

Abstract
The major suppressive immune cells in tumor sites are myeloid derived suppressor cells (MDSCs), tumor‐associated macrophages (TAMs), and Treg cells, and the major roles of these suppressive immune cells include hindering T‐cell activities and supporting tumor progression and survival. In this study, we analyzed the pattern of circulating MDSC subtypes in patients with non‐small cell lung cancer (NSCLC) whether those suppressive immune cells hinder T‐cell activities leading to poor clinical outcomes. First, we verified PMN‐MDSCs, monocytic‐MDSCs (M‐MDSCs), and Treg cells increased according to the stages of NSCLC, and MDSCs effectively suppressed T‐cell activities and induced T‐cell exhaustion. The analysis of NSCLC patients treated with anti‐PD‐1 immunotherapy demonstrated that low PMN‐MDSCs, M‐MDSCs, and CD39+CD8+ T cells as an individual and all together were associated with longer progression free survival and overall survival, suggesting PMN‐MDSCs, M‐MDSCs, and CD39+CD8+ T cells frequencies in peripheral blood might be useful as potential predictive and prognostic biomarkers.
Keywords: CD39, IL‐10, Immune checkpoint inhibitor, MDSC, Non‐small cell lung cancer
Pre‐existing PMN‐MDSCs, M‐MDSCs, and CD39+CD8+ T cells can be used as predictive biomarkers in anti‐PD‐1 immunotherapy targeting NSCLC. Together with MDSCs, IL‐10 possibly released by suppressive immune cells also leads poor clinical outcomes. Therefore, combinatorial strategies targeting MDSCs or IL‐10 should be investigated to improve outcomes of immune checkpoint inhibitors.
Introduction
Lung cancer is one of the leading causes of cancer‐related mortality worldwide; among the major subtypes, non‐small cell lung cancer (NSCLC) accounts for approximately 80‐85% of all lung cancer cases [1, 2]. Recently, immune checkpoint inhibitors of the programmed death‐1 (PD‐1) and programmed death‐ligand 1 (PD‐L1) interaction have emerged as a new therapeutic approach for advanced stages of lung cancer. Three immune checkpoint inhibitors, pembrolizumab, nivolumab, and atezolizumab, are now approved for use in patients with advanced NSCLC [3, 4, 5, 6].
Although PD‐L1 is the most widely adopted predictor, not all PD‐L1‐positive patients are likely to respond to anti‐PD‐1 immunotherapy. More importantly, some patients with low PD‐L1 expression still respond, making it an insufficient biomarker for selecting patients [7, 8, 9]. One reason that a number of patients do not respond to this therapy is possibly due to immunosuppressive tumor microenvironment, which is mediated by various immune cells including myeloid‐derived suppressor cells (MDSCs), tumor‐associated macrophages (TAMs), and Treg cells, and suppressive cytokines including TGF‐β, IL‐10, IL‐6, or vascular endothelial growth factor (VEGF). In addition, MDSCs release ROS or nitrogen oxide (NO) to manipulate T cells, causing them to become exhausted and no longer recognize tumor cells [10, 11, 12]. As a consequence, elevated suppressive immune cells populations correlate with tumor burden and promote T‐cell dysfunction, favoring tumor cell progression and proliferation.
Human polymorphonuclear MDSCs (PMN‐MDSCs) are defined as Lin− CD15+ CD14− CD11b+ HLA‐DR−/low and monocytic MDSCs (M‐MDSCs) as Lin− CD15− CD14+ HLA‐DR−/low. The major action of PMN‐MDSCs is to suppress T cells, while M‐MDSCs tend to differentiate into TAMs at tumor sites [13, 14]. MDSCs have been identified as an important prognostic biomarker and contribute to immune checkpoint inhibitor resistance [11]. MDSCs can also modulate TAMs and Treg cells development and activation reprogramming immune suppressive tumor microenvironment which is one of the major challenges in immunotherapy for NSCLC [15, 16, 17]. As a consequence of the suppressive environment, T cells express high levels of exhaustion markers including PD‐1 and CD39. Furthermore, tumor‐infiltrating CD8+ T cells expressing high CD39 exhibit features of exhaustion such as reduced TNF and IL‐2 expression and inhibited IFN‐gamma production [18, 19]. Therefore, in an immunosuppressive microenvironment, CD8+ T‐cell functions are very limited. Given the dynamic changes of immune cells in the tumor tissue microenvironment, analysis of circulating immune cells offers an alternative source as a noninvasive method. Further, the role of suppressive immune cells related to anti‐PD‐1 immunotherapy in NSCLC patients has not been fully established.
