

Abstract
Cu(I) P-type ATPases are transmembrane primary active ion pumps that catalyze the extrusion of copper ions across cellular membranes. Their activity is critical in controlling copper levels in all kingdoms of life. Biochemical and structural characterization established the structural framework by which Cu-pumps perform their function. However, the details of the overall mechanism of transport (uniporter vs. cotransporter) and electrogenicity still remain elusive. In this work, we developed a platform to reconstitute the model Cu(I)-pump from E. coli (EcCopA) in artificial lipid bilayer small unilamellar vesicles (SUVs) to quantitatively characterize the metal substrate, putative counterions and charge translocation. By encapsulating in the liposome lumen fluorescence detector probes (CTAP-3, pyranine and oxonol VI) responsive to diverse stimuli (Cu(I), pH and membrane potential), we correlated substrate, secondary-ion translocation and charge movement events in EcCopA proteoliposomes. This platform centered on multiple fluorescence reporters allowed study of the mechanism and translocation kinetic parameters in real-time for wild-type EcCopA and inactive mutants. The maximal initial Cu(I) transport rate of 165 nmol Cu(I) mg–1 min–1 and KM,Cu(I) = 0.15 ± 0.07 μM was determined with this analysis. We reveal that Cu(I) pumps are primary-active uniporters and electrogenic. The Cu(I) translocation cycle does not require proton counter-transport resulting in electrogenic generation of transmembrane potential upon translocation of one Cu(I) per ATP hydrolysis cycle. Thus, mechanistic differences between Cu(I) pumps and other better characterized P-type ATPases are discussed. The platform opens the venue to study translocation events and mechanisms of transport in other transition metal P-type ATPase pumps.
Graphical Abstract
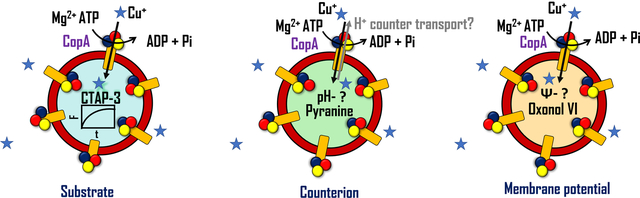
Real-time transport analyses define transmembrane Cu(I)-pumps as electrogenic uniporters
Transmembrane transporter proteins and ion pumps regulate the vectorial uptake and extrusion of metal ions across cellular membranes. Their activity plays a gatekeeper role in controlling metal concentrations within cells to ensure correct metal homeostasis and active extrusion of toxic metals1, 2. Copper P1B-type ATPases are transmembrane primary-active pumps belonging to the P-type ATPase superfamily, conserved throughout all kingdoms of life, which utilize the energy generated by ATP hydrolysis to drive Cu(I) transport across biological membranes against electrochemical gradients2–6.
As a consequence of the intracellular reducing environment, which favors the Cu(I) oxidation state, and the potentially harmful redox-active nature of copper ions, which are present in μM levels in cells (e.g. approx. 70 μM in yeast)7, no free Cu(I) is present in cells and a sophisticated network of Cu(I)-binding proteins and metallochaperones allow targeted delivery to cuproenenzymes and transporters to guarantee physiological copper homeostasis2. Cu(I)-pumps constitute an essential system to catalyze the selective translocation and export of Cu(I) ions thereby controlling the intracellular Cu(I) levels2–6. Their activity tightly balances the biogenesis and integrity of copper centers in vital enzymes to non-toxic intracellular copper levels2–6. In addition, their activity in eukaryotic cells is essential to provide Cu(I) in the sarco-endoplasmic reticulum lumen for maturation of transmembrane and membrane-bound cuproenzymes8, 9. In humans, mutations resulting in defects in the Cu(I)-transporting P1B-ATPases ATP7A and ATP7B lead to Menkes syndrome and Wilson disease, respectively10, 11. The activities of ATPP7A/B in controlling Cu(I) concentrations and their role in enzyme maturation has been linked to a number of biological processes contributing to pathological conditions such as lipoatrophy, carcinogenesis, neurological disorders and neurodegeneration12–16.
The metal substrate transport cycle in Cu(I)-pumps (belonging to the P1B-type ATPase class) follows the Post-Albers scheme characteristic of the P-type ATPases superfamily (Figure 1)4, 17, 18. P-type pumps alternate between two states, E1 and E2, that allow ion accessibility on opposite sides of the lipid bilayer, coupled to conformational changes in cytoplasmic domains induced by ATP binding, hydrolysis, phosphorylation and dephosphorylation17, 18. Substrate ion(s) bind to the transmembrane site(s) (E1 state), are occluded within the membrane upon ATP hydrolysis and phosphorylation (E1P), and then released from E2P to the opposite side of the membrane, resulting in dephosphorylation to regenerate E117, 18. Primary substrate translocation is generally coupled to a different secondary counter-ion transport that is translocated in the opposite direction during the E2P-E2-E1 transition. Counter-ion binding and transport has been shown to be a strict requirement for several characterized P-type ATPases, e.g. the sarco/endoplasmic reticulum Ca2+/H+-ATPase, SERCA, the plasma membrane Ca2+ ATPase, PMCA, the gastric H+/K+ ATPase and the Na⁺/K⁺-ATPase18–24.
Figure 1:
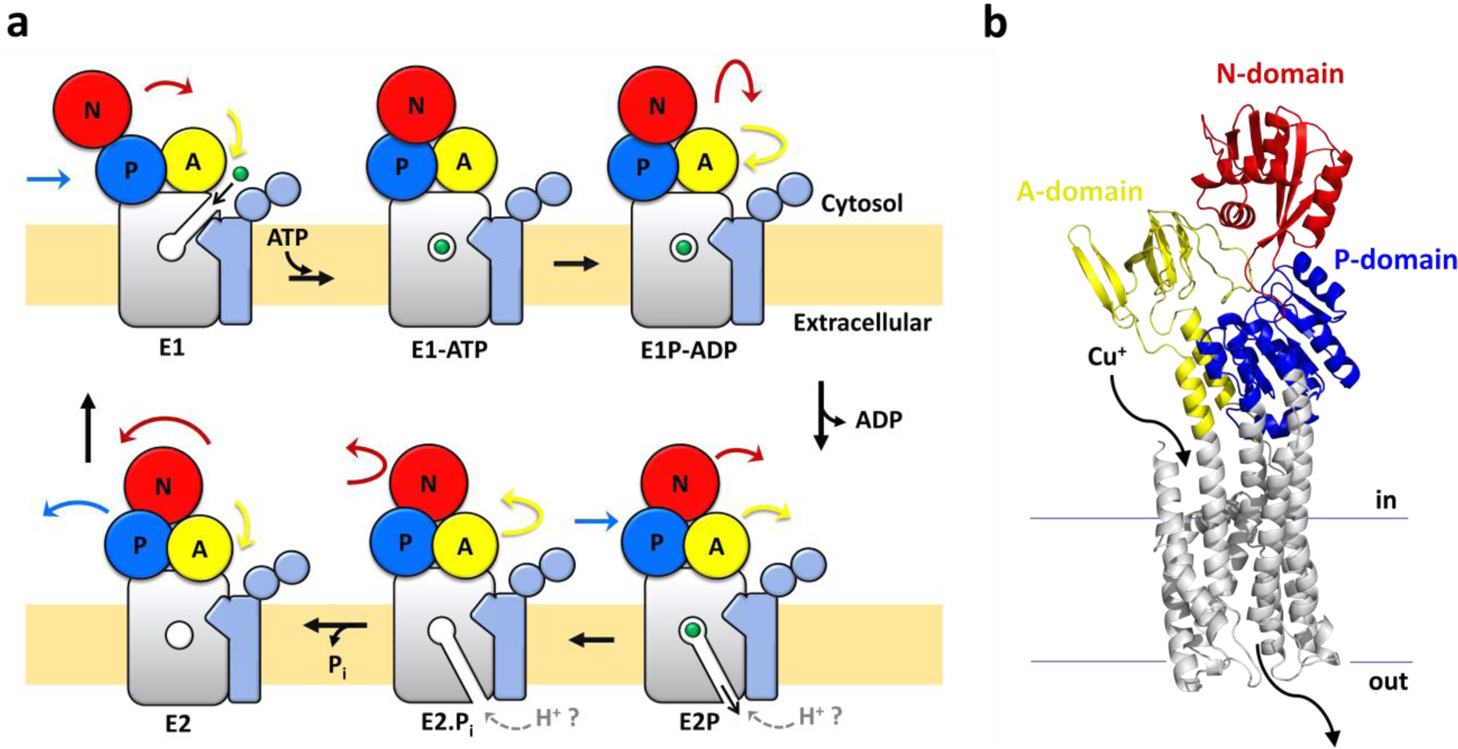
Cu(I)-pumps catalytic cycle and structure. (a) Schematic representation of the catalytic Post-Albers cycle in Cu(I)-pumps (based on the EcCopA topology). The transmembrane domain harbouring the transmembrane high-affinity Cu(I) binding-site is shown in grey colour. The A-, P- and N-domains are colored in yellow, blue and red, respectively. The two N-terminal helices (TM-A and -B) characteristic of P1B-type ATPases and the two soluble N-terminal metal binding domains are depicted in light blue. The colored arrows indicate the movements of the corresponding domains based on the catalytic cycle of SERCA and the known structures of CopA (in E2P state and transition state of dephosphorylation, E2.Pi) (b) EcCopA 3D homology model calculated with MEDELLER based on the structure of Legionella pneumophila CopA in the E2 state (PDB: 3RFU). The two N-terminal cytosolic metal binding domains are omitted in the model as the relative orientation and contacts to other domains are unknown and not resolved in the LpCopA structure.
This Post-Albers alternating access cycle is accomplished by the presence of a soluble ATP binding domain (N-domain), a phosphorylation domain (P-domain, for catalytic auto-phosphorylation) and an actuator module (A-domain) that tightly couples the energy from ATP hydrolysis to transmembrane conformational changes responsible for metal translocation17, 18.
Biochemical characterization of Cu(I)-pumps revealed the presence of specific motifs essential for Cu(I) recognition and translocation3, 25–28. Cu(I)-pumps belong to the P1B-1-type subclass of transition metal P-type ATPases. They are characterized by the presence of a conserved CPC motif in transmembrane helix 4 (TM4) and a MXXXS motif in TM6, which are both essential signature sequences providing ligands for metal coordination within the transmembrane domain. Several characterized Cu(I) pumps also possess one or multiple soluble ferredoxin-like N-terminal metal binding domains (MBDs) each carrying CXXC motifs involved in Cu(I) binding (two in E. coli CopA and 6 in human Cu(I) ATPases ATP7A/B)6, 29–35. However, these regulatory cytoplasmic metal binding domains present in P1B-1-type ATPases (MBDs) appear to possess a regulatory function and are not essential to confer metal selectivity and transport28. To note, while P1B-3-type ATPase subfamily members have been initially proposed to act as Cu(II)-pumps, recent investigations revisited their role in copper transport and revealed that also this subclass can act as Cu(I)-pumps. However, their predominant distribution in genomes of Archaea and extremophiles suggest a more limited and specific evolutionary role in cellular copper metabolism36–38.
