

Abstract
Skeletal stem/progenitor cells (SSPC) are critical regulators of bone homeostasis by providing a continuous supply of osteoblasts throughout life. In response to inductive signals, SSPC proliferate before osteoblast differentiation. Proliferation requires the duplication of all cellular components before cell division. This imposes a unique biosynthetic requirement for amino acids that can be used for biomass production. Thus, the ability to sense and respond to amino acid availability is likely a major determinant for proliferation. Using a cellular and genetic approach, we demonstrate the amino acid sensor GCN2 is required to support the robust proliferative capacity of SSPC during bone homeostasis. GCN2 ablation results in decreased postnatal bone mass due primarily to reduced osteoblast numbers. Decreased osteoblast numbers is likely attributed to reduced SSPC proliferation as loss of GCN2 specifically affected proliferation in cultured bone marrow stromal cells (BMSCs) without impacting osteoblast differentiation in vitro. Mechanistically, GCN2 regulates proliferation by increasing amino acid uptake downstream of the transcriptional effector ATF4. Collectively, these data suggest amino acid sensing through the GCN2/ATF4 pathway is indispensable for robust SSPC proliferation necessary for bone homeostasis.
Keywords: AMINO ACIDS, ATF4, GCN2, PROLIFERATION, SKELETAL STEM/PROGENITOR CELLS
Introduction
Bone is a dynamic tissue undergoing constant remodeling, which is controlled in part by both the number and activity of the bone-forming osteoblasts. Osteoblasts are responsible for secreting the collagen type 1–rich extracellular matrix and accessory proteins required for bone mineralization during both embryonic development and the postnatal bone remodeling critical for bone homeostasis.(1) Defects in osteoblast generation or activity can result in declining bone mass and increased fragility, hallmarks of human osteopenia and osteoporosis. In adult bone, osteoblasts may differentiate from multipotent skeletal stem/progenitor cells (SSPC) that reside in the bone marrow and are tightly associated with the vasculature.(2) SSPC are thought to arise postnatally and are enriched in the CD45−Ter119−PDGFRα+ Sca-1+ bone marrow fraction in vivo.(2–5) SSPC are generally thought to be quiescent in vivo but rapidly proliferate in response to osteoinductive signals or injury.(2) Conversely, with age, the SSPC may gradually lose its replicative capacity, which could result in diminished osteoblast formation, decreased bone mass, and reduced regenerative potential.(6) Thus, a comprehensive understanding of the mechanisms regulating SSPC proliferation will provide potential strategies to maintain or expand the SSPC population in order to promote bone formation and regeneration.
Cell division is associated with unique nutrient requirements to support biomass production. Although quiescent cells can obtain sufficient nutrients to maintain cellular homeostasis, proliferating cells must increase nutrient consumption to provide the biosynthetic building blocks necessary to duplicate their mass.(7) These biosynthetic demands impart unique metabolic requirements on proliferating cells. For example, proliferating cells increasingly consume and metabolize many nutrients, including amino acids.(8–11) Amino acids support mitosis by providing carbon and nitrogen for protein, nucleotide, and lipid biosynthesis and are responsible for the majority of cell mass production in proliferating cells.(9,12) Supporting this concept, blocking amino acid uptake and metabolism is being used to restrain cell proliferation in diverse cancers.(13,14) Although it is known SSPC increase glutamine consumption and require glutamine metabolism to support proliferation,(15) more broadly, the molecular mechanisms underpinning the regulation of amino acid consumption and how this regulates proliferation in SSPC are unknown.
Eukaryotic cells respond to insufficient amino acids by activating the integrated stress response (ISR) to restore cellular homeostasis.(16) The core event in this pathway is the phosphorylation of the alpha subunit of the eukaryotic translation initiation factor 2 (eIF2α) by the eIF2α kinase general control nonderepressible 2 (GCN2) (encoded by Eif2ak4). GCN2 is activated by uncharged transfer RNA (tRNA) when free amino acids are limited.(17,18) Activated GCN2 phosphorylates serine 51 of eIF2α, resulting in decreased translational initiation of most mRNAs; however, translation of the transcription factor ATF4 is increased. ATF4 then transcriptionally regulates a host of genes encoding both amino acid transporters and amino acid synthases to increase amino acid acquisition to mitigate amino acid depletion.(19–22) It is important to note that ATF4 is also a critical regulator of osteoblast differentiation and bone formation in part by regulating amino acid consumption to facilitate protein synthesis.(23–25) Indeed, GCN2 and ATF4 are required to support pathological bone formation in response to hyperactive WNT signaling.(21) In contrast, the role of GCN2 during bone homeostasis is incompletely understood. While GCN2 knockout mice are viable, bone abnormalities and/or proliferation defects under physiological conditions were not fully investigated.(26) Thus, it is unclear if amino acid sensing by GCN2 contributes to maintaining postnatal bone homeostasis or if GCN2 is required to support the robust proliferative capacity of SSPC.
Here, we define the contribution of Eif2ak4/GCN2-mediated amino acid sensing to proliferation and bone homeostasis. Using a genetic and cell biological approach, we demonstrate the GCN2-ATF4 pathway is dispensable for osteoblast differentiation. Rather, GCN2 activates ATF4, which transcriptionally regulates the expression of amino acid transporters that provide amino acids to support SSPC proliferation. Collectively, these data highlight a previously unknown role for the GCN2-ATF4 pathway in regulating cellular proliferation and bone homeostasis.
Materials and Methods
Mouse strains
The Eif2ak4−/− mouse strain is as previously described.(26) C57Bl/6J and Eif2ak4−/− mouse strains were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). The Animal Studies Committee at Duke University approved all mouse procedures. The initial animal work was approved by the committee at Washington University in St. Louis.
Mouse analyses
Bone phenotypes were analyzed at 2, 3, and 4 months of age. All animal studies were performed in a blinded and coded manner. Both male and female mice were analyzed. Radiographs of mouse skeleton were generated using a Faxitron X-ray system (Faxitron X-ray Corp, Tucson, AZ, USA) with 20-second exposure under 25 kV. Micro-computed tomography (VivaCT 80, Scanco Medical AG, Bruttisellen, Switzerland) was used for three-dimensional reconstruction and quantification of bone parameters (threshold set at 320) from 200 slices underneath the growth plate. Bone histomorphometry was performed on femurs fixed in 10% buffered formalin overnight at 4°C followed by 2-week decalcification in 14% EDTA. After decalcification, bones were embedded in paraffin and sectioned at a thickness of 5 μm. Alcian Blue/Picrosirius Red (AB/PSR) staining was performed following standard protocols. For dynamic histomorphometry, mice were injected with calcein (25 mg/kg) and alizarin red (75 mg/kg) intraperitoneally at 7 and 2 days before euthanization, respectively. Freshly isolated bones were embedded in OCT for fresh unfixed frozen sections using CryoJane Tape-Transfer System (Electron Microscopy Sciences, Hatfield, PA, USA). Both static and dynamic histomorphometry were quantified using ImageJ. Immunostaining was performed on 5-μm paraffin sections for osteocalcin (OCN) (Millipore, Burlington, MA, USA; AB10911). Antigen retrieval was performed by incubating bone sections in 10 μg/mL proteinase K for 10 minutes followed by incubation in 3% H2O2 (v/v in methanol) for 10 minutes.
Cell culture
Primary bone marrow stromal cells (BMSCs) were isolated from 4-month-old male and female mice. Briefly, the femur and tibias were isolated and all connective tissue was removed. The epiphyses were removed and marrow was collected by centrifugation. Red blood cells were lysed by red blood cell lysis buffer for 3 minutes. For colony-forming efficiency assays, 1 × 106 bone marrow cells were plated in a T25 flask. Three hours after plating, non-adherent cells were removed by washing vigorously with media. Cells were then cultured in 2% O2 for 7 days before colonies were stained with Crystal Violet for 15 minutes after fixation in 4% paraformaldehyde. For high-density BMSC cultures, bone marrow cells were plated and cultured in 2% O2 for 7 days. On days 3 and 6, plates were washed to remove non-adherent cells. BMSC were maintained as a monolayer in α-MEM with 15% fetal bovine serum, 100 U/mL penicillin G, and 100 μg/mL streptomycin under a humidified atmosphere of 5% CO2 at 37°C. BMSC were passaged at day 7 and all experiments were carried out at a seeding density of 21,000 cells/cm2. Primary murine calvarial osteoblasts (cOB) were isolated from 3-day-old neonates by collagenase P digestion. Calvarial bones were incubated in PBS containing 1.8 mg/mL collagenase P (Sigma-Aldrich, St. Louis, MO, USA; 11213873001) in a shaker at 37°C. Four sequential Collagenase P digestions were performed for 10 minutes each. The first digestion was discarded, and subsequent three digestions were collected. Cells released from digestion were plated in α-MEM. Osteogenic differentiation was initiated when cells were 95% confluent by replacing the medium with α-MEM supplemented with 50 mg/mL ascorbic acid (Sigma-Aldrich, A4544) and 10 mM β-glycerophosphate (Sigma-Aldrich, G9422) for 14 days with a change of media every 2 days. Alkaline phosphatase staining was performed using the one-step nitro-blue tetrazolium (NBT) and 5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt (BCIP) solution. Mineralization was assessed by Von Kossa or alizarin red staining.
