

Highlights
-
•
Cortical signatures selective to AD could provide an early MRI biomarker.
-
•
Autosomal dominant Alzheimer disease (ADAD) may model an ideal AD signature.
-
•
ADAD and late-onset maps overlap in parietal cortex but contain unique features.
-
•
Signatures predicted increasing amyloid within their own, but not across cohorts.
-
•
These results indicate atrophy in AD can take multiple spatial patterns.
Keywords: Alzheimer disease, Autosomal dominant Alzheimer disease, Preclinical, Cortical signature, Amyloid, Cortical thickness
Abbreviations: AD, Alzheimer disease; ADAD, autosomal dominant Alzheimer disease; APOE, apolipoprotein E; APP, amyloid precursor protein; AUROC, area under the receiver operating characteristic; AV-45, florbetapir; CDR, clinical dementia rating; CNADRC/DIAN, cognitively normal controls; DIAN, Dominantly Inherited Alzheimer Network; HCV, total hippocampal volume; Knight ADRC, Knight Alzheimer Disease Research Center; LOAD, late-onset Alzheimer disease; mcSUVR, mean cortical standardized uptake value ratio; MRI, magnetic resonance imaging; PCADRC, preclinical Alzheimer disease; PCDIAN, preclinical autosomal dominant Alzheimer disease; PET, positron emission tomography; PiB, Pittsburg compound-B; PSEN1, Presenilin 1; PSEN2, Presenilin 2; ROC, receiver operating characteristic; ROI, region of interest; SUVR, standardized uptake value ratio
Abstract
Defining a signature of cortical regions of interest preferentially affected by Alzheimer disease (AD) pathology may offer improved sensitivity to early AD compared to hippocampal volume or mesial temporal lobe alone. Since late-onset Alzheimer disease (LOAD) participants tend to have age-related comorbidities, the younger-onset age in autosomal dominant AD (ADAD) may provide a more idealized model of cortical thinning in AD. To test this, the goals of this study were to compare the degree of overlap between the ADAD and LOAD cortical thinning maps and to evaluate the ability of the ADAD cortical signature regions to predict early pathological changes in cognitively normal individuals.
We defined and analyzed the LOAD cortical maps of cortical thickness in 588 participants from the Knight Alzheimer Disease Research Center (Knight ADRC) and the ADAD cortical maps in 269 participants from the Dominantly Inherited Alzheimer Network (DIAN) observational study. Both cohorts were divided into three groups: cognitively normal controls (nADRC = 381; nDIAN = 145), preclinical (nADRC = 153; nDIAN = 76), and cognitively impaired (nADRC = 54; nDIAN = 48). Both cohorts underwent clinical assessments, 3T MRI, and amyloid PET imaging with either 11C-Pittsburgh compound B or 18F-florbetapir.
To generate cortical signature maps of cortical thickness, we performed a vertex-wise analysis between the cognitively normal controls and impaired groups within each cohort using six increasingly conservative statistical thresholds to determine significance. The optimal cortical map among the six statistical thresholds was determined from a receiver operating characteristic analysis testing the performance of each map in discriminating between the cognitively normal controls and preclinical groups. We then performed within-cohort and cross-cohort (e.g. ADAD maps evaluated in the Knight ADRC cohort) analyses to examine the sensitivity of the optimal cortical signature maps to the amyloid levels using only the cognitively normal individuals (cognitively normal controls and preclinical groups) in comparison to hippocampal volume.
We found the optimal cortical signature maps were sensitive to early increases in amyloid for the asymptomatic individuals within their respective cohorts and were significant beyond the inclusion of hippocampus volume, but the cortical signature maps performed poorly when analyzing across cohorts. These results suggest the cortical signature maps are a useful MRI biomarker of early AD-related neurodegeneration in preclinical individuals and the pattern of decline differs between LOAD and ADAD.
1. Introduction
Alzheimer disease (AD) is defined pathologically by the accumulation of amyloid plaques and neurofibrillary tangles. Amyloid pathology is detectable almost 20 years before symptom onset and is followed by limbic and neocortical neurofibrillary tangles (Bateman et al., 2012, Hardy and Higgins, 1992, Jack et al., 2013). Cognitively, AD manifests as a cortical dementia. Cerebral atrophy occurs nearer to onset and is more closely related, spatially and temporally, to cognitive decline. (Aschenbrenner et al., 2018, Brier et al., 2016a, Ossenkoppele et al., 2019). Estimates of volume and cortical thickness from magnetic resonance imaging (MRI) are one of the most common measures of neurodegeneration and have been incorporated into recommendations for using biomarkers to study AD (Jack et al., 2018). Determining the specific brain regions most susceptible to early neurodegeneration in AD would help identify individuals most at risk of cognitive decline.
Guided by neuropathological studies (Braak and Braak, 1995), much of AD research has focused on atrophy of the mesial temporal lobe and the hippocampus in particular (Apostolova et al., 2010, Bouwman et al., 2007, Convit et al., 1997, Gordon et al., 2016, Huijbers et al., 2015, Martin et al., 2010, Ridha et al., 2006, Vos et al., 2016). Late-onset Alzheimer disease (LOAD) is the most common form of AD and typically develops in individuals aged 65 and older. However, older healthy and AD individuals are known to have a high frequency of comorbidities, which can affect brain volumetrics (e.g. diabetes, hypertension, cerebrovascular disease) including hippocampal size (e.g., hippocampal sclerosis) (Faraco and Iadecola, 2013, Jagust et al., 2008, Moran et al., 2013, Zarow et al., 2008). These comorbidities make the hippocampus a relatively nonspecific structure for measuring neurodegeneration.
Looking beyond the hippocampus, previous studies have defined a composite of brain regions susceptible to AD-related cortical atrophy, known as AD cortical signatures (Dickerson et al., 2009, Jack et al., 2017, Wang et al., 2015) and have shown cortical signatures relate to cognitive performance in AD beyond the effects of age (Bakkour et al., 2013). Voxel or vertex-wise approaches (Dickerson et al., 2009, Wang et al., 2015) with modest sample sizes often led to statistical maps with a series of peak effects spread throughout cortical and subcortical areas. Approaches using a larger region of interest (ROI) derived from programs such as FreeSurfer avoid the small focal effects but may incorporate areas of the brain not affected by AD. We therefore propose that an ideal signature region should be large enough to be stable across multiple cohorts, yet specific enough that it does not incorporate non-informative regions.
Although AD cortical signatures include numerous brain regions, the age-related comorbidities in the LOAD population may still limit the sensitivity to detect AD-related neurodegeneration in asymptomatic individuals. Autosomal dominant Alzheimer disease (ADAD) is a rare form of AD caused by a mutation in one of three genes: amyloid precursor protein (APP), presenilin1 (PSEN1), or presenilin2 (PSEN2). ADAD has virtually complete penetrance and a consistent age of onset within families for each individual mutation (Bateman et al., 2010). Since ADAD has an earlier age of onset compared to LOAD (Ryman et al., 2014), ADAD individuals typically do not have age-related comorbidities. Prior work looking at atrophy in ADAD cohorts using ROI (Benzinger et al., 2013, Gordon et al., 2018a, Weston et al., 2016) and voxel-wise approaches (Scahill et al., 2013, Scahill et al., 2002) found a robust atrophy signature. Therefore, ADAD may provide a less biased depiction needed to identify changes in the preclinical stages of AD.
In this study, we sought to produce a refined AD cortical atrophy signature by 1) creating a signature without ROI restrictions and with a large cohort for more reliable regions, 2) comparing the degree of overlap between the ADAD and LOAD signatures, and 3) comparing the abilities of the ADAD and LOAD signatures to predict early amyloid pathological changes in the cognitively normal groups within and across cohorts.
2. Materials and methods
2.1. Participants
Two datasets were used to define and analyze AD cortical signatures of cortical thickness. For both datasets, dementia severity was defined by the global Clinical Dementia Rating (CDR) (Morris, 1993). The LOAD participants were selected from ongoing longitudinal studies of memory and aging at the Knight Alzheimer Disease Research Center (Knight ADRC) at Washington University in Saint Louis. All participants from the 12th semiannual Knight ADRC data release that had at least one 3 Tesla (3T) MRI scan, an amyloid positron emission tomography (PET) scan within a year of the MRI, a clinical assessment within two years of the MRI, and demographic data including age, sex, and apolipoprotein E (APOE) genotype were included in the study (n = 703). The date of the MRI scan was considered the baseline visit. Participants were subsequently divided into three groups: cognitively normal controls (CNADRC, CDR 0 and amyloid negative for all visits, n = 381), preclinical LOAD (PCADRC, CDR 0 and amyloid positive at baseline visit, n = 153), and cognitively impaired (LOAD, CDR 0.5 and amyloid positive at baseline visit and remains CDR 0.5 or higher at subsequent visits, n = 54). 115 participants were excluded as follows: 43 participants were CDR 0.5 and amyloid negative; 37 participants were CDR 0 and amyloid negative at baseline visit but did not remain so for all other visits; 26 participants were CDR > 0.5 at baseline visit; 6 participants were CDR 0.5 and amyloid positive at baseline visit but did not remain CDR 0.5 or higher for subsequent visits; 3 participants had a non-AD diagnosis.
ADAD participants were selected from the Dominantly Inherited Alzheimer Network (DIAN) observational study (Morris et al., 2012, Moulder et al., 2013). Participants are recruited from families having an ADAD pathogenic mutation in APP, PSEN1, or PSEN2 genes. Participant enrollment and procedures have been previously defined (Morris et al., 2012). For this study, participants from the 11th DIAN annual data release were required to have the same criteria as the Knight ADRC cohort (n = 386). Participants were then divided into three groups: cognitively normal mutation non-carrier controls (CNADAD, CDR 0 and amyloid negative for all time points, n = 145), preclinical ADAD mutation carriers (PCADAD, CDR 0 and amyloid positive at baseline visit, n = 76), and cognitively impaired mutation carriers (ADAD, CDR 0.5 and amyloid positive at baseline visit and remains CDR 0.5 or higher at subsequent visits, n = 48). 117 participants were excluded as follows: 65 participants were CDR 0 and amyloid negative mutation carriers; 29 participants were CDR > 0.5 at baseline visit; 18 participants were CDR 0.5 and amyloid negative; 2 participants were CDR 0 and amyloid negative mutation non-carriers at baseline visit but did not remain so for all other visits; 2 participants were CDR 0 and amyloid positive mutation non-carriers; 1 participant was CDR 0.5 and an amyloid positive mutation carrier but did not remain CDR 0.5 or higher for subsequent visits.