High-resolution molecular structures of a Cu(I)-transporting P1B-ATPase in Cu(I)–free conformational states of the catalytic cycle (E2P and E2.Pi, from Legionella Pneumophila, LpCopA) established the structural framework by which Cu(I)-pumps perform their function39, 40. In combination with a suite of structural and biophysical approaches, the bioinorganic chemistry properties of CopA established a model for Cu(I) translocation. In LpCopA Cu(I) is selected by a high-affinity transmembrane-binding site through a trigonal-planar coordination with the Cys residues of the conserved CPC motif of TM4 (C382 and C384 in L. pneumophila LpCopA, corresponding to C479 and C481 in E. coli EcCopA) and the conserved Met residue of TM6 (M717 of the MXXXS motif in LpCopA corresponding to M813 in EcCopA)41, 42. These residues are also essential for transport. Additionally, critical roles of other conserved intramembranous polar residues in facilitating copper uptake from the intracellular Cu(I) chaperones (Cu(I) entry site)41, binding to the high-affinity site and subsequent release through the Cu(I) exit pathway have been postulated39–41, 43–45. High topological and key-residues conservation reveals common mechanism of ion recognition and transport in all prokaryotic and eukaryotic Cu(I) P-type ATPases (Figure 1b)34.
The coupling of solute transport with a secondary ion flux in a specific stoichiometry might constitute a strict requirement for driving metal substrate transport in P1B-type ATPases. As an example, extensive biochemical and structural characterization of SERCA and PMCA established that their catalytic cycle requires proton counter-transport for the transport cycle to complete20, 24, 46.
By reconstituting E. coli CopA in giant unilamellar vesicles, a direct correlation between ATPase activity and Cu(I) transport was also demonstrated47. However, details of the overall mechanism of transport remain still elusive in Cu(I)-pumps. Moreover, it remains unknown whether the catalytic cycle mimics that of other P-type ATPases, and whether the pumps function as uniporters or co-transporters. Consequently, their electrogenicity also remains to be established. The aspects of the ATP-dependent Cu(I) translocation have not been fully addressed in the past because of the lack of molecular tools that would allow real-time monitoring of substrate translocation, counter-ion transport, and charge movement of pumps re-constituted in a native-like membrane environment.
To address these questions, we developed a biophysical platform to study substrate, secondary-ion translocation events, and charge movement in real-time upon E. coli CopA (EcCopA, utilized as a model system for Cu(I) pumps, Figure 1b) reconstitution in small unilamellar liposomes.
By encapsulating a diverse set of fluorescence probes in the lumen of liposomes, we performed and correlated real-time measurements of Cu(I) transport with time-dependent measurements of putative proton secondary ion translocation (pH sensor pyranine)48 and membrane potential evolution (membrane potential sensor oxonol VI)49. This platform provides a novel experimental setup with multiple fluorescence reporters that permit the study of substrate transport mechanisms and the determination of the underlying kinetic parameters.
By utilizing the lipid-compatible Cu(I)-selective fluorescent probe CTAP-350, we determined kinetic rates of Cu(I) translocation and demonstrated that Cu(I)-ATPases, in contrast to other P-type ATPase classes (e.g. Ca2+/H+ pump), are electrogenic uniporters without proton counter-flux. Consequently, Cu(I)-pumps are capable of building up a transmembrane potential by translocating one positive charge per ATP hydrolysis cycle. Our work reveals mechanistic differences with other, better characterized P-type ATPases. Moreover, the developed platform opens the avenue to study translocation events and mechanisms of transport in other transition metal P-type pumps.
Results and discussion
E. coli CopA expression and functional reconstitution in SUVs
We initially established protocols for recombinant expression and purification of full-length wt EcCopA in detergent micelles (n-Dodecyl-ß-D-maltoside, DDM) and subsequent reconstitution in artificial lipid bilayers. CopA was purified by STREP-tag II affinity chromatography (Strep-Tactin resin) followed by size exclusion chromatography (SEC) to >95–98% purity, as determined by SDS-PAGE analysis (Figure 2). SEC analysis revealed monodisperse CopA-DDM micelles complexes with no significant aggregation. Based on the elution volume and calibration with proteins of known Stokes radius, we extrapolated that EcCopA is purified as a monomeric protein in complex with DDM micelles. In purified P1B-type ATPases the rate of ATP hydrolysis is stimulated depending on the presence of the selected metal substrates. Cu(I)-dependent stimulation of ATPase activity of wild-type EcCopA in DDM in the presence of Mg2+/ATP revealed a hyperbolic Michaelis-Menten-type kinetics as a function of Cu(I), thus confirming that our purification protocols maintained the protein in a functional form (KM,Cu(I)= 0.11 ± 0.02 μM, and Vmax of 7.73 ± 0.19 nmol (mg min)−1) (Figure 3).
Figure 2:
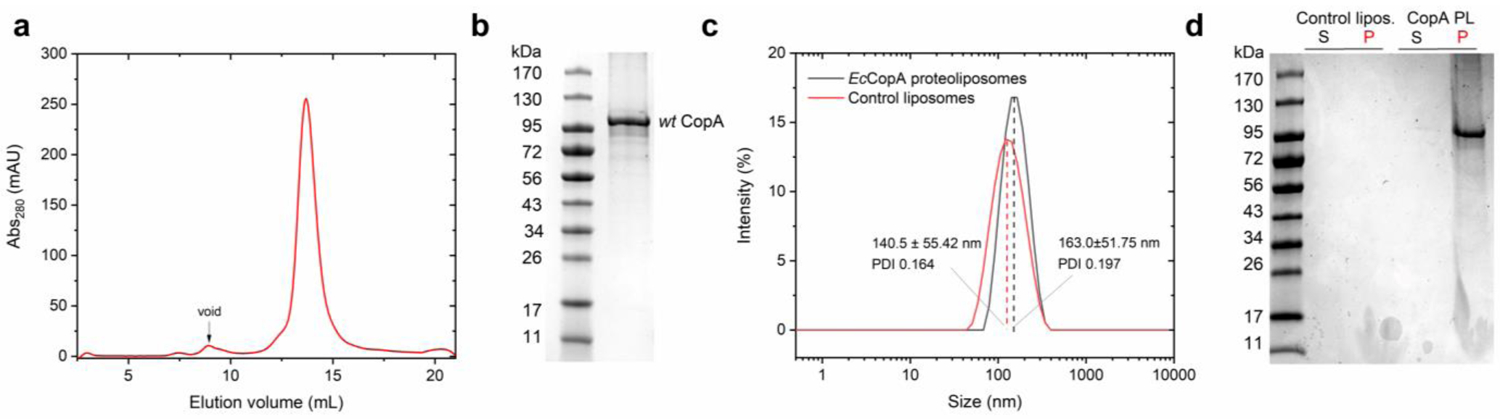
EcCopA purification and reconstitution in proteoliposomes. (a) Size Exclusion Chromatogram (SEC) elution profile of wt EcCopA purified in DDM micelles, and (b) corresponding SDS-PAGE of purified EcCopA. (c) Dynamic light scattering (DLS) analysis of control liposomes and EcCopA proteoliposomes revealing monodisperse vesicle size distribution. (d) SDS-PAGE analysis of EcCopA incorporation in isolated proteoliposomes (P, pellet) in comparison to non-incorporated protein (S, supernatant), and corresponding analysis of control liposomes.
Figure 3:
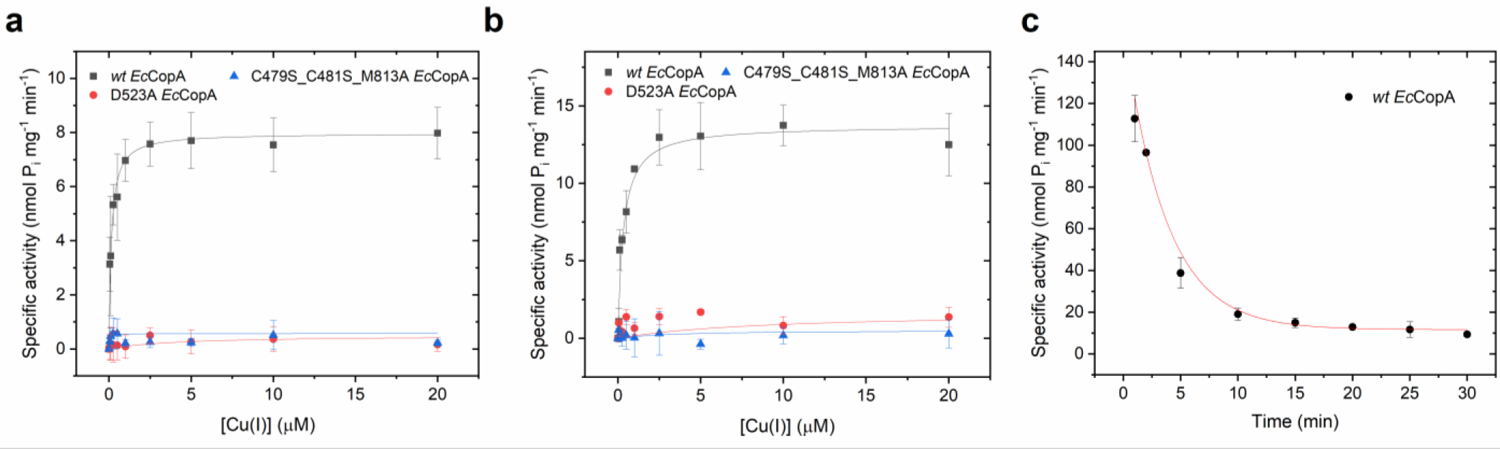
Cu(I)-dependent ATPase activity. (a) Cu(I)-dependent specific ATPase activity determined in DDM micelles for wt EcCopA (black), and the inactive mutants D523A EcCopA (red) and C479S_C481S_M813A EcCopA (blue), as a function of Cu(I) concentration (0 – 20 μM, supplied by in situ reduction of CuCl2 with ascorbic acid). Data were fitted with a Michaelis-Menten-type equation to determine KM,Cu(I) and VMax; v= (VMax* [Cu(I)])/ (KM,Cu(I) + [Cu(I)]). (b) Corresponding apparent specific ATPase activity in wt EcCopA proteoliposomes and EcCopA mutants. (c) Determination of Cu(I)-dependent specific ATPase activity in EcCopA proteoliposomes as a function of incubation time at saturating Cu(I) concentrations (10 μM). Data were fitted with an exponential decay function to derive the maximal specific ATPase activity at t = 0 min. For all ATPase activity measurements, the ATP hydrolysis rates were corrected for background values obtained in control liposomes, in which protein was not incorporated. All data are mean ± s.d. (n=3).
