

Highlights
-
•
Patient tumor subclones that survive chemotherapy acquire primitive cell traits.
-
•
HDAC inhibitors can reverse chemo-acquired stemness states and abolish self-renewal abilities.
-
•
Belinostat promotes stem cell differentiation and inhibits HDAC and MYC pathways.
-
•
A ‘one-two punch’, chemotherapy-HDAC inhibitor combination strategy reverses chemo-induced resistant phenotypes.
Keywords: CSL, Chemotherapy, HDAC, Single-cell RNA-Seq, MYC, Chemoresistance
Abbreviations: ALDH, Aldehyde dehydrogenases; AXL, Axl receptor tyrosine kinase; CBP, CREB-binding protein; CDK 4/6, Cyclin-Dependent Kinase 4/6; CDK 8, Cyclin-Dependent Kinase 8; CNV, Copy number variations; CSC, cancer stem cell; CSL, cancer stem cell-like; EGFR, epidermal growth factor receptor; EMT, Epithelial to mesenchymal transition; ER, estrogen receptor; FGFR1, Fibroblast growth factor receptor; HDAC, Histone deacetylase; HDACi, Histone deacetylase inhibitor; HER2, human epidermal growth factor receptor 2; MET, mesenchymal-to-epithelial transition; PR, progesterone receptor; scRNA-Seq, single cell RNA sequencing; ssGSEA, Single-sample Gene Set Enrichment Analysis; TNBC, triple negative breast cancer; WES, whole exome sequencing; WGS, whole genome sequencing
Abstract
Cancer cell phenotypes evolve during a tumor's treatment. In some cases, tumor cells acquire cancer stem cell-like (CSL) traits such as resistance to chemotherapy and diminished differentiation; therefore, targeting these cells may be therapeutically beneficial. In this study we show that in progressive estrogen receptor positive (ER+) metastatic breast cancer tumors, resistant subclones that emerge following chemotherapy have increased CSL abundance. Further, in vitro organoid growth of ER+ patient cancer cells also shows that chemotherapy treatment leads to increased abundance of ALDH+/CD44+ CSL cells. Chemotherapy induced CSL abundance is blocked by treatment with a pan-HDAC inhibitor, belinostat. Belinostat treatment diminished both mammosphere formation and size following chemotherapy, indicating a decrease in progenitor CSL traits. HDAC inhibitors specific to class IIa (HDAC4, HDAC5) and IIb (HDAC6) were shown to primarily reverse the chemo-resistant CSL state. Single-cell RNA sequencing analysis with patient samples showed that HDAC targets and MYC signaling were promoted by chemotherapy and inhibited upon HDAC inhibitor treatment. In summary, HDAC inhibition can block chemotherapy-induced drug resistant phenotypes with ‘one-two punch’ strategy in refractory breast cancer cells.
Graphical abstract
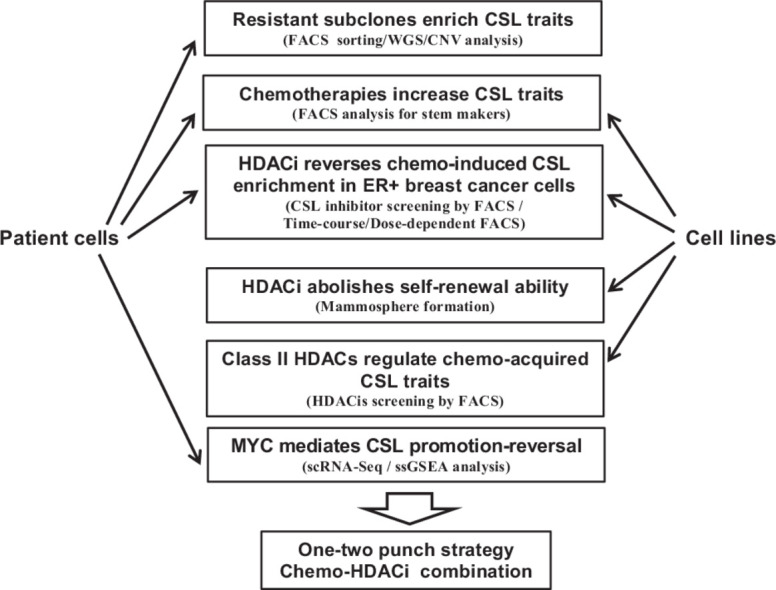
Introdcution
Breast cancer is the most frequently diagnosed and second leading cause of cancer-related deaths in women [1]. Chemotherapeutics are often standard of care in clinical oncology because of their effectiveness in reducing tumor burden and improving survival [2, 3]. Nevertheless, some patients will recur with metastatic progression, which has a 90% of cancer mortality [4], resulting in a 23% 5-year survival rate for these breast cancer patients [1].
Tumors are composed of heterogeneous populations of cells, thought to have a hierarchical organization driven by cancer stem cells (CSCs). CSCs are a small therapy-resistant sub-population of cells within tumors that possess the capacity of self-renewal and are capable of promoting a refractory state in patients following chemotherapeutic treatment due to their inherent chemoresistance [5, 6]. Breast CSCs exhibit a CD44 high/CD24 low phenotype with high ALDH1 expression [7]. Tumor cells can also acquire stem cell like characteristics, and may represent a de-differentiated state similar to CSCs and reflect a more primitive tumor cell progenitor [8].
Based on their mechanisms of action, chemotherapies can be divided into three major groups: antimetabolites; genotoxic agents (eg. doxorubicin serving as alkylating agent, which inhibits DNA topoisomerase II and induces DNA damage and apoptosis; carboplatin serving as intercalating agent, which binds in the grooves in the DNA helix and interfering with polymerase activity during replication/transcription); and mitotic spindle inhibitors (eg. paclitaxel, which disrupts mitosis by affecting the formation/function of spindle microtubule fibers required for chromosome alignment) [9]. Chemoresistance can be acquired by altered membrane transport through ABCB1 (P-gp or MDR1) for doxorubicin and paclitaxel [9], and enhanced DNA repair through increased level of excision repair cross-complementing protein (ERCC1) for carboplatin [10]. Current chemotherapeutic regimens target the bulk of tumor cells and may benefit from also targeting resistant cells, such as CSCs or cancer cells that have stem-like traits such as drug resistance or de-differentiated states [11]. Failure to eliminate these cells can lead to drug resistance, subsequent recurrence and metastasis [12], suggesting that targeting these populations may be necessary to improve outcomes [5,13]. Multiple strategies have been proposed to combat CSCs; however, clinical implementation has remained elusive [7, 14]. A strategy combining chemotherapy and anti-CSC compounds could increase efficacy in reducing the risk of breast cancer relapse and metastasis [7]. The drug-tolerant phenotype within a small subpopulation of cancer cells have been found transiently acquired and reversible, which could be selectively ablated by chromatin-modifying agents, such as HDAC inhibitor, suggesting a potential therapeutic opportunity with ‘one-two punch’ strategy [15].
