

Abstract
Adult cardiac hypoxia as a crucial pathogenesis factor can induce detrimental effects on cardiac injury and dysfunction. The global transcriptome and translatome reflecting the cellular response to hypoxia have not yet been extensively studied in myocardium. In this study, we conducted RNA sequencing (RNA-seq) and ribosome profiling technique (polyribo-seq) in rat heart tissues and H9C2 cells exposed to different periods of hypoxia stress in vivo and in vitro. The temporal gene-expression profiling displayed the distinction of transcriptome and translatome, which were mainly concentrated in cell apoptosis, autophagy, DNA repair, angiogenesis, vascular process, and cardiac cell proliferation and differentiation. A large number of genes such as GNAI3, SEPT4, FANCL, BNIP3, TBX3, ESR2, PTGS2, KLF4, and ADRB2, whose transcript and translation levels are closely correlated, were identified to own a common RNA motif “5′-GAAGCUGCC-3′” in 5′ UTR. NCBP3 was further determined to recognize this RNA motif and facilitate translational process in myocardium under hypoxia stress. Taken together, our data show the close connection between alterations of transcriptome and translatome after hypoxia exposure, emphasizing the significance of translational regulation in related studies. The profiled molecular responses in current study may be valuable resources for advanced understanding of the mechanisms underlying hypoxia-induced effect on heart diseases.
Keywords: translatome hypoxia cardiomyocytes, ribosome, NCBP3
Graphical Abstract
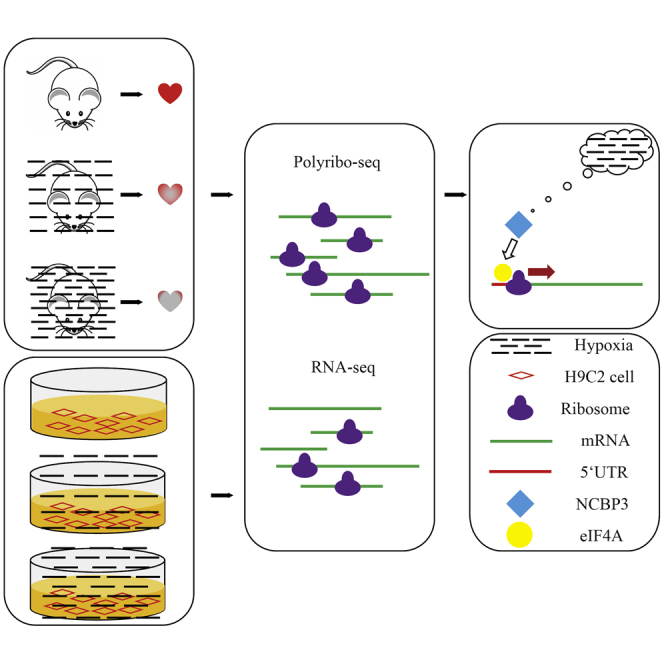
Shen et al. characterize a landscape of translatome and transcriptome in hypoxia-induced cardiomyocytes by RNA-seq and polysome profiling in vivo and in vitro. RNA motif and NCBP3 in 5′ UTR of genes with a good correlation of RNA abundance are identified for translational facilitation responding to hypoxia.
Introduction
Multiple clinical scenarios including respiratory system disorder and arterial vasculature interruption can cause the reduced global oxygen delivery. The heart is one of the most vulnerable organs to restrictions in oxygen supply. The systolic and diastolic function is highly dependent on constant oxygen support because mammalian heart muscle per se cannot produce enough energy under anaerobic conditions to maintain essential cellular processes.1 The role of oxygen and oxygen-associated processes in the heart is complex. As a crucial pathogenesis factor, severe or intermittent hypoxia can induce detrimental effects on cardiac dysfunction and failure,2 while mild or temporary hypoxia contribute to adult myocardial proliferation and regeneration.3 Therefore, an advanced understanding of molecular mechanisms responding to hypoxia stress is beneficial to preventing from the hypoxia-induced injury of cardiomyocytes.
RNA sequencing (RNA-seq) provides an unbiased way to investigate the genome-wide transcriptome profiling, and it can help construct the complicated gene regulatory network in the dynamic progression of human diseases. Previous studies have revealed novel gene signatures to accurately illustrate cardiac failure status and provide a large number of potential therapeutic targets.4,5 However, during the imposed “remodeling” of gene expression, transcription level alterations of certain mRNA do not closely correlate with those of the encoded proteins, which could partially depend on the differential recruitment of mRNAs to translating ribosomes.6 Besides, in response to environmental stimuli, the coordination of multiple RNA binding proteins (RBPs) bridging with ribosomes and translational initiation factors contribute to determining mRNA availability for fluctuations of the final translation levels.7 Ribosome profiling technique (polyribo-seq) based on sucrose-gradient separation of polysome-associated RNAs is allowed to assess their coding potential.8 To date, one similar study on cardiomyocyte hypertrophy has widely uncoupled the diffused independency of variations in mRNA abundances and their engagement on polysomes through the parallel comparison between total mRNAs (the transcriptome) and the polysome-associated mRNAs (the translatome).9 The given data shows that 5′ untranslated regions (UTRs) play an essential role in promoting mRNA recruitment to polysomes for translation enhancement in many species.10,11 However, the mechanism underlying the connection between translational regulation and hypoxia-induced stress in cardiomyocytes has not yet been elucidated.
