between the meal and dosing led to the lowest minimum value of the objective function (MVOF); therefore, this time window was imputed for all phase 3 patients. The phase 1 model with all parameters fixed described the phase 3 data moderately well. However, CLP in phase 3 patients was on average overpredicted; therefore, some model parameters were estimated by fitting the phase 1 model to the phase 3 data. It appeared that typical values for CLP, and the variances of IIV of CLP and of the residual error, could be estimated, whereas all other parameters were fixed to their original values. This was the base model used for the regorafenib covariate analysis (Section 3.3.2). The estimated typical value for CLP decreased from 4.0 L h−1 in phase 1, to 2.9 L h−1 in phase 3, whereas the variance of IIV of CLP increased.
3.3.2. Phase 3 covariate regorafenib model
In a univariate GAM analysis (covariates tested and baseline values are listed in Appendix Table A2 in the Supporting information), the following covariates were found to be statistically significantly associated with CLP: race (Asian vs rest of world), sex, BMI, baseline alanine aminotransferase (ALT), baseline haemoglobin, baseline bilirubin, baseline total protein, tumour type, baseline estimated glomerular filtration rate, Child−Pugh score, UGT1A9 inducer received during treatment period and weight. Selected covariates (Appendix Section 3 in the Supporting information) were tested in the model‐based covariate analysis, and of these, including sex as a covariate on CLP led to the largest drop in MVOF, followed by BMI. In a second step, BMI on CLP led to a significant drop in MVOF for the model with sex included. The addition of other covariates led to non‐significant drops in MVOF. Compared with the base model, the inclusion of the covariate effects sex and BMI led to an absolute decrease of the variance of IIV on CLP of 2%, and it appeared that women had on average a 17% lower CLP than men (full data in Appendix Section 3 in the Supporting information).
3.3.3. Phase 3 regorafenib–metabolite model
The phase 3 base regorafenib–metabolite model was developed by combining the phase 3 covariate regorafenib and phase 1 regorafenib–metabolite models. It was necessary to fix most metabolite parameters to those of the phase 1 regorafenib–metabolite model, and regorafenib parameters were fixed to the values of the phase 3 covariate model (Appendix Section 3 in the Supporting information).
3.3.4. Phase 3 covariate regorafenib–metabolite model
The regorafenib–metabolite base model was used to perform a covariate selection with univariate GAM analysis. For CLM‐2, the following covariate effects were found to be statistically significant: sex, baseline albumin, ALT, aspartate aminotransferase, haemoglobin and weight. For CLM‐5, the significant covariates were sex, weight, baseline ALT, hepatic function category, albumin, bilirubin and total protein. Because of long run times and model instability, a full model‐based covariate analysis for M‐2 and M‐5 analogous to the parent covariate analysis was not performed (Appendix Section 3 in the Supporting information), and only the most significant covariate effect for M‐2 and M‐5 was included, i.e. sex on CLM‐2 and CLM‐5. Parameter values for the final phase 3 regorafenib–metabolite model are shown in Table 3. All parameters could be estimated reliably by fitting the model to the phase 3 dataset for all three analytes simultaneously. Both the IIV on CL of parent and metabolites appeared to be unbiased (mean approximately zero) and independent of sex, indicating that the covariate effect included in the model was able to adequately take a sex‐dependent CL of parent and metabolites into account. Only weak correlations between IIV of CL of parent and metabolites and BMI were observed, indicating that the included covariate effect of BMI on CLP and the absence of a covariate effect directly on CLM‐2 and CLM‐5 was justified. The ETA shrinkage 28 was 18% for IIV on CLP and 16% for IIV on CLM‐2 and CLM‐5.
TABLE 3.