Serum measurement
Blood samples were collected from Eif2ak−/− and wild-type littermate mice through cardiac puncture. Serum P1NP and CTX-I were measured by using P1NP ELISA kit (Immunodiagnostic Systems, Boldon, UK; AC-33F1) and CTX-I ELISA kit (Immunodiagnostic Systems, AC-02F1), respectively, according to the manufacturer’s instructions.
Flow cytometry
Bone marrow cells were isolated from 4-month-old male and female mice as described above. After red blood cell lysis, the cells were washed with PBS, counted, and resuspended at 5 × 106 cells/mL in ice-cold PBS containing 2% FBS. Fluorescent conjugate primary antibodies (2 μg/mL) were added. Antibodies used were: anti-CD45-FITC (BD Biosciences, San Jose, CA, USA; 553772), anti-Sca1-Alexa Fluor 647 (BD Biosciences, 565355), anti-PDGFRα-PE-Cy7 (BD Biosciences, 562776). Cells were incubated with antibody for 45 minutes at 4°C. The cells were then washed with PBS three times before collection by centrifugation at 400g for 5 minutes. Cells were resuspended in 500 μL ice-cold PBS, 10% FBS, for analysis using a FACSCanto II flow cytometer (BD Biosciences). Data were analyzed using FlowJo (version 10).
EdU incorporation and TUNEL assays
EdU incorporation was performed using Click-iT EdU Cell Proliferation Kit (Invitrogen, Carlsbad, CA, USA; C10337) according to manufacturer’s instructions. Briefly, bone marrow cells were plated at clonal density and were incubated with EdU (5-ethynyl-2′-deoxyuridine, 10 μM) for 6 hours; then the cells were permeabilized with Triton X-100 (0.5%, v/v in PBS) after fixation in 4% paraformaldehyde. EdU incorporation was detected through incubation with Click-iT reaction cocktail for 30 minutes at room temperature. TUNEL staining was performed using the in situ Cell Death Detection Kit (Roche, Mannheim, Germany; 11684795910). Briefly, BMSC were permeabilized by Triton X-100 (0.1%, v/v in 0.1% sodium citrate) for 2 minutes on ice after fixation in 4% paraformaldehyde. Then the permeabilized cells were incubated with TUNEL reaction mixture for 60 minutes at 37°C. All image analysis was performed in a blinded and coded manner.
Amino acid consumption assays
Confluent cells were rinsed twice with PBS and once with Krebs Ringers HEPES buffer (KRH), pH 8.0 (120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 25 mM NaHCO3, 5 mM HEPES, 1 mM D-glucose). Cells were then incubated for 5 minutes in KRH containing 4 μCi/mL 3H-labeled amino acid. The reaction was terminated by washing three times with ice-cold KRH. Cells were then lysed with 1 mL 1% SDS and collected in 8 mL scintillation cocktail. Radioactivity was measured by Tri-Carb liquid scintillation analyzer. Radiolabeled amino acids used in this study were L-[2,3,4-3H]-glutamine (PerkinElmer, Waltham, MA, USA; NET323250UC), L-[3,4-3H]-glutamic acid (NET490001MC), L-[2,3-3H]-proline (NET551250UC), and L-[2,3,4-3H]-arginine (NET1123250UC).
Protein and collagen metabolic labeling assay
Confluent cells were rinsed twice with PBS and once with KRH. Cells were then incubated for 3 hours in KRH containing 4 μCi/mL 3H-labeled proline at 37°C. After incubation, cells were rinsed twice with ice-cold KRH and then lysed with 150 μL RIPA and centrifuged at 400g for 10 minutes. Next, 100 μL supernatant was mixed with 10 μL 100% trichloroacetic acid (TCA) and centrifuged at 400g for 5 minutes to precipitate proteins and remove unincorporated proline. The protein pellet was washed twice with 5% TCA and resuspended in 1 mL 0.25 nM NaOH. The sample was then split into three tubes. Tube I containing 100 μL supernatant was used to directly measure the radioactivity in total protein. Tube II containing 400 μL supernatant was incubated with 200 μL 60 μM HEPES containing 15 mg collagenase (Roche, 11249002001) at 37°C for 3 hours to digest collagenous protein. Tube III containing 400 μL supernatant was incubated with 200 μL 60 μM HEPES at 37°C for 3 hours. After incubation, protein was precipitated by adding 60 μL 100% TCA followed by centrifugation at 400g for 10 minutes. The supernatant was transferred into new tubes. The radioactivity of the supernatant from tube I, tube II, and tube III were measured by Tri-Carb liquid scintillation analyzer. Proline incorporation into total protein is determined from the radioactivity of tube I. The proline incorporation into collagen is determined by subtracting the radioactivity from the mock digested (tube III) from collagenase digested samples (tube II).
Virus production for shRNA knockdowns and ATF4 overexpression
The lentiviral vector pLKOpuro was modified to express shRNAs targeting ATF4, GCN2, or RFP as negative control as previously published.(21,25,27) Lentiviral expression vectors for V5 tagged luciferase (Luc) or ATF4 were obtained from the DNASU plasmid repository (www.dnasu.org). To make virus, the indicated vector was co-transfected with plasmids pMD2.g and psPax2 into 293 cells. Virus-containing media was filtered and collected. For infection, BMSC or ST2 cells were plated at 13,000 cells/cm2 and infected for 24 hours, then recovered for 24 hours in regular media before puromycin (5 μg/mL, for shRNA knockdown experiments) or blasticidin S-HCl (10 μg/mL, for expression experiments) selection for 48 hours. Cells were then recovered for 24 hours before further analyses as indicated. The shRNA sequences are listed in Supplemental Table S2.
RNA isolation and qPCR
Total RNA from bone tissue or cells was extracted using Trizol reagent. First-strand cDNA was synthesized from 500 ng of total RNA with the iScript cDNA synthesis kit. RT-qPCR was performed in technical and biological triplicates in a 96-well format on an ABI Quant Studio 3, using SYBR green chemistry. The PCR programs were 95°C for 3 minutes followed by 40 cycles of 95°C for 10 seconds and 60°C for 30 seconds. Gene expression was normalized to Actb mRNA and relative expression was calculated using the 2−ΔΔCt method. The primer sequences used in this study are listed in Supplemental Table S3.
Western blotting
Cells were lysed in lysis buffer containing 50 mM Tris–HCl, pH 7.4, 15 mM NaCl, 1% NP-40, and 0.1% SDS supplemented with a protease inhibitor mix. For isolating protein from bone tissue, femurs were freshly dissected and washed in ice-cold PBS. Both ends of the femur were cut off and bone marrow was flushed out by centrifugation at 8000g at 4°C for 10 seconds. Then, bone shaft was vigorously chopped 100 times in lysis buffer containing 50 mM Tris-HCl, pH 7.4, 15 mM NaCl, 1% NP-40, and 0.1% SDS supplemented with a protease inhibitor mix. Protein fractions were collected by centrifugation at 15,000g at 4°C for 10 minutes. Protein concentration was estimated by the BCA method. Protein (25 μg) was loaded onto 4% to 15% SDS-PAGE, and then transferred onto nitrocellulose membranes. The membranes were blocked with 5% BSA and then incubated with specific primary antibodies overnight at 4°C. The primary antibodies were from the following sources: anti-RUNX2 (Abcam, Cambridge, MA, USA; ab23981), anti-ATF4 (Santa Cruz Biotechnology, Dallas, TX, USA; sc-200), anti-ASCT2 (Cell Signaling Technology, Danvers, MA, USA; 5345), anti-β-actin (Cell Signaling Technology, 4970), anti-p-Eif2α (Cell Signaling Technology, 3398), total Eif2aα (Cell Signaling Technology, 5324). Membranes were then washed three times using TBS/Tween 0.1% and further incubated with the secondary antibody, HRP goat anti-rabbit or HRP anti-mouse, in 5% BSA (TBS/Tween 0.1%) for 1 hour at room temperature. All secondary antibodies were diluted 1:2000 in blocking solution. All blots were developed using either the Clarity ECL substrate or the Super Signal West Femto substrate. Each experiment was repeated for a minimum of three times with three independently prepared protein samples.