To address this issue, the left ventricular myocardial tissues of adult rat and rat cardiac myoblast H9C2 cell line exposed to hypoxic stress were carried out the RNA-seq and polyribo-seq. The differential expressed genes (DEGs) of high-throughput sequencing data were studied the enriched functions and signaling pathways. The genes having translation priority were concluded with a RNA motif at 5′ UTR. Finally, we indicated that one RBP might be associated with modulation of hypoxia-induced gene translational alterations. This study demonstrated the great importance of polyribo-seq in understanding the molecular pathogenesis of myocardial ischemia, underscoring the necessity of translatome analysis in future studies.
Results
Hypoxic Model Evaluation In Vivo and In Vitro
Initially, adult male SD rats (6- to 8-week old, 337.58 ± 29.84 g) were subjected to acute normobaric hypoxia at 10% oxygen12 of different periods (5 min, 10 min, 20 min, 30 min, and 1 h). The levels of arterial SaO2 and SOD in sera were significantly decreased while MDA was increased in 10 min compared to normoxia control (NC). Moreover, SaO2 and SOD reached the highest while MDA reached the lowest level in 30 min and 1 h compared to the other conditions (Figure S1), indicating two possible conditions of mild hypoxia (MH; 10 min) and severe hypoxia (SH; 30 min) in our study. The myocardial ischemic injury was validated by triphenyltetrazolium chloride (TTC) and Masson staining, the infarct area was observed in 10 min, and appeared larger at 30 min (Figure S2). Likewise, rat H9C2 cardiomyocytes were treated with the culture condition (1% O2, 94% N2, and 5% CO2) for MH (8 h) and SH (24 h) as previously described.13 The activity of HIF signaling pathway and the downstream targets such as VEGFA, SLC2A1 (also known as GLUT1), and LDHA were investigated to validate the hypoxic H9C2 model (Figure S3).
Transcriptome and Translatome Profiling of Rat Cardiomyocytes Exposed to Hypoxia
To broadly assess the alteration of translatome in hypoxic cardiomyocytes, we conducted RNA-seq and polyribo-seq in rat cardiomyocytes in border zone of infarct and H9C2 cells with mild and severe hypoxia, which were further integrated with the studies on the enrichment of RBP for translational regulation. Table S2 showed a high percentage alignment rate of reads for RNA-seq (89.4% in average) and polyribo-seq (89.6% in average). The biological replicates of each same group displayed strong correlations (Figure S4). Principal-component analysis (PCA) majorly indicated two distinct clusters of samples separated by the first three components (Figure 1A). Cluster 1 was identified as untreated cardiomyocytes and H9C2 cells and was separated from severe hypoxia (cluster 2), as well as mild and partial severe hypoxia (cluster 3). Unexpectedly, we found that the gene translation signatures of MH and gene transcription of SH were located much closer to make the obscure boundary between cluster 2 and 3 and distinguished more sub-clusters from these two clusters. Heatmap also showed that the transcriptome and translatome between MH and SH were clustered together compared to other groups (Figure 1B). Collectively, these data show the overall landscape between hypoxic cardiomyocytes in vivo and in vitro.
Figure 1.

Overview of RNA-Seq and Rib-Seq in Cardiomyocytes Exposed Hypoxia
(A) Principal-component analysis (PCA) of transcriptome and translatome of each sample. PC1, PC2, and PC3 represent the top three dimensions that explain 35.8%, 18.2%, and 9.7% of the variance among all samples, respectively. (B) Heatmap of the global differentially expressed genes (DEGs). Gene expression values are standardized with Z score and the color represents the standardized values as shown in the color bar. NC, normoxia control; MH, mild hypoxia; SH, severe hypoxia.
Differentially Expressed Genes Identified by RNA-Seq and Polyribo-Seq
Next, DEGs identified by transcriptome and translatome were studied. In cardiomyocytes, RNA-seq revealed 4,102 DEGs (2,319 upregulated and 1,783 downregulated) in MH compared to NC, and 1,353 DEGs (628 upregulated and 725 downregulated) in SH compared to MH with |log2(fold change [FC])| ≥1 and p <0.05. Likewise, the presence of 2,925 DEGs (1,527 upregulated and 1,398 downregulated) in MH compared to NC, as well as 1,759 DEGs (764 upregulated and 995 downregulated) in SH compared to MH was observed in H9C2 cells (Figure 2A). We found 498 upregulated and 357 downregulated genes in the DEGs intersection between MH versus NC and SH versus MH in vivo and in vitro including VEGFA, PTGS2, LDHA, and SLC2A1 (Table S3). Translatome data was analyzed in the same way as transcriptome. 956 upregulated genes and 753 downregulated genes in MH compared to NC, as well as 572 upregulated genes and 504 downregulated genes in SH compared to MH, were found in cardiomyocytes. 536 upregulated genes and 429 downregulated genes in MH compared to NC, as well as 384 upregulated genes and 361 downregulated genes in SH compared to MH, were found in H9C2 cells (Figure 2B). Here, we majorly focused on the coding genes in DEGs and observed 225 upregulated and 222 downregulated genes in the DEG intersection between MH versus NC and SH versus MH in vivo and in vitro (Table S4).