Single-cell sequencing techniques have been leveraged to identify resistant cancer evolution upon chemotherapy treatment [16, 17, 18, 19]. Previously, we performed whole-genome sequencing (WGS) and single-cell RNA-Seq (scRNA-Seq) on four patients’ matched pre-treatment (chemo-sensitive) and post-treatment (chemo-resistant) samples to investigate the mechanisms of acquired chemoresistance in breast cancer. Three of four patients demonstrated increased post-treatment stem cell-like properties, which may have promoted acquired drug resistance in these patients [19]. Based on this data, we sought to understand how multiple different chemotherapies impact CSL state and determine therapy approaches to prevent emergence of CSL cells. Here, we find that chemotherapies with different mechanisms of action can select for subclones with primitive traits through transcriptional changes. Belinostat, a pan-HDAC inhibitor, was found to prevent this selection and reverse CSL signaling, such as MYC and dedifferentiation pathways. The specific Class II HDAC inhibitors, LMK-235 and CAY10603, which target to HDAC4/5 and HDAC6 respectively, exhibited the similar reversal effect on chemo-treatment acquired stemness.
In total, this research provides the following unique findings: 1) a novel chemo-belinostat combination therapy strategy to target chemo-treatment acquired stemness, 2) the ability of belinostat to reverse CSL traits following chemotherapy treatment, 3) the specificity of HDAC inhibitors to target the stemness phenotypic shift and not just a change in tumor cell viability, 4) the use of patient tumor samples to improve translatability for the conclusion that stemness enrichment in subclonal populations of patients is linked to their survival during chemotherapy, 5) an understanding of the transcriptional changes in single tumor cells upon CSL phenotype reversal, and 6) the role of class II HDACs on chemoresistance through their reversal of primitive traits. Taken together, these findings could help to build a new strategy consisting of sequential chemotherapy and HDAC inhibitor combination treatment to combat refractory breast cancer.
Martials and methods
See all Martials and Methods in Supplementary Data
Results
Resistant subclones are enriched in CSL cells
We interrogated patient tumor cells for sublonal CSL states using copy-number variations (CNV) from WGS [19]. The treatment history for these four ER+ patients and the time points at which analysis was performed are shown in Fig. S1A. To obtain CSL and non-CSL populations, tumor cells were isolated to four quadrants (ALDH+/CD44+ cells, ALDH+/CD44- cells, ALDH-/CD44+ cells, and ALDH-/CD44- cells) (Fig. S1B) with the CSL population defined as ALDH+/CD44+ cells [20]. DNA from cells belonging to each of these quadrants was isolated and subjected to low-coverage WGS (2X to 10X) in order to infer CNVs so that we could identify tumor subclone cell proportions during treatment (Fig. S1C). The evolution of subclones is shown in Fig. S1D, E, F, G and described in the Methods. Each subclone was then analyzed for the presence of CSL vs. non-CSL populations. In two of four patients that are progressing on chemotherapy (Patient#1 and Patient#2), the progressing subclone that survives treatment and becomes resistant to chemotherapy has increased CSL abundance compared to dying subclones (Fig. 1A), suggesting genetic selection can enrich CSL cells in post-treatment resistant subclones in ER+ breast cancer.
Fig. 1.

Genetic selection and chemotherapy both enrich CSL subclones. (A) The proportion of each subclone with CSL vs. non-CSL populations in four patients. Cells were sorted via FACS using ALDEFLUOR and CD44 staining and DNA was isolated from each population, followed by low-coverage WGS to infer CNV and determine levels of subclonal CNVs. Heat maps for FACS mean values of ALDH, CD44 and CSL cells/Live upon chemotherapy treatment in cultured patient cells (B) and breast cancer cell lines (C) 3D organoids of patient cells and breast cancer cell lines were incubated with doxorubicin (0.1 µM), carboplatin (0.1 mM) and paclitaxel (1 µM) for 72 h, followed by ALDEFLUOR/CD44/DAPI staining and FACS to identify the FACS mean values of ALDH and CD44 in live populations and CSL cells/Live. The vehicle controls (DMSO treatment) were set as fold one. All the chemotherapy treatments were expressed as the fold change relative to the control. The mean values of triplicate tests were shown in the heat maps with color indicated as the legends for each treatment. Significance is marked with * for P<0.05, ** for P<0.01, *** for P<0.001.
Chemotherapy-induced feedback increases CSL traits in patient tumor cells
To further test the effect of chemotherapy on CSL promotion, we used additional patient cancer cells obtain from malignant pleural effusions or ascites. These samples were treated with three different chemotherapies of various mechanisms of action (doxorubicin, carboplatin, and paclitaxel) for 72 h, followed by flow cytometry to measure the CSL abundance using the FACS gating strategy shown in Fig. S2A. In the five ER+ patient cultures (Patient#5–9), all three chemotherapies increased CSL cells in the majority of samples (Fig. 1B and Fig. S2B). Interestingly, for three TNBC (triple-negative breast cancer) patient samples, the abundance of CSL/Live (live CSL cells versus all live cells) and CSL/Total (live CSL cells all live and dead cells) were generally decreased. CSL/Live represents the proportion of live CSL cells versus all live cells in the organoid, while CSL/Total represents the proportion of live CSL cells versus all live and dead cells in the organoid. The CSL/Total values are usually consistent with ALDH and CD44. In contrast, the CSL/Live cell counts are not always consistent to ALDH, CD44, or CSL/Total change trends because the drug treatments can dramatically reduce viability of cells. In summary, chemotherapy can induce CSL traits in ER+ breast cancer.
Chemotherapy-induced CSL states are recapitulated in breast cancer cell lines
To further investigate these phenomena, we tested four ER+ breast cancer cell lines (CAMA-1, T47D, MCF-7, ZR-75–1) and five TNBC cell lines (MDA-MB-231, BT549, HCC38, HCC1395, HCC1143). As shown in Fig. 1C and Fig. S2C, chemotherapy increased the abundance of ALDH, CD44, CSL/Live and CSL/Total in most ER+ cell lines, although T47D and MCF7 have been reported as ALDH-negative [21]. This promotion was again less present in TNBC cell lines following chemotherapy, and showed only increased abundance of CD44. The CSL enrichment by chemotherapy in ER+ cells and TNBC is shown with relative folds of ALDH and CD44 compared to DMSO control, which are highlighted for ER+ patient cells (Fig. S3A), TNBC patient cells (Fig. S3B), ER+ cell lines (Fig. S3C), and TNBC cell lines (Fig. S3D). TNBC patients and cell lines showed a lesser extent of primitive cell traits compared to ER+ cells.