Statistics
Statistical analyses were performed using GraphPad (La Jolla, CA, USA) Prism 6 software. All data are visualized using box and whisker plots with the median represented by the line. All data points are displayed. In cell culture studies, statistical significance was determined by an unpaired 2-tailed Student’s t test. For μCT and CFU studies, statistical significance was determined by a paired 2-tailed Student’s t test comparing paired littermate controls. A p value < 0.05 is considered statistically significant.
Results
Eif2ak4−/− mice have reduced postnatal bone mass accrual
To determine if Eif2ak4/GCN2 plays a role during bone development and homeostasis, we evaluated bone phenotypes in Eif2ak4-deficient mice. Mice homozygous for a null Eif2ak4 allele (Eif2ak4−) were born at Mendelian ratios and had no discernible defects in skeletal development or bone mass up to 2 months of age (Fig. 1A, B; Supplemental Fig. S1; Supplemental Table S1).(26,28,29) However, micro-computed tomography (μCT) of isolated femurs identified a progressive reduction in bone accrual manifest at 3 months of age in Eif2ak4−/− mice (Fig. 1). Quantification of the μCT analyses demonstrated that Eif2ak4 deletion resulted in significant reductions in trabecular bone volume per tissue volume (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), and bone mineral density (BMD), while increasing trabecular separation (Tb.Sp) at 4 months of age (Table 1). Eif2ak4−/− mice were also characterized with defects in cortical bone highlighted by decreased cortical thickness (Ct. Th) in both males and females with no effect on the total cortical bone area in males and reduced total cortical bone area in females (Table 1). Similarly, reduced bone mass was also observed in the Eif2ak4−/− tibias at 4 months of age (Supplemental Fig. S1C). Collectively, these data indicate that Eif2ak4−/− mice are characterized by reduced postnatal bone mass accrual compared with wild-type littermate controls.
Fig 1.
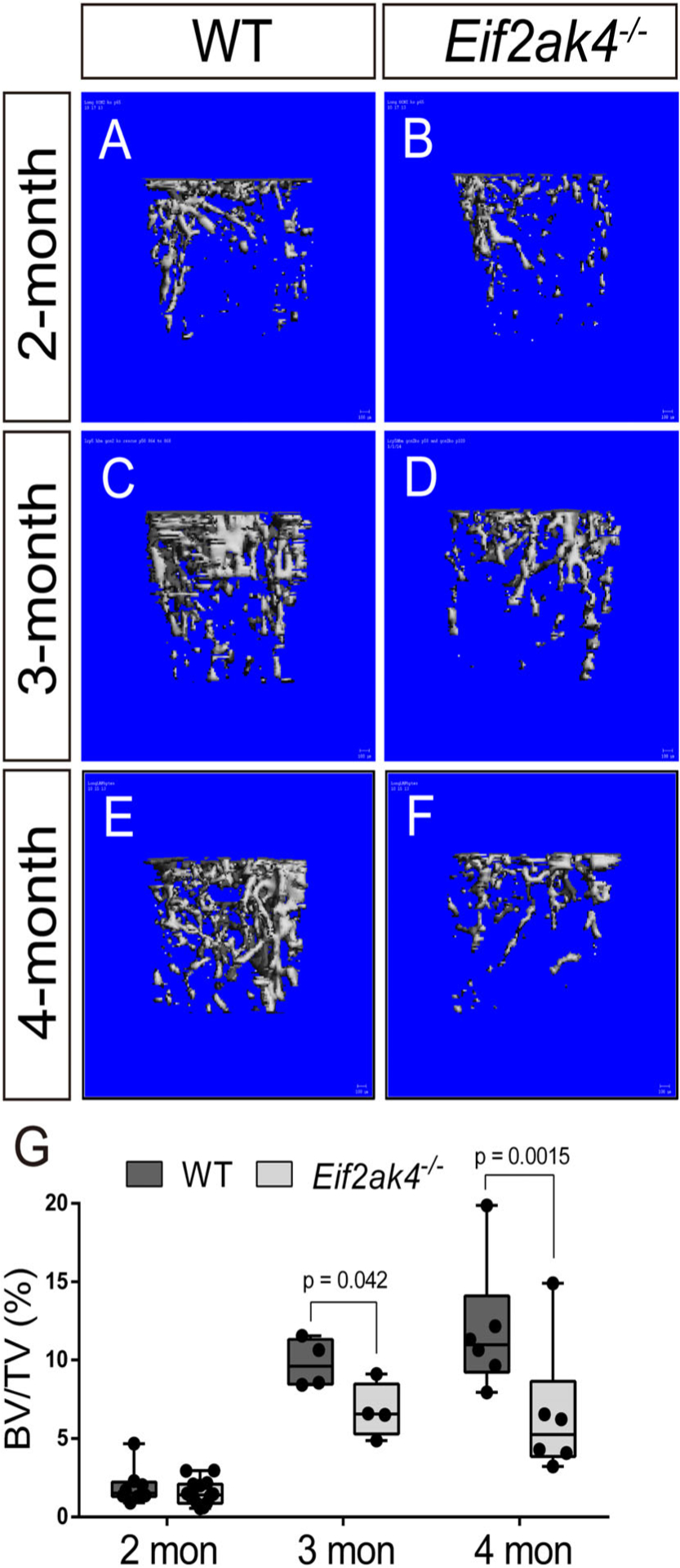
Eif2ak4/GCN2 deficiency reduces postnatal bone mass accrual. (A–F) Representative μCT images of the trabecular bone in the distal femur of 2- (A, B), 3- (C, D), or 4-month-old (E, F) wild-type (WT) (A, C, E) or Eif2ak4−/− (B, D, F) male mice. (G) Quantification of μCT images. BV/TV = bone volume/tissue volume.
Table 1.
Bone Parameters in Eif2ak4−/− Mice
| 4-month-old | ||
|---|---|---|
| wild type | Eif2ak4−/− | |
| Male (n) | 6 | 6 |
| Weight (g) | 24.4 ± 0.7 | 24.1 ± 1.7 |
| BV/TV | 0.122 ± 0.053 | 0.065 ± 0.048a |
| Tb.N (1/mm) | 2.94 ± 0.10 | 2.10 ± 0.47a |
| Tb.Th (mm) | 0.074 ± 0.009 | 0.062 ± 0.011a |
| Tb.Sp (mm) | 0.35 ± 0.01 | 0.51 ± 0.12a |
| BMD (mg HA/ccm) | 228 ± 23 | 183 ± 33a |
| Ct.BV/TV | 0.44 ± 0.04 | 0.43 ± 0.005 |
| Ct.Th (mm) | 0.18 ± 0.002 | 0.17 ± 0.001a |
| T.Ar (mm) | 1.89 ± 0.17 | 1.74 ± 0.08 |
| TMD (mg HA/ccm) | 1073 ± 38 | 1078 ± 14 |
| Female (n) | 10 | 10 |
| Weight (g) | 20.2 ± 1.5 | 19.6 ± 1.1 |
| BV/TV | 0.027 ± 0.018 | 0.015 ± 0.006a |
| Tb.N (1/mm) | 1.22 ± 0.08 | 1.08 ± 0.15 |
| Tb.Th (mm) | 0.068 ± 0.013 | 0.051 ± 0.008a |
| Tb.Sp (mm) | 0.83 ± 0.06 | 1.03 ± 0.14a |
| BMD (mg HA/ccm) | 140 ± 17 | 126 ± 11a |
| Ct.BV/TV | 0.45 ± 0.007 | 0.44 ± 0.0.016 |
| Ct.Th (mm) | 0.18 ±0.009 | 0.17 ± 0.012a |
| T.Ar (mm) | 1.67 ±0.14 | 1.58 ± 0.10a |
| TMD (mg HA/ccm) | 1093 ± 18 | 1081 ± 13 |
| Serum chemistry (n =5) (male) | ||
| P1NP (ng/mL) | 516.24 ± 167.16 | 287.47 ± 153.333 |
| CTX-I (ng/mL) | 29.05 ± 9.97 | 26.17 ± 10.58 |
| Dynamic histomorphometry (n = 5) (male) | ||
| MAR (μm2/d) | 0.72 ± 0.06 | 0.44 ± 0.03a |
| MS/BS (%) | 37.2 ± 5.0 | 25.7 ± 2.6a |
| BFR (μm3/μm2/d) | 0.27 ± 0.04 | 0.11 ± 0.01a |
| Static histomorphometry (n = 7) (male) | ||
| Ob.N/BS | 14.0 ± 0.3 | 7.4 ± 0.8a |
BV/TV = bone volume/tissue volume; Tb.N = trabecular number; Tb.Th = trabecular thickness; Tb.Sp = trabecular separation; BMD = bone mineral density; Ct.BV/TV = cortical bone volume/tissue volume; Ct.Th = cortical thickness; T.Ar = tissue area; TMD = tissue mineral density; P1NP = procollagen type 1 N propeptide; CTX-I = carboxy-terminal telopeptide of type I collagen; MAR = mineral apposition rate; MS/BS = mineralizing surface/bone surface; BFR = bone formation rate; Ob.N/BS = osteoblast number/bone surface.
p ≤ 0.05 compared with wild type.