Figure 2.

The DEGs and Their Enriched Functions and Signaling Pathways Responding to Hypoxia
(A and B) Venn diagram showing DEGs of the transcriptome (A) and translatome (B) among different conditions. (C) Dotplot for the enriched Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of DEGs. GeneRatio is calculated by the number of DGEs in the pathway divided by the total number of genes in the pathway. The size and the color of each dot represent the number of DEGs in the pathway and the p value respectively. NC, normoxia control; MH, mild hypoxia; SH, severe hypoxia.
Based on the union of DEGs, enrichment analysis with Gene Ontology (GO) terms in the category of biological process and Kyoto Encyclopedia of Genes and Genomes (KEGG) were carried out and identified top 10 hypoxia-related biological functions and pathways including hypoxia, autophagy, apoptosis, and myocardium differentiation (p < 0.05; Figure 2C). Besides the high overlap of these terms within transcriptome and translatome, the genes associated with some functions such as apoptosis and autophagy might be correlated with the translation processes compared to transcription, indicating that the gene expressed progression induced by hypoxia was likely to be different between transcription and translation process.
Divergence between Transcriptome and Translatome
To investigate the potential connection between gene transcription and translation upon hypoxia, we calculated Pearson correlation coefficient (r) of the aggregated DEGs between translatome and transcriptome (Figure 3A). Non-positive correlations (0 < r < 0.3) and negative correlations (r < 0) were observed for 3,662 genes. 603 genes with positive correlations (r ≥ 0.3) were further divided into two parts, in which the increasing trend of translation levels were higher or lower than transcription levels (log2k [slope of fitted line of each gene expression value of the samples of NC, MH, and SH] ≥ 1 or ≤ −1). Here, 177 genes with significantly different express change trend (DECTGs) between transcription and translation (Figure 3B). These DECTGs enriched in the signaling pathways of autophagy, apoptosis, and DNA repair including GNAI3, SEPT4, FANCL, BNIP3, and ADRB2 had higher translation increased trend compared to transcription, while other DECTGs such as TBX3, ESR2, PTGS2, and KLF4 enriched in the functions on angiogenesis, vascular process, and cardiac cell proliferation and differentiation were opposite (Figure 3C). The results above suggested that the translation levels rather than transcription of some specific genes might represent the truly gene expression responding to hypoxia stress.
Figure 3.

Correlation Analysis between Transcriptome and Translatome
(A) The correlation of gene expression between translatome and transcriptome. Each dot represents a Pearson correlation coefficient calculated with the expression levels of a gene in control, mild hypoxia (MH) and severe hypoxia (SH). (B) Volcano plots showing the significantly different express change trend genes (DECTGs) with significant positive correlation (r ≥ 0.3). (C) GO and KEGG analysis of DECTGs.
The Enriched “5′-GAAGCUGCC-3′” Motif in 5′ UTR Region of DECTGs
In general, the initiation of mRNA translation is modulated by specific proteins that can interact with the UTRs located on either side of the open reading frame (ORF).14 To further study the regulatory mechanism of DECTGs translation in myocardium upon hypoxic stress, we harvested the 5′ and 3′ UTRs of DECTGs and analyzed the role of RNA sequence (Figures 4A and 4B). “5′-GAAGCUGCC-3′” (abbreviate as DECTG RNA motif) at 5′ UTR ranked the top RNA motif implied a potential preferable sequence of 5′ UTR responding to hypoxia in myocardium. 86.3% genes with higher levels of translation than transcription in MH and SH were likely to enrich DECTG motif in their 5′ UTR, while genes with lower level of translation than transcription had no apparent patterns in 5′ or 3′ UTRs (Figure 4C).
Figure 4.

RNA Motif of DECTGs
(A and B) The potential consensus sequence of DECTGs at 5′ (A) and 3′ (B) UTRs. (C) The gene ratio of each motif.
To verify this hypothesis, we conducted CRISPR-Cas9 to knock out (KO) the DECTG motif at 5′ UTR of BNIP3 (“5′-CAAGCUGCCC-3′”) and ADRB2 (“5′-GAAGCUUCCA-3′”) in H9C2 cells. Upregulation of BNIP315 and ADRB216 in hypoxia condition displayed a more robust translational process compared to transcription (for BNIP3, log2FC = 1.87, p < 0.001, in cardiomyocytes with SH, log2FC = 2.05, p < 0.001 in H9C2 with SH; for ADRB2, log2FC = 1.59, p < 0.001 in cardiomyocytes with SH, log2FC = 2.46, p < 0.001 in H9C2 with SH). New generated sequences by the following non-homologous end-joining repair were aligned (Figure S5). The expressions of BNIP3 and ADRB2 in H9C2 cells treated with SH were detected using quantitative PCR (qPCR) and western blot (WB). We observed that the protein levels of BNIP3 and ADRB2 were significantly reduced in H9C2 of DECTG motif KO compared to wild-type (WT) both under the SH condition (Figures 5A and 5B), while mRNA levels stayed unchanged (Figure 5C). Furthermore, the occupancy of eukaryotic initiation factor 4, subunit A (eIF4A) was investigated on BNIP3 and ADRB2 using RNA immunoprecipitation (RIP)-qPCR. Consistently, the enrichments of eIF4A on the transcripts of BNIP3 and ADRB2 were diminished in DECTG motif KO compared to WT H9C2 cells with SH condition. Unexpectedly, the occupancy of eIF4A showed no significant difference between KO and WT in normoxia (Figures 5D and 5E), suggesting that the regulatory role of DECTG motif in translation process was dependent on oxygen concentration. Taken together, we revealed that RNA motif “5′-GAAGCUGCC-3′” in 5′ UTR might be associated with gene translation process in myocardium with hypoxia condition.