To verify the robustness of our approach, we also generated and purified inactive EcCopA mutants as validation of the functional characterization (Figure S1). To this end, we generated a mutant (D523A EcCopA) in the conserved aspartate phosphorylation site present in all P-type ATPases (P-domain) and a mutant lacking the transmembrane Met and Cys residues in transmembrane helices 4 and 6 (TM 4–6) essential for high-affinity Cu(I) binding and transport (C479S_C481S_M813A EcCopA). In agreement with the importance of these residues, analysis of the Cu(I)-stimulated ATPase hydrolysis rates confirmed complete inactivation of both mutants. The results confirmed the strict requirement of phosphorylation in the P-domain (critical in the Post-Albers scheme) and the S-based transmembrane Cu(I) recognition and translocation pathway to confer CopA catalytic activity (Figure 3a).
We sought to develop a strategy to investigate the Cu(I) translocation properties and kinetics in real-time by reconstituting purified EcCopA micelles into artificial lipid bilayers (Figure 4).
Figure 4:
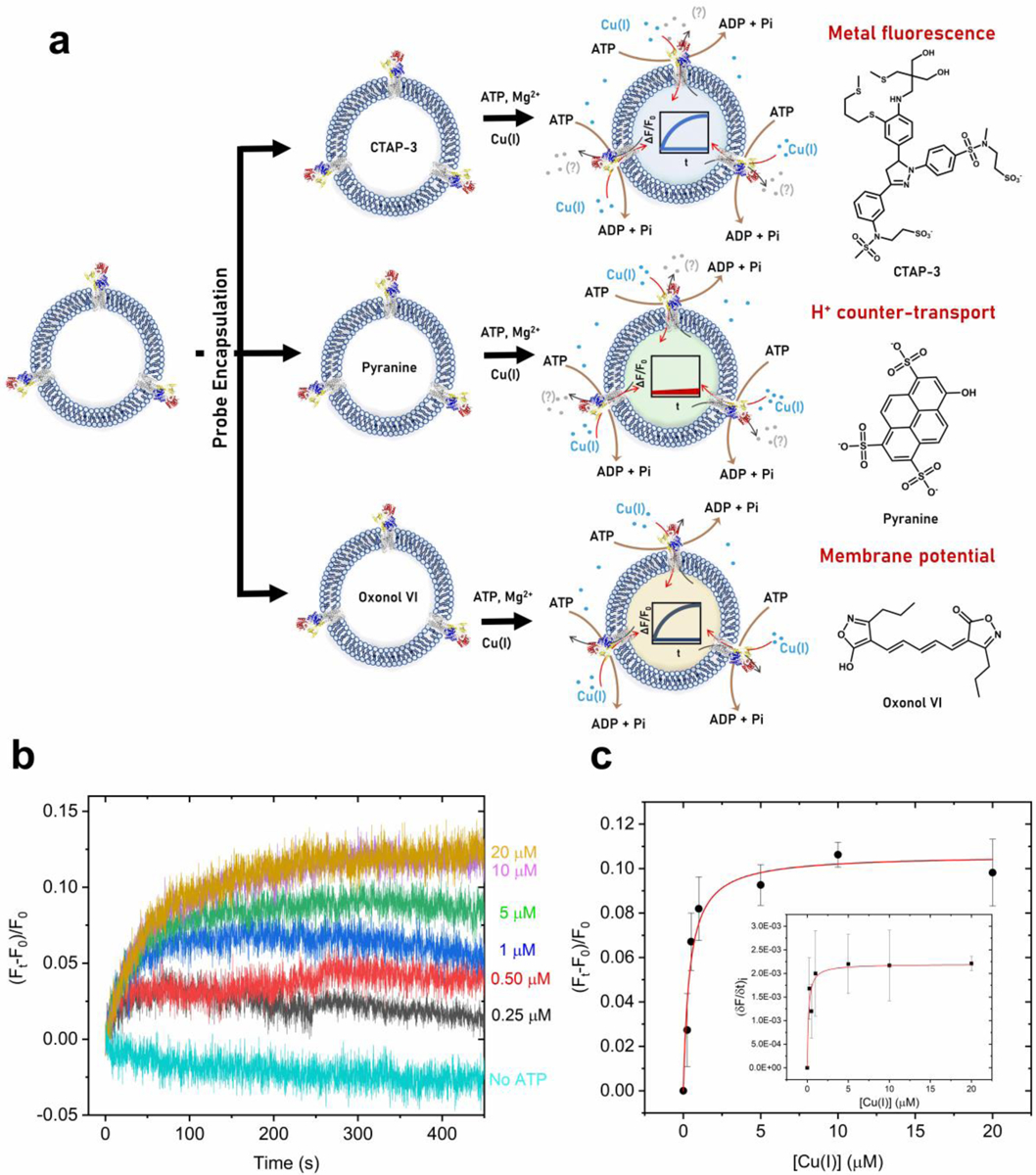
(a) Schematic representation of the experimental approach utilized to determine in real-time: i) Cu(I) transport in EcCopA proteoliposomes by encapsulation in the lumen the fluorescent probe CTAP-3 (λex= 365 nm; λem= 450 nm); ii) putative H+ counter- transport by encapsulating the pH probe pyranine (λex= 450nm; λem= 515 nm); iii) transmembrane membrane potential by encapsulating the membrane potential probe Oxonol VI (λex= 580 nm; λem= 660 nm). (b) Real-time fluorescent traces detecting Cu(I) transport in wt EcCopA proteoliposomes with CTAP-3 (20 μM) encapsulated in the lumen as a function of increasing Cu(I) concentrations (0–20 μM). Differential fluorescence at time t (Ft − F0) was normalized to fluorescence at t = 0 s before Cu(I) addition (F0) and corrected for the signals from control liposomes. (c) Michaelis−Menten type analysis fitting the maximum F change (ΔF/F0) as a function of Cu(I) concentrations (KM, Cu(I)= 0.40 ± 0.10 μM and (ΔF/F0)Max= 0.11 ± 0.01). Inset: Maximal initial Cu(I) transport rates in EcCopA proteoliposomes and corresponding fit with a Michaelis−Menten equation: (δF/δt) = (δF/δt)Max × [Cu(I)]/(KM+ [Cu(I)]). Values obtained are: KM, Cu(I)= 0.15 ± 0.07 μM and (δF/δt)Max= (2.2 ± 0.2) 10−3 s–1.
Purified EcCopA in DDM micelles was reconstituted in unilamellar liposomes via freeze-thaw cycles and subsequent extrusion through 200 nm filters. Membrane incorporation was achieved via detergent-based liposome destabilization coupled to detergent removal by incubation with Biobeads. EcCopA incorporation was quantified by SDS-PAGE through band-intensity analysis upon liposome separation by ultracentrifugation, followed by EcCopA quantification in the soluble and proteoliposome fractions (Figure 2d). The results revealed >95% incorporation in the generated proteoliposomes. Size distribution and polydispersity indexes of control liposomes and EcCopA proteoliposomes were determined by dynamic light scattering. Monodisperse size distributions (PDI: 0.164 and 0.197, respectively) and average diameters of 141 ± 55 nm and 163 ± 52 nm, respectively were obtained (Figure 2c). These data are consistent with optimal EcCopA reconstitution efficiencies corresponding to the generation of protein-free and protein-embedded SUVs. Based on the protein and lipid concentration (1:25 protein-to-lipids ratio, w/w), and the size of the EcCopA proteoliposomes, we estimated that each vesicle (SUV) contains an average of ~75 EcCopA molecules.
To verify the EcCopA functionality upon incorporation in proteoliposome the Cu(I)-dependent stimulation of ATPase activity was determined by quantification of released inorganic phosphate generated by catalytic ATP-hydrolysis turnover.
In agreement with the functional reconstitution of wt EcCopA proteoliposomes an apparent hyperbolic Michaelis–Menten-like Cu(I)-dependent ATPase activity was obtained (Figure 3b). The orientation of EcCopA insertion cannot be controlled in the reconstitution procedure51. Thus, the catalytic parameters were calculated based on a statistically-controlled random orientation in the artificial lipid bilayers and corrected for the samples extraluminal volume (ATP, ADP and inorganic phosphate are impermeable to the lipid bilayer due to their negative charge). An apparent KM,Cu(I) of 0.25 ± 0.05 μM, and Vmax of 13.55 ± 0.05 nmol (mg min)−1 were obtained. The reconstitution in lipid environment appears to better stabilize the protein resulting in higher catalytic activities compared to detergent micelles. Similar analysis on the D523A or C479S_C481S_M813A mutants confirmed complete inactivation of Cu(I)-stimulated ATPase activity validating the results obtained in detergent micelles (Figure 3b). We postulated that Cu(I) translocation in the lumen during the assays could result in the formation of a transmembrane potential that would counteract Cu(I) transport and thus result in attenuated apparent ATP hydrolysis rates during the timeframe of the measurements. To obtain an estimate for the maximal initial Cu(I) translocation velocity, we determined apparent ATPase rates as a function of incubation time at saturating Cu(I) concentrations (Figure 3c). In agreement with above hypothesis, the apparent specific ATPase rates decayed as a function of the incubation time. Fitting of the time-dependent data revealed a maximal specific initial ATPase rate of approximately 150 nmol mg−1 min−1, which is in agreement with previous specific activities (and KM,Cu(I)) determined in this and other prokaryotic CopA proteins (e.g.: CopA from E. coli determined in four independent studies: Vmax= 190 nmol mg−1 min−1, KM,Cu(I) = 1.5 μM/Vmax= 125 nmol mg−1 min−1, KM,Cu(I) = 5.4 μM /Vmax= 620 nmol mg−1 min−1, KM,Cu(I) = 5.4 μM/ Vmax= 27 nmol mg−1 min−1, KM,Cu(I) = 1.5 μM; CopA from Archeoglobus Fulgidus: Vmax= 1000 nmol mg−1 min−1, KM,Cu(I) = 2.7 μM; CopA1 from Pseudomonas aeruginosa: Vmax= 86 nmol mg−1 min−1, KM,Cu(I) = 26.2 μM; CopA from Legionella pneumophila: Vmax= 17.5 nmol mg−1 min−1, KM,Cu(I) = 0.6 μM)25, 41, 45, 47, 52–54.