In this study, we analyzed various circulating suppressive immune cell types, including PMN‐MDSCs, M‐MDSCs, and Treg cells to examine whether frequencies of those cells correlated with disease stage and clinical outcomes, including progression‐free survival (PFS), and overall survival (OS) in NSCLC patients treated with anti‐PD‐1 therapy. Moreover, we analyzed T‐cell exhaustion marker CD39 to evaluate suppressive influences of MDSCs on T‐cell activities. We found that PMN‐MDSCs, M‐MDSCs, and CD39+CD8+ T cells in the peripheral blood both individually or collectively might be useful as noninvasive biomarkers to predict prolonged PFS and OS in advanced NSCLC treated with either pembrolizumab or nivolumab. Further, our findings indicate that immune checkpoint inhibitor resistance mediated by suppressive immune cells makes these cells a promising target for combination therapy with immune checkpoint inhibitors to improve outcomes.
Results
Accumulation of suppressive immune cells and CD39+CD8+ T cells associated with more advanced stages
To elucidate whether circulating suppressive immune cells reflect tumor stages, we first compared PMN‐MDSCs, M‐MDSCs, and Treg cells in lung cancer patients from stage I to IV prior to any therapy. We found that PMN‐MDSCs, M‐MDSCs, and Treg cells were higher in stage III and IV compared with stage I and II patients (Supporting information Fig. 2A‐C). In addition, CD39 expression on CD8+ T cells, which is known to be a T‐cell exhaustion marker, was higher in advanced‐stage patients (Supporting information Fig. 2D). These results indicate that in advanced‐stage patients, circulating suppressive immune cells, including PMN‐MDSCs, M‐MDSCs, and Treg cells, are increased, as T cells express high CD39. The characteristics of 59 patients are summarized in Supporting information Table 1.
Clinical outcomes associated with pre‐existing MDSCs and CD39 expression on CD8+ T cells
MDSCs have been known to contribute to immune checkpoint inhibitor resistance and modulate TAMs and Treg cells development and activation in the tumor microenvironment [11, 15]. To evaluate whether circulating suppressive immune cells and exhausted T cells play a prognostic role in NSCLC patients treated with anti‐PD‐1 therapy, peripheral blood prior to anti‐PD‐1 therapy was collected. Patient characteristics are summarized in Table 1. Lin− CD15+ CD14− CD11b+ HLA‐DR−/low cells were defined as PMN‐MDSCs. Lin− CD15− CD14+ HLA‐DR−/low cells were defined as M‐MDSCs (Supporting information Fig. 1A), and CD39+ cells were gated from CD8+ T cells (Suppporting information Fig. 1B).
Table 1.
Characteristics of the patients under anti PD‐1 immunotherapy
| Characteristics | Discovery cohort (n = 83) | Validation cohort (n = 49) |
|---|---|---|
| Age (year) | ||
| Median | 62 | 62 |
| Range | 39‐88 | 34‐82 |
| Gender no. (%) | ||
| Male | 68 (81.9) | 37 (75.5) |
| Female | 15 (18.1) | 12 (24.5) |
| ECOG performance‐status score — no. (%) | ||
| 0 | 0 (0) | 2 (4) |
| 1 | 74 (89.2) | 39 (79.6) |
| ≥2 | 9 (10.8) | 8 (16.4) |
| Tumor histologic type — no. (%) | ||
| Adenocarcinoma | 48 (57.8) | 25 (51) |
| Squamous cell carcinoma | 26 (31.3) | 13 (26.5) |
| Others | 9 (10.9) | 11 (22.5) |
| PD‐L1 expression level — no. (%) | ||
| <1% | 15 (18) | 8 (16.3) |
| ≥1% | 57 (68.7) | 27 (55.1) |
| Unknown | 11 (13.3) | 14 (28.6) |
ECOG, Eastern Cooperative Oncology Group.