Figure 5.

The Gene Expression after the Motif “5′-GAAGCUGCC-3′” Deletion
(A–E) WB validation (A) with gray level difference analysis (B), mRNA (C), and RIPA of eIF4A enrichment on 5′ UTR of BNIP3 (D) and ADRB2 (E) after the RNA motif at their 5′ UTR deletion by CRISPR-Cas9. Data are presented as the mean ± standard error of the mean of three individual experiments. ∗p < 0.05 versus KO group. WT, wild-type; NC, normoxia control; KO, RNA motif at their 5′ UTR deletion by CRISPR-Cas9; SH, severe hypoxia.
Translation Process Facilitated by DECTG Motif Binding Protein NCBP3 in Hypoxia
Given the DECTG motif, the potential binding proteins were predicted using the databases of EuRBPDB (http://eurbpdb.syshospital.org/index.php),17 RegRNA 2.0 (http://regrna2.mbc.nctu.edu.tw/index.html),18 and ATtRACT (https://attract.cnic.es/).19 Nuclear cap-binding subunit 3 (NCBP3), EWS RNA binding protein 1 (EWSR1), and developmentally regulated GTP binding protein 2 (DRG2) were identified as the candidates with top three confidential scores (Figure 6A) and also as DEGs in MH and SH compared to NC in vivo and in vitro (Table S3). The expression of these candidate proteins was verified in cardiomyocytes and H9C2 cells using qPCR and WB. We observed that NCBP3 was significantly upregulated while EWSR1 and DRG2 were slightly increased in SH compared to NC (Figures 6B and 6C), suggesting that NCBP3 might be a target protein for translational regulation upon hypoxia stress. We cloned 50 bp biotin labeled RNA fragments containing “5′-GAAGCUGCC-3′” and other three similar motifs, and transfected into H9C2 cells, followed by streptavidin pull down. The substantial interaction of NCBP3 with RNA fragments containing DECTG motif compared to the one without DECTG motif and control was observed (Figure 6D).
Figure 6.

The Association of NCBP3 with “5′-GAAGCUGCC-3′” Motif
(A) The list of predicted RBPs for RNA motif. (B and C) mRNA (B) and protein (C) levels of NCBP3, EWSR1, and DRG2 in cardiomyocytes exposed hypoxia in vivo and in vitro. (D) Pull down validation of NCBP3 and eIF4A enrichments on RNA fragments with or without RNA motif in vitro. Data is presented as the mean ± standard error of the mean of three individual experiments. ∗p < 0.05 versus NC group. NC, normoxia control; SH, severe hypoxia.
Next, H9C2 cell with NCBP3 deficiency prepared by RNA interference were further conducted RNA-seq and polyribo-seq. Less than 10 DEGs were observed in NCBP3 knockdown H9C2 cells compared to NC either in normoxia or hypoxia (|log2FC| ≥ 1 and p < 0.05), suggesting that NCBP3 knockdown failed to impact gene transcription (Figure 7A). Moreover, NCBP3 knockdown did not impact whole translatome in normoxic H9C2 cells, however, it produced 1,263 downregulated genes and 341 upregulated genes in hypoxic H9C2 cells (Figure 7B). Additionally, comparison between transcriptome and translatome of hypoxic H9C2 cells with NCBP3 deficiency showed very few DECTGs (9 genes including NALCN, HK2, BZW2, MAP2K6, CTCF, MYL2, API5, PLOD1, and RPA1), which indicated that loss of NCBP3 weakened the hypoxia-induced translational facilitation. GO and KEGG analysis indicated that the genes whose translation was affected by NCBP3 knockdown were closely connected with programmed cell death and autophagy (Figure 7C).
Figure 7.

Transcriptome and Translatome Patterns of H9C2 Cells with NCBP3 Knockdown
(A and B) DEGs of transcriptome (A) and translatome (B) in H9C2 cells with NCBP3 knockdown. (C) GO and KEGG analysis of DEGs.
All in all, these results have revealed that NCBP3 can promote the gene translation process induced by hypoxic effect via interacting with a “5′-GAAGCUGCC-3′” motif in 5′ UTR in cardiomyocytes.