CSL phenotypes are reversed with an HDAC inhibitor
We sought to find an inhibitor that could reverse CSL trait induction following chemotherapy. We used two ER+ breast cancer cell lines (CAMA-1 and T47D) and two TNBC cell lines (MDA-MB-231 and BT549). Potential CSL inhibitors were selected based multiple lines of data: 1) potential strategies to suppress CSCs summarized by Lin et al.[14]. and others, which include targeting pathways that regulate EMT such as hedgehog (vismodegib), Wnt/β-catenin (ICG-001) or CSC markers [disulfiram (DSF) as an ALDH inhibitor], 2) drugs targeting HDACs (belinostat and entinostat), 3) growth factor receptor pathway inhibitors including AXL inhibitors (BGB324 and TP-0903), FGFR inhibitor (AZD4547), EGFR inhibitors (afatinib and gefitinib), and 4) cyclin-dependent kinases inhibitors (senexin A, ribociclib and abemaciclib), which are reported to play indispensable roles in stem cell self-renewal [22]. Three dimensional organoids of each cell line were treated with chemotherapy for 72 h, followed by inhibitors for another 72 h, and subjected to FACS analysis for CSL populations. To identify the CSL inhibitor, we considered both the reversal of mean values of ALDH and CD44 by FACS analysis, and the reduction of CSL/Live and CSL/Total. As shown in Fig. 2A, B and Fig. S4A, B, the pan-HDAC inhibitor belinostat consistently reversed ALDH levels promoted by three chemotherapy treatments in all four cell lines and reduced CD44 levels promoted by the three chemotherapy treatments in the majority of cell lines. In contrast to ER+ and TNBC cell lines, the Class I HDAC inhibitor entinostat only exhibited a reversal effect in one ER+ cell line (T47D). Additionally, an increased cytotoxicity of belinostat was also observed in the viability heat maps for all cell lines at the doses for CSL reversal effect (Fig. 2A, B and Fig. S4A, B). Taken together, these data indicate that belinostat may serve as a general anti-chemo-induced CSL modifier in breast cancer.
Fig. 2.

Belinostat reduced chemotherapy-promoted CSL state. The FACS mean values of ALDH and CD44 in live populations, CSL/Live, CSL/Total, and viability for CSL reversal drugs screening were expressed in heat maps for CAMA-1 (A) and MDA-MB-231 (B) cell lines. Cells were cultured in medium (3D) and treated for 72 h with doxorubicin (0.1 µM for CAMA-1, 0.5 µM for MDA-MB-231), carboplatin (50 µM for all both lines), and paclitaxel (1 µM for both cell lines), and then treated with potential CSL inhibitors as indicated doses for 72 h before ALDEFLUOR/CD44 staining. The vehicle controls (DMSO+DMSO) were set as fold one. All the other chemotherapy plus DMSO/inhibitor combinations were expressed as the fold change relative to the controls. The Chemo plus DMSO served as the positive controls. The mean values of triplicate tests were showed with color indicated as the legends. In heat maps of ALDH, CD44, CSL/Live, and CSL/Total, # is chemo plus DMSO significantly higher (p<0.05) than no treatment control, and in heat maps of viability, # is chemo plus DMSO significantly lower (p<0.05) than no treatment control. In all heat maps, * is chemo plus inhibitor significantly lower (p<0.05) than chemo plus DMSO. The potential inhibitors are highlighted with red blocks as its reversal effect for ALDH, CD44, CSL/Live, and CSL/Total.
To further interrogate the effect of belinostat on CSL cells, we tested belinostat alone treatment together with chemotherapy-belinostat combination treatments for four ER+ cell lines CAMA-1, T47D, MCF-7, ZR-75–1, and two TNBC cell lines MDA-MB-231 and BT549 (Fig. S5). As shown, belinostat alone can significantly reduce CSL/Total in all six cell lines and decreased CSL/Live in two ER+ cell lines (T47D and MCF7) and two TNBC cell lines as evidenced by the reversal of ALDH or CD44. Belinostat alone also significantly reduced the viability of all cell lines except MCF-7. In addition, two additional ER+ cell lines MCF-7 and ZR-75–1 were tested, and show similar results to CAMA-1 and T47D with chemotherapy-belinostat combination treatments. Together, these studies confirm the reversal effect of belinostat following chemotherapy treatments in ER+ cell lines. Across the ER+ cell lines, at least three out of four cell lines showed the promotion of ALDH, CD44, CSL/Live and CSL/Total by chemotherapy and reversal by belinostat in all three chemotherapy treatment groups. Importantly, although chemotherapy promoted CSL traits in TNBC were less pronounced than ER+ cell lines, which was indicated by CSL/Total fold change in MDA-MB-231 and BT549, belinostat also reversed CSL/Total in all three chemotherapy treatment groups for both TNBC cell lines. These results suggested a global reversal effect of belinostat on both innate and chemotherapy promoted CSL traits in both ER+ and TNBC cells.
Belinostat reverses chemotherapy-derived CSL enrichment in ER+ breast cancer cells
To further evaluate the potential of belinostat to reverse chemo-induced CSLs, we performed a time-course FACS analysis from Day 1 to Day 6 to measure the CSL phenotype over time using the experiment strategy shown in Fig. 3A. As shown in Fig. 3B, FACS analysis of the ER+ cell line (CAMA-1) showed steadily increasing ALDH levels with all three chemotherapies from Day 1 to Day 6, while the CD44 levels showed steady increases with doxorubicin and paclitaxel. Overall, there was an increase in CSL/Live from Day 1 to Day 6 in all three chemotherapy groups . The overall viability continuously decreased each day following all three chemotherapy treatments, while belinostat further reduced viability compared to chemotherapy only (Fig. S6A), indicating an increase in cytotoxicity with belinostat treatment in addition to its CSL reversal effect. Importantly, belinostat reversed the upward trajectory of ALDH, CSL/Live (Fig. 3B), and CSL/Total (Fig. S6A) with a significant reduction in treated versus control at Day 5 and Day 6. Of note, CD44 was only reversed by belinostat following paclitaxel treatments, but not carboplatin and doxorubicin treatment (Fig. 3B), suggesting some differences in chemotherapy effects. Summarized results across chemotherapy treatments show a statistically significant reduction in ALDH, CSL/Live, and CSL/Total with belinostat treatment at Day 6 (Fig. 3B and Fig. 6A).
Fig. 3.

Belinostat inhibited chemotherapy-promoted CSL state in ER+ cell lines and patient cancer cell samples. (A) Cell culture strategy for CSL reversal drug screening with cancer breast cell lines after chemotherapy promotion. The FACS mean values of ALDH, CD44, and CSL cells/live were shown in (B) for CAMA-1 and (C) for MDA-MB-231 with time-course curves. CAMA-1 cells and MDA-MB-231 cells were cultured in medium (3D) and treated for 72 h with doxorubicin (0.1 µM for CAMA-1, 0.5 µM for MDA-MB-231), carboplatin (50 µM), and paclitaxel (1 µM), and then replaced with medium containing belinostat (5 µM) for 72 h incubation. Samples were collected every day from Day 1 to Day 6, and followed by ALDEFLUOR/CD44/DAPI staining and FACS analysis. The differences between chemo plus DMSO (9 replicates from three chemo plus DSMO) and chemo plus belinostat (9 replicates from three chemo plus belinostat) at Day 6 were evaluated by student's T tests. The ALDH (D), CD44 (E), CSL cells/live (F) were expressed with curves for Patient#5, #6, and #7. Patient cells were cultured in Renaissance medium (3D) and treated with chemotherapy (doxorubicin 0.1 µM, carboplatin 0.1 mM, paclitaxel 1 µM) for 72 h, and then treated with DMSO/belinostat (1 µM or 2.5 µM) for another 72 h, followed by ALDEFLUOR/CD44/DAPI staining and FACS analysis. Column graphs were generated with averages from three patients for each treatment to evaluate the changes in ALDH (G), CD44 (H), and CSL/Live (I). In Figures B-I, DMSO treatment only in each day served as controls and was set as fold one. All the other treatments were expressed as the fold values relative to the controls. Each spot represents the mean values of triplicate tests. The statistical analysis was performed with student's t-tests between the indicated groups. Significance is marked with * for P<0.05, ** for P<0.01, *** for P<0.001, **** for P<0.0001. .