To analyze whether MDSC subtypes and CD39+CD8+ T‐cell frequencies low and high correlate with clinical outcomes, we separated each cell types by low and high group by median cut off. For discovery cohort, with a median follow‐up duration of months (8.4), patients with low PMN‐MDSCs (6.5 vs. 3.4 months; p = 0.03) and M‐MDSCs (5.2 vs. 2.3 months; p = 0.04) were associated with longer PFS. Patients with low PMN‐MDSCs (7 vs. 4.3 months; p = 0.04) and M‐MDSCs (5.8 vs. 4.3 months; p = 0.005) showed better OS (Fig. 1A and B). CD39+ T cells (gated with CD8+ T cells) only correlated with OS (6.5 vs. 7 months; p = 0.05) (Fig. 1C).
Figure 1.
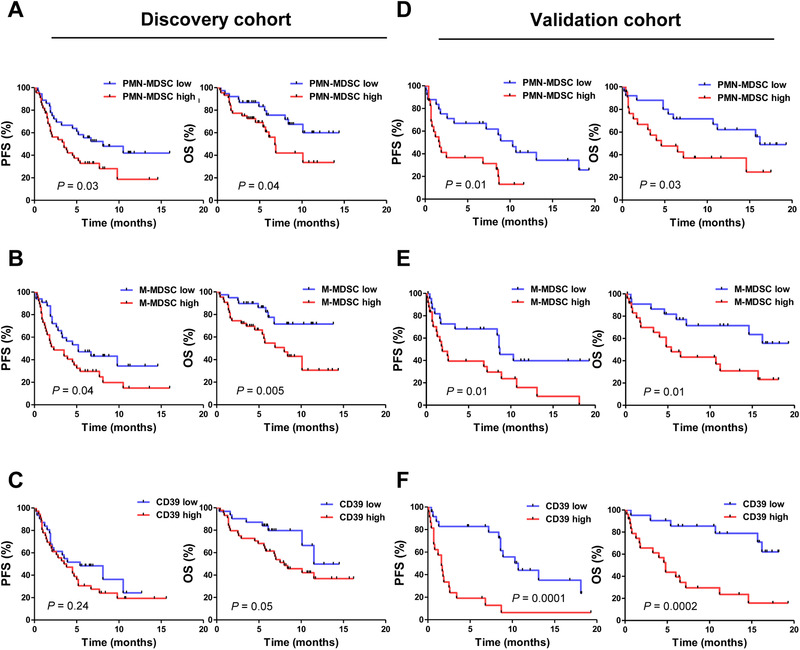
Progression‐free survival and overall survival depending on MDSCs and CD39 expression on CD8+ T cells (discovery cohort, n = 83; validation cohort, n = 49). PFS and OS of the patients with NSCLC depending on frequencies (%) of (A) PMN‐MDSCs, (B) M‐MDSCs, and (C) CD39+CD8+ T cells for discovery cohort. (D) PMN‐MDSCs, (E) M‐MDSCs, and (F) CD39+CD8+ T cells for validation cohort. All data of PMN‐MDSCs and M‐MDSCs were combined from individual experiments with one to two patient samples each time for MDSCs were analyzed from fresh PBMC prior to cryopreservation. CD39+CD8+ T cell data were combined from eight experiments with 10 to 12 patient samples each time for discovery cohort (n = 83) (A‐C) and five experiments with 9‐10 patient samples for validation cohort (n = 49) (D‐F). Kaplan–Meier survival curves were plotted with patients by median cutoff. Statistical significance was determined by log‐rank (Mantel–Cox) regression analysis, with the level of significance at p ⩽ 0.05.