Discussion
In general, transcriptomics studies majorly focus on change of global RNA levels in response to environmental stimuli or in some diseases. However, mRNAs associated with polysomes may truly reflect the final protein levels. Previous studies on human cells indicated a distinction between global transcription and translation of some certain mRNAs.20,21 Consistently, the omics data in our study provide a large number of DEGs between RNA-seq and polyribo-seq (Figures 2A and 2B). Interestingly, the global patterns of gene profiling show that the translatome in MH and transcriptome in SH more similar (Figures 1B and 1C). We explain that the rapid gene expression is required to respond to hypoxia stress, but the lack of enough transcripts cannot support essential amounts of proteins in MH, and the facilitation of translational process is able to compensate for this deficiency. As the hypoxia duration increases (in SH), transcription levels are elevated to maintain sufficient protein translation, therefore, the translatome in MH is an earlier regulatory event than transcriptome in SH. For example, a major part of disproportionately expressed mRNAs preferentially encode proteins associated with the functions of autophagy, apoptosis, and cell proliferation (Figure 2C), suggesting that infarcted cell reprogramming death and new cell replaced regeneration are likely to be a matter of urgency and priority in myocardial cells’ response to hypoxic stress. On contrary, those polysome-enriched genes associated with HIF-1 signaling pathway as the beginning event responding to hypoxia may gradually reduce the increasement of protein in SH. The comparison between transcription and translation indicates a dynamic and meticulous gene expression regulation in hypoxia-induced cardiomyocytes.
A further point that only 14% genes have the positive correlation between their transcription and translation (Figure 3A), suggesting that the degree of translational regulation following exposure to hypoxia may have been underestimated. More coding genes are blocked from the translational process despite their abundant mRNAs. This can be interpreted in two aspects. First, translational process is likely to depend more on a set of enzymes in polysome and ribonucleoprotein complex rather than mRNA levels. Second, the intracellular signaling pathways activated by hypoxia can determine the function of enzymes for translational process.
Given the identified RNA motif of DECTGs, structures in 5′ UTR are likely to be a crucial region for the translation of those genes being sensitive to hypoxia, which implies that cap-dependent translation initiation may participate in translational process in this system. Although eIF4A has substantially bound to BNIP3 and ADRB2, eIF4-mediated translation initiation may be conservative in normoxic environment and it is not supposed to be able to facilitate translation under the hypoxia condition (Figure 5C).
Additionally, NCBP3 is highly fitted to the identified RNA motif according to the possible RBP list (Figure 6A). The highly conserved nuclear cap-binding complex (CBC) interacting with the RNA cap structures can orchestrate RNA biogenesis processes such as pre-mRNA splicing, 3′ end processing, nonsense-mediated decay, nuclear-cytoplasmic transport, and recruitment of translation factors in the cytoplasm.22 The canonical CBC is composed of a heterodimer formed by NCBP2 and adaptor NCBP1, while NCBP3, as a pivotal and essential cap-binding protein under the stress-state conditions, can compensate for NCBP2 and form an alternative CBC with NCBP1.23 Translational control of gene expression plays a crucial role in the stress response. In particular, translation of mRNAs by the classical cap-dependent mechanism is silenced, whereas alternative translation mechanisms allow enhanced expression of a small group of messengers that are involved in the control of cell survival.24,25 Our data indicates that NCBP3 can remarkably facilitate eIF4A recruitment (Figure 6D) and contribute to enhancing the activity of hypoxia-response required specific pathways (Figure 7C).
Through studying the relative effect on transcriptional versus translational regulation, we reveal the unique gene expression landscape on translational level and determine a novel mechanism of RBP NCBP3 that may play a role in translational modulation in myocardium responding to hypoxia stress. The translatome is largely uninvestigated in cardiomyocytes and is underinvestigated in other cells, but similar studies in future may prove essential in understanding the role of the corresponding complex at 5′ UTR in translational regulation. Further work is clearly required to establish the mechanisms by how NCBP3 influence mRNA recruitment to cardiomyocyte polysomes.
Materials and Methods
Experimental Animals
Adult male Sprague-Dawley (SD) rats (age: 6–8 weeks, weight: 337.58 ± 29.84 g, n = 12) were purchased from SLAC Laboratory Animal Company (Shanghai, China). Rats were maintained at 60.0% ± 10.0% humidity and 23.0°C ± 2.0°C under pathogen-free conditions. A standard diet and free drinking water were given for adaptive feeding more than 2 weeks before the experiment. Rats were randomly divided into three groups (4 rats in each group) including normal control, mild, and severe hypoxia. Chamber (Baker Ruskinn InvivO2-1000, Ruskinn Technology, Bridgend, UK) was used to create a normobaric hypoxia environment (10% oxygen). Peripheral blood from tail vein was used to detect superoxide dismutase (SOD), malonaldehyde (MDA), and oxygen saturation (SaO2). Kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China) were used to evaluate the oxidative stress. SaO2 was detected by blood gas analyzer ABL520 (Radiometer, Copenhagen, Denmark). Rats were sacrificed by decapitation, and isolated the non-infarct left ventricular myocardial tissues for the subsequent experiments.
All procedures were performed in accordance with standard guidelines as described in the Guide for the Care and Use of Laboratory Animals (US National Institutes of Health 85-23, revised 1996). All animal protocols were agreed with the local Institutional Animal Care and Use Committee of XiangYa School of Medicine.
Masson’s Trichrome Staining
Myocardial infarct size was measured in order to determine the extent of IRI. 4 μm paraffin slides were conducted Masson’s staining following the manufacturer’s instructions (Solarbio, Beijing, China) to assess the myocardial infarct size. The extent of the necrotic area was imaged by an Olympus BX-51 microscope (Olympus Corporation, Tokyo, Japan) and measured by computerized planimetry (ImageJ 1.4; National Institutes of Health, Bethesda, MD, USA). Infarct size was expressed as the percentage of the total weight of the area at risk of the left ventricle. The silk-like fibers indicated the early phase, whereas the appearance of collagen deposition indicated the late phase of myocardial infarction. The muscle fibers were red and the collagen fibers were blue.