Real-time fluorescence detection of EcCopA Cu(I) translocation events with CTAP-3
Characterization of transmembrane translocation kinetics and mechanism of transport in Cu(I) transporters have been so far limited due to the inherent challenges of generating functional proteoliposomes where Cu(I) translocation can be monitored in real-time and the lack of Cu(I)-responsive fluorescent probes with binding affinities and dynamic ranges that could be utilized in lipid-rich solutions. For these reasons, previous analysis of Cu(I) transport in CopA relied on the determination of final Cu(I) accumulated in giant unilamellar vesicles by ICP-MS, which prevented real-time detection of Cu(I) translocation.
Searching for a fluorescent probe that retains a large dynamic range when encapsulated in liposomes, we selected CTAP-3 to study EcCopA-mediated Cu(I) transport in SUVs50 CTAP-3 is a water-soluble Cu(I)-selective fluorescent probe that is membrane impermeant. Due to its large fluorescence contrast and quantum yield upon Cu(I) saturation, the probe offers a limit of detection in the sub-part-per-trillion range.50. Functionalized with two negatively charged sulfonate groups, CTAP-3 interacts only weakly with lipid bilayers and maintains a high contrast ratio when encapsulated in proteoliposomes, even in the presence of adventitious copper contaminations that are expected to reduce the apparent turn-on response.
To study EcCopA ATP-dependent catalytic Cu(I) transmembrane translocation across lipid bilayers, CTAP-3 was encapsulated in control and EcCopA SUV lumen by freeze-thaw membrane fracture. Control and EcCopA proteoliposomes were sequentially supplemented with ATP and Mg2+ (required for ATP binding and hydrolysis in the N-domain) and exposed to buffered, oxygen-free solutions containing Cu(I) in the presence of ascorbate (prepared under inert atmosphere in an anaerobic glovebox). Cu(I) fluxes were monitored by fluorescence upon rapid mixing with the proteoliposomes at the CTAP-3-Cu(I) emission maximum (λexc= 365 nm; λem= 450nm). Kinetic traces in EcCopA proteoliposomes revealed a time-dependent development of the fluorescence signal, which reached saturation in 4–6 min indicative of real-time detection of CopA-mediated Cu(I) translocation across the proteoliposome lipid bilayer (Figure 4b).
Recordings of ΔF/F0 rose exponentially as a function of Cu(I) concentrations (ranging from 0 to 20 μM) upon rapid mixing of EcCopA proteoliposomes with extravesicular Cu(I). Control experiments conducted on the same proteoliposomes in the absence of Mg2+ or ATP did not elicit significant fluorescence response. The net Cu(I) transport into CopA proteoliposomes was estimated as ΔF/F0 as a function of time, with F0 the background fluorescence recorded before Cu(I) addition. Subtractions for any background leakage in control liposomes exposed to Cu(I) and ATP concentrations were performed, and background ion permeation in control liposomes was negligible. We determined initial transport rates (δF/δt) via exponential fitting of the Cu(I) transport traces (Figure 4c). Analysis of the initial velocities as a function of Cu(I) concentrations revealed a hyperbolic dependency in agreement with saturation-dependent Cu(I) transport (i.e. transporter-mediated). The curve was fitted with a Michaelis-Menten-like equation resulting in KM,Cu(I) = 0.15 ± 0.07 μM and a (δF/δt)MAX = 0.0022 ± 0.0002 s−1 (Figure 4c inset). The ΔF/F0 values at equilibrium were also analyzed as a function of Cu(I) concentrations revealing a similar hyperbolic dependency and apparent KM,Cu(I) =0.40 ± 0.10 μM and (ΔF/F0)MAX = 0.11 ± 0.01 s−1 (Figure 4c). By analyzing the transport traces at saturating Cu(I) concentrations (10 μM) and extrapolating the fluorescence response, we estimated the maximal apparent initial velocity. We determined the CopA concentrations in the CTAP-3-encapsulated proteoliposomes by SDS-PAGE using band densitometry analysis. We subsequently calibrated the CTAP-3 turn-on response (ΔF/F0) under the experimental condition used for transport assays by encapsulating known amounts of CTAP-3-Cu(I) complexes in the liposome lumen (Figure S2). This allowed for estimating the maximal initial Cu(I) transport rate at v ≃ 165 nmol Cu(I) mg–1 min–1, which corresponds to ≃ 15 ions/min per CopA molecule. The estimated maximal translocation rate is consistent with a Cu(I) translocation mechanism involving conformational changes and is in agreement with rates expected for primary-active ATP-driven transport systems55. By determining the ratio between the Cu(I) transport and the ATP hydrolysis rates we obtained a value of 1.1, which is consistent with translocation of 1 Cu(I) per ATP hydrolysis cycle, in agreement with previous studies47.
Real-time fluorescence detection of counter-ion transport using pyranine
Extensive biochemical and structural studies of the SERCA Ca2+-pump has revealed a strict requirement of proton counter-transport for ion substrate translocation. In SERCA1a the vectorial translocation of 2 Ca2+ per ATP is coupled to counter-transport of 2–3 H+, where the stoichiometry varies with pH56. Thus, SERCA act as a Ca2+/H+ antiporter24.
Structural investigations and solid supported membranes (SSM) experiments on pH-dependency, charge transfer, and mutations elucidated the existence of “H+-gated pathway” in SERCA allowing luminal Ca2+ release by Ca2+/H+ exchange56–58.
However, ATP-dependent charge transfer experiments on SSM for the human Cu(I) ATPases (ATP7A/ATP7B) and SERCA indicated distinctive features of catalytic and transport mechanisms. No variation of net charge transfer upon pH changes was observed in Cu(I) ATPases and no evidence of Cu+/H+ exchange required for copper release was evident. Thus, mechanistic differences in the catalytic cycles between Cu(I)-pumps and SERCA are plausible59, 60.
However, whether Cu(I)-pumps requires proton counter-transport to complete the catalytic cycle and allow the E2-E1 transition remains to be determined.
We analyzed putative H+ counter-transport in EcCopA proteoliposomes by encapsulating the fluorescent pH indicator pyranine. Pyranine is a water-soluble, membrane-impermeable arylsulfonate derivative (8-hydroxy-1,3,6-pyrenetrisulfonate) whose fluorescence intensity (at 515 nm) depends on the protonation state of the 8-hydroxyl group (pKa ≃ 7.2)48. Pyranine is thus a pH indicator that can be incorporated in the lumen of proteoliposomes as it does not bind to lipidic membranes as a consequence of its negative charge. Deprotonation of the hydroxyl group at increasing pH results in a significant increase of the fluorescence. Thus, when encapsulated in a proteoliposome lumen, the time-dependent fluorescence increase/decrease is reflective of proton efflux or influx, respectively.
Pyranine was encapsulated in the lumen of control and EcCopA SUVs by freeze-thaw membrane fracture. The SUVs were supplemented with Mg2+ and ATP, and Cu(I) substrate translocation was initiated by rapid mixing with buffered solutions containing Cu(I) as described for CTAP-3 experiments. Fluorescence traces were recorded at the pyranine emission maximum (λexc= 450 nm; λem= 515 nm) in similar timeframes utilized to study Cu(I) translocation with CTAP-3.
Analysis of the kinetic traces revealed no detectable change in pyranine fluorescence, and thus pH, in EcCopA proteoliposomes supplemented with ATP, Mg2+ and Cu(I) (Figure 5a). Traces were identical to the one obtained in control liposomes as well as to the one obtained with the inactive D523A or C479S_C481S_M813A mutants which in turn show a complete abolishment of Cu(I) transport activity (Figure 6a–b). The ability of this approach to detect small changes in luminal pH was demonstrated under similar buffering capacity (20 mM MOPS) experimental conditions for the bacterial Ca2+/H+ pump LMCA, for which ATP and Ca2+-dependent proton counter-transport was unambiguously detected61. Thus, these measurements reveal that CopA is not a Cu(I)/H+ antiporter, a key feature that differentiates Cu(I)-pumps form other well characterized P-type classes as SERCA or PMCA.
Figure 5:
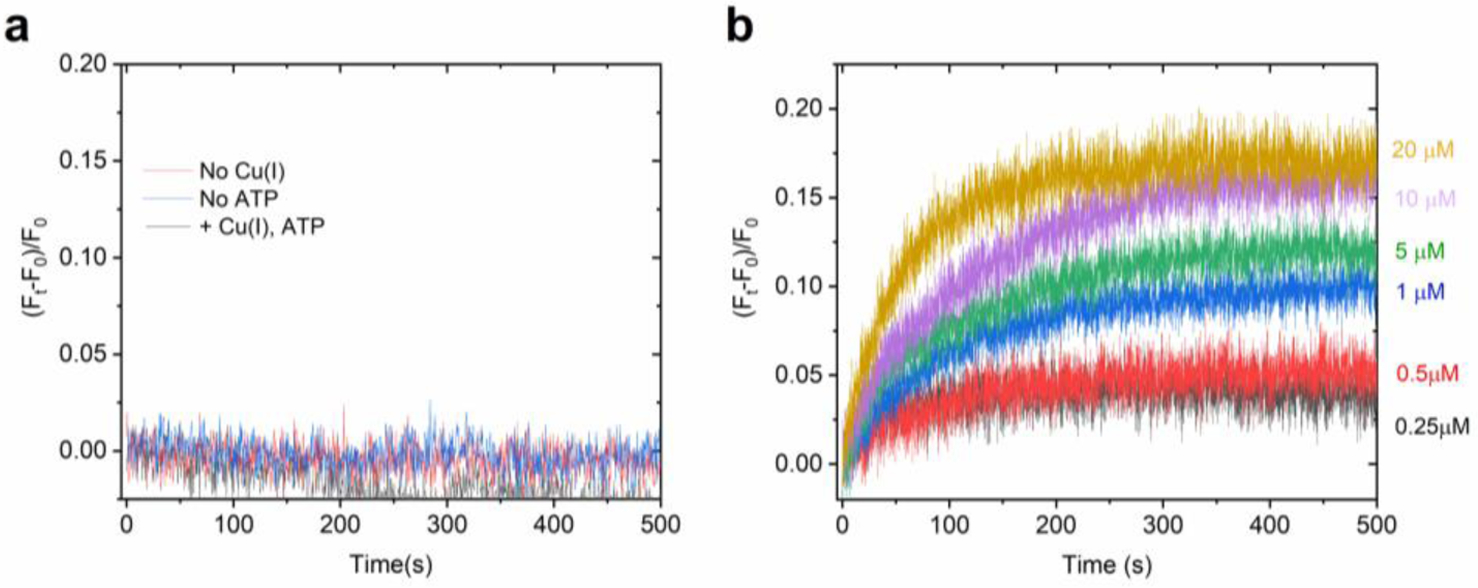
Investigation of H+ counter transport and electrogenicity in wt EcCopA proteoliposomes. (a) Determination of H+ counter-ion transport in wt EcCopA proteoliposome by monitoring changes in lumenal pH as a function of time with the pH-indicator pyranine under conditions for Cu(I) transport (Cu(I) = 10 μM). Negative control traces were recorded in the absence of Cu(I) or ATP. Differential fluorescence at time t, (Ft − F0)/F0, was normalized to the fluorescence before Cu(I) or ATP addition and corrected for the signals in control liposomes. (b) Real-time measurements of transmembrane potential generation in wt EcCopA proteoliposomes, under conditions of Cu(I) transport, at increasing Cu(I) concentrations (0–20 μM) measured upon encapsulating the membrane potential probe Oxonol VI in the proteoliposome lumen. Data are mean ± s.d. (n=2).