Similar to discovery cohort, with a median follow‐up duration of months (7), patients with low PMN‐MDSCs (7 vs. 1.7 months; p = 0.01), M‐MDSCs (8.4 vs. 1.9 months; p = 0.01), and CD39+ T cells (8.6 vs. 1.6 months; p = 0.0001) were associated with longer PFS. Patients with low PMN‐MDSCs (11.2 vs. 4.3 months; p = 0.03) M‐MDSCs (9.9 vs. 5.4 months; p = 0.01), and CD39+ T cells (11.6 vs. 4.0 months; p = 0.0002) also showed better OS (Fig. 1D‐F).
Clinical outcomes associated with MDSCs and CD39+CD8+ T cells after anti‐PD‐1 immunotherapy
Our group reported that a higher fold‐change of Ki‐67 7 days after anti‐PD‐1 therapy predicted clinical outcomes in patients with thymic epithelial cell tumors and NSCLC [20]. Therefore, we collected baseline blood samples and 7 days after the therapy and analyzed PFS and OS depending on MDSC subtypes and CD39+CD8+ T cells. For discovery cohort, only CD39+CD8+ T cells correlated with PFS (p = 0.04), and PMN‐MDSCs (p = 0.02), and M‐MDSCs (p = 0.006) were correlated with OS (Supporting information Fig. 3A, C). For validation cohort, M‐MDSCs (p = 0.04) were correlated with PFS, and PMN‐MDSCs (p = 0.05) and CD39+CD8+ T cells (p = 0.04) were correlated with OS (Supporting information Fig. 3B, D). We also analyzed MDSCs and CD39+CD8+ T cells decrease or increase after 7 days compared with baseline, but increased or decreased group of the patients showed no differences when compared PFS and OS (Supporting information Fig. 4A‐D).
Effect of PMN‐MDSCs on T‐cell function and fate of M‐MDSCs
PMN‐MDSCs suppress T‐cell function and M‐MDSCs are known to be differentiated into TAMs in tumor sites [21]. We next evaluated whether PMN‐MDSCs and T cells coculture results in decreased T‐cell activities. First, we isolated PMN‐MDSCs and T cells from the same patient peripheral blood and cocultured them with different ratios, and then analyzed Ki‐67 expression in CD3+ T cells. At the presence of PMN‐MDSCs, T‐cell proliferation decreased (p < 0.0001) (Supporting informatiom Fig. 2E) but CD39 expression on CD8+ T cells increased (p = 0.03), indicating T‐cell activities possibly diminished by PMN‐MDSCs (Fig. 2A). Also, when the patients were grouped into both PMN‐MDSCs and CD39+ T cells low and high group, both low group showed higher response rate (92.8%, 13/14) compared with both high group (25%, 3/12) (Fig. 2B). Then, to confirm PMN‐MDSCs and CD39+ T‐cells correlation, CD39 expression was analyzed from PMN‐MDSCs low and high group. PMN‐MDSCs low group of the patients showed low CD39 expression on CD8+ T cells, and when PMN‐MDSCs were high, CD39 expression on T cells was high (p = 0.03) (Fig. 2C). Also, PMN‐MDSCs and CD39+ T‐cells correlation coefficient showed high PMN‐MDSCs associated with high CD39 expression on CD8+ T cells (p = 0.04) (Fig. 2D).
Figure 2.