Cell Culture
Rat cardiac myoblast H9C2 cell line was obtained from Cell Bank of Shanghai Institutes of Biological Sciences (Shanghai, China). Cells were cultured in DMEM medium with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA) at the incubator with 37°C, 5% CO2, and 100% humidity. Nucleotides of NCBP3 small interfering RNA (siRNA; 5′-UGUUCUUUCUUUUCAAUUGCU-3′, 5′-CAAUUGAAAAGAAAGAACAGC-3′), the guide RNA for BNIP3 (5′-GCTTGCGCCGCTCAGCTCTCGCGG-3′), and ADRB2 (5′-GCCGTCCGGGGGCGGACTCCTGG-3′) were synthetized by GenePharma (Shanghai, China). siRNAs or plasmids were transfected into H9C2 cells using Neon transfection system (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cells were harvested after 48-h transfection for the consequent experiments. Guide RNAs were cloned into pU6gRNACas9puro (GenePharma) and transfected into H9C2 cells, followed by 5 μg/mL puromycin screening for 3 more days. The cell clones were picked up and identified by PCR.
TTC Staining
2 mm freshly isolated heart tissues were added into 0.5% TTC/0.1 M phosphate buffer (pH = 7.5) solution at 37°C for 30 min in dark, then quenched by 2 M H2SO4. Tissues were washed by PBS for 5 min and pictured. ImageJ 1.4 was used to measure the infarct area.
qPCR Assay
1 mg left ventricular myocardial tissues excluded infarct area added 1 mL Trizol were squashed within liquid nitrogen. 10ˆ6 H9C2 cells could be directly dissolved by Trizol. 200 μL chloroform were added and vortexed for 15 s followed by 13,000 rpm centrifuge at 4°C. The upper supernatant was transferred to a new tube and added the same volume of isoproponal with 15 s vortex. The precipitation was washed by pure and 75% ethanol, then dissolved in the appropriate volume of diethylpyrocarbonate (DEPC) water. First-Strand Synthesis System for reverse transcription (Thermo Fisher Scientific) was used to synthesize cDNA from 1.5 μg total RNA according to the oligo(dT) version of the protocol. qPCR was performed using CFX fast real-time PCR system (Bio-Rad Laboratories, Hercules, CA, USA). The following cycle parameters were used for all experiments: 30 s at 94°C for pre-denaturation, 20 s at 94°C, 30 s at 60°C, and 30 s at 72°C for total 45 cycles. The relative levels of mRNA for each specific gene were normalized to GAPDH. Table S1 shows the sequences for all primer sets used in these experiments.
WB Assay
1 μg left ventricular myocardial tissues excluded infarct area or 10ˆ6 H9C2 cells were cut into small pieces and treated with radioimmunoprecipitation assay (RIPA) buffer containing 1% SDS and protease inhibitors. Then, the lysate was subjected to SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad Laboratories). The membrane was blocked with 5% fat-free milk in PBST for 30 min, followed by incubation overnight at 4°C with final dilutions of primary antibodies against HIF-1α (#14179, Cell Signaling Technology, Beverly, MA, USA), VEGFA (ab231260, Abcam, Cambridge, MA, USA), GLUT1 (21829-1, Proteintech Group, Rosemont, IL, USA), LDHA (#3558, Cell Signaling Technology), BNIP3 (#3769, Cell Signaling Technology), ADRB2 (#13096-1, Proteintech Group), NCBP3 (ab91556, Abcam), EWSR1 (#55191-1, Proteintech Group), DRG2 (#14743-1, Proteintech Group), eIF4A2 (#11280-1, Proteintech Group), and GAPDH (#60004-1, Proteintech Group). After that, the membrane was washed three times and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Proteintech Group). The blotting bands were developed with ECL plus immunoblotting detection reagents (Thermo Fisher Scientific) and captured using ImageJ.
RIPA
RIPA is followed as previously described.26 Briefly, 1 × 10ˆ7 H9C2 cells were harvested, resuspended in nuclear isolation buffer (1.28 M sucrose, 40 mM Tris pH 7.5, 20 mM MgCl2, 4% Triton X-100) and kept on ice for at least 30 min with frequent mixing. The pellet nuclei were centrifuged with 14,000 rpm for 15 min, resuspended by wash buffer (150 mM KCl, 25 mM Tris pH 7.4, 5 mM EDTA, 0.5 mM DTT, 0.5% NP40, 100 U/mL RNAase inhibitor (Solarbio), 1 × Protease inhibitors cocktail (Solarbio)) and sheared the chromatin through sonication by high power, 5 s on, 30 s off for 30 cycles. After that, 90% nuclei were incubated with 1 μg NCBP3 or eIF4A antibodies overnight and 40 μL Protein A/G beads (Thermo Fisher Scientific) 2 h by gentle rotation at 4°C while the rest of 10% were harvested as input. The pellet beads were centrifuged by 3,000 rpm 3 min, washed three times. Both the input and pellet beads were purified by RNAiso plus (Takara, Kusatsu, Japan) and conducted reverse transcription using QuantiTect Reverse Transcription Kit (QIAGEN, Hilden, Germany).