Fig. 6.

Class II HDAC inhibitors reverse chemotherapy promoted CSL traits in ER+ and TNBC cells. Time-course curves of relative fold changes for FACS mean values of ALDH and CD44, and CSL/Live were shown for chemotherapy plus LMK235 and CAY10693 treatments in CAMA-1 (A, B) and MDA-MB-231 (C, D). Both cell lines were cultured in medium (3D) and treated for 72 h with doxorubicin (0.1 µM for CAMA-1, 0.5 µM for MDA-MB-231), carboplatin (50 µM), and paclitaxel (1 µM), and then replaced with medium containing LMK-235 (5 µM) or CAY10603 (5 µM) for 72 h incubation. Samples were collected every day from Day 1 to Day 6, and followed by ALDEFLUOR/CD44/DAPI staining and FACS analysis. DMSO treatment only in each day served as control and was set as fold one. All the other treatments were expressed as the fold values relative to the controls. Each spot represents the mean values of triplicate tests. The differences between chemo plus DMSO (9 replicates from three chemo plus DSMO) and chemo plus belinostat (9 replicates from three chemo plus belinostat) at Day 6 were evaluated by student's T tests. Significance is marked with * for P<0.05, ** for P<0.01, *** for P<0.001, **** for P<0.0001. .
Less consistent results were found in the TNBC cell line (Fig. 3C and Fig. S6B). CD44 (Fig. 3C), and CSL/Live (Fig. 3C) slightly increased with doxorubicin and carboplatin, but not paclitaxel. Further, ALDH, CD44, CSL/Live, and viability decreased with belinostat following all three chemotherapy treatments, exhibiting a statistically significant reduction across all chemotherapies in all of above at Day 6 (Fig. 3C and Fig. S6B). Overall, the CSL reversal following chemotherapy was less pronounced in TNBC than ER+ cells, as indicated by the reduction of CSL cells versus live cells in MDA-MB-231 (0.67–1.4 fold in chemotherapy alone versus 0.46–0.71 fold in chemotherapy plus belinostat) versus those in CAMA-1 (3.1–11.2 fold in chemotherapy alone versus 1.56–2.4 fold in chemotherapy plus belinostat), perhaps reflecting the lack of chemotherapy-promoted CSL traits in TNBC versus ER+ cells, and higher baseline CSL in TNBC cells [23].
As chemotherapy strongly promoted CSL state in ER+ breast cancer patient samples, with a less pronounced effect in TBNC patient samples (Fig. 1B), we further tested the reversal of CSL over time by belinostat in three ER+, PR+, Her2- breast cancer patient samples (Patient#5–7). As shown in Fig. 3D and F, FACS analysis indicated that ALDH and CSL/Live were generally increased by chemotherapy and reversed by belinostat. Note, CD44 decrease was found in the majority but not all of the patient cells (Fig. 3E). CSL/Total were reduced by belinostat, although induction of CSL state alter chemotherapy varied by patient, most likely due to prior treatments the patient received and various levels of response to chemotherapy (Fig. S6C). Importantly, we found all chemotherapies decreased the viability of cancer cells, and belinostat further decreased viability in a dose-dependent manner (Fig. S6D). Across all patient samples, ALDH was increased following chemotherapy treatments compared to vehicle controls, and significantly decreased by belinostat in dose-dependent manner (Fig. 3G). Note, CD44 levels did not show a significant common effect either on chemotherapy promotion or belinostat reversal, although the expected trends could be observed between indicated groups (Fig. 3H). CSL/Live (Fig. 3I) and CSL/Total (Fig. S6E) showed similar trends with ALDH (Fig. 3G), but were not significant due to the high variation of CD44 (Fig. 3H). The viability decreased with all chemotherapies, and further decreased with belinostat in doxorubicin and carboplatin treated cells with dose-dependent manner (Fig. S6F). Overall, these experiments validated the cell line studies with patient tumor samples and show that belinostat may serve as a general anti-CSL inhibitor following multiple chemotherapies in ER+ breast cancer, reflecting a one-two punch treatment strategy.
Belinostat inhibits chemotherapy induced mammosphere formation
We next tested the ability of belinostat to modulate CSL self-renewal and proliferation using a mammosphere formation assay [24]. Two cycles of mammosphere culture were performed to select stem-like cells with self-renewal ability and eliminate various transitions following chemotherapy +/- belinostat. Both ER+ (CAMA-1) cells and TNBC (MDA-MB-231) cells show a significantly diminished mammosphere formation following belinostat treatment after chemotherapy after two-cycle cultures (Fig. 4A, B, C, D).
Fig. 4.

Belinostat inhibited mammosphere formation of chemotherapy-treated breast cancer cell lines. CAMA-1 and MDA-MB-231 cells from spheroids with 3 days chemo treatment (DMSO, doxorubicin 0.1 µM for CAMA-1, 0.5 µM for MDA-MB-231, carboplatin 50 µM, and paclitaxel 1 µM) were collected, dissociated, and cultured for two 14 days periods with mammosphere assay medium containing DMSO/belinostat (5 µM) as described in the Methods. Mammosphere total numbers and total area of CAMA-1 are shown in (A) and (B). Mammosphere total numbers and total area of MDA-MB-231 are shown in (C) and (D). The treatments with DMSO only serve as the control in each figure and are set as fold one. All the other treatments are expressed as fold change relative to the control in each figure. The statistical analysis was performed with student's t-tests between the indicated groups with triplicate tests. # is p<0.05 relative to DMSO plus DMSO only, * is p<0.05 relative to chemo/DMSO plus DMSO in its group.