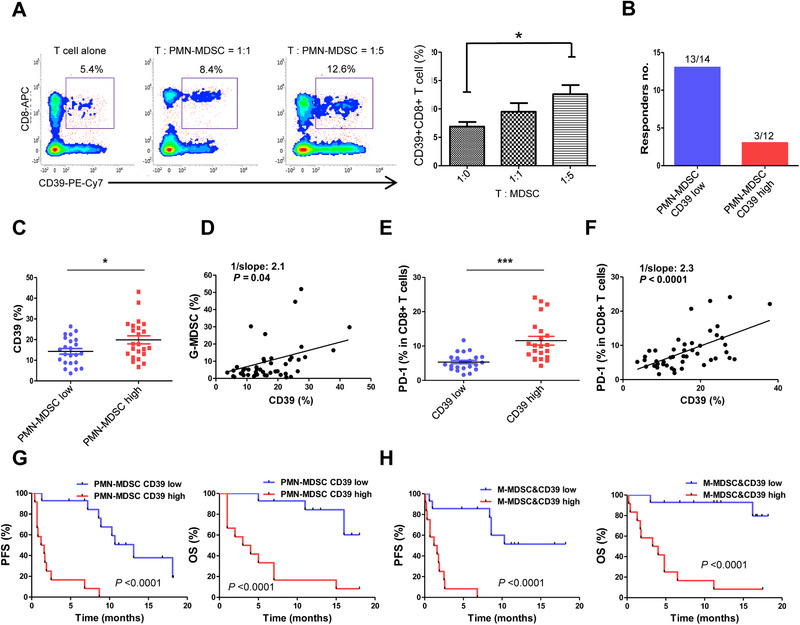
The role of MDSCs and clinical outcomes depending on MDSCs and CD39+CD8+ T cells (validation cohort, n = 49). (A) PMN‐MDSCs from PBMCs sorted with CD15 microbead were cocultured with CD3+ T cells from the same patient and CD39 expression on CD8+ T cells was examined in T cell alone group and T:MDSC (1:1 and 1:5) ratios by flow cytometry (n = 8). Data are combined from four individual experiments with two patient samples per experiment. (B) The number of responders depending on both PMN‐MDSCs and CD39+ T cells low (13/14) and high group (3/12) of the patients (total n = 49). (C) CD39 expression on CD8+ T cells depending on PMN‐MDSCs low and high group measured by flow cytometry. (D) CD39+ T‐cells frequencies (%) correlation with PMN‐MDSCs frequencies (%). (E) PD‐1 expression depending on CD39+ T cells low and high group measured by flow cytometry. (F) Correlation coefficient of PD‐1 expression (%) and CD39+ T cells (%). PFS and OS of (G) PMN‐MDSCs and CD39+ T cells, and (H) M‐MDSCs and CD39+ T cells collectively low and high groups. All data were combined from five experiments with 9‐10 patient samples each time for validation cohort (n = 49) (C‐H). The error bar represents standard deviation of the mean (A). The center value is Mean ± SEM (C and E). For correlation coefficient, a linear regression model was used. Kaplan–Meier survival curves were plotted by median cutoff. Statistical significance was determined by log‐rank (Mantel–Cox) regression analysis, with the level of significance at p ⩽ 0.05. *, p ⩽ 0.05; **, p ⩽ 0.01; ***, p ⩽ 0.001 (Mann–Whitney U test).
Besides CD39 expression on CD8+ T cells, PD‐1 expression is also a hallmark of T‐cell exhaustion. Therefore, we next compared PD‐1 expression depending on CD39 low and high group. As expected, CD39 expression low group showed low PD‐1 expression (p < 0.0001) (Fig. 2E). Also, PD‐1 expression was highly associated with CD39 expression on CD8+ T cells (p < 0.0001) (Fig. 2F). These results indicate that patients with high MDSCs at peripheral blood, CD39, and PD‐1 were highly expressed on CD8+ T cells causing T cells exhausted. The representative gating strategy for T cells is shown in Supporting information Fig. 1C.
Combined analysis of MDSCs and CD39 expression on T cells both low or high with clinical outcomes
Since both MDSC subtypes and CD39 expression on T cells correlated with PFS and OS, we performed combined analysis of both PMN‐MDSCs and CD39+ T cells, and M‐MDSCs and CD39+ T cells low and high group by median cutoff of each cell type. Patients who had low frequencies of both PMN‐MDSCs and CD39+ T cells (p OS < 0.0001; p PFS < 0.0001), and M‐MDSCs and CD39+ T cells (p OS < 0.0001; p PFS < 0.0001) low groups showed more prominent differences of PFS and OS compared to patients with all high groups (Fig. 2G and H).