We next sought to understand how Eif2ak4 deletion affects postnatal bone mass. Histomorphometric analysis of histological sections confirmed the age-specific reduction in trabecular bone volume in the distal femur of 4-month-old Eif2ak4−/− mice (Fig. 2A, B). Static histomorphometry quantified a significant reduction in osteoblast number as Eif2ak4−/− mice had significantly fewer osteoblasts per bone surface (Ob.N/BS) compared with littermate controls at 4 months of age (Table 1). Consistent with this, quantification of osteocalcin (OCN, encoded by Bglap) immunofluorescence staining confirmed the significant reduction in the overall number of OCN expressing osteoblasts in Eif2ak4−/− mice (Fig. 2C, D). We next evaluated the expression of GCN2 pathway components and select osteoblast markers in bone extracts from Eif2ak4−/− mice. As expected, we did not detect GCN2 protein expression in Eif2ak4−/− bones (Fig. 2E). Activated GCN2 phosphorylates serine 51 of EIF2α, which inhibits the translation of many proteins but specifically enhances translation of the transcription factor ATF4. Bone extracts from Eif2ak4−/− mutant mice were characterized by a significant reduction in EIF2α phosphorylation specifically at serine 51 (Fig. 2E). Consistent with this, ATF4 was reduced at both the mRNA and protein level (Fig. 2E, F). Likewise, the ATF4 target gene Ddit3 was significantly reduced in Eif2ak4−/− bones compared with wild-type controls (Fig. 2G). Moreover, Eif2ak4−/− bones were characterized by significantly less RUNX2 protein, while osteocalcin was significantly reduced at the mRNA level (eg, Bglap), confirming decreased osteoblast numbers (Fig. 2E, H). Consistent with this conclusion, dynamic histomorphometry revealed a significant reduction in osteoblast coverage as exemplified by the mineralized surface per bone surface (MS/BS) (Fig. 2I–K; Table 1). Osteoblast activity was affected by loss of Eif2ak4 in a sex-dependent manner as the mineral apposition rate (MAR) was unaffected in female mice (1.18 ± 0.2 versus 1.14 ± 0.15 for wild type and mutant, respectively) but significantly reduced in male mutant mice (0.72 ± 0.06 versus 0.44 ± 0.03 for wild type and mutant, respectively) (Table 1). Collectively, this resulted in significantly less bone-forming activity in Eif2ak4−/− mice as exemplified by significant reductions in the bone formation rate (BFR) (Table 1). Consistent with this, Eif2ak4−/− mice were characterized by a significant reduction in serum levels of procollagen type 1 N propeptide (P1NP), a direct readout of osteoblast activity and bone formation (Table 1). Conversely, serum C-terminal telopeptide type 1 collagen (CTX) levels, a direct readout of osteoclast activity, were unchanged in Eif2ak4−/− mice (Table 1). Overall, these data indicate Eif2ak4 regulates postnatal bone accrual primarily through regulation of osteoblast number and activity.
Fig 2.
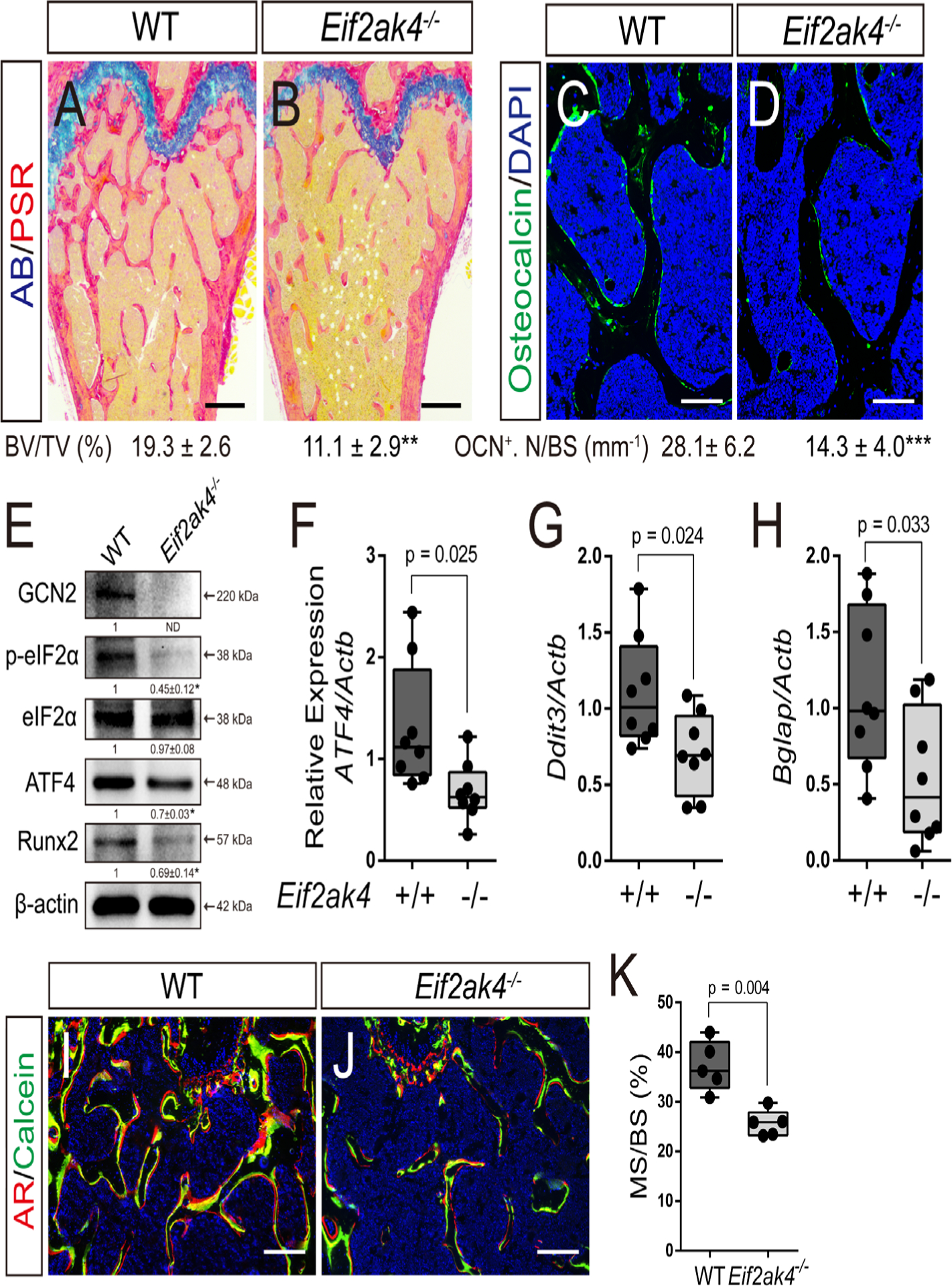
Decreased bone mass is attributed to reduced osteoblast numbers in Eif2ak4−/− mice. (A, B) Representative Alcian blue Picrosirius red–stained distal femur of 4-month-old mice. Scale bar = 100 μm. BV/TV = bone volume/tissue volume. (C, D) Representative osteocalcin (OCN) immunofluorescent staining of 4-month-old femur trabecular bone. Scale bar = 25 μm. Western blot (E) or RT-qPCR (F–H) analyses of GCN2 pathway components and osteoblast markers in femur extracts isolated from 4-month-old mice. Phosphorylated EIF2α normalized to total EIF2α; others normalized to β-actin. (I, J) Representative calcein and alizarin red double-labeled sections of the distal femur from 4-month-old mice. Scale bar = 50 μm. (K) Quantification of mineralized surface per bone surface (MS/BS) derived from double labeling. n ≥ 5.