Model parameter estimates of the final phase 3 parent–metabolite enterohepatic circulation model, showing parameter estimates and precision
| Parameter | Value | SE | RSE % | L95% CI | U95% CI |
|---|---|---|---|---|---|
| Fixed effects | |||||
| k a, h−1 | 0.482 | Fixed | ‐ | ‐ | ‐ |
| FRM2 | −0.355 | Fixed | ‐ | ‐ | ‐ |
| CLP/(1‐FRM2), L h−1 | 3.05 | Fixed | ‐ | ‐ | ‐ |
| Sex on regorafenib clearance | 0.169 | Fixed | ‐ | ‐ | ‐ |
| BMI on regorafenib clearance | −0.363 | Fixed | ‐ | ‐ | ‐ |
| VCP/(1‐FRM2), L | 10.7 | Fixed | ‐ | ‐ | ‐ |
| k GE, h−1 | 100 | Fixed | ‐ | ‐ | ‐ |
| Breakfast, h | 08:00 | Fixed | ‐ | ‐ | ‐ |
| Lunch, h | 12:00 | Fixed | ‐ | ‐ | ‐ |
| Dinner, h | 18:00 | Fixed | ‐ | ‐ | ‐ |
| QP/(1‐FRM2), L/h−1 | 11.0 | Fixed | ‐ | ‐ | ‐ |
| VPP/(1‐FRM2), L | 162 | Fixed | ‐ | ‐ | ‐ |
| k CG, h−1 | 0.141 | Fixed | ‐ | ‐ | ‐ |
| DGE, h | 0.5 | Fixed | ‐ | ‐ | ‐ |
| CLM‐2, L h−1 | 1.99 | 0.00508 | 0.255 | 1.98 | 2.00 |
| Sex on M‐2 clearance | 0.380 | 0.0192 | 5.06 | 0.342 | 0.418 |
| CLM‐5, L h−1 | 1.42 | 0.0532 | 3.76 | 1.31 | 1.52 |
| Sex on M‐5 clearance | 0.761 | 0.0387 | 5.08 | 0.685 | 0.837 |
| FRM5 | −1.09 | Fixed | ‐ | ‐ | ‐ |
| Factor IIV CLM‐5 | 2.21 | 0.0752 | 3.41 | 2.06 | 2.35 |
| Individual random effects | |||||
| ω2 k a | 0.127 | Fixed | ‐ | ‐ | ‐ |
| ω2CLP | 0.189 | 0.0123 | 6.53 | 0.165 | 0.213 |
| ω2CLM‐2 | 0.385 | 0.0219 | 5.68 | 0.342 | 0.428 |
| ω2FRM2 | 0.156 | Fixed | ‐ | ‐ | ‐ |
| ω2FRM5 | 0.841 | Fixed | ‐ | ‐ | ‐ |
| ω CLP, CLM‐2/M‐5 | 0.206 | 0.0168 | 8.16 | 0.173 | 0.238 |
| Residual error | |||||
| Parent Prop. error, SD | 0.543 | 0.00124 | 0.229 | 0.540 | 0.545 |
| M‐2 Add. error, SD | 0.001 | Fixed | ‐ | ‐ | ‐ |
| M‐2 Prop. error, SD | 0.485 | 0.00938 | 1.93 | 0.467 | 0.504 |
| M‐5 Add. error, SD | 0.001 | Fixed | ‐ | ‐ | ‐ |
| M‐5 Prop. error, SD | 1.14 | 0.113 | 9.93 | 0.919 | 1.36 |
FRM2 and FRM5 on logit scale corresponds with 41% and 25%, respectively. The estimated factor for IIV on CLM‐5 corresponds with a variance of 0.385.(2.212) = 1.88.
Volumes, clearances and intercompartmental flow are relative to oral bioavailability (Foral), which is not listed for clarity.
Add., additive; BMI, body mass index; CG, central compartment to gallbladder; CI, confidence interval; CL, apparent total body clearance; DGE, duration of gallbladder emptying; FRM2/5, fraction of formation of metabolite M‐2/M‐5; GE, gallbladder emptying; k, rate constant; k a, absorption rate constant; L, lower; P, parent (e.g. in CLP); Prop., proportional; Q, intercompartmental clearance; RSE, relative standard error; SD, standard deviation; SE, standard error; U, upper; VC, central compartment volume; VP, peripheral compartment volume.
3.3.5. Model application
The contributions of covariates to the variability of exposure as predicted by the population PK model are shown in forest plots (Figure 4). For parent regorafenib, the ratio and confidence interval of median AUCs for male:female is <1, but remains within the bioequivalence boundaries (Figure 4A). For BMI, the ratios of median AUCs are also within these boundaries. For the unbound (protein binding corrected) total AUCs, the ratio of median AUCs for male:female are <1 and the confidence is outside the lower boundary for bioequivalence, indicating a possible difference between males and females (Figure 4B). This observation may be driven by a limited number of outlying individuals who were best described with a very low individual CLM‐5 (and thus a large AUC), and also resulted in a very high estimate of the variance for IIV on CLM‐5.