For the ER+ cells, doxorubicin and paclitaxel both increased mammosphere counts (up to 174.1% and 131.1%), and mammosphere total areas (up to 257.9% and 211.9%) compared to DMSO control (set to 100% for each condition). Strikingly, belinostat dramatically reduced the mammosphere counts (to 2.9–3.7%) and mammosphere total areas (to 2.3–6.7%) following all chemotherapy treatments. Similarly, for the TNBC cells, all chemotherapies strongly increased mammosphere counts (up to 490.6%, 330.8%, and 353.8%), and mammosphere total areas (up to 736.2%, 320.8%, and 366.6%) compared to DMSO control. Belinostat again reduced the mammosphere counts (42.7–51.3%) and mammosphere total areas (21.5–71.9%) following all chemotherapy treatments. Thus, the general anti-CSL effect of belinostat in combination with chemotherapy is a one-two treatment with the ability to inhibit CSL self-renew and proliferation with mammosphere assays.
Class II HDACs are involved in chemo-acquired CSL reversal
As the pan-HDAC inhibitor belinostat, but not the Class I HDACs inhibitor, entinostat [25, 26] reversed CSL states following chemotherapy treatments in all test cell lines, HDAC inhibitors targeting specific HDAC isoforms were tested in order to define HDAC proteins essential for this activity using the FACS strategy described above (Fig. 2). Tested were Class I HDACs inhibitors SantacruzanateA (HDAC2i) [27], RGFP966 (HDAC3i) [26], PCI-34,051 (HDAC8i) [25]; Class IIa HDACs inhibitor LMK-235 (HDAC4/5i) [28]; and Class IIb HDACs inhibitor CAY10603 (HDAC6i) [29, 30]. As shown in Fig. 5A and 5B, the HDAC4/5 inhibitor LMK-235 and HDAC6 inhibitor CAY10603 consistently reversed ALDH levels, CSL/Live, and CSL/Total promoted by all three chemotherapy treatments in both cell lines. Both inhibitors reversed CD44 levels in all three chemotherapy treatments in MDA-MB-231 and doxorubicin and carboplatin induced CD44 levels in CAMA-1. Other specific HDAC inhibitors did not reverse these primitive traits. These results suggest that HDAC class IIa and IIb regulate HDAC mediated CSL traits plasticity.
Fig. 5.

Specific HDAC isoforms target chemotherapy-acquired CSL state. The FACS mean values of ALDH and CD44 in live populations, CSL/Live, CSL/Total, and viability for CSL reversal drugs screening were expressed in heat maps for CAMA-1 (A) and MDA-MB-231 (B) cell lines. Cells were cultured in medium (3D) and treated for 72 h with doxorubicin (0.1 µM for CAMA-1, 0.5 µM for MDA-MB-231), carboplatin (50 µM for all cell lines), and paclitaxel (1 µM for all cell lines), and then treated with specific HDAC isoforms inhibitors as indicated doses for 72 h before ALDEFLUOR/CD44 staining. The vehicle controls (DMSO+DMSO) were set as fold one. All the other chemotherapy plus DMSO/inhibitor combinations were expressed as the fold change relative to the controls. The Chemo plus DMSO served as the positive controls. The mean values of triplicate tests were showed with color indicated as the legends. In heat maps of ALDH, CD44, CSL/Live, and CSL/Total, # is chemo plus DMSO significantly higher (p<0.05) than no treatment control, and in heat maps of viability, # is chemo plus DMSO significantly lower (p<0.05) than no treatment control. In all heat maps, * is chemo plus inhibitor significantly lower (p<0.05) than chemo plus DMSO. The potential specific HDAC isoforms inhibitors target to CSL cells are highlighted with red blocks as its reversal effect for both ALDH and CD44.
To further test the CSL state reversal effect of Class II HDACs inhibitors, a time-course FACS analysis was performed with CAMA-1 and MDA-MB-231 cell lines treated with LMK-235 or CAY10603 following three chemotherapies (Fig. 6A-D, and Fig. S7A-D). Both Class II HDAC inhibitors decreased cell viability compared to chemotherapy treatment alone, and exhibited strong CSL reversal activity similar to belinostat, including decreases in ALDH levels and statistically significant diminished levels of both CSL/Live and CSL/Total following chemotherapy. Detailed FACS plots at Day 6 (Fig. S8A and S8B) exhibit the significant reduction in the number of CSL cells constrained in the ALDH+, CD44+ positive quadrants with chemotherapy followed by class II HDAC inhibitors compared to chemotherapy alone. Taken together, these results indicate that class II HDACs (HDAC4/5/6) regulate chemo-acquired stemness states in one-two punch strategy.
MYC pathways mediated CSL promotion/reversal upon chemotherapy/belinostat treatment
To understand the mechanism of how belinostat reverses the chemotherapy-induced CSL properties, we performed single-cell RNA-Seq analysis with two ER+ patient samples (Patient#5 and Patient#6). The unbiased clustering of the gene expression profiles of the Patient#6 and Patient#5 cells indicate changes in the gene expression profiles of the cells after chemotherapy treatments and combination chemotherapies plus belinostat (Fig. 7A, B). The Patient#6 cells exhibited relatively similar transcriptional programs to all chemotherapies, and also to the combination treatments. Chemotherapy generated a distinctive cell population that was further changed by belinostat (Fig. 7A), indicating that belinostat can reverse the stem cell phenotypes, but not all aspects of the chemotherapy. In contrast, the Patient#5 cells showed a drug-specific response where each chemotherapy led to distinct changes in the cell populations, and relatively little changes in the transcriptional programs were induced by belinostat (Fig. 7B) .
Fig. 7.

Chemotherapy and belinostat reprogrammed transcriptome of patient cells. Unbiased UMAPs for ER+ patient sample Patient#6 (A) and Patient#5 (B) under treatment (Tx). Patient cells were cultured in Renaissance medium (3D) and treated with chemotherapy (doxorubicin 0.1 µM, carboplatin 0.1 mM, paclitaxel 1 µM) for 72 h, and then treaded with DMSO/belinostat (1 µM) for 72 h. The cells were collected, purified with Dead Cell Removal Kit, and subjected to ICELL8Ⓡ Single-Cell System as described in Methods. For each patient, the UMAPs for all treatments were shown in the top, and the UMAPs for each individual treatment were shown in the bottom with different colors indicated as the legends. Violin plots with GSEA pathways enrichment scores related to HDAC targets, stem cell differentiation and MYC pathway were presented in (C) for Patient#6 and (D) for Patient#5. The statistical analysis was performed with student's t-test between the indicated groups, and significance is marked with * for P<0.05, ** for P<0.01, *** for P<0.001, **** for P<0.0001, *****for P<0.00001.