2.2. Ethics statement
All participants or their legal caregivers provided written informed consent approved by their local institution’s review board. Each site’s institutional review board approved all study procedures. For the DIAN study, the institutional review board at Washington University in St. Louis provided supervisory review and human studies approval.
2.3. MRI acquisition and processing
For the Knight ADRC cohort, T1-weighted images were acquired using a magnetization-prepared rapid gradient-echo sequence on either the Siemens Biograph mMR (n = 172) or the Siemens 3T TIM Trio (n = 416) scanner. Structural T1 scans for the Biograph mMR were acquired with 1 × 1 × 1.2 mm resolution, 2300 ms repetition time, 2.95 ms echo time, 9 degree flip angle, 176 frames, and a 240 × 256 field of view in sagittal orientation. Structural T1 scans for the TIM Trio were acquired with a 1 × 1 × 1 mm resolution, 2400 ms repetition time, 3.16 echo time, 8 degree flip angle, 176 frames, and a 256 × 256 field of view in sagittal orientation. For the DIAN cohort, the T1-weighted images were acquired using the Alzheimer Disease Neuroimaging Initiative structural MRI protocol (Jack et al., 2010) using either a Siemens (n = 236) or a Phillips (n = 33) 3T scanner and the images underwent quality control assessments for protocol compliance. Structural T1 scans had a resolution of 1 × 1 × 1.2 mm. Images from both cohorts were processed with FreeSurfer 5.3-HCP (Fischl et al., 2002) and were resampled to 1 × 1 × 1 mm resolution for volumetric segmentation and cortical reconstruction. The total hippocampal volume (HCV) was derived from the sum of the left and right hemisphere hippocampal volume from the automated FreeSurfer regions.
2.4. PET acquisition and processing
Amyloid PET imaging was performed with 11C-Pittsburgh compound B (PiB) in the DIAN cohort and either PiB or 18F-florbetapir (AV-45) in the Knight ADRC cohort. All PET scans were processed with the PET Unified Pipeline (PUP, https://github.com/ysu001/PUP) using FreeSurfer derived ROIs (Su et al., 2015, Su et al., 2013) to calculate the standardized uptake value ratio (SUVR) using the cerebellar cortex as a reference region. A 30–60 min post-injection window for PiB and a 50–70 min post-injection window for AV-45 were used in the Knight ADRC cohort and a 40–70 min post injection window for PiB was used in the DIAN cohort. Partial volume correction was performed using a geometric transfer matrix (Rousset et al., 1998, Su et al., 2015). To measure amyloid burden, we calculated the mean cortical SUVR (mcSUVR) by averaging the partial volume corrected SUVRs from the FreeSurfer ROIs in the lateral orbitofrontal, mesial orbitofrontal, rostral mesial frontal, superior frontal, superior temporal, mesial temporal, and precuneus regions as previously defined (Su et al., 2013).
To standardize the PiB and AV-45 tracers, the mcSUVR values for the Knight ADRC cohort were converted into Centiloid units (Klunk et al., 2015) using a linear transformation from previously defined equations (Su et al., 2019, Su et al., 2018). The value of zero on the Centiloid scale represents the mean amyloid burden for healthy participants and the value of 100 on the Centiloid scale represents the mean amyloid burden for AD participants. For statistical analysis, we utilized Centiloid for the Knight ADRC cohort and mcSUVR for the DIAN cohort. Amyloid positivity was defined as an mcSUVR ≥ 1.42 for PiB scans (Brier et al., 2016b, Sutphen et al., 2015) and an mcSUVR ≥ 1.22 for AV-45 scans (Mishra et al., 2017).
2.5. Creation of the cortical maps
For each hemisphere, cortical thickness spatial maps were created via vertex-wise analyses using a general linear model in FreeSurfer. The LOAD cortical maps were generated by computing cortical thickness differences between the LOAD and CNADRC groups while adjusting for age and sex. Similarly, the ADAD cortical maps were generated by calculating cortical thickness differences between the ADAD and CNDIAN groups while accounting for the effects of age and sex.
Generating a robust cortical signature requires a balance between specificity and sensitivity; it is a tradeoff between having a large enough cortical map which adequately captures the most informative regions but not so large it incorporates regional information with limited predictive value. For this reason, Monte Carlo simulations were performed using a selection of vertex-wise thresholds (p < 0.05, p < 0.01, p < 0.005, p < 0.001, p < 0.0005 and p < 0.0001) followed by a p < 0.001 cluster-wise threshold to correct for multiple comparisons. Each Monte Carlo vertex-wise threshold generated a different ROI, which is identified as the significant cluster of vertices, and were generated separately for the Knight ADRC and DIAN cohorts (Supplementary Fig. 1, Supplementary Fig. 2). The LOAD and ADAD ROIs were mapped to participant space within and across cohorts. For each hemisphere, the mean cortical thickness values were obtained for each cortical map (Supplementary Fig. 3).
To identify the ROI cortical maps which represent the optimal “cortical signature”, we compared the ability of the mean cortical thickness in each ROI to discriminate between the CN and PC groups by calculating the area under the receiver operating characteristic (AUROC) curve using the pROC package (Robin et al., 2011) in R. The left and right hemisphere ROIs which exhibited the best performance (i.e. those with the highest AUROC values) were identified as the optimal cortical signature and defined separately for the Knight ADRC and DIAN cohorts. Subsequently, the thickness values from the optimal cortical maps were averaged across hemispheres and weighted by the number of vertices in the ROI to obtain the cortical signature thickness. The optimal ADAD and LOAD cortical signatures are available online at https://github.com/benzinger-icl/ADcortsig-roi.
To verify that the cortical signature thickness performed with similar results to the receiver operating characteristic (ROC) analysis, a within-cohort analysis was done examining the cortical signature thickness performance from the ADAD and LOAD maps in predicting PCDIAN versus CNDIAN and PCADRC versus CNADRC groups, respectively. Second, we performed a cross-cohort analysis examining the cortical signature thickness performance from the ADAD and LOAD maps in predicting PCADRC versus CNADRC and PCDIAN versus CNDIAN groups, respectively. As atrophy is a biomarker that occurs before but close to symptom onset, we also repeated the cross-cohort analysis in a subset of the PCADRC (n = 39) and PCDIAN (n = 19) participants who were in the top quartile in terms of amyloid burden.
2.6. Statistical analysis
All statistical tests were conducted in R version 3.6.0 (R Core Team, 2019). Logistic regression models were used to examine the ability of cortical signature thickness to discriminate PC versus CN groups with amyloid status as the dependent term and the cortical signature thickness as the independent term while adjusting for age and sex. Additionally, we performed linear regression models to examine the ability of cortical signature thickness to predict amyloid burden in the PC and CN groups. The amyloid burden measure was the dependent term, defined in Centiloid units for the Knight ADRC cohort and mcSUVR for the DIAN cohort, while the cortical signature thickness measure was the independent term with adjustments made to account for age and sex variability. The purpose of the linear models is to examine the amyloid levels immediately above and below the amyloid status cutoff as these values are meaningful and may provide a more sensitive measure to detect the PC and CN groups. The logistic and linear models were again run including HCV as a covariate as a direct comparison of MRI biomarkers. Due to the prevalence of comorbidities in LOAD, we additionally examined the association of obesity, measured with body mass index (BMI), and white matter hyperintensity (WMH) burden, measured with the log transformed WMH volume, against the LOAD cortical signature thickness for a subset of the CNADRC and PCADRC groups (nBMI = 374; nWMH = 237) by calculating the Pearson correlation coefficient (r). Finally, we were interested in quantifying demographic differences within each cohort. For our continuous variables, age and education, ANOVAs were run and follow-up pair-wise t-tests were conducted for any significant outcomes. Similarly, for our categorical variables, APOE ε4status and sex, chi-square tests were run and follow-up pair-wise chi-square tests were conducted for significant outcomes.
3. Results
3.1. Demographics
The demographics from the DIAN and Knight ADRC cohorts are presented in Table 1.
Table 1.
Participant demographics.
| DIAN Cohort |
Knight ADRC Cohort |
|||||
|---|---|---|---|---|---|---|
| CNDIAN | PCDIAN | ADAD | CNADRC | PCADRC | LOAD | |
| N | 145 | 76 | 48 | 381 | 153 | 54 |
| Age, years | 38.5 (11.3)b | 37.2 (9.0)c | 44.8 (10.6) | 65.5 (9.4)a, b | 73.1 (7.1)c | 76.5 (7.1) |
| EYO | −9.9 (11.8) | −9.9 (7.8) | 3.0 (2.7) | – | – | – |
| Male, n (%) | 58 (40.0) | 37 (48.7) | 18 (37.5) | 150 (39.4) | 66 (43.1) | 29 (53.7) |
| Education, years | 15.0 (2.7)b | 14.7 (2.9)c | 13.4 (2.5) | 16.0 (2.4) | 16.0 (2.4) | 15.1 (2.9) |
| APOE ε4+, n (%) | 42 (29.0) | 26 (34.2) | 15 (31.2) | 106 (27.8)a, b | 88 (57.5)c | 41 (75.9) |
| PSEN1/PSEN2/APP, n | 97 / 18 / 30 | 59 / 11 / 6 | 39 / 2 / 7 | – | – | – |
| CDR 0/0.5, n | 145 / 0 | 76 / 0 | 0 / 48 | 381 / 0 | 153 / 0 | 0 / 54 |
| Aβ-/Aβ+, n | 145 / 0 | 0 / 76 | 0 / 48 | 381 / 0 | 0 / 153 | 0 / 54 |
| ADAD cortical signature thickness, mm | 2.34 (0.11) | 2.31 (0.13) | 2.09 (0.17) | 2.22 (0.12) | 2.16 (0.12) | 2.08 (0.12) |
| LOAD cortical signature thickness, mm | 2.74 (0.12) | 2.73 (0.12) | 2.60 (0.15) | 2.60 (0.12) | 2.53 (0.12) | 2.36 (0.13) |
| PiB-PET SUVR | 1.05 (0.07) | 2.05 (0.61) | 2.89 (1.08) | – | – | – |
| Centiloid | −0.4 (3.1) | 44.8 (27.5) | 82.5 (48.6) | 0.1 (6.2) | 48.2 (26.6) | 82.5 (27.0) |
Mean (standard deviation) unless otherwise noted.