Eif2ak4/GCN2 is required for cell proliferation
We next determined how Eif2ak4/GCN2 affected osteoblast numbers. To test if Eif2ak4 ablation affects osteoblast differentiation, we first performed differentiation assays in primary cOB, which are thought to represent committed osteoblast progenitors. Eif2ak4−/− cOB were able to undergo osteoblast differentiation and displayed normal alkaline phosphatase (ALP) staining, collagen matrix deposition, and mineralization and osteoblast marker gene induction (Supplemental Fig. S2). We next assayed differentiation in high-density BMSC cultures. BMSC consist of SSPC and other stromal cells and are thought to be less committed to the osteoblast lineage compared with cOB. Similar to the cOB, BMSC isolated from Eif2ak4−/− mice underwent normal osteoblast differentiation and displayed normal ALP staining, collagen matrix deposition, and mineralization (Fig. 3A). In addition, we observed no effect on the expression or induction of a number of osteoblast marker genes (eg, Runx2, Sp7, Akp2, Ibsp, and Bglap) (Fig. 3B). Collectively, this suggests that osteoblast differentiation is not affected by loss of Eif2ak4. Because Eif2ak4 ablation did not affect osteoblast differentiation per se, we evaluated the effects on the SSPC population using colony-forming efficiency (CFE) assays. Eif2ak4−/− mice displayed a significant reduction in overall CFE, indicating Eif2ak4 ablation may reduce the number of SSPC (Fig. 4A, B). SSPC are enriched in the CD45−PDGFRα+Sca-1+ bone marrow fractions.(2–5) Direct analysis of the CD45−PDGFRα+Sca-1+ bone marrow population using flow cytometry demonstrated no difference in the overall number of CD45−PDGFRα+Sca-1+ SSPC in Eif2ak4−/− mice, indicating the CFE defect is not the result of loss of the SSPC population (Fig. 4C, D). To test this hypothesis, we performed CFE assays on cells isolated from wild-type C57Bl/6 mice plated in duplicate and cultured in the presence or absence of the GCN2 inhibitor A-92 (referred to as GCN2i from hereon). Acute treatment with GCN2i prevented increased Eif2a phosphorylation in response to amino acid starvation (Supplemental Fig. S5A) and significantly reduced the overall number of colonies formed despite plating similar numbers of SSPC (Fig. 4E, F). Based on these data, we hypothesized that Eif2ak4 regulates either SSPC viability or proliferative capacity. To test this hypothesis, we evaluated the effect of Eif2ak4 ablation on both cell death and proliferation. Reduced CFE in Eif2ak4−/− mice was not the result of increased apoptosis because there was no difference in the number of TUNEL-positive cells in BMSC isolated from Eif2ak4−/− mice compared with wild-type controls (Supplemental Fig. S3). Rather, BMSC isolated from Eif2ak4−/− mice incorporated significantly less EdU compared with wild-type mice (Fig. 4G, H). To determine if decreased proliferation was directly or secondarily related to the result of loss of Eif2ak4/GCN2, we acutely inhibited Eif2ak4/GCN2. Importantly, inhibition of GCN2 using GCN2i or shRNA-mediated knockdown of GCN2 both significantly reduced proliferation similar to Eif2ak4 knockout in both primary BMSC and the mouse bone marrow cell line ST2 (Fig. 4I, J; Supplemental Figs. S4 and S5). Collectively, these data demonstrate Eif2ak4/GCN2 is directly required for proliferation and expansion of BMSC.
Fig 3.
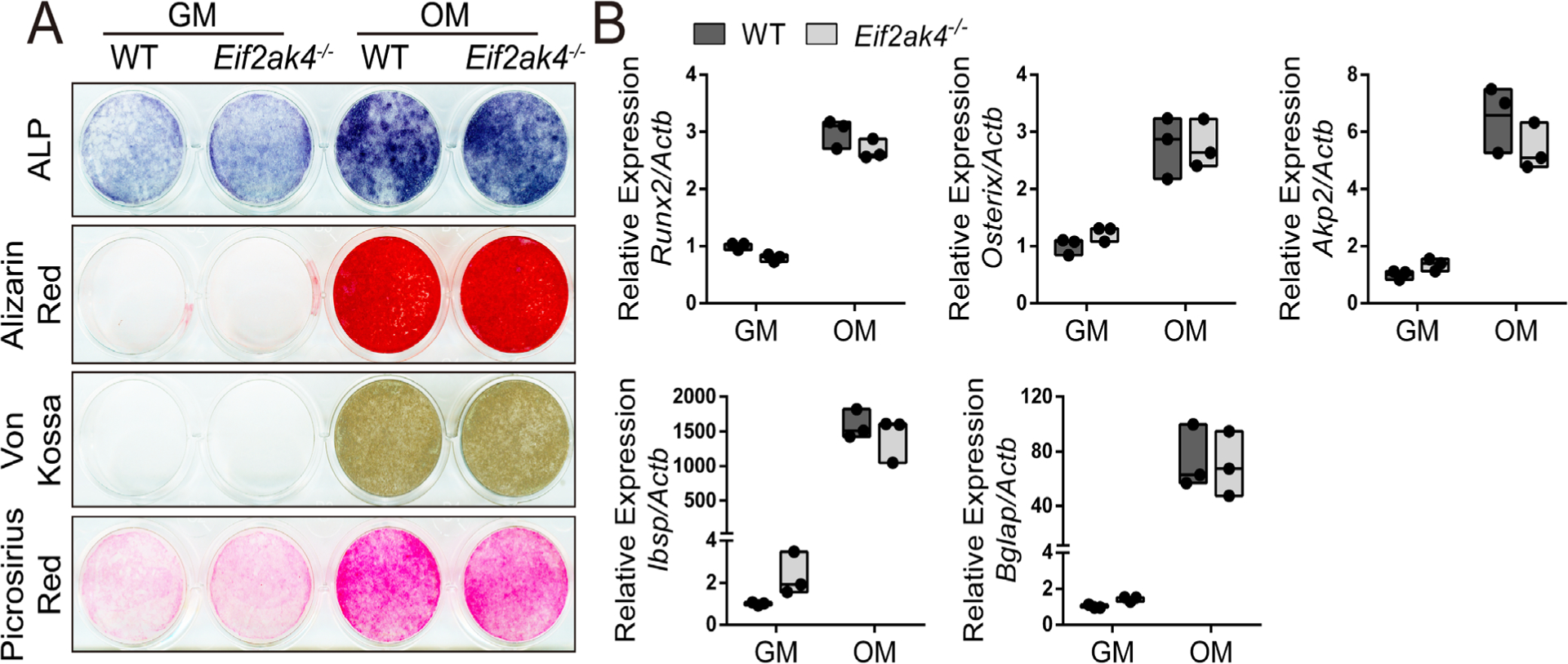
Eif2ak4 ablation has no effect on osteogenic differentiation. (A, B) Effect of Eif2ak4 deletion on alkaline phosphatase (ALP), alizarin red, Von Kossa, or Picrosirius red staining (A) or osteogenic gene expression (B) measured by RT-qPCR. Gene expression normalized to Actb mRNA. n = 3 individual mice. GM = growth media; OM = osteogenic media.
Fig 4.
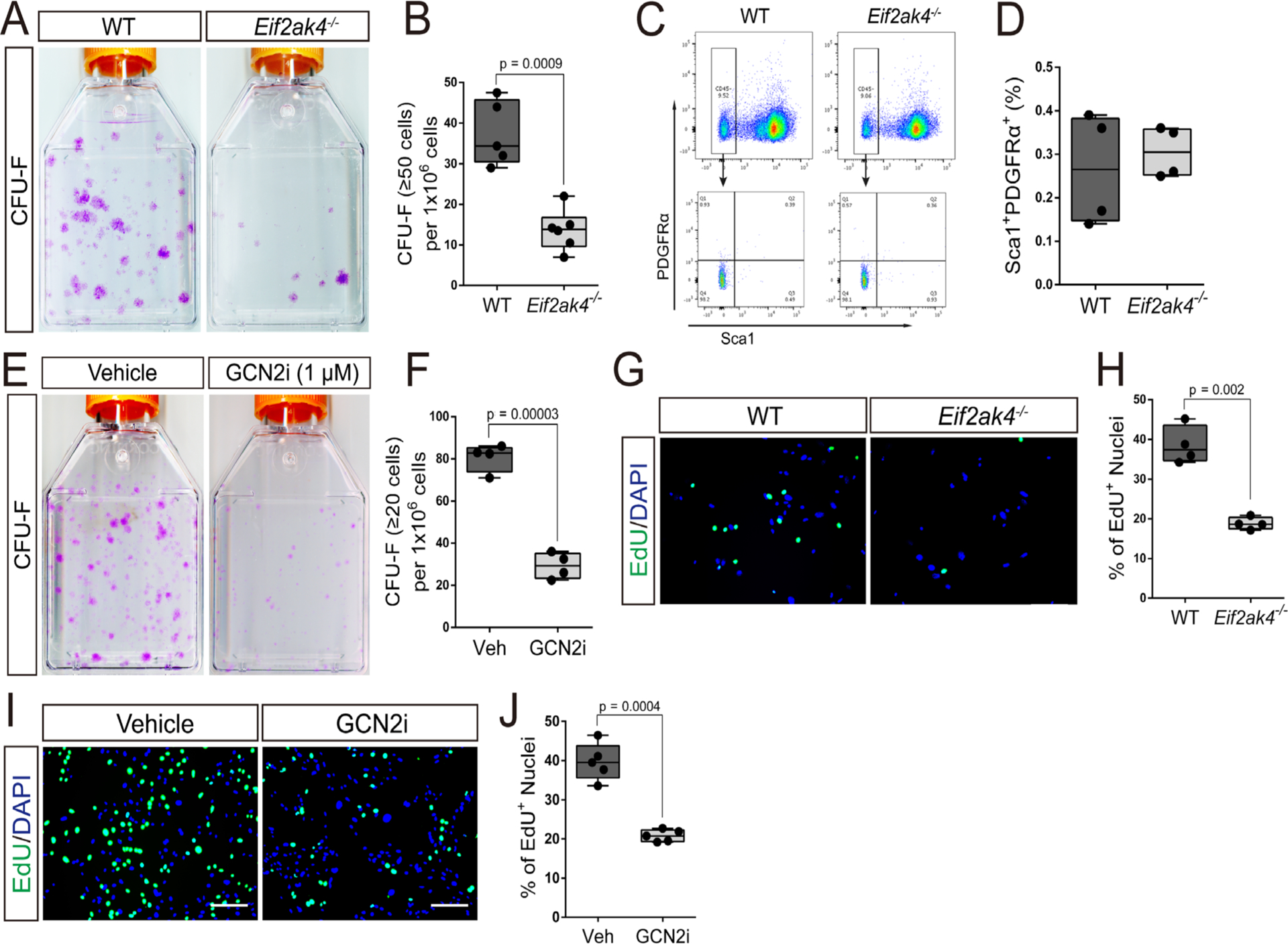
Eif2ak4/GCN2 activity is required for proliferation. (A) Representative images of colony-forming unit (CFU) assays stained with crystal violet. (B) Quantification of the number of colonies containing at least 50 cells isolated from 4-month-old Eif2ak4−/− or wild-type littermates. (C, D) Gating strategy to quantify the CD45−, PDGFRa+, Sca1+ population using flow cytometry. (E, F) CFU assay showing the effect of GCN2 inhibitor (GCN2i) treatment on the number of colonies containing at least 20 cells isolated from 4-month-old C57Bl/6 wild-type mice. (G–J) Effect of Eif2ak4 ablation (G, H) or GCN2i (I, J) on EdU incorporation in primary BMSC. Scale bar = 25 μm.