To focus on the specific (rather than overall) changes seen with drug treatment, we performed a pathway analysis to identify the pathways that are altered by chemotherapy or combination treatment. Shared reversal pathways were selected as described in Methods, and heat maps were generated based on the averages of ssGSEA enrichment scores for the shared reversal pathways for both Patient#6 (Fig. S9,and Fig. S10) and Patient#5 (Fig. S11 and Fig. S12). For each patient sample, we selected the pathways that were either increased by chemotherapy and decreased by belinostat or the inverse pattern (Fig. S9-S12). We found that HDAC pathway (HELLER_HDAC_TARGETS_DN) was up-regulated by all three chemotherapy treatments in both patient samples and down-regulated by belinostat except paclitaxel group in Patient#6 (Fig. 7C, D). This indicates that chemotherapy treatments promoted HDAC targets in this gene set (eg, CD44 as stemness marker, BCL2 as anti-apoptotic regulator), while belinostat reversed them; concomitant with the changes in CSL properties. We also observed that a stem cell differentiation signature (BOQUEST_STEM_CELL_CULTURED_VS_FRESH_UP) was reduced by all three chemotherapy treatments and promoted by belinostat in both patient samples (Fig. 7C, D). In another words, the CSL state of the survival cells were up-regulated by all chemotherapy treatments, and down-regulated by belinostat. Consistent with CSL state, the MYC pathway (BILD_MYC_ONCOGENIC_SIGNATURE) was increased by most of chemotherapy treatments, and inhibited by belinostat except paclitaxel group in both Patients. HDAC inhibition has been reported to down-regulate MYC expression transcriptionally and post-transcriptionally through regulation of MYC acetylation [31], in turn to up-regulate of CDKN1A/B (p21/CIP1/WAF1, p27/KIP1) to induce cell cycle arrest and autophagic cell death [32, 33]. Thus, MYC, as a key factor in CSC reprogramming and self-renewal in basal-like breast cancer [34], may contribute to reversal of the CSL promotion-reversal process mediated by chemotherapy- HDACi treatment.
Discussion and conclusion
Cancer stem cells (CSCs) are not targeted by conventional chemotherapy, initiating growth of new tumors as well as causing disease relapse. [35]. Therefore, there remains potential in developing treatment approaches that minimize CSC or CSL cells to delay or prevent refractory cancer development. Our study indicates that subclonal selection in patients during therapy promotes CSL state in breast cancer. Further, both flow cytometry and mammosphere assays show a reversal of CSL state following chemotherapy by the use of the HDAC inhibitor belinostat. Specifically, Class II HDACs are found to target chemo-acquired CSL phenotypes. Single-cell RNA sequencing analysis following the one-two punch treatment strategy indicated that the RNA transcriptome was strongly reprogramed by chemotherapy treatments, and reversed by belinostat, which promoted stem cell differentiation, and inhibited HDAC targets and MYC pathway.
Although HDAC inhibitors have been used to suppress the CSC population for their ability to block key signaling pathways pertinent to CSC maintenance[14, 36], how HDAC inhibitors reverse CSL state following standard of care chemotherapies is not well-defined. Previous clinical trials of HDACi target to CSL cells focused either on the monotherapy for refractory peripheral T-cell lymphoma, or concurrent combination of chemotherapy with HDACi on solid tumors [37, 38], assessing antitumor activity by apoptosis/viability test, more than targeting to acquired stemness. It has not been studied if specific chemotherapies or this class of drug generally, can induce CSL state, and if HDAC inhibitors can reverse CSL states driven by different chemotherapies. As detailed results above, our study addresses subclonal evolution of stem-like traits in patients as they acquire resistance to chemotherapy, as well as approaches to reverse this acquired resistant trait.
In this study, we initially tested multiple drugs published previously to block stem cell-like state. Only one was effective following chemotherapy induced stem-like states across cell lines and patient tumor cells. Although there was some heterogeneity in ALDH or CD44 expression level modulated by different chemotherapies, we generally saw an increase in CSL levels for ER+ tumor cells and to a lesser extent TNBC cells. Belinostat broadly blocked increased CSL cells following chemotherapy treatment based on flow cytometry and mammosphere assays. The mammosphere formation assays demonstrated that belinostat could abolish the self-renewal capability of survival cells after chemotherapy treatments, which has resulted in resistant CSL subtypes. Importantly, these results were consistent across different chemotherapies tested, suggesting a convergence on CSL resistance.
In our FACS strategy to identify the CSL cells, ALDH and CD44 were selected as CSL state markers rather than CD24 because ALDHhiCD44+ CSL breast cancer cells has been found to contribute to chemotherapy resistance [20]. Of note, the initial CSL state of ER+ CAMA-1 cells starts from very low primitive cell traits (indicated by low CD44 level, between 102 and 103) compared to TNBC MDA-MB-231 cells (indicated by high CD44 level, between 103 and 105), which might suggests higher potential for CSL state induction with chemotherapy for ER+ cells than TNBC, as TNBC already show high levels of CSL (see detailed FACS analysis following chemotherapies in Fig. S8A, B). Compared to the FACS analysis, the mammosphere assay tests the self-renewal ability of resistant cells rather than the changes of stemness makers. Cells with high level expression of CD44 are more resistant to chemotherapy and more likely to form a mammosphere [23], which would benefit TNBC more than ER+ cells due to their enrichment of progenitor cells (ALDH negative, CD44 high). In this case, the inhibition effect of belinostat on CD44+ cells could play a major role to abolish the self-renewal capability of chemo-resistant cells. Reversal of CD44 in ER+ cell lines/patient samples and inhibition of ALDH in TNBC cell lines were also identified by FACS analysis, and varied among cell lines and patient samples with different chemotherapies. Future research may help delineate cell populations sensitive to belinostat treatment. While this study focused on the ER+ subtype as we are targeting the chemo promoted CSL traits reversal, and the results from both cell lines and patient samples indicated that the chemotherapy promoted primitive traits are more pronounced in ER+ cells than TNBC cells, we do see the CSL reversal in all, suggesting a more global reversal effect. In fact, given the high levels of baseline CSL in TNBC, belinostat may be effective therapy in this population even without chemotherapy.
HDACs can be classified into four classes (class I to IV): Class I (HDAC1, HDAC2, HDAC3, and HDAC8). Class IIa (HDAC4, HDAC5, HDAC7, HDAC9) and IIb (HDAC 6 and 10), class III (sirtuins 1–7), and class IV (HDAC11) [39]. Class I HDACs only localize in nucleus, while Class II HDACs exhibit their ability to shuttle between nucleus and cytoplasm, and can deacetylate non-histone proteins in cytoplasm [36, 39]. Positive associations of high expression of Class II HDACs with cell proliferation and CSC differentiation were found in various cancer types for HDAC4 [40, 41], HDAC 5 [42, 43], and HDAC6 [44, 45, 46]. HDAC5 and HDAC6 transcriptional regulation on MYC has been reported to play a key role in these processes [30, 44, 46]. The HDAC4/5i LMK-235 and HDAC6i CAY10603 both exhibited their CSL reversal effect on reducing CSL abundance and additional cytotoxicity, suggesting their strong potential on clinical therapy to eliminate the chemo-promoted CSL cells. Further research is needed to dissect the role of class II HDACs in mediating CSL traits in chemotherapy-HDACi combination.
Taken together, our findings demonstrated inhibition on HDACs, especially Class II HDACs (HDAC4/5/6), reverses chemotherapy-induced acquisition of primitive cell traits and drug-resistance in ER+ breast cancer, and provided an effective ‘one-two punch’ strategy to combat refractory ER+ breast cancer with sequential chemotherapy and HDACi combination.