FIGURE 4.
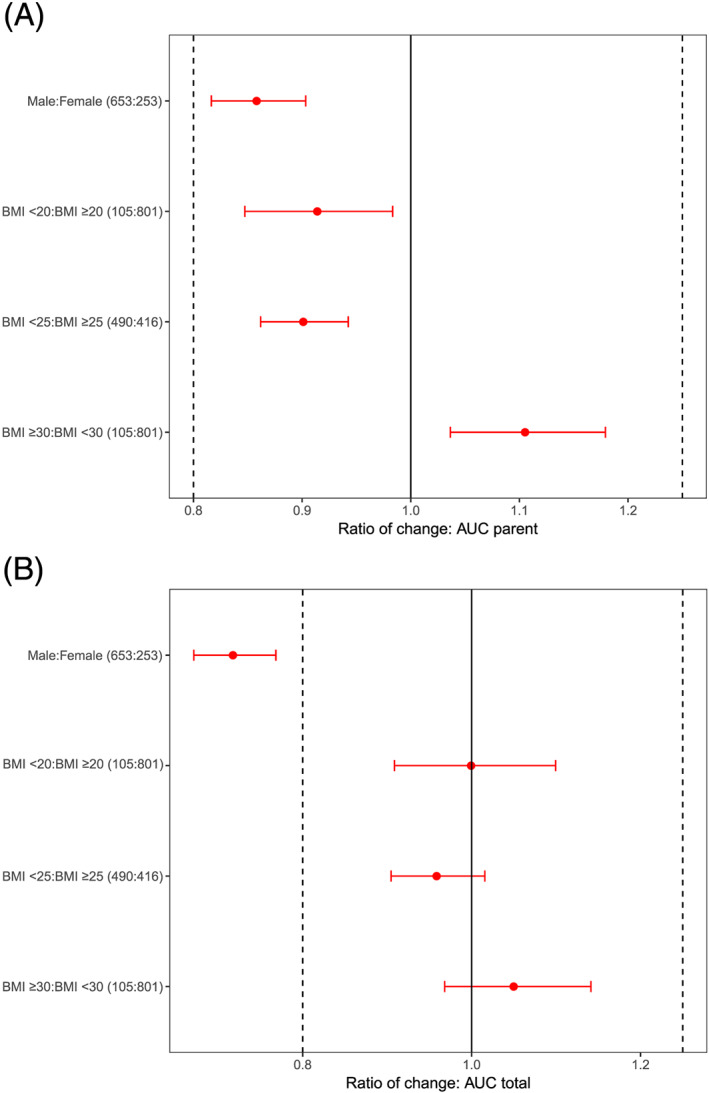
Contribution of sex and BMI covariates to the variability of exposure in phase 3 studies determined based on post hoc estimates from the final model and shown as a distribution of the exposure of (A) parent regorafenib AUC at steady state of nominal dosing (160 mg day−1), and (B) protein binding corrected total AUC (parent, M‐2 and M‐5). Horizontal red dots and bars are the median and 90% CI, respectively, of the ratio by dichotomized variable of AUC0‐24h,ss geometric means. The median and CI were obtained by sampling 10,000 replicate trials based on the individual AUC0‐24h,ss values with replacement, per group. Individual AUC0‐24h,ss were derived from the individual clearance estimates using the final PK model. Vertical lines are commonly used bioequivalence boundaries 0.8–1.25.
AUC, area under curve; BMI, body mass index; CI, confidence interval; PK, pharmacokinetic; ss, steady state
Prediction‐corrected VPC for the final model based on the four phase 3 studies and covariate analysis is shown in Figure 5. These data confirm that the model can adequately describe the central tendency and variability in the regorafenib and M‐2 data. The model appeared to over‐predict the observed variability in the M‐5 data at the lower end of the concentration range, and therefore caution should be exercised when applying the current variance estimates in the simulation of future populations.
FIGURE 5.