ADAD = autosomal dominant Alzheimer disease; LOAD = late-onset Alzheimer disease; CNDIAN = cognitively normal mutation non-carrier; CNADRC = cognitively normal controls; PCDIAN = preclinical ADAD; PCADRC = preclinical LOAD; EYO = estimated years to symptom onset; APOE = apolipoprotein E; PSEN1 = presenilin 1; PSEN2 = presenilin 2; APP = amyloid precursor protein; CDR = Clinical Dementia Rating; Aβ-/Aβ+ = amyloid negative/amyloid positive; PiB-PET = Pittsburgh compound B positron emission tomography.
Significant differences between CN and PC, p < 0.05.
Significant differences between CN and ADAD/LOAD, p < 0.05.
Significant differences between PC and ADAD/LOAD, p < 0.05.
Within the DIAN cohort, significant age and education differences were found between the CNDIAN versus ADAD groups and PCDIAN versus ADAD groups. However, there were no significant results for sex or APOE ε4 status from these analyses. In contrast, we report significant age and APOE ε4 status differences for all groups within the Knight ADRC cohort, but there were no significant differences in education and sex for these groups.
3.2. ADAD and LOAD cortical signatures
The ADAD cortical maps for each vertex-wise threshold are presented in Supplementary Fig. 1. The AUROC values from the ROC analysis discriminating PCDIAN and CNDIAN groups for each hemisphere and each vertex-wise significance threshold (p < 0.05, p < 0.01, p < 0.005, p < 0.001, p < 0.0005, p < 0.0001) are presented in Table 2.
Table 2.
AUROC values from the ROC analysis discriminating PC and CN participants for each ROI cortical map in their respective cohort.
| ADAD ROI Cortical Maps |
LOAD ROI Cortical Maps |
|||
|---|---|---|---|---|
| Monte Carlo Vertex-wise Thresholds | RH AUROC | LH AUROC | RH AUROC | LH AUROC |
| p < 0.05 | 0.4671 | 0.5416 | 0.6470 | 0.6455 |
| p < 0.01 | 0.5357 | 0.5397 | 0.6332 | 0.6487 |
| p < 0.005 | 0.5330 | 0.5431 | 0.6427 | 0.6518 |
| p < 0.001 | 0.5400 | 0.5329 | 0.6425 | 0.6443 |
| p < 0.0005 | 0.4601 | 0.5396 | 0.6422 | 0.6509 |
| p < 0.0001 | 0.4540 | 0.5469 | 0.6326 | 0.6379 |
Bold indicates the highest AUROC value for each ROI and hemisphere.
ADAD = autosomal dominant Alzheimer disease; LOAD = late-onset Alzheimer disease; AUROC = area under the receiver operating characteristic.
The highest AUROC value for the right hemisphere was 0.5400, which corresponded to the cortical map generated using the p < 0.001 threshold. For the left hemisphere, the highest AUROC value was 0.5469, which corresponded to the cortical map defined using the p < 0.0001 threshold. These two cortical maps define the optimal cortical signature for ADAD shown in Fig. 1A. The associated effect size maps are presented in Fig. 1B.
Fig. 1.

A. The optimal ADAD cortical signature depicting cortical thickness differences between the ADAD and CNDIAN groups including age and sex as covariates and using a p < 0.001 cluster-wise threshold. The vertex-wise threshold was found to be p < 0.001 for right hemisphere and p < 0.0001 for left hemisphere. Significant vertices are depicted in yellow highlighting the inferior parietal and precuneus. B. The cortical thickness effect size maps comparing ADAD and CNDIAN. The blue indicates areas where the ADAD group had lower cortical thickness values compared to the CNDIAN and the red indicates areas where the ADAD group had higher cortical thickness values compared to the CNDIAN group. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We also identified the approximate FreeSurfer regions which best match the vertex-wise signature by calculating the percentage of voxels in the signature map overlaying each FreeSurfer region. Specifically, the vertex-wise signature was converted into a volume and the percentage of overlapping voxels was determined by dividing the number of cortical signature voxels within a FreeSurfer region by the total number of voxels in that FreeSurfer region and multiplying by 100. The FreeSurfer region with the highest ADAD signature overlap for the right hemisphere is the inferior parietal region (60% overlap) and for the left hemisphere is the precuneus region (54% overlap).
Identical analyses were performed in the Knight ADRC cohort to determine the optimal cortical signature. The LOAD cortical maps for all vertex-wise thresholds are presented in Supplementary Fig. 2 and the AUROC values for each hemisphere and threshold are presented in Table 2. The best AUROC was 0.6470 for the right hemisphere, corresponding to the p < 0.05 vertex-wise threshold, and 0.6518 for the left hemisphere, corresponding to the p < 0.005 threshold. These two cortical maps are used to define the optimal LOAD cortical signature presented in Fig. 2A. The associated effect size maps of the cortical signature are found in Fig. 2B.
Fig. 2.

A. The optimal LOAD cortical signature depicting cortical thickness differences between the LOAD and CNADRC groups including age and sex as covariates and using a p < 0.001 cluster-wise threshold. The vertex-wise threshold was found to be p < 0.05 for right hemisphere and p < 0.005 for left hemisphere. Significant vertices are depicted in blue highlighting predominantly the temporal lobe. B. The cortical thickness effect size maps comparing LOAD and CNADRC. The blue indicates areas where the LOAD group had lower cortical thickness values compared to the CNADRC and the red indicates areas where the LOAD group had higher cortical thickness values compared to the CNADRC group. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The FreeSurfer region with the greatest LOAD signature overlap for the left and right hemisphere is the entorhinal cortex (85% right hemisphere overlap and 74% left hemisphere overlap). A list of additional FreeSurfer regions and the associated cortical signature overlap percentage are presented in Supplemental Table 1. We additionally compared the ADAD and LOAD cortical signatures (Fig. 3). The LOAD signature emphasized atrophy in the temporal lobe whereas ADAD is more prominent in the parietal and precuneus. There is also a noticeable overlap within the precuneus and parietal regions in both signatures.
Fig. 3.
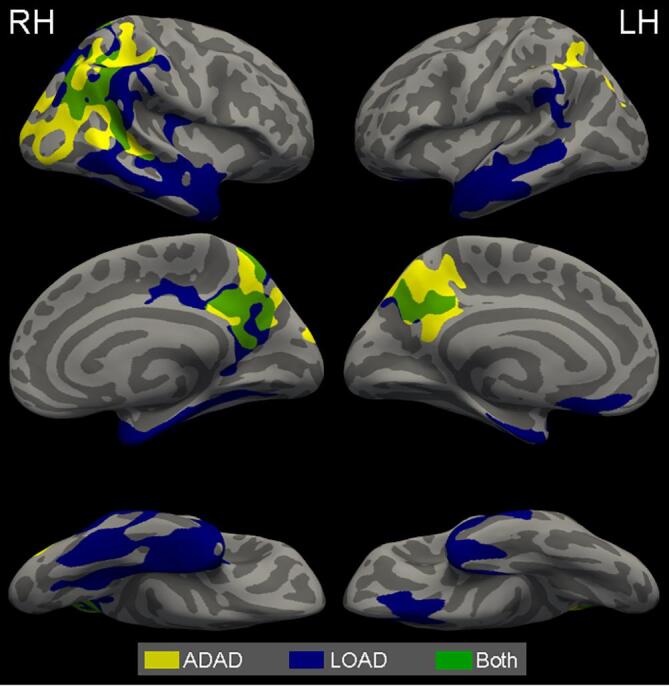
A comparison of the ADAD and LOAD optimal cortical signatures. Yellow indicates ADAD areas, blue indicates LOAD areas, and green indicates areas where both ADAD and LOAD cortical signatures overlap. Much of the overlap between the two signatures are in parts of the precuneus and parietal regions. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. Within-cohort analysis: association of the cortical signature thickness with amyloid
Although each hemispheric signature was optimized to discriminate between PC and CN groups, we wanted to confirm the cortical signatures’ capabilities within its own cohort and compare it to a more common MRI biomarker, HCV. We found that the ADAD cortical signature thickness marginally separated CNDIAN and PCDIAN (ADAD cortical signature thickness: β = −2.44, p = 0.048). While the ADAD cortical signature thickness discriminated CNDIAN and PCDIAN, the HCV did not reach statistical significance. (ADAD cortical signature thickness: β = −2.51, p = 0.044; HCV: β = 7.37e-5, p = 0.69). In the linear regression analysis, the ADAD cortical signature thickness predicted amyloid burden within the combined PCDIAN mutation carrier and CNDIAN mutation non-carrier groups (ADAD cortical signature thickness: β = −0.78, p = 0.025) whereas HCV was not able to discriminate groups (ADAD cortical signature thickness: β = −0.76, p = 0.03; HCV: β = −1.42e-5, p = 0.79).