Figure 6:
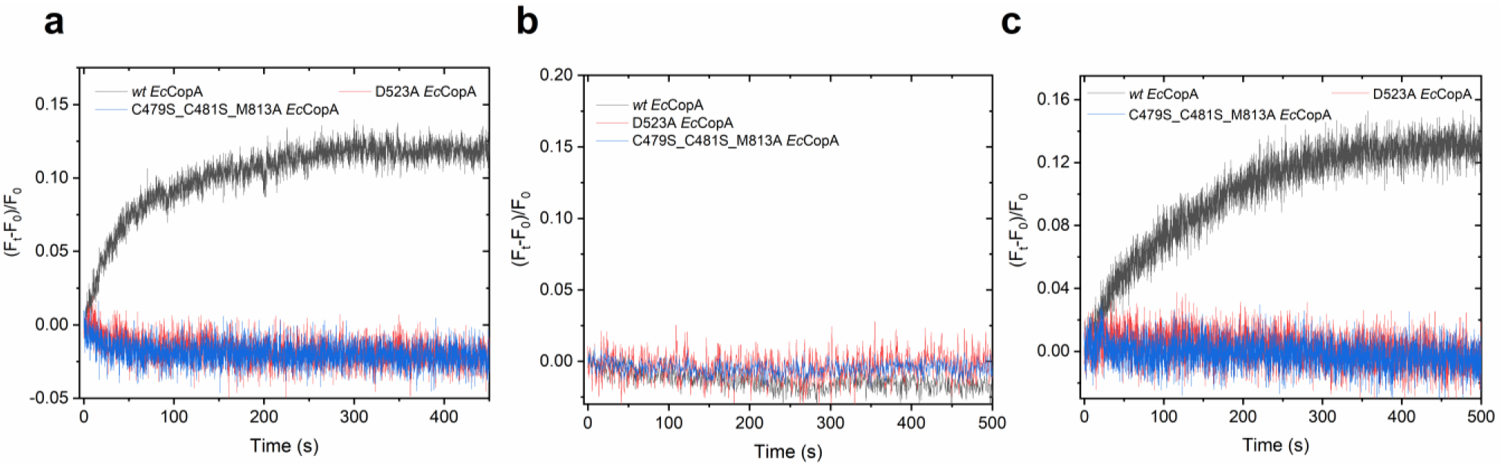
Real-time Cu(I) transport, H+ counter-transport and membrane potential development at saturating Cu(I) concentrations (10 μM) in wt EcCopA and mutants. (a) Real-time Cu(I) transport by wt EcCopA (black), D523A EcCopA (red) and C479S_C481S_M813A EcCopA (blue) measured by encapsulating CTAP-3 in the proteoliposome lumen. (b) H+ counter-ion transport by wt EcCopA (black), D523A EcCopA (red) and C479S_C481S_M813A EcCopA (blue) measured by encapsulating pyranine in the proteoliposome lumen (c) Development of membrane potential upon Cu(I) translocation in wt EcCopA (black), D523A EcCopA (red) and C479S_C481S_M813A EcCopA (blue) measured by encapsulating Oxonol VI in the proteoliposome lumen. Data are mean ± s.d. (n=2)
Real-time fluorescence detection of electrogenicity using oxonol VI
Our analysis demonstrated that CopA catalyzes energy-dependent Cu(I) vectorial translocation that is not coupled to proton counter-transport. The close conservation of critical residues in the ion translocation pathway with LpCopA39, 41 and our kinetic analysis indicates the existence of a single high-affinity transmembrane metal binding site in the E1-PCu(I) state that allows the translocation of one 1 Cu(I) per ATP hydrolysis in each E1–E2 cycle. We thus hypothesized that, since CopA does not require proton antiport, it must translocate a net positive charge resulting in electrogenic transport, unless Na+ (which is present in the transport buffer) would be counter-transported. To test this hypothesis, we performed real-time measurement of transmembrane potential in EcCopA proteoliposomes (Figure 5b). Oxonol VI (bis-(3-propyl-5-oxoisoxazol-4-yl) pentamethine oxonol) is a slow-response fluorescent membrane-potential probe that shows voltage-dependent partitioning between the aqueous phase and lipid bilayers. This partitioning results in a change of the relative fluorescence intensity. Oxonol VI responds to changes in potential more rapidly than other slow-response oxonols (e.g oxonol V) and is thus a better probe for measuring faster potential changes49, 62. Consequently, it can be utilized to study ion channels and electrogenic pump activity in cells and liposomes.
We encapsulated Oxonol VI in the EcCopA proteoliposome lumen and monitored changes in transmembrane potential upon Cu(I) transport by fluorescence. The CopA and control SUVs were supplemented with Mg2+ and ATP, and Cu(I) substrate translocation initiated by rapid mixing with buffered solutions containing Cu(I) (Cu(I)= 0–20 μM) under the same conditions utilized for Cu(I) transport recordings with CTAP-3. Analysis of the fluorescence kinetic traces at oxonol VI emission maximum (λexc= 580 nm; λem = 660 nm) revealed a concentration and time-dependent fluorescence increase (Figure 5b).
As the formation of a positive-inside membrane potential would result in an increase in the oxonol VI fluorescence signal we concluded that EcCopA catalyzed the net transfer of positive charges from solution into the proteoliposome lumen. Moreover, the traces recorded in proteoliposomes containing oxonol VI paralleled in timescales the corresponding CTAP-3 turn-on signal, indicating that the net translocation of positive charges correlates to the active translocation of Cu(I). As ATP is negatively charged at pH=7, and thus membrane-impermeable, only the CopA molecules possessing the N- and P- domain-oriented opposite to the lumen could be activated for Cu(I) transport.
The observed development in CopA proteoliposomes of a positive-inside potential that is dependent on the simultaneous presence of ATP, Mg2+ and Cu(I) clearly indicates that CopA is an electrogenic primary-active pump. These results were also validated by the analysis of the inactive D523A or C479S_C481S_M813A mutants under the same conditions. The development of oxonol VI fluorescence signal was completely abolished, confirming that no electrogenic transport occurred upon protein inactivation (Figure 6c).
Our multichannel platform demonstrates the CopA transports Cu(I) across the lipid bilayer and does not catalyze Cu(I)/H+ exchange, in agreement to the pyranine data. The existence of a single transmembrane metal binding site responsible for translocation must result in the generation of a positive inside membrane potential in the transport process, as validated by our oxonol VI analysis. Thus, overall our data conclude that CopA is a transmembrane electrogenic Cu(I) uniporter.
This work reveals unique features in Cu(I) pumps compared to other characterized P-type ATPases (Figure 7). In zinc pumps, also belonging to the P1B-type subfamily of transition metal pumps, the absence of proton counter-transport was demonstrated. However, structural analysis of ZntA from Shigella Sonnei in its phosphoenzyme ground state (E2P) and a dephosphorylation intermediate (E2.Pi) displayed, in the E2P state, a wide extracellular release pathway from the high-affinity site. This release conduit closes in the E2.Pi state, in which the metal binding residue (D714) interacts with a conserved residue (K693), acting as a built-in counter ion and stimulating Zn(II) release, as proposed for H+-ATPases. This built-in counter-ion appears responsible in preventing proton counter-transport and stabilize an occluded E2.Pi form. Thus, a mechanistic link between Zn(II)-ATPases and PIII-type H+-ATPases was proposed. However, at the same time, structural features of the release pathway also resemble PII-type ATPases such as the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) and Na+, K+-ATPase57, 63. In the latter, counterion binding (H+ and K+, respectively) result in occlusion in the E2P to E2.Pi transition. The metal substrate release mechanism is different in Cu(I) and Zn(II) pumps. The SsZntA built-in counterion residues are not conserved in CopA and a different mechanism should be in place to prevent H+ back transport in Cu(I) pumps as we demonstrated in our work. While the SsZntA structure in the E2P state shows a wide opening (as in SERCA), the exit conduit closes in the E2.Pi state61. The internal salt bridge allows no apparent need for counter-ion transport for completing the cycle. The LpCopA structure in the E2P state instead does not show a wide release pathway, and no change in the exit channel occurs in the E2P to E2.Pi transition40. A different release mechanism in Cu(I) pumps is likely as Cu(I) is transferred to acceptors to prevent its release in free form, in light of its redox nature. Thus, while Zn(II) release in the E2 state can be achieved through the wide exit channel, in Cu(I) pumps metal release is mediated by a narrow conduit that allows consecutive ligand exchange reactions. This might prevent the need of counter-ion translocation as we demonstrated in our study to close the exit conduit leading to completion of the catalytic cycle, and at the same time avoid the requirement of a built-in counter-ion. Thus, modalities of transport unique for Cu(I) pumps can be proposed.