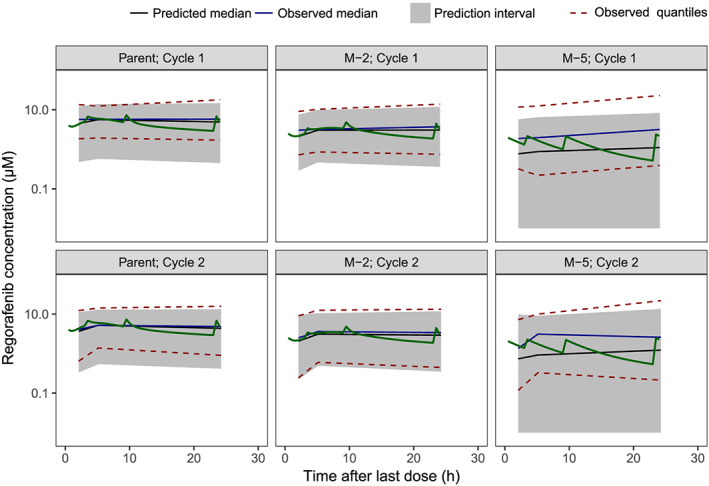
Prediction‐corrected VPC for the final model. Green lines represent the simulation of a dense PK profile of a typical male patient with reference body mass index (parent, M‐2, M‐5 and cycle specific). Plots overlaid with actual phase 3 observations are provided in Appendix Figure A2 in the Supporting information.
PK, pharmacokinetic; VPC, visual predictive check
4. DISCUSSION
Regorafenib and its metabolites have complex metabolic routes; increased understanding of the PK of regorafenib and its pharmacologically active metabolites will allow assessment of the potential effects of patient demographics and selected clinical characteristics on exposure. This is particularly important given the use of regorafenib in clinical practice and its investigation in a variety of cancers. Furthermore, the observation during the clinical development of regorafenib that EHC is a significant pathway indicated that this needed to be applied in a population PK model to adequately predict drug concentrations. This was particularly the case for pre‐dose and extended post‐dose levels, since regorafenib is administered within a certain period after a meal, leading to higher plasma levels in trough samples after repeated dosing due to gallbladder emptying.
To consider EHC in the modelling, an additional model compartment was required to represent the gallbladder as a collection and storage repository for regorafenib and its metabolites. When bile is secreted into the gut after food intake, 17 regorafenib is reabsorbed, potentially accounting for prolonged plasma exposure. 18 A model characterised by assumed first‐order absorption with two transit compartments between the dose and central compartment accounted for delayed uptake of regorafenib resulting in a plateau phenomenon, with its distribution described using a two‐compartment model and that includes a gallbladder compartment. A switch‐function release model, as used in other published studies, 25 was required to model the start and stop of regorafenib release from the gallbladder to the central compartment.
Plasma concentrations of regorafenib after a single dose and after multiple dosing in two phase 1 dose‐finding studies and four pivotal phase 3 studies were well described by the population PK model and provided estimates that were closer to those derived by noncompartmental analysis for dense sampling designs employed in phase 1 studies, compared with a two‐compartmental model that does not incorporate EHC. Population PK analyses of phase 3 studies of regorafenib, in which trough samples are usually taken directly before dosage (i.e. after a meal), require the incorporation of the EHC model. Recording actual mealtimes would be advantageous in studies of regorafenib, and detailed records in future clinical trials would facilitate more robust population PK estimates of exposure. However, exact mealtimes are not currently routinely available and/or recorded, and the period between food and dosage is likely to vary across studies and among individuals because of variable compliance. We found that assuming mealtimes were related to dosing time led to more reliable exposure estimates than assuming fixed times, and analyses of steady state AUC showed that the EHC model is able to estimate exposure adequately on the basis of sparse data without actual information on the time of food intake.
Parent−metabolite models were developed sequentially, and VPCs indicated that the PK of parent regorafenib and metabolites M‐2 and M‐5 was adequately described. It was necessary to simplify this model by fixing metabolite parameters to respective parent values for the apparent volume of distribution of the central and peripheral compartment, inter‐compartmental CL and the elimination rate from the central to the gallbladder compartment.