For the Knight ADRC, the LOAD cortical signature thickness did not significantly separate the CNADRC and PCADRC groups (LOAD cortical signature thickness: β = −0.89, p = 0.36), primarily due to the significant effect of age in the model (age: β = 0.09, p = 3.74e-10), and HCV performed similarly to the cortical signature (LOAD cortical signature thickness: β = −0.85, p = 0.41; HCV: β = −1.60e-5, p = 0.90). When treating amyloid as a continuous measure, the LOAD cortical signature thickness predicted amyloid burden (LOAD cortical signature thickness: β =−24.37, p = 0.016) beyond the covariates but HCV did not reach statistical significance (LOAD cortical signature thickness: β = –22.14, p = 0.036; HCV: β = −9.70e-4, p = 0.46). Amyloid as a continuous variable performed better in all statistical models when compared to analyzing amyloid as a categorical variable. The distributions for the cortical signature thickness and HCV within the DIAN and Knight ADRC cohorts are shown in Fig. 4.
Fig. 4.

The distributions of the ADAD and LOAD cortical signature thickness and hippocampal volume within the DIAN and Knight ADRC cohorts, respectively. For comparison, the cortical signature thickness and hippocampal volume were normalized relative to the cognitively normal control groups. The black circles indicate the mean for each group. CNDIAN = cognitively normal mutation non-carrier (dark green); PCDIAN = preclinical ADAD (orange); ADAD = autosomal dominant Alzheimer disease (purple); CNADRC = cognitively normal controls (pink); PCADRC = preclinical LOAD (green); LOAD = late-onset Alzheimer disease (yellow). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
When analyzing BMI and WMH volume against the LOAD cortical signature thickness in the CNADRC and PCADRC groups, no association was present between BMI and LOAD cortical signature thickness (r = −0.06). Notably, there was an association between the log transformed WMH volume and LOAD cortical signature thickness (r = −0.63), primarily driven by age (Supplementary Fig. 4).
3.4. Cross-cohort analysis: association of the cortical signature thickness with amyloid
Since there are known similarities between ADAD and LOAD, we examined whether the ADAD signature could differentiate between the PCADRC and CNADRC groups and whether the LOAD signature could differentiate between the PCDIAN and CNDIAN groups. We found the ADAD cortical signature thickness was unable to discriminate the PCADRC and CNADRC groups (ADAD cortical signature thickness: β = −0.61, p = 0.52) and likewise when including HCV into the model (ADAD cortical signature thickness: β = −0.55, p = 0.57, HCV: β = −3.47e-5, p = 0.79). The ADAD cortical signature thickness could not predict amyloid burden (ADAD cortical signature thickness: β = -12.46, p = 0.21) and HCV performed equally (ADAD cortical signature thickness: β = −9.94, p = 0.33; HCV: β = −0.0015, p = 0.25). Subsequently, the logistic regression analysis was repeated to only include the top quartile amyloid burden PC participants. In this analysis, the ADAD cortical signature thickness could not discriminate the groups (ADAD cortical signature thickness: β = −2.08, p = 0.18) and was unable to predict amyloid burden (ADAD cortical signature thickness: β = −16.13, p = 0.14).
The LOAD cortical signature thickness was unable to distinguish between the CNDIAN and PCDIAN groups (LOAD cortical signature thickness: β = −1.01, p = 0.42) and HCV performed similarly (LOAD cortical signature thickness: β = −1.10, p = 0.39; HCV: β = 5.56e-5, p = 0.77). Additionally, the LOAD cortical signature thickness could not predict amyloid burden (LOAD cortical signature thickness: β = -0.38, p = 0.29) and likewise for HCV (LOAD cortical signature thickness: β =−0.35, p = 0.34; HCV: β = −1.92e-5, p = 0.72). When restricting the PCDIAN group to the top quartile amyloid burden participants, the LOAD cortical signature thickness was unable to separate the groups (LOAD cortical signature thickness: β = −1.86, p = 0.38) or predict amyloid burden (LOAD cortical signature thickness: β = −0.57, p = 0.19).
4. Discussion
AD-related atrophy affects many brain regions beyond just the hippocampus alone. LOAD individuals are also likely to have age-related comorbidities which could make it difficult to characterize regions more prone to AD-specific pathology. Since individuals affected by ADAD tend to be younger and thus lack many age-related comorbidities, this cohort may provide a better cortical atrophy model specific to AD. The goals of this study were to create a refined and reliable cortical atrophy signature by using a large cohort and no ROI restrictions and to compare the ADAD and LOAD signature maps and their ability to detect amyloid burden in cognitively normal groups within and across cohorts. We defined signature maps of cortical thickness separately for LOAD and ADAD by comparing the cognitively impaired and CN groups. The ADAD and LOAD cortical signatures can be accessed online (https://github.com/benzinger-icl/ADcortsig-roi). These cortical signatures included the temporal lobe, parietal, and occipital regions. In the statistical analyses of amyloid, we analyzed only the PC and CN groups. We found the optimal signatures are sensitive in detecting amyloid levels in the cognitively normal groups within their respective cohorts and are significant beyond the inclusion of hippocampus volume, yet the signatures performed poorly when applied cross-cohort (e.g. ADAD signature evaluated in Knight ADRC cohort). In both the Knight ADRC and DIAN cohorts, the signature maps were better predictors when analyzing amyloid as a continuous variable rather than a categorical variable in all statistical models. This result suggests that amyloid levels immediately above and below the amyloid positivity cutoff are important and provide a more sensitive measure to detect the subtle atrophy differences between the CN and PC groups. Finally, by using a continuous variable over dichotomized, our models are optimized for greater statistical power and therefore more able to detect true effects where they exist.
To generate the cortical signatures, we used a vertex-wise approach on a substantial sample size and determined the optimal statistical threshold for multiple comparison correction for each hemisphere. Previous work defining AD cortical signatures used predefined large regions of interest (Dickerson et al., 2009, Jack et al., 2017, Weston et al., 2016) or a similar vertex-wise approach (Wang et al., 2015) with a modest sample size. The spatial extent of significant effects seen in vertex or voxel-wise analyses is highly dependent upon the sample size with a smaller sample creating small peaks which may not translate to other cohorts. Predefined regions of interest from programs like FreeSurfer avoid the small focal effects but incorporate areas that may not be susceptible to AD-related pathology. There is a compromise between having large enough ROIs that the region is stable but not so large it includes marginally predictive areas. By examining cortical maps defined by multiple significance thresholds, we found the optimal tradeoff between these two properties. Each statistical threshold for multiple comparisons correction generated a new cortical map with different ROIs with the strongest threshold (p < 0.0001) producing the smallest ROIs. We tested how well each statistical threshold for the left and right hemisphere discriminated the PC and CN groups within-cohort and defined optimal cortical signatures from the best performing thresholds. Since our primary interest is focused on individuals transitioning between cognitively normal to early stages of dementia, the selected Monte Carlo thresholds optimize the signatures to the earliest changes in neurodegeneration. We found the optimal selection varied between the left and right hemispheres as well as between the LOAD and ADAD cortical maps. More specifically, the AUROC values in the left hemisphere were overall higher than the right hemisphere for both the ADAD and LOAD cortical maps. Past work have shown hemispherical differences in Alzheimer disease (Kim et al., 2012, Long et al., 2013, Minkova et al., 2017, Toga and Thompson, 2003) and there is also evidence the left hemisphere has an earlier and more rapid progression of atrophy relative to the right hemisphere in AD (Damoiseaux et al., 2009, Thompson et al., 2007). This earlier progression may explain the higher AUROC values in the left hemisphere compared to the right hemisphere. Additionally, the AUROC values in LOAD were higher compared to ADAD, whereas in the regression analyses, the ADAD signature thickness outperformed LOAD. This is primarily due to the inclusion of the age and sex covariates in the regression models. These covariates were not included in the optimization ROC analyses. Since LOAD is largely driven by age, the covariates reduce the predictive value of the cortical signature. This variance shows using a single threshold for each hemisphere and between diseases would have included areas with marginal effect or removed vital areas. Our approach for defining cortical signatures has not, to our knowledge, been attempted previously. We identified the percentage overlap of the cortical signatures on FreeSurfer regions and found the cortical signatures did not encompass any FreeSurfer region entirely, with the highest overlap percentage being 85%. The cortical signature maps are a blend of the most significant areas within each FreeSurfer region, strengthening the point that a larger ROI may not represent the AD cortical thinning behavior. We tested whether different comorbidities, particularly obesity and WMH burden, influence the LOAD cortical signature thickness within the CNADRC and PCADRC groups. BMI had no association to cortical thickness whereas WMH volume had a moderate association, largely due to age. ADAD is also associated with increased WMH burden (Lee et al., 2016). Since the ADAD and LOAD cortical signatures are largely distinct from each other, this may further exemplify WMH burden is not influencing the cortical signatures.
In a comparison between the LOAD and ADAD cortical signatures, we found there is spatial overlap between the two cortical signatures in portions of the right hemisphere inferior and superior parietal regions and portions of the precuneus in both hemispheres (Fig. 3). This spatial overlap may be due to the interrelatedness of these regions on both the structural and functional networks. Further understanding of the underlying network structure in these regions is an important future direction. Examining the significant maps from all possible thresholds (Supplemental Fig. 1, 2) and the effect size maps (Figs. 1B, 2B) show atrophy in ADAD and LOAD is occurring in similar regions, but to a different magnitude. The LOAD signature emphasized atrophy in the temporal lobe regions, while the ADAD signature was most focused on parietal regions. This shift in spatial emphasis is consistent with previous work examining atrophy in LOAD, (La Joie et al., 2012, Singh et al., 2006, Thompson et al., 2003, Whitwell et al., 2007) and ADAD (Apostolova et al., 2011, Cash et al., 2013, Fortea et al., 2010, Gordon et al., 2018a, Knight et al., 2011). The observed atrophy spatial differences also mirror the differences found in the tau pathology patterns, rather than amyloid pathology. Previous work demonstrated tau pathology and cortical atrophy have a high association within the temporal, occipital, parietal, and frontal lobe for the LOAD participants (Gordon et al., 2018b). In ADAD, there are higher levels of tau pathology in the precuneus and a higher precuneus to inferior temporal tau pathology ratio when compared to LOAD (Gordon et al., 2019). Additionally, spatial differences are also seen in early-onset LOAD with previous work corroborating both atrophy (Frisoni, 2005, Möller et al., 2013) and tau PET deposition (Schöll et al., 2017) occur preferentially in the cortex. In contrast, the amyloid spatial pattern between the diseases are largely similar (Shepherd et al., 2009). ADAD having a more prevalent amyloid pathology (Ringman et al., 2016, Shepherd et al., 2009) as well as an increased amyloid tracer uptake in the basal ganglia (Benzinger et al., 2013, Gordon et al., 2019, Gordon et al., 2018a) are the major differences between the diseases.