Figure 7:
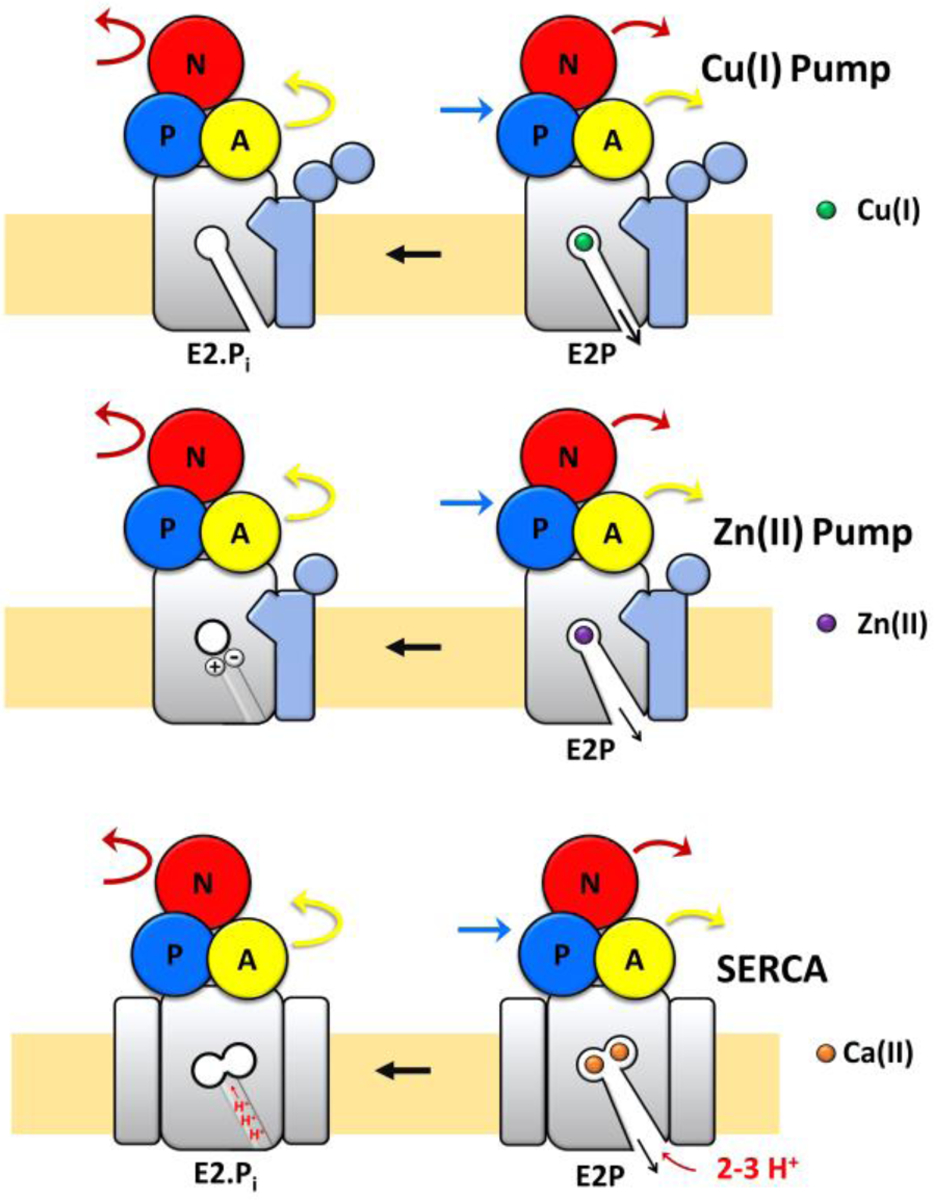
Differences in metal release and counterion transport in Cu(I), Zn(II) and Ca(II) (SERCA) P-type ATPases. In Cu(I)-pumps, substrate release from the high-affinity transmembrane coordinating residues occurs through a narrow conduit in the E2 state involving a series of ligand exchange reactions and no proton counter-transport. No major movements in the transmembrane helices occurs in the E2P to E2.Pi transition. In Zn(II)-pumps metal release state occurs in the E2 state through a wide-open conduit. The exit channel closes in E2P to E2.Pi transition into an occluded state with formation of a built-in counter-ion (between conserved Asp and Lys residues) and no proton counter-transport. In SERCA, Ca2+ substrate release takes place through a wide-open conduit in the E2 state and consequent re-occlusion in E2P to E2.Pi transition is coupled with proton counter-transport.
Conclusions
In this work, we developed a platform based on diverse fluorescent probes responsive to multiple stimuli to study metal substrate translocation and mechanism of transport in real-time in Cu(I) P-type ATPases. The strategy allowed us to study the transport properties of EcCopA in a native-like lipid-bilayer environment.
Our analysis revealed unique features in the mechanism of transport by CopA. CopA is an electrogenic uniporter in which Cu(I) translocation events are not coupled to counterion transport. By translocating a net positive charge per ATP hydrolysis cycle CopA is capable of generating a transmembrane potential. Overall, the work reveals distinct transport features in Cu(I) P-type ATPase pumps and mechanistic differences with other better characterized P-type ATPases.
The developed platform opens the avenue to study translocation events and mechanism of transport in real-time in other less-characterized transition metal P-type pumps that catalyze the transport of other essential and toxic transition metals.
Experimental
Expression and purification of wild-type CopA and mutants
For recombinant expression, the synthetic DNA (Genscript Inc.) encoding codon-optimized EcCopA (Uniprot accession number: Q59385) was cloned into a pET-52b(+) vector with an N-terminal STREP-tag II.
The expression plasmid was transformed into E. coli BL21(DE3) GOLD competent cells (Agilent Technologies). Cells were grown in Terrific Broth (TB) media supplemented with 1% glycerol (v/v) and ampicillin (50 μg mL−1). The overnight preculture was inoculated in fresh TB media and cells were grown at 37 °C under agitation until they reached OD600=2. Cells were cooled to 25 °C, and protein expression was induced by adding isopropyl thiogalactopyranoside (IPTG) to a final concentration of 0.3 mM. Cell cultures were subsequently incubated at 25 °C, under agitation for 18 h.
Cells were harvested by centrifugation (20 min, 4 °C, 14,000 xg; Thermo Scientific Sorvall LYNX 6000 centrifuge) and resuspended in lysis buffer (20 mM Tris/HCl pH 8, 150 mM NaCl, 5 mM MgCl2, 30 μg/mL deoxyribonuclease I from bovine pancreas (Sigma-Aldrich), supplemented with 2x EDTA-free protease inhibitor cocktail tablets (Thermo Scientific)). Cells were lysed in an ice-cold microfluidizer at 20000 psi by circulating three-times the cell suspension through a Z-shaped diamond chamber (Microfluidics M-110P). Cell debris was removed by centrifugation (20 min, 4 °C, 20,000 × g; Thermo Scientific Sorvall LYNX 6000 centrifuge). The membrane fraction was isolated by ultracentrifugation (1 h, 4 °C, 205,100 xg, Beckman Optima XPN80).
The membrane pellet was resuspended in buffer (20 mM Tris/HCl, pH 8, 500 mM NaCl, 1% (w/v) glycerol, EDTA-free protease inhibitor cocktail) to a final concentration of 1 g of cells mL−1. Membrane suspensions were flash-frozen in liquid nitrogen and stored at −80 °C.
All mutants (D523A EcCopA and C479S_C481S_M813A EcCopA) were generated by site-directed mutagenesis (Genscript Inc.), expressed and purified as described for wild-type EcCopA.
wtCopA and mutants were purified by affinity chromatography via the N-term STREP-tag II using a 5 mL StrepTrap affinity column (GE Healthcare).
In a typical purification, protein extraction was achieved by supplementing the membrane suspension (5 mL) with n-dodecyl-ß-D-maltoside (DDM) (Anatrace) in an ice-chilled extraction buffer (20 mM Tris/HCl, pH 8, 500 mM NaCl, 1 mM EDTA, 5 mM β-mercaptoethanol, 1% (w/v) DDM and EDTA-free protease inhibitor cocktail (Thermo Scientific)). The obtained suspension was vigorously stirred at 4 °C for 1 hour. Residual membranes and precipitated proteins were removed by ultracentrifugation (20 min, 4 °C, 205,100 xg; Beckman Optima XPN80). The supernatant containing detergent-solubilized EcCopA was loaded onto a pre-equilibrated StrepTrap column in wash buffer (20 mM Tris/HCl, pH 8, 500 mM NaCl, 1 mM EDTA, pH 8, 1 mM dithiothreitol (DTT), 0.05% (w/v) DDM) using an AKTA Pure FPLC system (GE Health care). The column was washed with 20 CV buffer. EcCopA bound to StrepTrap column was eluted in 6CV buffer (30 ml) containing D-desthiobiotin (20 mM Tris/HCl, pH 8, 500 mM NaCl, 1 mM EDTA, pH 8, 1 mM dithiothreitol (DTT), 0.05% (w/v) DDM and 2.5 mM D-desthiobiotin). The buffer was immediately exchanged using an HiPrep 26/10 desalting column (GE Healthcare) to desalting buffer (20 mM MOPS/NaOH, pH 7, 500 mM NaCl, 1 mM ascorbic acid, 0.05% (w/v) DDM). Purified protein was concentrated using 100,000 MWCO concentrators (Sartorious VIVASPIN 20) by centrifugation (2100 × g, 4 °C; Thermo scientific Sorvall ST8 centrifuge). Aggregated protein or impurities were removed by size exclusion chromatography on a Superdex 200 10/300 column (GE Healthcare) using desalting buffer. Protein purity was verified by SDS-PAGE (4–15% Tris-Glycine Mini-PROTEAN gels, BioRad). Purified protein was concentrated to ~ 6 mg mL−1 and concentration was determined by Abs280 (ε=70275 M−1 cm−1) or SDS-PAGE band densitometry analysis. Final purified protein was reconstituted in proteoliposomes without freezing.
Reconstitution of wtCopA and mutants in small unilamellar vesicle (SUVs) proteoliposomes
E. coli polar lipid extract and L-α-phosphatidylcholine (from chicken egg) were purchased from Avanti Polar Lipids. Purified wtCopA and mutants were reconstituted at a protein-to-phospholipid ratio of 1:25 (w/w). E. coli polar lipids and L-α-phosphatidylcholine dissolved in chloroform were mixed (3:1 ratio, w/w) in a pear-shaped flask and dried under nitrogen flow and continuous rotation to obtain a thin lipid film. Lipids were stored in a vacuum desiccator overnight to completely remove chloroform. The following day the lipid film was hydrated in MilliQ water containing 1 mM ascorbic acid (treated with Chelex resin to remove metal contaminations). The lipid suspension was buffered with 20 mM MOPS/NaOH, pH 7, 100 mM NaCl, 1 mM ascorbic acid, supplied from a concentrated Chelex-treated buffer stock, to a final lipid concentration of 25 mg mL−1.
For small unilamellar vesicle (SUVs) preparation, the lipid suspension was subjected to three freeze-thaw cycles in LN2 followed by 11 extrusions through polyether sulfone (PES) membranes with a 1 mL gas-tight syringe system (Avanti, Polar Lipids, Inc). PES membranes in sequential decreasing pore sizes (1 μm, 400 nm, and 200 nm) were used. SUVs were destabilized by DDM addition to final 0.02% (w/v), and tilted for 1 h, 25°C.