Cytokine expression correlates with MDSCs frequencies and clinical outcomes
As MDSCs are known to release small molecules and cytokines, including IL‐10, TGF‐β, and IL‐6 to induce TAM and Treg cell activities [21], we conducted IL‐10, TGF‐β, and IL‐6 ELISA assays using plasma collected from the patients prior to the therapy. For discovery cohort, patients with durable clinical benefit (DCB) showed IL‐10 low in plasma level (p = 0.06) (Fig. 3A) and also IL‐10 low group showed better PFS (6.1 vs. 4.9 months; p = 0.07) and OS (6.7 vs. 5.3 months; p = 0.02) compared with IL‐10 high group (Fig. 3B). For discovery cohort, IL‐10 low group showed more prominent response to the drug compared to IL‐10 high group (p = 0.0004) (Fig. 3C), and better PFS (8.6 vs. 1.3 months; p = 0.002) and OS (11.2 vs. 4.4 months; p = 0.002) compared with IL‐10 high group (Fig. 3D). TGF‐β did not correlate with PMN‐MDSCs or M‐MDSCs, whereas IL‐6 correlated with only M‐MDSCs (data not shown).
Figure 3.
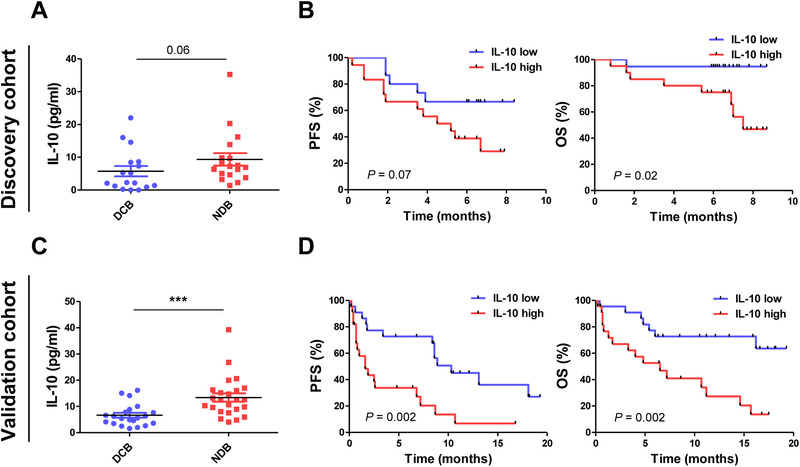
Cytokine protein expression (discovery cohort, n = 40; validation cohort, n = 49). (A) IL‐10 plasma level (pg/mL) from durable clinical benefiters (DCB) and nondurable clinical benefiters (NDB) measured by ELISA. (B) PFS and OS of the patients with NSCLC depending on low and high IL‐10 for discovery cohort. (C) IL‐10 from DCB and NDB, measured by ELISA. (D) PFS and OS of the patients with NSCLC depending on low and high IL‐10 for validation cohort. All data were from one experiment (ELISA) with 40 patient samples for discovery cohort and 49 patient samples for validation cohort. The center value is Mean ± SEM (A, C). *, p ⩽ 0.05; **, p ⩽ 0.01; ***, p ⩽ 0.001 (Mann–Whitney U test). Kaplan–Meier survival curves were plotted with patients by median cutoff. Statistical significance was determined by log‐rank (Mantel–Cox) regression analysis, with the level of significance at p ⩽ 0.05.
Since, MDSCs are known to release IL‐10, we analyzed both MDSCs and IL‐10 low and high group and compared their PFS and OS. For discovery cohort, both PMN‐MDSCs and IL‐10 (p PFS = 0.001, p OS = 0.06) (Fig. 4A), M‐MDSCs and IL‐10 (p PFS = 0.11, p OS = 0.002) (Fig. 4B) low group showed longer PFS and OS compared with both high groups. For validation cohort, both PMN‐MDSCs and IL‐10 (p PFS = 0.001, p OS = 0.006) (Fig. 4C), M‐MDSCs and IL‐10 (p PFS = 0.0004, p OS = 0.0008) (Fig. 4D) low group also showed longer PFS and OS. Next, we confirmed the correlation of MDSCs and IL‐10. IL‐10 low group showed low PMN‐MDSCs frequencies (Fig. 4E) and both PMN‐MDSCs and IL‐10 low group of the patients showed better response rate (80%, 12/15) compared with both high group (13.3%, 2/15) (Fig. 4F). IL‐10 low group also showed lower M‐MDSC frequencies compared with IL‐10 high group (Fig. 4G) and both M‐MDSC and IL‐10 low group of the patients showed better response rate (82.7%, 12/14) compared with both high group (15.3%, 2/13) (Fig. 4H).