The exact cause of the different atrophy spatial pattern is unknown but is potentially due to multiple factors. There is evidence that increasing age at dementia onset is tied to more temporal lobe predominant atrophy (Frisoni, 2005, Möller et al., 2013). The pattern may also be tied to the different drivers of pathology in the two forms of the disease. ADAD mutations lead to an overproduction of amyloid burden (Potter et al., 2013) while LOAD is a product of multiple genetic and lifestyle influences that affect both production (Ye et al., 2005) and clearance (Mawuenyega et al., 2010, Moore et al., 2016). Understanding more about this spatial divergence will elucidate unknown factors about AD pathobiology and potentially highlight new therapeutic mechanisms. These different degrees of atrophy between the diseases are most likely playing a role in the unsuccessful cross-cohort analysis in this study and suggests that cortical signature maps are not interchangeable.
This study has many strengths which include the relatively large sample from the Knight ADRC and DIAN cohorts and the application of the cortical signatures to preclinical stages of AD. This comparison between the ADAD and LOAD signatures and the cross-cohort analysis in relation to amyloid pathology has not been studied before to our knowledge. One limitation for this study is the use of cross-sectional data. Identifying regions where longitudinal changes in cortical thickness occur may provide more detail in the progression of the disease. Additionally, future work is needed to examine the performance of the cortical signature in predicting longitudinal cognitive decline. Another limitation is that the cortical signature is only for amnestic presentation due to the use of solely research-based cohorts. Literature suggests that atypical presentations, such as posterior cortical atrophy, might have different atrophy signatures (Lehmann et al., 2011). Clinical cohorts tend to include more participants with comorbidities compared to research cohorts and thus this study may not be translatable to a clinical setting. Finally, prior work suggests there may be differences in atrophy due to specific mutations (Scahill et al., 2013), however the infrequency of APP and PSEN2 prevent us from analyzing these differences. As studies of ADAD become more common, it would be of interest to investigate the effect of the different ADAD mutations.
5. Conclusion
The LOAD and ADAD cortical signatures are sensitive to amyloid burden in asymptomatic individuals in their respective cohorts. This finding illustrates that the cortical signature maps are an indicator of AD-related neurodegeneration in early stages of AD and are a useful MRI biomarker measure. However, the different spatial emphasis in atrophy between LOAD and ADAD may be responsible for the unsuccessful cross-cohort analysis. Therefore, the cortical signatures are not compatible across cohorts and should be optimized within the targeted disease.
Funding
This work was supported by The Dominantly Inherited Alzheimer Network (DIAN, UF1AG032438) and funded by the National Institutes of Health (U19AG03243808, P01AG003991, P01AG026276, P01AG005681, K01AG053474, R01AG03158, and P30AG019610), the German Center for Neurodegenerative Diseases (DZNE), XNAT (R01EB009352), Neuroimaging Informatics and Analysis Center (P30NS098577), the Center for High-Performance Computing (1S10RR022984-01A1 and 1S10OD018091-0), ADHS Grant No. CTR040636 (previously ADHS Grant No. ADHS14-052688), Alzheimer’s Association (AARG-17-532945), the BrightFocus Foundation (ADR A2017272S), Alzheimer Association International Research Program (AARFD-20-681815), and the Raul Carrea Institute for Neurological Research (FLENI). This work was also supported by the generous support of Barnes-Jewish Hospital, the Paula and Rodger O. Riney Fund, the Danial J Brennan MD Fund, the Fred Simmons and Olga Mohan Fund, and the Willman Fund, the Arizona Alzheimer’s Consortium, DHS of the State of Arizona, the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI). This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications.
CRediT authorship contribution statement
Aylin Dincer: Conceptualization, Formal analysis, Investigation, Writing - original draft, Writing - review & editing, Data curation. Brian A. Gordon: Conceptualization, Formal analysis, Investigation, Writing - review & editing. Amrita Hari-Raj: Resources, Data curation, Conceptualization. Sarah J. Keefe: Resources, Data curation, Conceptualization. Shaney Flores: Resources, Data curation, Conceptualization. Nicole S. McKay: Resources, Data curation, Conceptualization, Formal analysis. Angela M. Paulick: Resources, Data curation, Conceptualization. Kristine E. Shady Lewis: Resources, Data curation, Conceptualization. Rebecca L. Feldman: Resources, Data curation, Conceptualization. Russ C. Hornbeck: Resources, Data curation, Conceptualization. Ricardo Allegri: Project administration, Investigation. Beau M. Ances: Project administration, Investigation. Sarah B. Berman: Project administration, Investigation. Adam M. Brickman: Project administration, Investigation. William S. Brooks: Project administration, Investigation. David M. Cash: Project administration, Investigation. Jasmeer P. Chhatwal: Project administration, Investigation. Martin R. Farlow: Project administration, Investigation. Christian Fougère: Project administration, Investigation. Nick C. Fox: Project administration, Investigation. Michael J. Fulham: Project administration, Investigation. Clifford R. Jack: Project administration, Investigation. Nelly Joseph-Mathurin: Resources, Data curation, Conceptualization. Celeste M. Karch: Project administration, Investigation. Athene Lee: Project administration, Investigation. Johannes Levin: Project administration, Investigation. Colin L. Masters: Project administration, Investigation. Eric M. McDade: Project administration, Investigation. Hwamee Oh: Project administration, Investigation. Richard J. Perrin: Project administration, Investigation, Project administration, Investigation. Cyrus Raji: Formal analysis. Stephen P. Salloway: Project administration, Investigation. Peter R. Schofield: Project administration, Investigation. Yi Su: Project administration, Investigation. Victor L. Villemagne: Project administration, Investigation. Qing Wang: Resources, Data curation, Conceptualization. Michael W. Weiner: Project administration, Investigation. Chengjie Xiong: Project administration, Investigation. Igor Yakushev: Project administration, Investigation. John C. Morris: Funding acquisition, Project administration, Supervision. Randall J. Bateman: Funding acquisition, Project administration, Supervision. Tammie L.S. Benzinger: Funding acquisition, Project administration, Supervision, Writing - review & editing.
Declaration of Competing Interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: The following authors have no conflicts of interest regarding this study: AD, AHR, BAG, SJK, SF, AMP, KESL, RLF, RCH, NSM, SBB, MRF, CF, MJF, CMK, AL, HO, VLV, QW, BMA, DMC, NCF, JL, CLM, MWW, RJP, CX. RA reports grants from CONICET. AMB is a scientific consultant for Regeneron Pharmaceuticals and Cognition Therapeutics, Inc., owns stocks in Venus Medtech, which were awarded for past service on an advisory board for Keystone Heart Ltd, and has a patent (US patent #9867566) for technologies for white matter hyperintensity quantification. WSB paid for DIAN expenses from NIH and other grants as declared in funding from Washington University School of Medicine. He also received grants from the Roth Charitable Foundation. JPC is a consultant for Otsuka pharmaceuticals. CRJ has consulted for Lily, serves on an independent data monitoring board for Roche, and as a speaker for Eisai, but he receives no personal compensation from any commercial entity. He receives research support from NIH and the Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Clinic. SPS reports fees from Biogen, Lilly, Eisai, Genentech, Roche, Novartis, and Avid, outside the submitted work. PRS received grants from NIA, grants from Anonymous Foundation, grants from Roth Charitable Foundation, during the conduct of the study. YS is supported by the Arizona Alzheimer’s Consortium, DHS and the State of Arizona, ADHS Grant, the BrightFocus Foundation, and the Alzheimer’s Association. JCM received grants from NIH grants. IY reports personal fees from Piramal, Blue Earth Diagnostics, and ABC-CRO and received grants from German Research Foundation and Federal Ministry of Education and Research, outside the submitted work. RJB receives lab research funding from the National Institutes of Health, Alzheimer’s Association, BrightFocus Foundation, Rainwater Foundation Tau Consortium, Association for Frontotemporal Degeneration, the Cure Alzheimer’s Fund, the Tau SILK Consortium (AbbVie, Biogen, and Eli Lilly and Co.), and an anonymous foundation. Funding for clinical trials includes the National Institutes of Health, Alzheimer's Association, Eli Lilly and Co, Hoffmann-La Roche, Janssen, Avid Radiopharmaceuticals, GHR Foundation, and an anonymous foundation. RJB also receives research funding from the DIAN-TU Pharma Consortium (Abbvie, Biogen, Eisai, Eli Lilly and Co/Avid Radiopharmaceuticals, Hoffmann-La Roche/Genentech, Janssen, and United Neuroscience). RJB has received honoraria from Roche as an Advisory Board member. Washington University and RJB have equity ownership interest in C2N Diagnostics and receive royalty income based on technology (stable isotope labeling kinetics and blood plasma assay) licensed by Washington University to C2N Diagnostics. RJB receives income from C2N Diagnostics for serving on the scientific advisory board. Washington University, with RJB as co-inventor, has submitted the US nonprovisional patent application “Methods for Measuring the Metabolism of CNS Derived Biomolecules In Vivo” and provisional patent application “Plasma Based Methods for Detecting CNS Amyloid Deposition”. NJM is supported partly by the Alzheimer Association International Research Program. CR reports other from Brainreader ApS, other from Neurevolution LLC, other from Apollo Health, outside the submitted work. EMM reports serving on a Data Safety Committee for Eli Lilly and Alector; personal honorarium for Continuing Medical Education activities for Esai and Eli Lilly and UpToDate; Institutional grant support from Eli Lilly, Hoffman-La Roche and Janssen. TLSB has investigator initiated research funding from the NIH, the Alzheimer's Association, the Barnes-Jewish Hospital Foundation and Avid Radiopharmaceuticals (a wholly owned subsidiary of Eli Lilly). TLSB participates as a site investigator in clinical trials sponsored by Avid Radiopharmaceuticals, Eli Lilly, Biogen, Eisai, Jaansen, and Roche. She serves as an unpaid consultant to Eisai and Siemens. She is on the Speaker's Bureau for Biogen.