Eif2ak4/GCN2 is required for amino acid uptake in proliferating BMSC
We next sought to understand how Eif2ak4/GCN2 regulates BMSC proliferation. GCN2 responds to amino acid depletion by stimulating amino acid consumption and synthesis downstream of the transcriptional effector ATF4. Importantly, ATF4 was significantly reduced in both whole bone extracts and BMSC isolated from Eif2ak4−/− mice (Figs. 2E, F and 5A). To test if reduced Atf4 expression was important for the proliferation phenotype in Eif2ak4 mutant cells, we targeted Atf4 using shRNA (Fig. 5B). Reducing Atf4 expression significantly reduced proliferation as determined by decreased EdU incorporation in both primary BMSC and ST2 cells (Fig. 5C, D; Supplemental Fig. S6). These data suggest ATF4 is required for cell proliferation downstream of Eif2ak4/GCN2 in BMSC. ATF4 directly regulates the expression of genes involved in amino acid uptake and metabolism in response to cellular stress.(22,30,31) Indeed, qPCR analyses identified a significant downregulation of select amino acid transporters (eg, Slc1a1, Slc1a5, Slc7a5, and Slc38a2) in Eif2ak4−/− BMSC (Fig. 5E). Likewise, targeting GCN2 using short hairpin RNA or GCN2i resulted in significantly less amino acid transporter expression similar to Eif2ak4 mutant cells (Supplemental Figs. S4 and S5). Moreover, reducing Atf4 expression significantly reduced the expression of the majority of amino acid transporters similar to Eif2ak4 inhibition in both BMSC and ST2 cells (Fig. 5F; Supplemental Fig. S6). Because Eif2ak4 mutant BMSC are characterized by reduced amino acid transporter expression, we next sought to evaluate amino acid consumption in mutant BMSC. To do this, we took a targeted approach to evaluate amino acid uptake using radiolabeled amino acids. Eif2ak4 deletion significantly reduced the uptake of all amino acids evaluated (eg, glutamine, glutamate, proline, and arginine) (Fig. 5G). Likewise, acute knockdown or inhibition of GCN2 resulted in a similar reduction in amino acid consumption (Supplemental Figs. S4 and S5). Conversely, we observed a significant increase in both protein and collagen synthesis in Eif2ak4 mutant cells consistent with the known role of GCN2 to inhibit protein synthesis when activated (Fig. 5H, I). Decreased amino acid uptake in Eif2ak4 mutant cells was likely the result of decreased ATF4 expression as ATF4 knockdown significantly diminished amino acid uptake in both BMSC and ST2 cells (Fig. 5J; Supplemental Fig. S6). These data indicate Eif2ak4/GCN2 is required for amino acid consumption upstream of the transcriptional effector ATF4.
Fig 5.
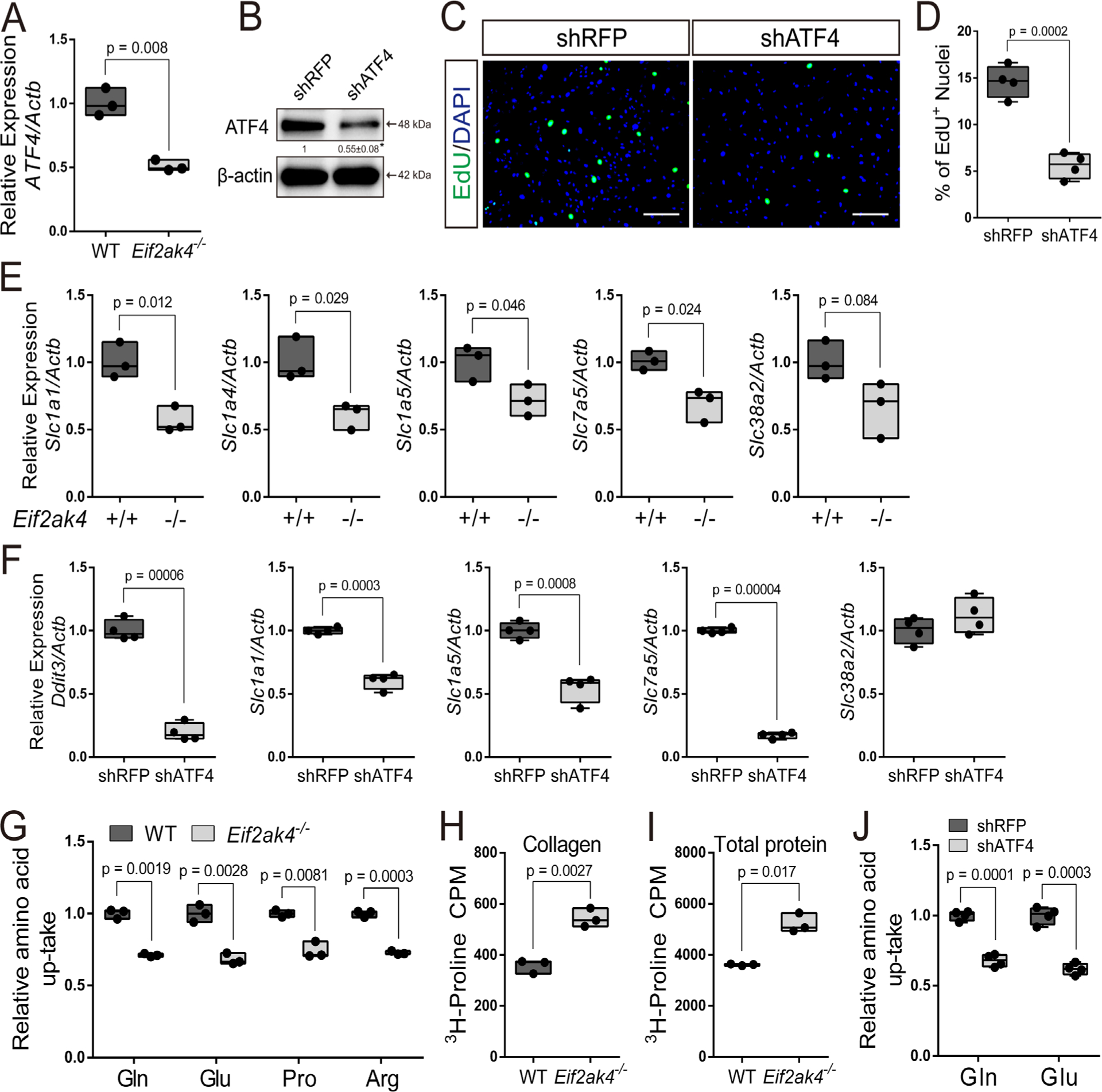
The GCN2/ATF4 pathway is required for robust amino acid consumption. (A) RT-qPCR analyses of Atf4 in BMSC isolated from wild-type (WT) or Eif2ak4−/− mice. (B) Validation of ATF4 knockdown by Western blot. ATF4 normalized to Actb. (C, D) Effect of ATF4 knockdown on EdU incorporation in BMSC. Scale bar = 25 μm. (E, F) RT-qPCR analyses of amino acid transporters in high-density BMSC cultures from wild-type (WT) or Eif2ak4−/− littermates (E) or ATF4 knockdown BMSC (F). (G) Radiolabeled amino acid uptake assays in BMSC isolated from Eif2ak4−/− or wild-type littermates. (H, I) Radiolabeled proline incorporation into collagen (H) or total protein (I) in wild-type (WT) or Eif2ak4−/− cOB. (J) Radiolabeled amino acid uptake assays in ATF4 knockdown BMSC. shRNA for RFP used as a negative control. Gene expression normalized to Actb mRNA levels. Each data point represents primary BMSC isolated from an individual mouse for each genotype. n ≥ 3.