Since PK data after intravenous administration of regorafenib and of the individual metabolites were not available, the absolute fraction of parent regorafenib that is converted into M‐2 and M‐5 could not be determined. Therefore, as described in other studies (e.g. Bertrand et al. 29 ), all parameters describing drug volumes of distribution for the metabolites were fixed to the value for the parent drug; VPCs indicated that this provided adequate and unbiased estimates for regorafenib, M‐2 and M‐5. Evaluation of continuous and categorical covariates indicated that sex and BMI have an effect on the PK (CLP) of regorafenib, with women having a lower CLP than men. The final model suggests that an increase in BMI leads to a decrease in CLP; this may be a result of lower CYP3A4 expression, resulting in lower CLP of CYP3A4 substrates, which has been reported in patients with high BMI. 30 For regorafenib, M‐2 and M‐5, sex was included as a covariate in the final model. Overall, all the covariate effects accounted for only a small portion of the overall variability in exposure (the inclusion of the covariate effects sex and BMI led to an absolute decrease of the variance of IIV on CLP of 2%), have limited clinical relevance and are not predicted to affect exposure to the extent that dose adjustments are required. However, the unexplained variability is rather high for regorafenib, M‐2 and M‐5, and there may be other unknown covariates that could explain a larger proportion of the variability compared with the preselected covariates in the analysis described here.
A limitation of our population PK model is that it assumes that M‐2 is only formed pre‐systemically by CYP3A4, whereas studies of the metabolic pathway indicate that M‐2 is also formed systemically. 13 In addition, the model assumes that M‐5 is formed exclusively from parent regorafenib, whereas published data on the metabolic pathways indicate that M‐5 can also be formed from M‐2. However, the population PK model does not allow these pathways to be distinguished and the final model assumes that the CL estimates for the parent and the two major metabolites are correlated. The identified covariate effects for parent regorafenib may thus also influence M‐5 if CLP also reflects the formation of M‐5. 13
During phase 3 studies, protocol‐directed guidance on dosing times relative to mealtimes are normally provided (e.g. after breakfast) and given the severity of the disease, it is reasonable to assume that patients follow this advice; nonetheless, it is subject to individual variation. This work demonstrates that the PK of regorafenib is highly influenced by EHC. A key strength of the study is that it provides a tool for reliably estimating individual exposure for a drug that is subject to extensive EHC in a sparse sampling setting by population PK modelling and allocating mealtimes relative to dose rather than fixed times. In this case, the phase 3 data were best described by taking the protocol‐directed guidance on dosing times relative to mealtimes into account, and assuming meals were taken 1 h before the dose. Analysis of the phase 1 data suggests that this method provides unbiased results that are comparable to using available mealtime data. Although this model was shown to estimate exposure adequately on the basis of sparse data without actual information on food intake, recording of actual mealtimes or PK sampling after a prolonged post‐prandial period are recommended.
In order to evaluate the findings of this population PK modelling study within a clinical perspective – in particular (i) improved estimation of exposure taking enterohepatic recirculation into account and (ii) the impact of covariates sex and BMI on regorafenib exposure – our current understanding of the exposure–response relationships for the efficacy and safety of regorafenib should be considered. A minimal target concentration of regorafenib to maintain efficacy in current clinical practice has been suggested by Verheijen et al. 31 who have proposed the use of mean trough concentration after 160 mg/day for 21 days. However, the current population PK analysis shows that individual pre‐dose levels of regorafenib vary considerably depending on the time of the last meal intake and that the observed trough concentration does not necessarily reflect actual exposure. Therefore, based on the data presented here, the use of target trough concentrations for estimating exposure measures should be re‐evaluated taking mealtime information into account, and by applying the EHC approach. Taking an additional PK sample preferably 3–4 hours post‐dose (and post‐mealtime) would further increase the robustness of the exposure estimate (Figure 4A).
In addition, we have recently published exposure–response modelling data (also including EHC) of parent regorafenib for efficacy (overall survival) and hand–foot skin reaction (HFSR), which is among the most common regorafenib‐related toxicities. 32 The probability of grade 3 HFSR was found to correlate with exposure (HFSR tended to be higher in patients with higher exposure – with a maximum of around 20% in the highest exposure quartile), which is unaffected by EHC, rather than maximum plasma concentration. It could be hypothesised that a longer time between gallbladder emptying (e.g. by skipping a meal) could result in a greater quantity of regorafenib in the gallbladder available for reabsorption and, as a result, a higher maximum concentration. However, since this would not alter regorafenib clearance, and therefore exposure, this would not increase the probability of grade 3 HFSR. It should be noted that a limitation of this analysis is that some food intake is assumed, and a more physiological‐based approach (e.g. as published by Guiastrennec et al. 33 ) to describe the EHC of total bile acids, would be required to extend the scope of the model and explore the possible effect of major changes in food intake pattern on exposure.