To reconstitute purified wt EcCopA and mutants in liposomes, a concentrated protein stock (~ 6 mg/mL) was added to the detergent-destabilized liposomes to a final ratio of 1:25 (w/w) protein:lipid, on ice. Control liposomes were generated with the same procedure using the corresponding buffer in place of protein stocks.
Mixtures were tilted at 4 °C for 1 h. Detergent was removed by addition of Bio-Beads (SM-2; Bio-Rad). Biobeads were first activated by sequential washing with methanol, ethanol and H2O. Beads were collected by filtration and added to the liposome solution to obtain a slurry (40 mg mL−1). Proteoliposomes and control liposomes were maintained under tilting (4 °C) and the detergent was completely removed via Bio-beads exchange after 1, 2, 16, and 18 h. Proteoliposomes and control liposomes were collected by ultracentrifugation (160,000 × g, 4 °C, 45 min; Sorvall mX120+ Micro-Ultracentrifuge). SUVs pellets were collected and resuspended in 20 mM MOPS/NaOH, pH 7, 100 mM NaCl, 1 mM ascorbic acid (treated with Chelex) to a final lipid concentration of 25 mg mL−1 (EcCopA =1 mg mL−1). Protein incorporation in proteoliposomes was determined by SDS-PAGE analysis of supernatant and the resuspended pellets after ultra-centrifugation (4–15% Tris-Glycine Mini-PROTEAN gels, BioRad). The proteoliposomes and control liposomes were stored at −80 °C after flash-freezing with LN2, until used for the activity and transport assays. The average number of EcCopA molecules per SUV was determined as described previously51.
Determination of specific ATPase activity of detergent- solubilized wt EcCopA protein and mutants.
All solutions were prepared in Chelex-treated miliQ water. CopA and mutants (34.4 μL) were placed in a 96-well plate and mixed with 1M MgCl2 (0.4 μL, 10 mM final concentration) and 100 mM ascorbic acid (0.4 μL, 1 mM final concentration). A CuCl2 stock (5 μM- 2 mM) was added to generate a final Cu(I) concentration series (0.05 μM- 20 μM). The reaction was initiated by adding 10 mM ATP (4 μL, 1mM final concentration). The reaction mixture was incubated at 37 °C for 20 minutes, with shaking (350 rpm). Released inorganic Pi was quantified with the Malachite green-phosphate assay kit (Sigma-Aldrich MAK307). Malachite green reagent was added to all reaction mixtures. The solutions were incubated for 10 minutes at RT for color development. The absorbance at 620 nm was measured using a Tecan Spark 20M plate reader. Controls were performed in the absence of MgCl2. The generated inorganic phosphate was calculated using a standard curve. The specific activity was calculated as nmol Pi mg−1 min−1.
Determination of specific ATPase activity of wtCopA and mutants in proteoliposomes.
Proteoliposomes and control liposomes were extruded sequentially through 1 μM, 0.4 μM and 0.2 μM membrane filters using gas-tight syringes prior to ATPase activity assay. Reaction mixtures were prepared in Eppendorf tubes similarly to detergent solubilized protein. Samples were incubated at 37 °C, 20 min, 350 rpm. After reaction completion, the mixtures were centrifuged (160,000 xg, 3 minutes, 4 °C, Sorvall mX120+ Micro-Ultracentrifuge) to pellet any aggregated lipid. The supernatant was placed in a 96-well plate and the Malachite green solution was added. The absorbance at 620nm was measured after 10 min as described above.
Determination of time-dependent ATPase activity assays.
ATPase activity assays were carried out as above by incubating proteoliposomes and control liposomes with the reaction mixtures in different time intervals (1 min – 30 min). Released Pi was determined with Malachite green-phosphate assay kit and specific activity determined as described above.
Real-time determination of Cu(I) transport using CTAP-3.
CTAP-3 was synthetized as described previously50. Proteoliposomes and control liposomes were diluted to a final lipid concentration of 12.5 mg mL−1 in 20 mM MOPS/NaOH pH=7.0, 100 mM NaCl, 1 mM ascorbate. A CTAP-3 stock (10 mM in H2O) was added to final concentration 20 μM. Dye encapsulation in the proteoliposomes lumen was obtained by 3 freeze-thaw cycles. Proteoliposomes were sequentially extruded through 1 μM, 0.4 μM and 0.2 μM filters using gas-tight syringes. Proteoliposome pellets were collected by ultra-centrifugation (160,000 xg, 45 minutes, 4 °C; Sorvall Mx 120+ micro-ultracentrifuge) and the supernatant containing excess CTAP-3 removed. CTAP-3 encapsulated proteoliposomes were washed in the same buffer (20 mM MOPS/NaOH pH=7.0, 100 mM NaCl, 1 mM ascorbate), collected by an additional ultra-centrifugation step and resuspended in buffer.
Buffer solutions (20 mM MOPS/NaOH pH=7.0, 100 mM NaCl and 1 mM ascorbate), were made oxygen-free on a Schlenk-line by three vacuum/nitrogen cycles. Copper stock solutions were freshly prepared prior to each experiment in an anaerobic glove box purged with constant N2 flow. To prepare the Cu(I) stock dilution series, a 100 mM solution was generated by dissolving [Cu(CH3CN)4]PF6 in 100% acetonitrile. 10x stock solutions were prepared by serial dilution in transport buffer, such that the final CH3CN in stock solutions was less than 4%. The Cu(I) transport assays were conducted at 37 °C in a sub-micro quartz cell (Starna Cells) using a Fluoromax-4 spectrofluorometer (Horiba Scientific). Transport assays were performed in the presence of 1 mM ATP (100 mM stock) and 10 mM MgCl2 (1 M stock) on 106 μL samples. The reaction was initiated by addition of 12 μL of Cu(I) stocks (final concentration: 0.25 – 20 μM). The time-dependent fluorescence change was monitored for 500 s in 0.1 s intervals (λexc= 365 nm, slit width= 2.00 nm; λem= 450 nm, slit width= 2.00 nm). (F-F0)/F0 was calculated using the fluorescence before addition of Cu(I) as F0. Assays were also carried out in the absence of ATP and Mg2+. Transport assays were conducted for EcCopA mutants and control liposomes using the same procedure.
Calibration of CTAP-3 response.
Liposomes were diluted to a final lipid concentration of 12.5 mg mL–1. All solutions were made oxygen-free on a Schlenk-line by three vacuum/nitrogen cycles and the preparation steps conducted in an anaerobic glovebox. CTAP-3 was added to the liposomes to a final concentration of 20 μM. CTAP-3 containing liposomes were divided into 500 μL fractions. Freshly prepared Cu(I) stocks (100x) in 20 mM MOPS/NaOH (pH 7.0, 100 mM NaCl, 1mM ascorbate) were added in each tube to obtain CTAP-3-Cu(I) complexes with final concentrations up to 50 μM. The samples were removed from the anaerobic chamber and the CTAP-3-Cu(I) complexes were encapsulated in the liposomes by 3 freeze-thaw cycles followed by extrusion through 0.2 μm membrane filters. Liposomes were collected by ultracentrifugation (160,000 xg, 45 min, 4°C, Sorvall Mx 120+ micro ultracentrifuge) and the supernatant discarded. The liposome pellet was washed and suspended in the transport buffer. Fluorescence response was recorded at λexc= 365 nm and λem= 450 nm. The ΔF/F0 values were normalized for the fluorescence response of 20 μM CTAP-3 encapsulated in liposomes (in the absence of Cu(I)).
Determination of H+ counter-transport with pyranine.
wtCopA and mutant (D523A EcCopA and C479S_C481S_M813A EcCopA) proteoliposomes were collected by ultracentrifugation (160,000 xg, 45 min, 4 °C; Sorvall Mx 120+ micro-ultracentrifuge) and the pellets resuspended in buffer containing 10 mM MOPS/NaOH pH 7, 10 mM MgCl2, 100 mM NaCl, 1 mM ascorbate. The fluorescent pH-indicator, pyranine (Alfa Aesar), was added to final 1 mM and encapsulated in the proteoliposome lumen by 3 freeze-thaw cycles followed by sequential extrusion through 1 μM, 0.4 μM and 0.2 μM membrane filters using gas-tight syringes. Proteoliposome pellets were collected by ultra-centrifugation and the supernatant containing excess pyranine discarded.
Pellets were washed, collected by ultracentrifugation and resuspended in the same buffer for H+ counter-transport assays. Assays were conducted similarly to Cu(I) transport assays in the presence of 1 mM ATP and 10 mM MgCl2. The reaction was initiated by adding Cu(I) stocks to final concentration of 10 μM. The time-dependent fluorescence change (ΔF/F0) was recorded for 500 s in 0.5 s intervals (λexc= 450 nm, slit width= 1.00 nm; λem= 515 nm, slit width= 1.00 nm). Control experiments were performed in the absence of ATP or Cu(I).
Determination of electrogenicity with the membrane potential probe oxonol VI.
Wt EcCopA and mutants (D523A EcCopA and C479S_C481S_M813A EcCopA) proteoliposomes and control liposomes were diluted 1:2 (v/v) in buffer (20 mM MOPS/NaOH pH 7.0, 100 mM NaCl, 1 mM ascorbate). Oxonol VI was added to a final concentration of 5 μM. Encapsulation in the proteoliposome lumen was obtained via three freeze-thaw cycles followed by sequential extrusion through 1 μM, 0.4 μM and 0.2 μM membrane filters using gas-tight syringes. Proteoliposomes were collected by ultracentrifugation and excess oxonol VI in supernatant removed. Proteoliposome pellets were washed and resuspended in the same buffer.
The reactions were conducted in the presence of 1 mM ATP 10mM MgCl2. The reactions were initiated by adding Cu(I) stock solutions (12 μL) freshly prepared in an anaerobic glove box (Final Cu(I): 0.25 μM −20 μM). Mutants were analyzed at 10 μM Cu(I). The time-dependent fluorescence change (ΔF/F0) was recorded for 500s in 0.1 s intervals (λexc= 580 nm, slit width= 4.00 nm; λem= 660 nm, slit width= 5.00 nm).
Determination of SUVs size distribution of wtCopA and the mutants by Dynamic light scattering (DLS).
Native and CTAP-3-encapsulated proteoliposomes and control liposomes were prepared as described previously. Size distributions of vesicles were analyzed by UV-Vis dynamic light scattering (DLS) with Zeta sizer Nano ZS (Malvern Panalytical). Samples were placed in a disposable polystyrene cuvette and the measurements conducted at 25.0° C at a scattering angle of 175° and a laser wavelength of 633 nm (refractive index of sample medium=1.33 and refractive index of material= 1.51).