Funding
This research was supported by the National Cancer Institute of the National Institutes of Health under U54 Support Award (1U54CA209978NIH) and Award Number P30CA042014. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. J.T.C. was supported by the Cancer Prevention Research Institute of Texas Core Facility Support Award RP170668.
Authors' contributions
Feng Chi: Methodology; Formal analysis; Investigation; Writing - Original Draft; Writing - Review & Editing. Jiayi Liu: Methodology; Software; Formal analysis; Investigation. Samuel W. Brady: Methodology; Software; Formal analysis; Investigation; Writing - Original Draft. Patrick A. Cosgrove: Methodology; Investigation. Aritro Nath: Methodology; Software; Investigation; Validation; Data Curation. Jasmine A. McQuerry: Formal analysis; Writing - Original Draft. Sumana Majumdar: Resources. Philip J. Moos: Resources. Jeffrey T. Chang: Methodology; Software; Validation; Data Curation; Writing - Original Draft; Funding acquisition. Michael Kahn: Methodology; Resources. Andrea H. Bild: Formal analysis; Validation; Visualization; Writing - Original Draft; Writing - Review & Editing; Supervision; Project administration; Funding acquisition.
Data and materials availability
The scRNA-Seq datasets generated and/or analysed during the current study are available in the Gene Expression Omnibus (GEO) database under accession GSE147326.
Ethics approval and consent to participate
Informed consent was obtained from all patients in this study. Protocols were approved by the University of Utah Institutional Review Board (IRB 20357) and the City of Hope Institutional Review Board (IRB 17334).
Declaration of competing interest
The authors declare that they have no competing interests
Acknowledgments
We thank the anonymous patients used in our study for their generous contributions to research. The AXL inhibitor BGB324 was provided by BerGenBio. Research reported in this publication utilized the High-Throughput Genomics and Bioinformatic Analysis Shared Resource at Huntsman Cancer Institute at the University of Utah
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.tranon.2020.100946.
Appendix. Supplementary materials
References
- 1.Gonzalez-Angulo A.M., Morales-Vasquez F., Hortobagyi G.N. Overview of resistance to systemic therapy in patients with breast cancer. Adv. Exp. Med. Biol. 2007;608:1–22. doi: 10.1007/978-0-387-74039-3_1. [DOI] [PubMed] [Google Scholar]
- 2.Malhotra V., Perry M.C. Classical chemotherapy: mechanisms, toxicities and the therapeutic window. Cancer Biol. Ther. 2003;2(4 Suppl 1):S2–S4. [PubMed] [Google Scholar]
- 3.Chabner B.A., Roberts T.G., Jr Timeline: chemotherapy and the war on cancer. Nat. Rev. Cancer. 2005;5(1):65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen D., Bos P.D., Massague J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer. 2009;9(4):274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 5.Valent P., Bonnet D., De Maria R., Lapidot T., Copland M., Melo J.V., Chomienne C., Ishikawa F., Schuringa J.J., Stassi G. Cancer stem cell definitions and terminology: the devil is in the details. Nat. Rev. Cancer. 2012;12(11):767–775. doi: 10.1038/nrc3368. [DOI] [PubMed] [Google Scholar]
- 6.Al-Hajj M., Wicha M.S., Benito-Hernandez A., Morrison S.J., Clarke M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palomeras S., Ruiz-Martínez S., Puig T. Targeting breast cancer stem cells to overcome treatment resistance. Molecules. 2018;23(9):E2193. doi: 10.3390/molecules23092193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lombardo Y., de Giorgio A., Coombes C.R., Stebbing J., Castellano L. Mammosphere formation assay from human breast cancer tissues and cell lines. J. Vis. Exp. 2015;(97) doi: 10.3791/52671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luqmani Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005;14(Suppl 1):35–48. doi: 10.1159/000086183. [DOI] [PubMed] [Google Scholar]
- 10.Chu G. Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair. J. Biol. Chem. 1994;269(2):787–790. [PubMed] [Google Scholar]
- 11.Creighton C.J., Li X., Landis M., Dixon J.M., Neumeister V.M., Sjolund A., Rimm D.L., Wong H., Rodriguez A., Herschkowitz J.I. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. U. S. A. 2009;106(33):13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groenendijk F.H., Bernards R. Drug resistance to targeted therapies: Deja vu all over again. Mol. Oncol. 2014;8(6):1067–1083. doi: 10.1016/j.molonc.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doherty M.R., Smigiel J.M., Junk D.J., Jackson M.W. Cancer stem cell plasticity drives therapeutic resistance. Cancers (Basel) 2016;8(1):E8. doi: 10.3390/cancers8010008. pii: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin P.C., Hsieh H.Y., Chu P.C., Chen C.S. Therapeutic opportunities of targeting histone deacetylase isoforms to eradicate cancer stem cells. Int. J. Mol. Sci. 2018;19(7):E1939. doi: 10.3390/ijms19071939. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma S.V., Lee D.Y., Li B., Quinlan M.P., Takahashi F., Maheswaran S., McDermott U., Azizian N., Zou L., Fischbach M.A. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim C., Gao R., Sei E., Brandt R., Hartman J., Hatschek T., Crosetto N., Foukakis T., Navin N.E. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell. 2018;173(4):879–893. doi: 10.1016/j.cell.2018.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sachs N., de Ligt J., Kopper O., Gogola E., Bounova G., Weeber F., Balgobind A.V., Wind K., Gracanin A., Begthel H. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172(1–2):373–386. doi: 10.1016/j.cell.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 18.Colacino J.A., Azizi E., Brooks M.D., Harouaka R., Fouladdel S., McDermott S.P., Lee M., Hill D., Madden J., Boerner J. Heterogeneity of human breast stem and progenitor cells as revealed by transcriptional profiling. Stem Cell Rep. 2018;10(5):1596–1609. doi: 10.1016/j.stemcr.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brady S.W., McQuerry J.A., Qiao Y., Piccolo S.R., Shrestha G., Jenkins D.F., Layer R.M., Pedersen B.S., Miller R.H., Esch A. Combating subclonal evolution of resistant cancer phenotypes. Nat. Commun. 2017;8(1):1231. doi: 10.1038/s41467-017-01174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Croker A.K., Allan A.L. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44⁺ human breast cancer cells. Breast Cancer Res. Treat. 2012;133(1):75–87. doi: 10.1007/s10549-011-1692-y. [DOI] [PubMed] [Google Scholar]