Simulations based on exposure–response modelling showed that a 25% reduction in dose, and therefore reduced exposure, is not expected to result in reduced efficacy. 32 Therefore, the 25% lower exposure of total AUC in female patients observed in this population PK study (Figure 4B) would not be expected to result in lower efficacy compared with male patients. Moreover, no difference in efficacy by gender was seen in our (multivariate) exposure–response analysis in 964 patients with mCRC from the phase 3 CONCUR and CORRECT studies. 32 A similar finding has recently been published based on a multivariate analysis of overall survival in a cohort of 78 patients with mCRC. 34
In conclusion, the population PK model incorporating EHC described in this analysis adequately describes the PK profiles of regorafenib and its two pharmacologically active metabolites, M‐2 and M‐5, in patients with sparse PK sampling. Overall, the model does not indicate that the selected covariate effects on exposure require adjustments to initial doses for individual patients. The model is useful for estimating individual patient's exposure to regorafenib based on sparse sampling both in clinical trials and in clinical practice, which is becoming increasingly important with the growing application of flexible dosing to optimise the duration of treatment and clinical benefit.
COMPETING INTERESTS
A.K., S.H. and H.J.D. were paid consultants for Bayer during the conduct of the analysis; J.Z. and B.A.P. are employees of Bayer AG, and own stocks in Bayer; A.C. is an employee of Bayer AG and owns stocks in Bayer and AstraZeneca.
CONTRIBUTORS
B.A.P., H.J.D. and A.K. planned the analysis. A.K. and S.H. performed the analysis, developed the models and created tables and figures. B.A.P., H.J.D., S.H. and A.K. wrote the manuscript. All authors were involved in defining the assumptions and objectives of the analysis. All authors contributed to writing and revising the manuscript.
Supporting information
Data S1. Supporting info item
ACKNOWLEDGEMENTS
Administrative and editorial assistance in the preparation of this manuscript was provided by Matthew Naylor of OPEN Health Medical Communications (Choice), with financial support from Bayer; the authors retained complete editorial control over the content. The authors acknowledge the following principal investigators of the phase 3 studies from which data were used for the analysis described in this manuscript: Axel Grothey, MD, West Cancer Center, University of Tennessee, Memphis, TN, USA (Study 14387 CORRECT, NCT01103323, EudraCT2009‐012787‐14); Eric Van Cutsem, MD, PhD, University Hospitals Gasthuisberg/Leuven & KU Leuven, Leuven, Belgium (Study 14387 CORRECT, NCT01103323, EudraCT2009‐012787‐14); George D. Demetri, MD, Ludwig Center at Dana‐Farber Cancer Institute and Harvard Medical School, Boston, MA, USA (Study 14874 GRID, NCT01271712, EudraCT2009‐017957‐37); Jin Li, MD, Department of Medical Oncology, Tongji University Shanghai East Hospital, and Department of Oncology, Shanghai Medical College, Tongji University, Shanghai, China (Study 15808 CONCUR, NCT01584830); Jordi Bruix, MD, PhD, BCLC Group, Liver Unit, Hospital Clinic, University of Barcelona, IDIBAPS, CIBEREHD, Barcelona, Spain (Study 15982 RESORCE, NCT01774344, EudraCT2012‐003649‐14). This study was funded by Bayer AG, Berlin, Germany.
Keunecke A, Hoefman S, Drenth H‐J, Zisowsky J, Cleton A, Ploeger BA. Population pharmacokinetics of regorafenib in solid tumours: Exposure in clinical practice considering enterohepatic circulation and food intake. Br J Clin Pharmacol. 2020;86:2362–2376. 10.1111/bcp.14334
Anne Keunecke and Sven Hoefman contributed equally.