Pull-Down Assay
Biotin labeled RNA fragments were synthetized by GenePharma and transfected into H9C2 cells using Neon transfection system. After 24 h, 20 μL streptavidin beads (Vazyme, Nanjing, Jiangsu, China) was incubated with cell lysis to capture the biotin RNA fragments for rotating 2 h at 4°C. Beads were washed by 0.5 M RNase-free LiCl three times, resuspended with 27 μL ddH2O, added 3 μL 10X protein loading buffer, and denatured within boiled water bath for 5 min. The samples could be stored at −80°C or detected by WB assay.
RNA-Seq
2 μg RNA of each sample was used for library preparation by NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA) following manufacturer’s recommendations and were sequenced on an Illumina Hiseq platform. The raw data was trimmed adaptors and filter out low quality reads using Trimmomatic,27 and checked the quality of clean reads using Fastqc.28 Next, clean reads were aligned to the latest rattus norvegicus genome assembly Rnor_6.0 using Hisat2.29 The transcripts were assembled and estimated the expression levels by FPKM values using the StringTie algorithm with default parameters.30 Differential mRNA and lncRNA expression among the groups were evaluated using a R package Ballgown,31 and computed the significance of differences by the Benjamini & Hochberg (BH) p value adjustment method. Gene annotation is described by Ensembl genome browser database (http://www.ensembl.org/index.html). The R package ClusterProfiler was used to annotate the differential genes with GO terms and KEGG pathways.32
Polyribo-Seq
Polyribo-seq was performed as previously described.33 One additional step was required for RNA isolation. After RNA was released from cells, 15%–40% linear sucrose-gradient centrifugation at 38,000 rpm for 2.5 h at 4°C by a SW41 Ti rotor (Beckman Coulter, Brea, CA, USA). Fractionation with the peaks of A254 absorbing material followed by free mRNA, 40S, 60S, and 80S successively were polysomal peaks. Polysomal peaks were harvested to extract RNA, which represented polysome profiles, namely translatome. Data analysis procedure was the same with RNA-seq.
Data Availability
Raw data of RNA-seq and polyribo-seq were submitted to ArrayExpress with the accession number of E-MTAB-9541.
Statistical Analysis
The results were presented as the mean ± SD. The significance of difference among the groups was assessed by Student’s t test. All analysis was processed by SPSS 20 software. p value less than 0.05 was considered as statistical significance.
Acknowledgments
This work is supported by National Natural Science Foundation of China (grant number 81870303).
Author Contributions
Z.S. performed all experiments and analyzed the data. L.Z. undertook all bioinformatic studies. Z.Z. drafted and revised the manuscript.
Declaration of Interest
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.10.019.
Supplemental Information
References
- 1.Giordano F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu J., Stefaniak J., Hafner C., Schramel J.P., Kaun C., Wojta J., Ullrich R., Tretter V.E., Markstaller K., Klein K.U. Intermittent Hypoxia Causes Inflammation and Injury to Human Adult Cardiac Myocytes. Anesth. Analg. 2016;122:373–380. doi: 10.1213/ANE.0000000000001048. [DOI] [PubMed] [Google Scholar]
- 3.Kimura W., Nakada Y., Sadek H.A. Hypoxia-induced myocardial regeneration. J Appl Physiol (1985) 2017;123:1676–1681. doi: 10.1152/japplphysiol.00328.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y., Morley M., Brandimarto J., Hannenhalli S., Hu Y., Ashley E.A., Tang W.H., Moravec C.S., Margulies K.B., Cappola T.P., Li M., MAGNet consortium RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics. 2015;105:83–89. doi: 10.1016/j.ygeno.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J., Wang Y., Duan Z., Hu W. Hypoxia-induced alterations of transcriptome and chromatin accessibility in HL-1 cells. IUBMB Life. 2020;72:1737–1746. doi: 10.1002/iub.2297. [DOI] [PubMed] [Google Scholar]
- 6.Halbeisen R.E., Gerber A.P. Stress-dependent coordination of transcriptome and translatome in yeast. PLoS Biol. 2009;7:e1000105. doi: 10.1371/journal.pbio.1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tebaldi T., Re A., Viero G., Pegoretti I., Passerini A., Blanzieri E., Quattrone A. Widespread uncoupling between transcriptome and translatome variations after a stimulus in mammalian cells. BMC Genomics. 2012;13:220. doi: 10.1186/1471-2164-13-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Popa A., Lebrigand K., Paquet A., Nottet N., Robbe-Sermesant K., Waldmann R., Barbry P. RiboProfiling: a Bioconductor package for standard Ribo-seq pipeline processing. F1000Res. 2016;5:1309. doi: 10.12688/f1000research.8964.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Markou T., Marshall A.K., Cullingford T.E., Tham L., Sugden P.H., Clerk A. Regulation of the cardiomyocyte transcriptome vs translatome by endothelin-1 and insulin: translational regulation of 5′ terminal oligopyrimidine tract (TOP) mRNAs by insulin. BMC Genomics. 2010;11:343. doi: 10.1186/1471-2164-11-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuura H., Shinmyo A., Kato K. Preferential translation mediated by Hsp81-3 5′-UTR during heat shock involves ribosome entry at the 5′-end rather than an internal site in Arabidopsis suspension cells. J. Biosci. Bioeng. 2008;105:39–47. doi: 10.1263/jbb.105.39. [DOI] [PubMed] [Google Scholar]