Acknowledgements
We acknowledge the altruism of the participants and their families and contributions of the DIAN and the Knight ADRC research and support staff at each of the participating sites for their contributions to this work. Without the generous contribution and time the participants gave for these studies, this work would not be possible.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nicl.2020.102491.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Supplementary figure 1.

Supplementary figure 2.

Supplementary figure 3.

Supplementary figure 4.

References
- Apostolova L.G., Hwang K.S., Medina L.D., Green A.E., Braskie M.N., Dutton R.A., Lai J., Geschwind D.H., Cummings J.L., Thompson P.M., Ringman J.M. Cortical and hippocampal atrophy in patients with autosomal dominant familial Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2011;32(2):118–125. doi: 10.1159/000330471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova L.G., Mosconi L., Thompson P.M., Green A.E., Hwang K.S., Ramirez A., Mistur R., Tsui W.H., de Leon M.J. Subregional hippocampal atrophy predicts Alzheimer's dementia in the cognitively normal. Neurobiol. Aging. 2010;31(7):1077–1088. doi: 10.1016/j.neurobiolaging.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschenbrenner A.J., Gordon B.A., Benzinger T.L.S., Morris J.C., Hassenstab J.J. Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology. 2018;91(9):e859–e866. doi: 10.1212/WNL.0000000000006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkour A., Morris J.C., Wolk D.A., Dickerson B.C. The effects of aging and Alzheimer's disease on cerebral cortical anatomy: specificity and differential relationships with cognition. NeuroImage. 2013;76:332–344. doi: 10.1016/j.neuroimage.2013.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman R.J., Aisen P.S., De Strooper B., Fox N.C., Lemere C.A., Ringman J.M., Salloway S., Sperling R.A., Windisch M., Xiong C. Autosomal-dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimers Res. Ther. 2010;3(1):1. doi: 10.1186/alzrt59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman R.J., Xiong C., Benzinger T.L.S., Fagan A.M., Goate A., Fox N.C., Marcus D.S., Cairns N.J., Xie X., Blazey T.M., Holtzman D.M., Santacruz A., Buckles V., Oliver A., Moulder K., Aisen P.S., Ghetti B., Klunk W.E., McDade E., Martins R.N., Masters C.L., Mayeux R., Ringman J.M., Rossor M.N., Schofield P.R., Sperling R.A., Salloway S., Morris J.C. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 2012;367(9):795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzinger T.L.S., Blazey T., Jack C.R., Koeppe R.A., Su Y., Xiong C., Raichle M.E., Snyder A.Z., Ances B.M., Bateman R.J., Cairns N.J., Fagan A.M., Goate A., Marcus D.S., Aisen P.S., Christensen J.J., Ercole L., Hornbeck R.C., Farrar A.M., Aldea P., Jasielec M.S., Owen C.J., Xie X., Mayeux R., Brickman A., McDade E., Klunk W., Mathis C.A., Ringman J., Thompson P.M., Ghetti B., Saykin A.J., Sperling R.A., Johnson K.A., Salloway S., Correia S., Schofield P.R., Masters C.L., Rowe C., Villemagne V.L., Martins R., Ourselin S., Rossor M.N., Fox N.C., Cash D.M., Weiner M.W., Holtzman D.M., Buckles V.D., Moulder K., Morris J.C. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc. Natl. Acad. Sci. 2013;110(47):E4502–E4509. doi: 10.1073/pnas.1317918110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman F.H., Schoonenboom S.N.M., van der Flier W.M., van Elk E.J., Kok A., Barkhof F., Blankenstein M.A., Scheltens P.h. CSF biomarkers and medial temporal lobe atrophy predict dementia in mild cognitive impairment. Neurobiol. Aging. 2007;28(7):1070–1074. doi: 10.1016/j.neurobiolaging.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol. Aging. 1995;16(3):271–278. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- Brier M.R., Gordon B., Friedrichsen K., McCarthy J., Stern A., Christensen J., Owen C., Aldea P., Su Y.i., Hassenstab J., Cairns N.J., Holtzman D.M., Fagan A.M., Morris J.C., Benzinger T.L.S., Ances B.M. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl. Med. 2016;8(338):338ra66. doi: 10.1126/scitranslmed.aaf2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier M.R., McCarthy J.E., Benzinger T.L.S., Stern A., Su Y.i., Friedrichsen K.A., Morris J.C., Ances B.M., Vlassenko A.G. Local and distributed PiB accumulation associated with development of preclinical Alzheimer's disease. Neurobiol. Aging. 2016;38:104–111. doi: 10.1016/j.neurobiolaging.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cash D.M., Ridgway G.R., Liang Y., Ryan N.S., Kinnunen K.M., Yeatman T., Malone I.B., Benzinger T.L.S., Jack C.R., Thompson P.M., Ghetti B.F., Saykin A.J., Masters C.L., Ringman J.M., Salloway S.P., Schofield P.R., Sperling R.A., Cairns N.J., Marcus D.S., Xiong C., Bateman R.J., Morris J.C., Rossor M.N., Ourselin S., Fox N.C. The pattern of atrophy in familial Alzheimer disease: volumetric MRI results from the DIAN study. Neurology. 2013;81(16):1425–1433. doi: 10.1212/WNL.0b013e3182a841c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convit A., De Leon M.J., Tarshish C., De Santi S., Tsui W., Rusinek H., George A. Specific hippocampal volume reductions in individuals at risk for Alzheimer’s disease. Neurobiol. Aging. 1997;18(2):131–138. doi: 10.1016/S0197-4580(97)00001-8. [DOI] [PubMed] [Google Scholar]
- Damoiseaux J.S., Smith S.M., Witter M.P., Sanz-Arigita E.J., Barkhof F., Scheltens P., Stam C.J., Zarei M., Rombouts S.A.R.B. White matter tract integrity in aging and Alzheimer's disease. Hum. Brain Mapp. 2009;30(4):1051–1059. doi: 10.1002/hbm.20563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson, B.C., Bakkour, A., Salat, D.H., Feczko, E., Pacheco, J., Greve, D.N., Grodstein, F., Wright, C.I., Blacker, D., Rosas, H.D., Sperling, R.A., Atri, A., Growdon, J.H., Hyman, B.T., Morris, J.C., Fischl, B., Buckner, R.L., 2009. The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb. Cortex 19, 497–510. https://doi.org/10.1093/cercor/bhn113. [DOI] [PMC free article] [PubMed]
- Faraco G., Iadecola C. Hypertension: a harbinger of stroke and dementia. Hypertension. 2013;62(5):810–817. doi: 10.1161/HYPERTENSIONAHA.113.01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B., Salat D.H., Busa E., Albert M., Dieterich M., Haselgrove C., van der Kouwe A., Killiany R., Kennedy D., Klaveness S., Montillo A., Makris N., Rosen B., Dale A.M. Whole brain segmentation. Neuron. 2002;33(3):341–355. doi: 10.1016/S0896-6273(02)00569-X. [DOI] [PubMed] [Google Scholar]
- Fortea J., Sala-Llonch R., Bartrés-Faz D., Bosch B., Lladó A., Bargalló N., Luis Molinuevo J., Sánchez-Valle R., Molinuevo J.L., Sánchez-Valle R. Increased cortical thickness and caudate volume precede atrophy in PSEN1 mutation carriers. J. Alzheimer’s Dis. 2010;22:909–922. doi: 10.3233/JAD-2010-100678. [DOI] [PubMed] [Google Scholar]
- Frisoni G.B. Structural correlates of early and late onset Alzheimer's disease: voxel based morphometric study. J. Neurol. Neurosurg. Psychiatry. 2005;76(1):112–114. doi: 10.1136/jnnp.2003.029876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, B.A., Blazey, T., Su, Y., Fagan, A.M., Holtzman, D.M., Morris, J.C., Benzinger, T.L.S., 2016. Longitudinal β-amyloid deposition and hippocampal volume in preclinical Alzheimer disease and suspected non-Alzheimer disease pathophysiology. JAMA Neurol. 73, 1192–1200. https://doi.org/10.1001/jamaneurol.2016.2642. [DOI] [PMC free article] [PubMed]
- Gordon, B.A., Blazey, T.M., Christensen, J., Dincer, A., Flores, S., Keefe, S., Chen, C., Su, Y., McDade, E.M., Wang, G., Li, Y., Hassenstab, J., Aschenbrenner, A., Hornbeck, R., Jack, C.R., Ances, B.M., Berman, S.B., Brosch, J.R., Galasko, D., Gauthier, S., Lah, J.J., Masellis, M., van Dyck, C.H., Mintun, M.A., Klein, G., Ristic, S., Cairns, N.J., Marcus, D.S., Xiong, C., Holtzman, D.M., Raichle, M.E., Morris, J.C., Bateman, R.J., Benzinger, T.L.S., 2019. Tau PET in autosomal dominant Alzheimer’s disease: relationship with cognition, dementia and other biomarkers. Brain 142, 1063–1076. https://doi.org/10.1093/brain/awz019. [DOI] [PMC free article] [PubMed]
- Gordon, B.A., Blazey, T.M., Su, Y., Hari-Raj, A., Dincer, A., Flores, S., Christensen, J., McDade, E., Wang, G., Xiong, C., Cairns, N.J., Hassenstab, J., Marcus, D.S., Fagan, A.M., Jack, C.R., Hornbeck, R.C., Paumier, K.L., Ances, B.M., Berman, S.B., Brickman, A.M., Cash, D.M., Chhatwal, J.P., Correia, S., Förster, S., Fox, N.C., Graff-Radford, N.R., la Fougère, C., Levin, J., Masters, C.L., Rossor, M.N., Salloway, S., Saykin, A.J., Schofield, P.R., Thompson, P.M., Weiner, M.M., Holtzman, D.M., Raichle, M.E., Morris, J.C., Bateman, R.J., Benzinger, T.L.S., 2018a. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol. 17, 211–212. https://doi.org/10.1016/S1474-4422(18)30028-0. [DOI] [PMC free article] [PubMed]