The authors confirm that there is no Principal Investigator for this paper. The specialised analysis and model development presented in this paper uses data from phase 3 studies, which have been previously published elsewhere.
DATA AVAILABILITY STATEMENT
Availability of the data underlying this publication will be determined according to Bayer's commitment to the European Federation of Pharmaceutical Industries and Associations (EFPIA) and Pharmaceutical Research and Manufacturers of America (PhRMA) “Principles for responsible clinical trial data sharing”. This pertains to scope, time point and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers, patient‐level clinical trial data, study‐level clinical trial data and protocols from clinical trials in patients for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after 1 January 2014. Interested researchers can use www.clinicalstudydatarequest.com to request access to anonymised patient‐level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the “Study sponsors section” of the portal. Data access will be granted to anonymised patient‐level data, protocols and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.
REFERENCES
- 1. Abou‐Elkacem L, Arns S, Brix G, et al. Regorafenib inhibits growth, angiogenesis, and metastasis in a highly aggressive, orthotopic colon cancer model. Mol Cancer Ther. 2013;12(7):1322‐1331. [DOI] [PubMed] [Google Scholar]
- 2. Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73‐4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245‐255. [DOI] [PubMed] [Google Scholar]
- 3. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second‐line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open‐label, phase II safety study. Eur J Cancer. 2013;49(16):3412‐3419. [DOI] [PubMed] [Google Scholar]
- 4. Eisen T, Joensuu H, Nathan PD, et al. Regorafenib for patients with previously untreated metastatic or unresectable renal‐cell carcinoma: a single‐group phase 2 trial. Lancet Oncol. 2012;13(10):1055‐1062. [DOI] [PubMed] [Google Scholar]
- 5. George S, Wang Q, Heinrich MC, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol. 2012;30(19):2401‐2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mross K, Frost A, Steinbild S, et al. A phase I dose‐escalation study of regorafenib (BAY 73‐4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(9):2658‐2667. [DOI] [PubMed] [Google Scholar]
- 7. Strumberg D, Scheulen ME, Schultheis B, et al. Regorafenib (BAY 73‐4506) in advanced colorectal cancer: a phase I study. Br J Cancer. 2012;106(11):1722‐1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sunakawa Y, Furuse J, Okusaka T, et al. Regorafenib in Japanese patients with solid tumors: phase I study of safety, efficacy, and pharmacokinetics. Invest New Drugs. 2014;32(1):104‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2017;389(10064):56‐66. [DOI] [PubMed] [Google Scholar]
- 10. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet. 2013;381(9863):295‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet. 2013;381(9863):303‐312. [DOI] [PubMed] [Google Scholar]
- 12. Li J, Qin S, Xu R, et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol. 2015;16(6):619‐629. [DOI] [PubMed] [Google Scholar]
- 13. Gerisch M, Hafner FT, Lang D, et al. Mass balance, metabolic disposition, and pharmacokinetics of a single oral dose of regorafenib in healthy human subjects. Cancer Chemother Pharmacol. 2018;81(1):195‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bayer HealthCare Pharmaceuticals Inc . Stivarga Prescribing Information . 2019.
- 15. Tlemsani C, Huillard O, Arrondeau J, et al. Effect of glucuronidation on transport and tissue accumulation of tyrosine kinase inhibitors: consequences for the clinical management of sorafenib and regorafenib. Expert Opin Drug Metab Toxicol. 2015;11(5):785‐794. [DOI] [PubMed] [Google Scholar]
- 16. Zopf D, Fichtner I, Bhargava A, et al. Pharmacologic activity and pharmacokinetics of metabolites of regorafenib in preclinical models. Cancer Med. 2016;5(11):3176‐3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mackie CR, Baxter JN, Grime JS, Hulks G, Cuschieri A. Gall bladder emptying in normal subjects—a data base for clinical cholescintigraphy. Gut. 1987;28(2):137‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fu Q, Chen M, Anderson JT, et al. Interaction between sex and organic anion‐transporting polypeptide 1b2 on the pharmacokinetics of regorafenib and its metabolites regorafenib‐N‐oxide and regorafenib‐glucuronide in mice. Clin Transl Sci. 2019;12(4):400‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bekaii‐Saab TS, Ou FS, Ahn DH, et al. Regorafenib dose‐optimisation in patients with refractory metastatic colorectal cancer (ReDOS): a randomised, multicentre, open‐label, phase 2 study. Lancet Oncol. 2019;20(8):1070‐1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beal S, Sheiner LB, Boeckmann A, Bauer RJE. NONMEM 7.4 users guides (ICON plc, Gaithersburg, MD, 1989–2018). https://nonmem.iconplc.com/nonmem743/guides. Accessed 10 February 2020.