We next sought to determine if increasing ATF4 expression was sufficient to rescue the Eif2ak4−/− cellular phenotype. To do this, we ectopically expressed either luciferase (Luc) or human Atf4 (hsATF4) in Eif2ak4−/− BMSC (Fig. 6A, B). Importantly, we observed increased expression of the ATF4 target gene Ddit3/Chop, demonstrating the functionality of hsATF4 overexpression (Fig. 6C). Moreover, hsATF4 expression significantly increased amino acid transporter expression to a similar extent and rescued amino acid uptake in Eif2ak4−/− BMSC (Fig. 6D, E). Finally, hsATF4 expression was able to rescue proliferation in Eif2ak4−/− BMSC (Fig. 6F, G). Collectively, these data indicate that intact GCN2/ATF4 signaling is critical for maintaining amino acid transporter expression and amino acid consumption necessary for BMSC proliferation (Fig. 6H).
Fig 6.
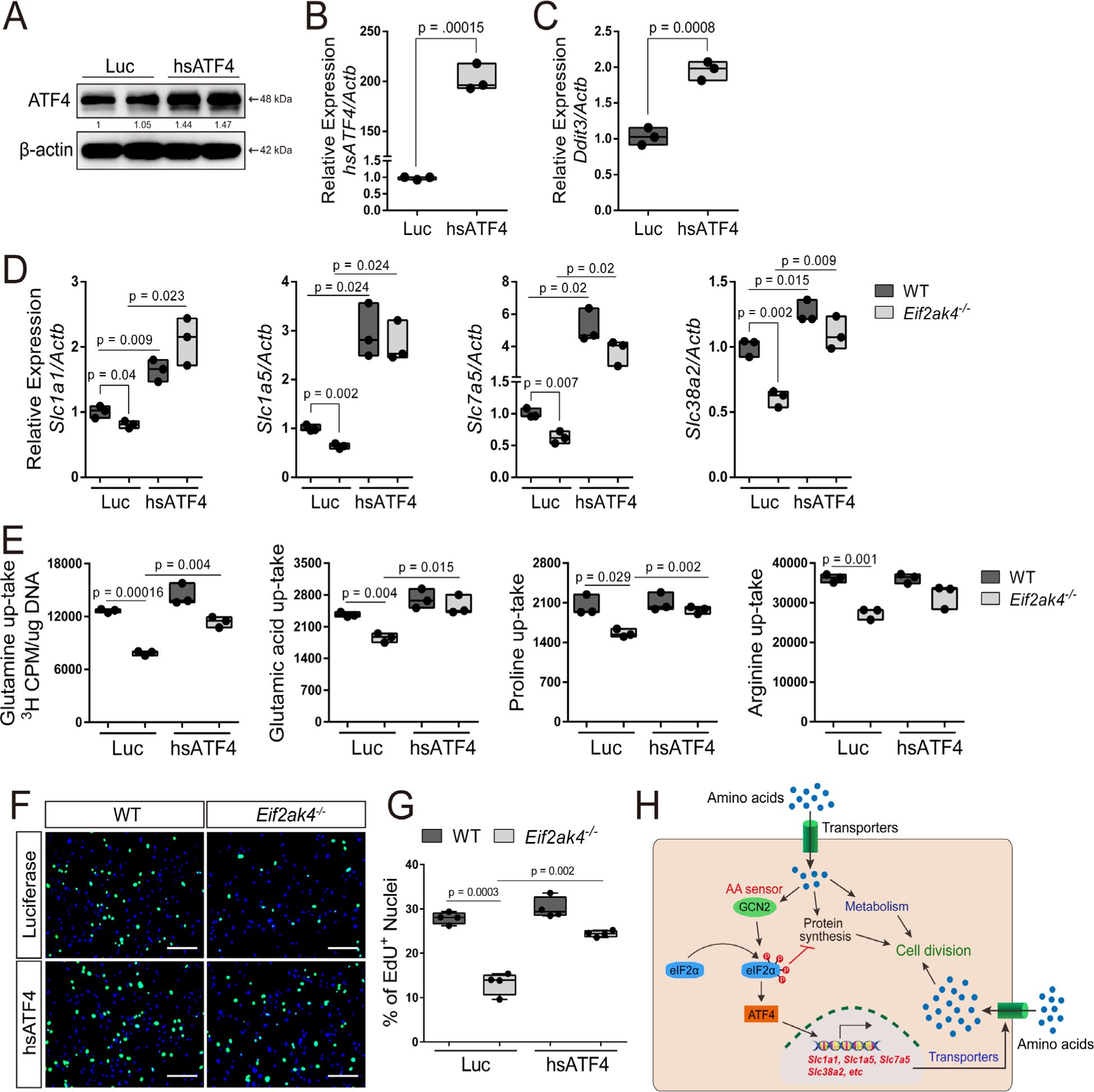
ATF4 rescues amino acid uptake and proliferation in the absence of GCN2. (A–C) Validation of Atf4 and Ddit3 expression in primary BMSC measured by Western blot (A) or qPCR (B, C). Effect of Atf4 expression on amino acid transporter expression (D), radiolabeled amino acid uptake (E), or EdU incorporation (F, G). Scale bar = 25 μm. Each data point represents primary BMSC isolated from an individual mouse for each genotype. n ≥ 3. Gene expression normalized to β-actin mRNA levels. (H) Model for how GCN2-mediated amino acid sensing facilitates amino acid uptake and cell division.
Discussion
We show here that the amino acid sensor Eif2ak4/GCN2 is a critical regulator of postnatal bone mass accrual. Genetically ablating Eif2ak4 results in a progressive reduction in bone accrual. Mechanistically, the decreased bone mass results from a reduction in overall osteoblast numbers in both male and female mice and reduced individual osteoblast activity in male mice only. Decreased osteoblast numbers is not the result of defective osteoblast differentiation; rather, it is likely attributed to decreased proliferation in Eif2ak4-deficient cells. Because of technical challenges, we were unable to evaluate proliferation in SSPC directly. However, we observed reduced proliferation in BMSC cultures, which contain both SSPC and their progeny among other cells. Importantly, we observed no difference in the overall number of CD45−PDGFRα+Sca-1+ SSPC in Eif2ak4−/− animals. These data suggest the proliferation defect we observe is likely in a cell derived from the CD45−PDGFRα+Sca-1+ SSPC. Thus, we conclude that decreased proliferation in Eif2ak4−/− SSPC or their descendants is the underlying cause of reduced osteoblast numbers and diminished bone formation. Further, our data indicate that Eif2ak4/GCN2 activates ATF4 in order to increase amino acid consumption necessary for proliferation. Collectively, these data provide the first example of the critical role for GCN2-dependent amino acid sensing to regulate proliferation and postnatal bone accrual in mice.
Eif2ak4/GCN2 was recently shown to be required for osteoblast differentiation and increased bone-forming activity in response to pathological WNT signaling.(21) It is intriguing then that osteoblast differentiation was unaffected by the loss of Eif2ak4 at least in vitro (Fig. 3; Supplemental Fig. S2). Taken together, these data suggest Eif2ak4/GCN2 may be dispensable for osteoblast differentiation in vivo. A possible explanation for this observation would be compensation by another EIF2α kinase in vivo. Indeed, the EIF2α kinase, Eif2ak3, encoding double-stranded RNA-activated protein kinase (PKR)-like endoplasmic reticulum kinase (PERK), is required in osteoblasts to increase anabolic capacity to facilitate bone matrix production and promote terminal osteoblast differentiation and maturation.(32,33) This suggests individual EIF2α kinases play distinct roles during different stages of osteoblast differentiation. In proliferating cells, amino acid depletion likely activates the GCN2-ATF4 pathway, where ATF4 stimulates amino acid uptake to facilitate proliferation. Conversely, during osteoblast differentiation, collagen synthesis and secretion rapidly increase. This in turn results in an accumulation of unfolded proteins in the endoplasmic reticulum and activation of the PERK-ATF4 pathway to increase genes involved in amino acid uptake, oxidative protein folding, and terminal osteoblast differentiation (eg, Bglap).(22,33) Thus, different stages of the osteoblast differentiation continuum likely experience unique metabolic stresses and respond accordingly. In proliferating SSPC, the predominant stress appears to be amino acid depletion, whereas in osteoblasts the predominant stress is unfolded protein in the ER. Thus, Eif2ak4/GCN2 activation is critical for proliferation in osteoblast progenitors but can be compensated for by the activation of Eif2ak3/PERK in osteoblasts.