- Gordon B.A., McCullough A., Mishra S., Blazey T.M., Su Y.i., Christensen J., Dincer A., Jackson K., Hornbeck R.C., Morris J.C., Ances B.M., Benzinger T.L.S., Wolk D., Villemagne V., Dickerson B. Cross‐sectional and longitudinal atrophy is preferentially associated with tau rather than amyloid β positron emission tomography pathology. Alzheimer’s Dement. Diagnosis, Assess. Dis. Monit. 2018;10(1):245–252. doi: 10.1016/j.dadm.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J., Higgins G. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science:1566067. [DOI] [PubMed] [Google Scholar]
- Huijbers, W., Mormino, E.C., Schultz, A.P., Wigman, S., Ward, A.M., Larvie, M., Amariglio, R.E., Marshall, G.A., Rentz, D.M., Johnson, K.A., Sperling, R.A., 2015. Amyloid-β deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain 138, 1023–1035. https://doi.org/10.1093/brain/awv007. [DOI] [PMC free article] [PubMed]
- Jack C.R., Jr., Bennett D.A., Blennow K., Carrillo M.C., Dunn B., Haeberlein S.B., Holtzman D.M., Jagust W., Jessen F., Karlawish J., Liu E., Molinuevo J.L., Montine T., Phelps C., Rankin K.P., Rowe C.C., Scheltens P., Siemers E., Snyder H.M., Sperling R., Elliott C., Masliah E., Ryan L., Silverberg N. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimer's Dement. 2018;14(4):535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C.R., Jr., Bernstein M.A., Borowski B.J., Gunter J.L., Fox N.C., Thompson P.M., Schuff N., Krueger G., Killiany R.J., DeCarli C.S., Dale A.M., Carmichael O.W., Tosun D., Weiner M.W. Update on the magnetic resonance imaging core of the Alzheimer's disease neuroimaging initiative. Alzheimer's Dement. 2010;6(3):212–220. doi: 10.1016/j.jalz.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C.R., Knopman D.S., Jagust W.J., Petersen R.C., Weiner M.W., Aisen P.S., Shaw L.M., Vemuri P., Wiste H.J., Weigand S.D., Lesnick T.G., Pankratz V.S., Donohue M.C., Trojanowski J.Q. Update on hypothetical model of Alzheimer’s disease biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C.R., Jr., Wiste H.J., Weigand S.D., Therneau T.M., Lowe V.J., Knopman D.S., Gunter J.L., Senjem M.L., Jones D.T., Kantarci K., Machulda M.M., Mielke M.M., Roberts R.O., Vemuri P., Reyes D.A., Petersen R.C. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimer's Dement. 2017;13(3):205–216. doi: 10.1016/j.jalz.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W.J., Zheng L., Harvey D.J., Mack W.J., Vinters H.V., Weiner M.W., Ellis W.G., Zarow C., Mungas D., Reed B.R., Kramer J.H., Schuff N., DeCarli C., Chui H.C. Neuropathological basis of magnetic resonance images in aging and dementia. Ann. Neurol. 2008;63(1):72–80. doi: 10.1002/ana.21296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.H., Lee J.W., Kim G.H., Roh J.H., Kim M.-J., Seo S.W., Kim S.T., Jeon S., Lee J.-M., Heilman K.M., Na D.L. Cortical asymmetries in normal, mild cognitive impairment, and Alzheimer's disease. Neurobiol. Aging. 2012;33(9):1959–1966. doi: 10.1016/j.neurobiolaging.2011.06.026. [DOI] [PubMed] [Google Scholar]
- Klunk W.E., Koeppe R.A., Price J.C., Benzinger T.L., Devous M.D., Sr., Jagust W.J., Johnson K.A., Mathis C.A., Minhas D., Pontecorvo M.J., Rowe C.C., Skovronsky D.M., Mintun M.A. The Centiloid project: standardizing quantitative amyloid plaque estimation by PET. Alzheimer's Dement. 2015;11(1):1–15.e4. doi: 10.1016/j.jalz.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight W.D., Kim L.G., Douiri A., Frost C., Rossor M.N., Fox N.C. Acceleration of cortical thinning in familial Alzheimer's disease. Neurobiol. Aging. 2011;32(10):1765–1773. doi: 10.1016/j.neurobiolaging.2009.11.013. [DOI] [PubMed] [Google Scholar]
- La Joie R., Perrotin A., Barre L., Hommet C., Mezenge F., Ibazizene M., Camus V., Abbas A., Landeau B., Guilloteau D., de La Sayette V., Eustache F., Desgranges B., Chetelat G. Region-specific hierarchy between atrophy, hypometabolism, and β-amyloid (Aβ) load in Alzheimer’s disease dementia. J. Neurosci. 2012;32:16265–16273. doi: 10.1523/JNEUROSCI.2170-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Viqar F., Zimmerman M.E., Narkhede A., Tosto G., Benzinger T.L.S., Marcus D.S., Fagan A.M., Goate A., Fox N.C., Cairns N.J., Holtzman D.M., Buckles V., Ghetti B., McDade E., Martins R.N., Saykin A.J., Masters C.L., Ringman J.M., Ryan N.S., Förster S., Laske C., Schofield P.R., Sperling R.A., Salloway S., Correia S., Jack C., Jr., Weiner M., Bateman R.J., Morris J.C., Mayeux R., Brickman A.M. White matter hyperintensities are a core feature of Alzheimer's disease: evidence from the dominantly inherited Alzheimer network: White Matter Hyperintensities in Familial AD. Ann. Neurol. 2016;79(6):929–939. doi: 10.1002/ana.24647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M., Crutch S.J., Ridgway G.R., Ridha B.H., Barnes J., Warrington E.K., Rossor M.N., Fox N.C. Cortical thickness and voxel-based morphometry in posterior cortical atrophy and typical Alzheimer's disease. Neurobiol. Aging. 2011;32(8):1466–1476. doi: 10.1016/j.neurobiolaging.2009.08.017. [DOI] [PubMed] [Google Scholar]
- Long X., Zhang L., Liao W., Jiang C., Qiu B. Distinct laterality alterations distinguish mild cognitive impairment and Alzheimer's disease from healthy aging: statistical parametric mapping with high resolution MRI: laterality alterations in Aging, MCI and AD. Hum. Brain Mapp. 2013;34(12):3400–3410. doi: 10.1002/hbm.22157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.B., Smith C.D., Collins H.R., Schmitt F.A., Gold B.T. Evidence that volume of anterior medial temporal lobe is reduced in seniors destined for mild cognitive impairment. Neurobiol. Aging. 2010;31(7):1099–1106. doi: 10.1016/j.neurobiolaging.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega K.G., Sigurdson W., Ovod V., Munsell L., Kasten T., Morris J.C., Yarasheski K.E., Bateman R.J. Decreased clearance of CNS b-amyloid in Alzheimer’s disease. Science (80-.) 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkova L., Habich A., Peter J., Kaller C.P., Eickhoff S.B., Klöppel S. Gray matter asymmetries in aging and neurodegeneration: a review and meta-analysis: VBM-ALE analysis of GM asymmetries. Hum. Brain Mapp. 2017;38(12):5890–5904. doi: 10.1002/hbm.23772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S., Gordon B.A., Su Y.i., Christensen J., Friedrichsen K., Jackson K., Hornbeck R., Balota D.A., Cairns N.J., Morris J.C., Ances B.M., Benzinger T.L.S. AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: defining a summary measure. NeuroImage. 2017;161:171–178. doi: 10.1016/j.neuroimage.2017.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller C., Vrenken H., Jiskoot L., Versteeg A., Barkhof F., Scheltens P., van der Flier W.M. Different patterns of gray matter atrophy in early- and late-onset Alzheimer’s disease. Neurobiol. Aging. 2013;34(8):2014–2022. doi: 10.1016/j.neurobiolaging.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Moore K.M., Girens R.E., Larson S.K., Jones M.R., Restivo J.L., Holtzman D.M., Cirrito J.R., Yuede C.M., Zimmerman S.D., Timson B.F. A spectrum of exercise training reduces soluble Aβ in a dose-dependent manner in a mouse model of Alzheimer's disease. Neurobiol. Disease. 2016;85:218–224. doi: 10.1016/j.nbd.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Moran C., Phan T.G., Chen J., Blizzard L., Beare R., Venn A., Munch G., Wood A.G., Forbes J., Greenaway T.M., Pearson S., Srikanth V. Brain atrophy in Type 2 diabetes: regional distribution and influence on cognition. Diab. Care. 2013;36(12):4036–4042. doi: 10.2337/dc13-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J.C. The clinical dementia rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412. doi: 10.1212/WNL.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Morris J.C., Aisen P.S., Bateman R.J., Benzinger T.LS., Cairns N.J., Fagan A.M., Ghetti B., Goate A.M., Holtzman D.M., Klunk W.E., McDade E., Marcus D.S., Martins R.N., Masters C.L., Mayeux R., Oliver A., Quaid K., M Ringman J., Rossor M.N., Salloway S., Schofield P.R., Selsor N.J., Sperling R.A., Weiner M.W., Xiong C., Moulder K.L., Buckles V.D. Developing an international network for Alzheimer’s research: the Dominantly Inherited Alzheimer Network. Clinical Investigation. 2012;2(10):975–984. doi: 10.4155/cli.12.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder K.L., Snider B., Mills S.L., Buckles V.D., Santacruz A.M., Bateman R.J., Morris J.C. Dominantly inherited Alzheimer Network: facilitating research and clinical trials. Alzheimers Res. Ther. 2013;5(5):48. doi: 10.1186/alzrt213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R., Smith R., Ohlsson T., Strandberg O., Mattsson N., Insel P.S., Palmqvist S., Hansson O. Associations between tau, Aβ, and cortical thickness with cognition in Alzheimer disease. Neurology. 2019;92(6):e601–e612. doi: 10.1212/WNL.0000000000006875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter, R., Patterson, B.W., Elbert, D.L., Ovod, V., Kasten, T., Sigurdson, W., Mawuenyega, K., Blazey, T., Goate, A., Chott, R., Yarasheski, K.E., Holtzman, D.M., Morris, J.C., Benzinger, T.L.S., Bateman, R.J., 2013. Increased In Vivo Amyloid-β42 production, exchange, and loss in presenilin mutation carriers. Sci. Transl. Med. 5, 189ra77–189ra77. https://doi.org/10.1126/scitranslmed.3005615. [DOI] [PMC free article] [PubMed]