Supplementary Material
Acknowledgements
The work was supported by funds from The University of Texas at Dallas (G.M.), the Robert A. Welch Foundation (AT-1935-20170325 to G.M.) and by the National Institute of General Medical Sciences of the National Institutes of Health (R35GM128704 to G.M.; R01GM067169 and R35GM136404 to C.J.F). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Electronic Supplementary Information (ESI) available: [Supplementary Figures 1–2]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Ma Z, Jacobsen FE and Giedroc DP, Chem. Rev, 2009, 109, 4644–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boal AK and Rosenzweig AC, Chem. Rev, 2009, 109, 4760–4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arguello JM, Raimunda D and Gonzalez-Guerrero M, J. Biol. Chem, 2012, 287, 13510–13517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenzweig AC and Arguello JM, Curr. Top. Membr, 2012, 69, 113–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplan JH and Lutsenko S, J. Biol. Chem, 2009, 284, 25461–25465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barry AN, Shinde U and Lutsenko S, J. Biol. Inorg. Chem, 2010, 15, 47–59. [DOI] [PubMed] [Google Scholar]
- 7.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC and O’Halloran TV, Science, 1999, 284, 805–808. [DOI] [PubMed] [Google Scholar]
- 8.Lutsenko S, Barnes NL, Bartee MY and Dmitriev OY, Physiol. Rev, 2007, 87, 1011–1046. [DOI] [PubMed] [Google Scholar]
- 9.Lutsenko S, Metallomics, 2016, 8, 840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lutsenko S and Petris MJ, J. Membr. Biol, 2003, 191, 1–12. [DOI] [PubMed] [Google Scholar]
- 11.Lutsenko S, Washington-Hughes C, Ralle M and Schmidt K, J. Biol. Inorg. Chem, 2019, 24, 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt K, Ralle M, Schaffer T, Jayakanthan S, Bari B, Muchenditsi A and Lutsenko S, J. Biol. Chem, 2018, 293, 20085–20098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gualandi F, Sette E, Fortunato F, Bigoni S, De Grandis D, Scotton C, Selvatici R, Neri M, Incensi A, Liguori R, Storbeck M, Karakaya M, Simioni V, Squarzoni S, Timmerman V, Wirth B, Donadio V, Tugnoli V and Ferlini A, Neuromuscul. Disord, 2019, 29, 776–785. [DOI] [PubMed] [Google Scholar]
- 14.Tao C, Wang Y, Zhao Y, Pan J, Fan Y, Liang X, Cao C, Zhao J, Petris MJ, Li K and Wang Y, Diabetologia, 2019, 62, 2340–2353. [DOI] [PubMed] [Google Scholar]
- 15.Shanbhag V, Jasmer-McDonald K, Zhu S, Martin AL, Gudekar N, Khan A, Ladomersky E, Singh K, Weisman GA and Petris MJ, Proc. Natl. Acad. Sci. USA, 2019, 116, 6836–6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mercer SW, Wang J and Burke R, J. Biol. Chem, 2017, 292, 4113–4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhlbrandt W, Nat. Rev. Mol. Cell. Biol, 2004, 5, 282–295. [DOI] [PubMed] [Google Scholar]
- 18.Bublitz M, Morth JP and Nissen P, J. Cell. Sci, 2011, 124, 2515–2519. [DOI] [PubMed] [Google Scholar]
- 19.Moller JV, Olesen C, Winther AM and Nissen P, Q. Rev. Biophys, 2010, 43, 501–566. [DOI] [PubMed] [Google Scholar]
- 20.Toyoshima C, Biochim. Biophys. Acta, 2009, 1793, 941–946. [DOI] [PubMed] [Google Scholar]
- 21.Faxen K, Andersen JL, Gourdon P, Fedosova N, Morth JP, Nissen P and Moller JV, J. Biol. Chem, 2011, 286, 1609–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong D, Chi X, Ren K, Huang G, Zhou G, Yan N, Lei J and Zhou Q, Nat. Commun, 2018, 9, 3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abe K, Irie K, Nakanishi H, Suzuki H and Fujiyoshi Y, Nature, 2018, 556, 214–218. [DOI] [PubMed] [Google Scholar]
- 24.Dyla M, Kjaergaard M, Poulsen H and Nissen P, Annu. Rev. Biochem, 2019, 89. [DOI] [PubMed] [Google Scholar]
- 25.Mandal AK, Yang Y, Kertesz TM and Arguello JM, J. Biol. Chem, 2004, 279, 54802–54807. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez-Guerrero M, Eren E, Rawat S, Stemmler TL and Arguello JM, J. Biol. Chem, 2008, 283, 29753–29759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arguello JM, Gonzalez-Guerrero M and Raimunda D, Biochemistry, 2011, 50, 9940–9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sitsel O, Gronberg C, Autzen HE, Wang K, Meloni G, Nissen P and Gourdon P, Biochemistry, 2015, 54, 5673–5683. [DOI] [PubMed] [Google Scholar]
- 29.Banci L, Bertini I, Ciofi-Baffoni S, Huffman DL and O’Halloran TV, J. Biol. Chem, 2001, 276, 8415–8426. [DOI] [PubMed] [Google Scholar]
- 30.Banci L, Bertini I, Cantini F, DellaMalva N, Herrmann T, Rosato A and Wuthrich K, J. Biol. Chem, 2006, 281, 29141–29147. [DOI] [PubMed] [Google Scholar]
- 31.Yu CH, Lee W, Nokhrin S and Dmitriev OY, Sci. Rep, 2018, 8, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banci L, Bertini I, Ciofi-Baffoni S, Gonnelli L and Su XC, J. Mol. Biol, 2003, 331, 473–484. [DOI] [PubMed] [Google Scholar]
- 33.Allen GS, Wu CC, Cardozo T and Stokes DL, Structure, 2011, 19, 1219–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gourdon P, Sitsel O, Lykkegaard Karlsen J, Birk Moller L and Nissen P, Biol. Chem, 2012, 393, 205–216. [DOI] [PubMed] [Google Scholar]
- 35.Agarwal S, Hong D, Desai NK, Sazinsky MH, Arguello JM and Rosenzweig AC, Proteins, 2010, 78, 2450–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mana-Capelli S, Mandal AK and Arguello JM, J. Biol. Chem, 2003, 278, 40534–40541. [DOI] [PubMed] [Google Scholar]
- 37.Meloni G, Zhang L and Rees DC, ACS Chem. Biol, 2014, 9, 116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purohit R, Ross MO, Batelu S, Kusowski A, Stemmler TL, Hoffman BM and Rosenzweig AC, Proc. Natl. Acad. Sci. U S A, 2018, 115, 2108–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gourdon P, Liu XY, Skjorringe T, Morth JP, Moller LB, Pedersen BP and Nissen P, Nature, 2011, 475, 59–64. [DOI] [PubMed] [Google Scholar]
- 40.Andersson M, Mattle D, Sitsel O, Klymchuk T, Nielsen AM, Moller LB, White SH, Nissen P and Gourdon P, Nat. Struct. Mol. Biol, 2014, 21, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattle D, Zhang L, Sitsel O, Pedersen LT, Moncelli MR, Tadini-Buoninsegni F, Gourdon P, Rees DC, Nissen P and Meloni G, EMBO Rep, 2015, 16, 728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzalez-Guerrero M and Arguello JM, Proc. Natl. Acad. Sci. USA, 2008, 105, 5992–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mattle D, Sitsel O, Autzen HE, Meloni G, Gourdon P and Nissen P, J. Mol. Biol, 2013, 425, 2299–2308. [DOI] [PubMed] [Google Scholar]
- 44.Padilla-Benavides T, McCann CJ and Arguello JM, J. Biol. Chem, 2013, 288, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Padilla-Benavides T, George Thompson AM, McEvoy MM and Arguello JM, J. Biol. Chem, 2014, 289, 20492–20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toyoshima C, Arch. Biochem. Biophys, 2008, 476, 3–11. [DOI] [PubMed] [Google Scholar]
- 47.Wijekoon CJ, Udagedara SR, Knorr RL, Dimova R, Wedd AG and Xiao Z, J. Am. Chem. Soc, 2017, 139, 4266–4269. [DOI] [PubMed] [Google Scholar]
- 48.Clement NR and Gould JM, Biochemistry, 1981, 20, 1534–1538. [DOI] [PubMed] [Google Scholar]
- 49.Apell HJ and Bersch B, Biochim. Biophys. Acta, 1987, 903, 480–494. [DOI] [PubMed] [Google Scholar]
- 50.Morgan MT, McCallum A and Fahrni CJ, Chem. Sci, 2016, 7, 1468–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abeyrathna SS, Abeyrathna NS, Thai NK, Sarkar P, D’Arcy S and Meloni G, Biochemistry, 2019, 58, 4337–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fan B and Rosen BP, J Biol Chem, 2002, 277, 46987–46992. [DOI] [PubMed] [Google Scholar]
- 53.Gonzalez-Guerrero M, Raimunda D, Cheng X and Arguello JM, Mol. Microbiol, 2010, 78, 1246–1258. [DOI] [PubMed] [Google Scholar]
- 54.Drees SL, Beyer DF, Lenders-Lomscher C and Lubben M, Mol. Microbiol, 2015, 97, 423–438. [DOI] [PubMed] [Google Scholar]
- 55.Ashcroft F, Gadsby D and Miller C, Philos. Trans. R. Soc. Lond. B Biol. Sci, 2009, 364, 145–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Inesi G and Tadini-Buoninsegni F, J. Cell. Commun. Signal, 2014, 8, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olesen C, Picard M, Winther AM, Gyrup C, Morth JP, Oxvig C, Moller JV and Nissen P, Nature, 2007, 450, 1036–1042. [DOI] [PubMed] [Google Scholar]
- 58.Bublitz M, Musgaard M, Poulsen H, Thogersen L, Olesen C, Schiott B, Morth JP, Moller JV and Nissen P, J. Biol. Chem, 2013, 288, 10759–10765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tadini-Buoninsegni F, Bartolommei G, Moncelli MR, Pilankatta R, Lewis D and Inesi G, FEBS Lett, 2010, 584, 4619–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lewis D, Pilankatta R, Inesi G, Bartolommei G, Moncelli MR and Tadini-Buoninsegni F, J. Biol. Chem, 2012, 287, 32717–32727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang K, Sitsel O, Meloni G, Autzen HE, Andersson M, Klymchuk T, Nielsen AM, Rees DC, Nissen P and Gourdon P, Nature, 2014, 514, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clarke RJ and Apell HJ, Biophys. Chem, 1989, 34, 225–237. [DOI] [PubMed] [Google Scholar]
- 63.Morth JP, Pedersen BP, Toustrup-Jensen MS, Sorensen TL, Petersen J, Andersen JP, Vilsen B and Nissen P, Nature, 2007, 450, 1043–1049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.