Proliferating cells have unique metabolic needs compared with nonproliferating cells. For example, proliferating cells must synthesize all of the cellular components necessary to duplicate their mass. Amino acids fulfill these biosynthetic requirements by providing carbon and nitrogen for biomass production in proliferating cells.(9,34–36) It is intuitive then that proliferation would be tightly coupled to amino acid availability.(37,38) In the absence of certain amino acids, including arginine, glutamine, isoleucine, etc., cell proliferation is halted in late G1 due to loss of cyclin D1 and persistent P21 expression.(39–43) These nutrient-mediated metabolic checkpoints are postulated to function to ensure the availability of adequate raw materials before committing to genome duplication and cell division.(41) Amino acid utilization in proliferating cells likely depletes intracellular amino acid pools activating the GCN2/pEIF2α/ATF4 signaling cascade. ATF4 then induces a transcriptional response to enhance amino acid acquisition and biosynthesis necessary for proliferation.(22) In cells lacking either Eif2ak4 or Atf4, we observed a significant reduction in DNA synthesis (Figs. 5 and 6). This is attributed to reduced amino acid consumption predicted to mimic the effects of amino acid withdrawal on cell cycle progression. Although we were unable to directly evaluate cell cycle kinetics, our data indicate GCN2 and ATF4 function to increase amino acid consumption to ensure an adequate supply of glutamine, arginine, and other amino acids necessary to exit these metabolic checkpoints and proceed to S phase (Fig. 6H). Further studies are warranted to determine the precise temporal requirements of Eif2ak4 and Atf4 during cell cycle progression.
In summary, our findings provide new insights into the regulation of BMSC proliferation. We identify Eif2ak4/GCN2-dependent amino acid sensing as a critical regulator of proliferation necessary for osteoblast endowment and postnatal bone mass accrual. These data indicate activating the GCN2/ATF4 signaling cascade or stimulating amino acid consumption may provide a therapeutic approach to stimulate proliferation to either maintain or expand the SSPC population in aged individuals.
Supplementary Material
Acknowledgments
The authors thank all members of the Karner Lab for critical comments on this manuscript. This work was supported by NIH R01 grants R01 AR060456 to FL and R01 AR071967 to CMK. The initial mouse work was performed at Washington University in St. Louis School of Medicine.
Footnotes
Disclosures
All authors state that they have no conflicts of interest.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4091.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423(6937):349–55. [DOI] [PubMed] [Google Scholar]
- 2.Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15(2): 154–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morikawa S, Mabuchi Y, Kubota Y, et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med. 2009;206(11):2483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Omatsu Y, Sugiyama T, Kohara H, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33(3):387–99. [DOI] [PubMed] [Google Scholar]
- 5.Park D, Spencer JA, Koh BI, et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell. 2012;10(3):259–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Wu Q, Wang Y, Li L, Bu H, Bao J. Senescence of mesenchymal stem cells (review). Int J Mol Med. 2017;39(4):775–82. [DOI] [PubMed] [Google Scholar]
- 7.Palm W, Thompson CB. Nutrient acquisition strategies of mammalian cells. Nature. 2017;546(7657):234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Bermudez J, Baudrier L, La K, et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol. 2018;20(7):775–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hosios AM, Hecht VC, Danai LV, et al. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev Cell. 2016;36(5):540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104(49):19345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. [DOI] [PubMed] [Google Scholar]
- 12.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones CL, Stevens BM, D’Alessandro A, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34(5):724–40.e4. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhutia YD, Babu E, Ramachandran S, Ganapathy V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015;75(9):1782–8. . [DOI] [PubMed] [Google Scholar]
- 15.Yu Y, Newman H, Shen L, et al. Glutamine metabolism regulates proliferation and lineage allocation in skeletal stem cells. Cell Metab. 2019;29(4):966–78.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34(Pt 1): 7–11. [DOI] [PubMed] [Google Scholar]
- 17.Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–99. . [DOI] [PubMed] [Google Scholar]
- 18.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. [DOI] [PubMed] [Google Scholar]
- 19.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17(10):1374–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karner CM, Esen E, Okunade AL, Patterson BW, Long F. Increased glutamine catabolism mediates bone anabolism in response to WNT signaling. J Clin Invest. 2015;125(2):551–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han J, Back SH, Hur J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15(5):481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rached MT, Kode A, Xu L, et al. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 2010;11(2):147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elefteriou F, Benson MD, Sowa H, et al. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 2006;4(6):441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karner CM, Lee SY, Long F. Bmp induces osteoblast differentiation through both Smad4 and mTORC1 signaling. Mol Cell Biol. 2017;37(4):e00253–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang P, McGrath BC, Reinert J, et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol. 2002;22(19):6681–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karner CM, Esen E, Chen J, Hsu FF, Turk J, Long F. Wnt protein signaling reduces nuclear acetyl-CoA levels to suppress gene expression during osteoblast differentiation. J Biol Chem. 2016;291(25): 13028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orsini H, Araujo LP, Maricato JT, et al. GCN2 kinase plays an important role triggering the remission phase of experimental autoimmune encephalomyelitis (EAE) in mice. Brain Behav Immun. 2014;37: 177–86. [DOI] [PubMed] [Google Scholar]
- 29.Jiang HY, Wek RC. GCN2 phosphorylation of eIF2alpha activates NF-kappaB in response to UV irradiation. Biochem J. 2005;385(Pt 2): 371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Geldermalsen M, Wang Q, Nagarajah R, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. 2016;35(24):3201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren P, Yue M, Xiao D, et al. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J Pathol. 2015;235(1):90–100. [DOI] [PubMed] [Google Scholar]
- 32.Wei J, Sheng X, Feng D, McGrath B, Cavener DR. PERK is essential for neonatal skeletal development to regulate osteoblast proliferation and differentiation. J Cell Physiol. 2008;217(3):693–707. [DOI] [PubMed] [Google Scholar]
- 33.Saito A, Ochiai K, Kondo S, et al. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J Biol Chem. 2011;286(6):4809–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Labuschagne CF, van den Broek NJ, Mackay GM, Vousden KH, Maddocks OD. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014;7(4):1248–58. [DOI] [PubMed] [Google Scholar]
- 35.Jain M, Nilsson R, Sharma S, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336 (6084):1040–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alkan HF, Walter KE, Luengo A, et al. Cytosolic aspartate availability determines cell survival when glutamine is limiting. Cell Metab. 2018;28(5):706–20.e6. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eagle H Nutrition needs of mammalian cells in tissue culture. Science. 1955;122(3168):501–14. [DOI] [PubMed] [Google Scholar]
- 38.Eagle H The specific amino acid requirements of a mammalian cell (strain L) in tissue culture. J Biol Chem. 1955;214(2):839–52. [PubMed] [Google Scholar]
- 39.Ley KD, Tobey RA. Regulation of initiation of DNA synthesis in CHINESE hamster cells: II. Induction of DNA synthesis and cell division by isoleucine and glutamine in G(1)-arrested cells in suspension culture. J Cell Biol. 1970;47(2):453–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nelsen CJ, Rickheim DG, Tucker MM, et al. Amino acids regulate hepatocyte proliferation through modulation of cyclin D1 expression. J Biol Chem. 2003;278(28):25853–8. [DOI] [PubMed] [Google Scholar]
- 41.Saqcena M, Menon D, Patel D, Mukhopadhyay S, Chow V, Foster DA. Amino acids and mTOR mediate distinct metabolic checkpoints in mammalian G1 cell cycle. PLoS One. 2013;8(8):e74157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tobey RA. Production and characterization of mammalian cells reversibly arrested in G1 by growth in isoleucine-deficient medium. Methods Cell Biol. 1973;6:67–112. . [DOI] [PubMed] [Google Scholar]
- 43.Tobey RA, Ley KD. Isoleucine-mediated regulation of genome repliction in various mammalian cell lines. Cancer Res. 1971;31(1):46–51. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.