- R Core Team . R Found. Stat. Comput; Vienna, Austria: 2019. R: A Language and Environment for Statistical Computing [WWW Document] https://www.r-project.org/ [Google Scholar]
- Ridha B.H., Barnes J., Bartlett J.W., Godbolt A., Pepple T., Rossor M.N., Fox N.C. Tracking atrophy progression in familial Alzheimer’s disease: a serial MRI study. Lancet Neurol. 2006;5:828–834. doi: 10.1016/S1474-4422(06)70550-6. [DOI] [PubMed] [Google Scholar]
- Ringman J.M., Monsell S., Ng D.W., Zhou Y., Nguyen A., Coppola G., Van Berlo V., Mendez M.F., Tung S., Weintraub S., Mesulam M.-M., Bigio E.H., Gitelman D.R., Fisher-Hubbard A.O., Albin R.L., Vinters H.V. Neuropathology of autosomal dominant Alzheimer disease in the national Alzheimer coordinating center database. J. Neuropathol. Exp. Neurol. 2016;75(3):284–290. doi: 10.1093/jnen/nlv028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.-C., Müller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12(1) doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset O.G., Ma Y., Evans A.C. Correction for partial volume effects in PET: principle and validation. J. Nucl. Med. 1998;39:904–911. [PubMed] [Google Scholar]
- Ryman D.C., Acosta-Baena N., Aisen P.S., Bird T., Danek A., Fox N.C., Goate A., Frommelt P., Ghetti B., Langbaum J.B.S., Lopera F., Martins R., Masters C.L., Mayeux R.P., McDade E., Moreno S., Reiman E.M., Ringman J.M., Salloway S., Schofield P.R., Sperling R., Tariot P.N., Xiong C., Morris J.C., Bateman R.J. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology. 2014;83(3):253–260. doi: 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scahill R.I., Ridgway G.R., Bartlett J.W., Barnes J., Ryan N.S., Mead S., Beck J., Clarkson M.J., Crutch S.J., Schott J.M., Ourselin S., Warren J.D., Hardy J., Rossor M.N., Fox N.C. Genetic influences on atrophy patterns in familial Alzheimer's disease: a comparison of APP and PSEN1 mutations. JAD. 2013;35(1):199–212. doi: 10.3233/JAD-121255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scahill R.I., Schott J.M., Stevens J.M., Rossor M.N., Fox N.C. Mapping the evolution of regional atrophy in Alzheimer's disease: unbiased analysis of fluid-registered serial MRI. Proc. Natl. Acad. Sci. 2002;99(7):4703–4707. doi: 10.1073/pnas.052587399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöll, M., Ossenkoppele, R., Strandberg, O., Palmqvist, S., Jögi, J., Ohlsson, T., Smith, R., Hansson, O., 2017. Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer’s disease. Brain 140, 2286–2294. https://doi.org/10.1093/brain/awx171. [DOI] [PubMed]
- Shepherd C., McCann H., Halliday G.M. Variations in the neuropathology of familial Alzheimer’s disease. Acta Neuropathol. 2009;118(1):37–52. doi: 10.1007/s00401-009-0521-4. [DOI] [PubMed] [Google Scholar]
- Singh, V., Chertkow, H., Lerch, J.P., Evans, A.C., Dorr, A.E., Kabani, N.J., 2006. Spatial patterns of cortical thinning in mild cognitive impairment and Alzheimer’s disease. Brain 129, 2885–2893. https://doi.org/10.1093/brain/awl256. [DOI] [PubMed]
- Su Y.i., Blazey T.M., Snyder A.Z., Raichle M.E., Marcus D.S., Ances B.M., Bateman R.J., Cairns N.J., Aldea P., Cash L., Christensen J.J., Friedrichsen K., Hornbeck R.C., Farrar A.M., Owen C.J., Mayeux R., Brickman A.M., Klunk W., Price J.C., Thompson P.M., Ghetti B., Saykin A.J., Sperling R.A., Johnson K.A., Schofield P.R., Buckles V., Morris J.C., Benzinger T.L.S. Partial volume correction in quantitative amyloid imaging. NeuroImage. 2015;107:55–64. doi: 10.1016/j.neuroimage.2014.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, Y., D’Angelo, G.M., Vlassenko, A.G., Zhou, G., Snyder, A.Z., Marcus, D.S., Blazey, T.M., Christensen, J.J., Vora, S., Morris, J.C., Mintun, M.A., Benzinger, T.L.S., 2013. Quantitative analysis of PiB-PET with FreeSurfer ROIs. PLoS One 8, e73377. https://doi.org/10.1371/journal.pone.0073377. [DOI] [PMC free article] [PubMed]
- Su Y.i., Flores S., Hornbeck R.C., Speidel B., Vlassenko A.G., Gordon B.A., Koeppe R.A., Klunk W.E., Xiong C., Morris J.C., Benzinger T.L.S. Utilizing the Centiloid scale in cross-sectional and longitudinal PiB PET studies. NeuroImage: Clin. 2018;19:406–416. doi: 10.1016/j.nicl.2018.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.i., Flores S., Wang G., Hornbeck R.C., Speidel B., Joseph‐Mathurin N., Vlassenko A.G., Gordon B.A., Koeppe R.A., Klunk W.E., Jack C.R., Jr., Farlow M.R., Salloway S., Snider B.J., Berman S.B., Roberson E.D., Brosch J., Jimenez‐Velazques I., Dyck C.H., Galasko D., Yuan S.H., Jayadev S., Honig L.S., Gauthier S., Hsiung G.-Y., Masellis M., Brooks W.S., Fulham M., Clarnette R., Masters C.L., Wallon D., Hannequin D., Dubois B., Pariente J., Sanchez‐Valle R., Mummery C., Ringman J.M., Bottlaender M., Klein G., Milosavljevic‐Ristic S., McDade E., Xiong C., Morris J.C., Bateman R.J., Benzinger T.L.S. Comparison of Pittsburgh compound B and florbetapir in cross‐sectional and longitudinal studies. Alzheimer’s Dement. Diagnosis, Assess. Dis. Monit. 2019;11(1):180–190. doi: 10.1016/j.dadm.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutphen C.L., Jasielec M.S., Shah A.R., Macy E.M., Xiong C., Vlassenko A.G., Benzinger T.L.S., Stoops E.E.J., Vanderstichele H.M.J., Brix B., Darby H.D., Vandijck M.L.J., Ladenson J.H., Morris J.C., Holtzman D.M., Fagan A.M. Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol. 2015;72(9):1029. doi: 10.1001/jamaneurol.2015.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson P.M., Hayashi K.M., de Zubicaray G., Janke A.L., Rose S.E., Semple J., Herman D., Hong M.S., Dittmer S.S., Doddrell D.M., Toga A.W. Dynamics of gray matter loss in Alzheimer's disease. J. Neurosci. 2003;23(3):994–1005. doi: 10.1523/JNEUROSCI.23-03-00994.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson P.M., Hayashi K.M., Dutton R.A., Chiang M.-C., Leow A.D., Sowell E.R., de Zubicaray G., Becker J.T., Lopez O.L., Aizenstein H.J., Toga A.W. Tracking Alzheimer's disease. Ann. N. Y. Acad. Sci. 2007;1097(1):183–214. doi: 10.1196/annals.1379.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toga A.W., Thompson P.M. Mapping brain asymmetry. Nat. Rev. Neurosci. 2003;4(1):37–48. doi: 10.1038/nrn1009. [DOI] [PubMed] [Google Scholar]
- Vos S.J.B., Gordon B.A., Su Y.i., Visser P.J., Holtzman D.M., Morris J.C., Fagan A.M., Benzinger T.L.S. NIA-AA staging of preclinical Alzheimer disease: discordance and concordance of CSF and imaging biomarkers. Neurobiol. Aging. 2016;44:1–8. doi: 10.1016/j.neurobiolaging.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Benzinger T.L., Hassenstab J., Blazey T., Owen C., Liu J., Fagan A.M., Morris J.C., Ances B.M. Spatially distinct atrophy is linked to β-amyloid and tau in preclinical Alzheimer disease. Neurology. 2015;84:1254–1260. doi: 10.1212/WNL.0000000000001401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston P.S.J., Nicholas J.M., Lehmann M., Ryan N.S., Liang Y., Macpherson K., Modat M., Rossor M.N., Schott J.M., Ourselin S., Fox N.C. Presymptomatic cortical thinning in familial Alzheimer disease: a longitudinal MRI study. Neurology. 2016;87(19):2050–2057. doi: 10.1212/WNL.0000000000003322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell J.L., Przybelski S.A., Weigand S.D., Knopman D.S., Boeve B.F., Petersen R.C., Jack C.R. 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer’s disease. Brain. 2007;130:1777–1786. doi: 10.1093/brain/awm112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S., Huang Y., Mullendorff K., Dong L., Giedt G., Meng E.C., Cohen F.E., Kuntz I.D., Weisgraber K.H., Mahley R.W. Apolipoprotein (apo) E4 enhances amyloid peptide production in cultured neuronal cells: ApoE structure as a potential therapeutic target. Proc. Natl. Acad. Sci. 2005;102:18700–18705. doi: 10.1073/pnas.0508693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarow C., Sitzer T.E., Chui H.C. Understanding hippocampal sclerosis in the elderly: epidemiology, characterization, and diagnostic issues. Curr. Neurol. Neurosci. Rep. 2008;8:363–370. doi: 10.1007/s11910-008-0057-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.