- 21. Hafner FT, Werner D, Kaiser M. Determination of regorafenib (BAY 73‐4506) and its major human metabolites BAY 75‐7495 (M‐2) and BAY 81‐8752 (M‐5) in human plasma by stable‐isotope dilution liquid chromatography‐tandem mass spectrometry. Bioanalysis. 2014;6(14):1923‐1937. [DOI] [PubMed] [Google Scholar]
- 22. Leech RM, Worsley A, Timperio A, McNaughton SA. Understanding meal patterns: definitions, methodology and impact on nutrient intake and diet quality. Nutr Res Rev. 2015;28(1):1‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baxter JN, Grime JS, Critchley M, Shields R. Relationship between gastric emptying of solids and gall bladder emptying in normal subjects. Gut. 1985;26(4):342‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marciani L, Cox EF, Hoad CL, et al. Effects of various food ingredients on gall bladder emptying. Eur J Clin Nutr. 2013;67(11):1182‐1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Okour M, Brundage RC. A gallbladder‐based enterohepatic circulation model for pharmacokinetic studies. Eur J Drug Metab Pharmacokinet. 2019;44(4):493‐504. [DOI] [PubMed] [Google Scholar]
- 26. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol. 2017;145(Suppl 1):S272‐S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2018;46(D1):D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Savic RM, Karlsson MO. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11(3):558‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bertrand J, Laffont CM, Mentre F, Chenel M, Comets E. Development of a complex parent‐metabolite joint population pharmacokinetic model. AAPS J. 2011;13(3):390‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brill MJ, Diepstraten J, van Rongen A, van Kralingen S, van den Anker JN, Knibbe CA. Impact of obesity on drug metabolism and elimination in adults and children. Clin Pharmacokinet. 2012;51(5):277‐304. [DOI] [PubMed] [Google Scholar]
- 31. Verheijen RB, Yu H, Schellens JHM, Beijnen JH, Steeghs N, Huitema ADR. Practical recommendations for therapeutic drug monitoring of kinase inhibitors in oncology. Clin Pharmacol Ther. 2017;102(5):765‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grothey A, Hoefman S, Ruppert M, et al. Expoloration of efficacious alternative regorafenib regimens to manage hand‐foot skin reaction (HFSR). Ann Oncol 2019, 30 (suppl 5);v198‐v252. Poster presentation at European Society for Medical Oncology, Barcelona, Spain. Poster 658P. https://register.event-works.com/elsevier/esmo2019/ps/pv/id/78609/. Accessed 10 February 2020. [Google Scholar]
- 33. Guiastrennec B, Sonne DP, Bergstrand M, Vilsboll T, Knop FK, Karlsson MO. Model‐based prediction of plasma concentration and enterohepatic circulation of total bile acids in humans. CPT Pharmacometrics Syst Pharmacol. 2018;7(9):603‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aljubran A, Elshenawy MA, Kandil M, et al. Efficacy of regorafenib in metastatic colorectal cancer: a multi‐institutional retrospective study. Clin Med Insights Oncol. 2019;13:1179554918825447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting info item
Data Availability Statement
Availability of the data underlying this publication will be determined according to Bayer's commitment to the European Federation of Pharmaceutical Industries and Associations (EFPIA) and Pharmaceutical Research and Manufacturers of America (PhRMA) “Principles for responsible clinical trial data sharing”. This pertains to scope, time point and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers, patient‐level clinical trial data, study‐level clinical trial data and protocols from clinical trials in patients for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after 1 January 2014. Interested researchers can use www.clinicalstudydatarequest.com to request access to anonymised patient‐level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve patient care. Information on the Bayer criteria for listing studies and other relevant information is provided in the “Study sponsors section” of the portal. Data access will be granted to anonymised patient‐level data, protocols and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that patient privacy is safeguarded.