- 11.Desai P.R., Lengeler K., Kapitan M., Janßen S.M., Alepuz P., Jacobsen I.D., Ernst J.F. The 5′ Untranslated Region of the EFG1 Transcript Promotes Its Translation To Regulate Hyphal Morphogenesis in Candida albicans. MSphere. 2018;3:e00280-18. doi: 10.1128/mSphere.00280-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayward L.F., Castellanos M., Noah C. Cardiorespiratory variability following repeat acute hypoxia in the conscious SHR versus two normotensive rat strains. Auton. Neurosci. 2012;171:58–65. doi: 10.1016/j.autneu.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Hantelys F., Godet A.C., David F., Tatin F., Renaud-Gabardos E., Pujol F., Diallo L.H., Ader I., Ligat L., Henras A.K. Vasohibin1, a new mouse cardiomyocyte IRES trans-acting factor that regulates translation in early hypoxia. eLife. 2019;8:e50094. doi: 10.7554/eLife.50094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sajjanar B., Deb R., Raina S.K., Pawar S., Brahmane M.P., Nirmale A.V., Kurade N.P., Manjunathareddy G.B., Bal S.K., Singh N.P. Untranslated regions (UTRs) orchestrate translation reprogramming in cellular stress responses. J. Therm. Biol. 2017;65:69–75. doi: 10.1016/j.jtherbio.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 15.Li Y., Ren S., Xia J., Wei Y., Xi Y. EIF4A3-Induced circ-BNIP3 Aggravated Hypoxia-Induced Injury of H9c2 Cells by Targeting miR-27a-3p/BNIP3. Mol. Ther. Nucleic Acids. 2020;19:533–545. doi: 10.1016/j.omtn.2019.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grisanti L.A., Gumpert A.M., Traynham C.J., Gorsky J.E., Repas A.A., Gao E., Carter R.L., Yu D., Calvert J.W., García A.P. Leukocyte-Expressed β2-Adrenergic Receptors Are Essential for Survival After Acute Myocardial Injury. Circulation. 2016;134:153–167. doi: 10.1161/CIRCULATIONAHA.116.022304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao J.Y., Yang B., Zhang Y.C., Wang X.J., Ye Y., Peng J.W., Yang Z.Z., He J.H., Zhang Y., Hu K. EuRBPDB: a comprehensive resource for annotation, functional and oncological investigation of eukaryotic RNA binding proteins (RBPs) Nucleic Acids Res. 2020;48(D1):D307–D313. doi: 10.1093/nar/gkz823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang T.H., Huang H.Y., Hsu J.B., Weng S.L., Horng J.T., Huang H.D. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinformatics. 2013;14(Suppl 2):S4. doi: 10.1186/1471-2105-14-S2-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giudice G., Sanchez-Cabo F., Torroja C., Lara-Pezzi E. ATtRACT-a database of RNA-binding proteins and associated motifs. Database. 2016;2016:baw035. doi: 10.1093/database/baw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grolleau A., Bowman J., Pradet-Balade B., Puravs E., Hanash S., Garcia-Sanz J.A., Beretta L. Global and specific translational control by rapamycin in T cells uncovered by microarrays and proteomics. J. Biol. Chem. 2002;277:22175–22184. doi: 10.1074/jbc.M202014200. [DOI] [PubMed] [Google Scholar]
- 21.Rajasekhar V.K., Viale A., Socci N.D., Wiedmann M., Hu X., Holland E.C. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol. Cell. 2003;12:889–901. doi: 10.1016/s1097-2765(03)00395-2. [DOI] [PubMed] [Google Scholar]
- 22.Gonatopoulos-Pournatzis T., Cowling V.H. Cap-binding complex (CBC) Biochem. J. 2014;457:231–242. doi: 10.1042/BJ20131214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gebhardt A., Habjan M., Benda C., Meiler A., Haas D.A., Hein M.Y., Mann A., Mann M., Habermann B., Pichlmair A. mRNA export through an additional cap-binding complex consisting of NCBP1 and NCBP3. Nat. Commun. 2015;6:8192. doi: 10.1038/ncomms9192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holcik M., Sonenberg N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 25.James C.C., Smyth J.W. Alternative mechanisms of translation initiation: An emerging dynamic regulator of the proteome in health and disease. Life Sci. 2018;212:138–144. doi: 10.1016/j.lfs.2018.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao H., He Y., Li H., Zhu A., Ye Y., Liu G., Zhao C., Zhang X. The opposite role of alternatively spliced isoforms of LINC00477 in gastric cancer. Cancer Manag. Res. 2019;11:4569–4576. doi: 10.2147/CMAR.S202430. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrews S. 2013. FastQC A Quality Control tool for High Throughput Sequence Data. [Google Scholar]
- 29.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frazee A.C., Pertea G., Jaffe A.E., Langmead B., Salzberg S.L., Leek J.T. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 2015;33:243–246. doi: 10.1038/nbt.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chassé H., Boulben S., Costache V., Cormier P., Morales J. Analysis of translation using polysome profiling. Nucleic Acids Res. 2017;45:e15. doi: 10.1093/nar/gkw907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data of RNA-seq and polyribo-seq were submitted to ArrayExpress with the accession number of E-MTAB-9541.