- 21.Charafe-Jauffret E., Ginestier C., Iovino F., Wicinski J., Cervera N., Finetti P., Hur M.H., Diebel M.E., Monville F., Dutcher J. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69(4):1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim S., Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140(15):3079–3093. doi: 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- 23.Holiday D.L., Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13(4):215. doi: 10.1186/bcr2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw F.L., Harrison H., Spence K., Ablett M.P., Simões B.M., Farnie G., Clarke R.B. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J. Mammary Gland Biol. Neoplasia. 2012;17(2):111–117. doi: 10.1007/s10911-012-9255-3. [DOI] [PubMed] [Google Scholar]
- 25.Schölz C., Weinert B.T., Wagner S.A., Beli P., Miyake Y., Qi J., Jensen L.J., Streicher W., McCarthy A.R., Westwood N.J., Choudhary C. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat. Biotechnol. 2015;33(4):415–423. doi: 10.1038/nbt.3130. [DOI] [PubMed] [Google Scholar]
- 26.Beyer M., Romanski A., Mustafa A.M., Pons M., Büchler I., Vogel A., Pautz A., Sellmer A., Schneider G., Bug G. HDAC3 activity is essential for human leukemic cell growth and the expression of β-catenin, MYC, and WT1. Cancers (Basel) 2019;11(10):E1436. doi: 10.3390/cancers11101436. 26pii: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou H., Cai Y., Liu D., Li M., Sha Y., Zhang W., Wang K., Gong J., Tang N., Huang A. Pharmacological or transcriptional inhibition of both HDAC1 and 2 leads to cell cycle blockage and apoptosis via p21Waf1/Cip1 and p19INK4d upregulation in hepatocellular carcinoma. Cell Prolif. 2018;51(3):e12447. doi: 10.1111/cpr.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xue Y., Lian W., Zhi J., Yang W., Li Q., Guo X., Gao J., Qu H., Lin W., Li Z. HDAC5-mediated deacetylation and nuclear localisation of SOX9 is critical for tamoxifen resistance in breast cancer. Br. J. Cancer. 2019;121(12):1039–1049. doi: 10.1038/s41416-019-0625-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma X.J., Xu G., Li Z.J., Chen F., Wu D., Miao J.N., Zhan Y., Fan Y. HDAC-selective inhibitor Cay10603 has single anti-tumour effect in Burkitt's lymphoma cells by impeding the cell cycle. Curr. Med. Sci. 2019;39(2):228–236. doi: 10.1007/s11596-019-2024-4. [DOI] [PubMed] [Google Scholar]
- 30.Bitler B.G., Wu S., Park P.H., Hai Y., Aird K.M., Wang Y., Zhai Y., Kossenkov A.V., Vara-Ailor A., Rauscher F.J., III ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017;19(8):962–973. doi: 10.1038/ncb3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nebbioso A., Carafa V., Conte M., Tambaro F.P., Abbondanza C., Martens J., Nees M., Benedetti R., Pallavicini I., Minucci S. c-MYC modulation and acetylation is a key HDAC inhibitor target in cancer. Clin. Cancer Res. 2017;23(10):2542–2555. doi: 10.1158/1078-0432.CCR-15-2388. [DOI] [PubMed] [Google Scholar]
- 32.Poli V., Fagnocchi L., Fasciani A., Cherubini A., Mazzoleni S., Ferrillo S., Miluzio A., Gaudioso G., Vaira V., Turdo A. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat. Commun. 2018;9(1):1024. doi: 10.1038/s41467-018-03264-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klieser E., Urbas R., Stättner S., Primavesi F., Jäger T., Dinnewitzer A., Mayr C., Kiesslich T., Holzmann K., Di Fazio P. Comprehensive immunohistochemical analysis of histone deacetylases in pancreatic neuroendocrine tumors: HDAC5 as a predictor of poor clinical outcome. Hum. Pathol. 2017;65:41–52. doi: 10.1016/j.humpath.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Yang A., Qin S., Schulte B.A., Ethier S.P., Tew K.D., Wang G.Y. MYC inhibition depletes cancer stem-like cells in triple-negative breast cancer. Cancer Res. 2017;77(23):6641–6650. doi: 10.1158/0008-5472.CAN-16-3452. Sulaiman A, McGarry S, Han X, Liu S, Wang L. CSCs in Breast Cancer-One Size Does Not Fit All: Therapeutic Advances in Targeting Heterogeneous Epithelial and Mesenchymal CSCs. Cancers (Basel). 2019;11(8): E1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu N., Li S., Wu N., Cho K.S. Acetylation and deacetylation in cancer stem-like cells. Oncotarget. 2017;8(51):89315–89325. doi: 10.18632/oncotarget.19167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poole R.M. Belinostat: first global approval. Drugs. 2014;74(13):1543–1554. doi: 10.1007/s40265-014-0275-8. [DOI] [PubMed] [Google Scholar]
- 37.Hii L.W., Chung F.F., Soo J.S., Tan B.S., Mai C.W., Leong C.O. Histone deacetylase (HDAC) inhibitors and doxorubicin combinations target both breast cancer stem cells and non-stem breast cancer cells simultaneously. Breast Cancer Res. Treat. 2020;179(3):615–629. doi: 10.1007/s10549-019-05504-5. [DOI] [PubMed] [Google Scholar]
- 38.Seto E., Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014;6(4) doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang Z.H., Wang C.Y., Zhang W.L., Zhang J.T., Yuan C.H., Zhao P.W., Lin Y.Y., Hong S., Li C.Y., Wang L. Histone deacetylase HDAC4 promotes gastric cancer SGC-7901 cells progression via p21 repression. PLoS ONE. 2014;9(6):e98894. doi: 10.1371/journal.pone.0098894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song B., Wang Y., Xi Y., Kudo K., Bruheim S., Botchkina G.I., Gavin E., Wan Y., Formentini A., Kornmann M. Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene. 2009;28(46):4065–4074. doi: 10.1038/onc.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li A., Liu Z., Li M., Zhou S., Xu Y., Xiao Y., Yang W. HDAC5, a potential therapeutic target and prognostic biomarker, promotes proliferation, invasion and migration in human breast cancer. Oncotarget. 2016;7(25):37966–37978. doi: 10.18632/oncotarget.9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao M., Li L., Zhou J., Cui X., Tian Q., Jin Y., Zhu Y. MiR-2861 behaves as a biomarker of lung cancer stem cells and regulates the HDAC5-ERK System genes. Cell Reprogram. 2018;20(2):99–106. doi: 10.1089/cell.2017.0045. [DOI] [PubMed] [Google Scholar]
- 43.Hsieh T.H., Tsai C.F., Hsu C.Y., Kuo P.L., Lee J.N., Chai C.Y., Wang S.C., Tsai E.M. Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J. 2012 Feb;26(2):778–787. doi: 10.1096/fj.11-191742. [DOI] [PubMed] [Google Scholar]
- 44.Yang W., Liu Y., Gao R., Yu H., Sun T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018 Feb 28;415:164–176. doi: 10.1016/j.canlet.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Li Y., Zhang X., Polakiewicz R.D., Yao T.P., Comb M.J. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J. Biol. Chem. 2008;283(19):12686–12690. doi: 10.1074/jbc.C700185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.North B.J., Almeciga-Pinto I., Tamang D., Yang M., Jones S.S., Quayle S.N. Enhancement of pomalidomide anti-tumor response with ACY-241, a selective HDAC6 inhibitor. PLoS ONE. 2017;12(3) doi: 10.1371/journal.pone.0173507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.