Figure 4.
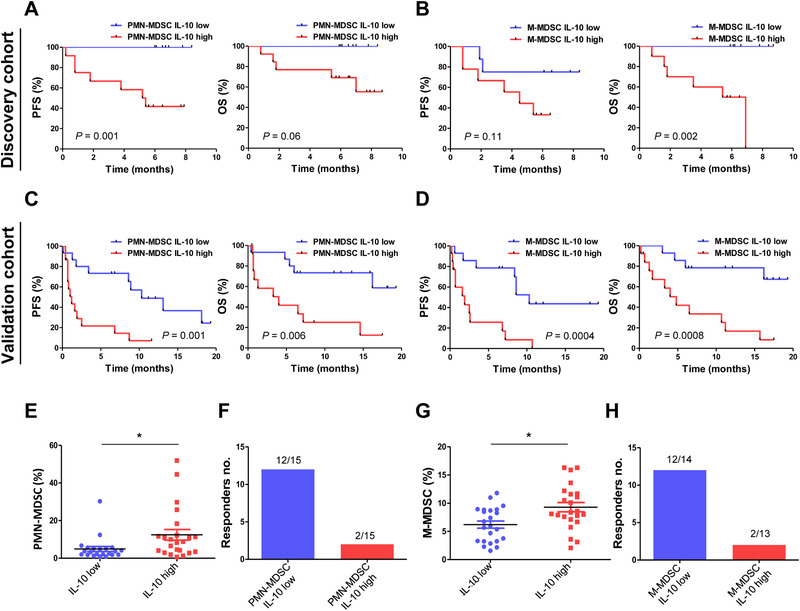
Cytokine protein expression combined with MDSCs (discovery cohort, n = 40; validation cohort, n = 49). PFS and OS of the patients depending on (A) PMN‐MDSCs and IL‐10, (B) M‐MDSCs and IL‐10 low and high groups for discovery cohort, and PFS and OS of the patients depending on (C) PMN‐MDSCs and IL‐10, (D) M‐MDSCs and IL‐10 low and high groups for validation cohort. (E) PMN‐MDSCs frequencies (measured by flow cytometry) depending on IL‐10 (measured by ELISA) low and high groups. (F) The number of responders depending on both PMN‐MDSCs and IL‐10 low (12/15) and high (2/15) groups (total n = 49). (G) M‐MDSCs frequencies (measured by flow cytometry) depending on IL‐10 (measured by ELISA) low and high groups. (H) The number of responders depending on both M‐MDSCs and IL‐10 low (12/14) and high (2/13) groups in validation cohort (n = 49). All data were from one experiment with 40 patient samples for discovery cohort and 49 patient samples for validation cohort. The center value is Mean ± SEM (E, G). Kaplan–Meier survival curves were plotted with patients by median cutoff of each cell types. Statistical significance was determined by log‐rank (Mantel–Cox) regression analysis, with the level of significance at p ⩽ 0.05. *, p ⩽ 0.05; **, p ⩽ 0.01; ***, p ⩽ 0.001 (Mann–Whitney U test).
For further evaluation of these cytokines at mRNA levels, we analyzed IL‐10, TGF‐β, and IL‐6 mRNA levels prior to therapy to test their correlation with suppressive immune cells and clinical outcomes. When we compared IL‐10 low and high group of the patients, IL‐10 low group showed longer OS (7.1 vs. 5.7 months; p = 0.001) (Supporting information Fig. 5A). High frequencies of PMN‐MDSCs (statistically not significant) and M‐MDSCs (p = 0.03) correlated with high IL‐10 mRNA expression (Supporting information Fig. 5B). Then, we analyzed whether suppressive immune cells and IL‐10 both low and high group correlated with clinical outcomes. Low PMN‐MDSCs and IL‐10 (8 vs. 3.7 months; p = 0.007), low M‐MDSCs and IL‐10 (8 vs. 5 months; p = 0.005) showed better OS compared with both high groups (Supporting information Fig. 5C).
Discussion
In this study, we found that circulating suppressive immune cells, including PMN‐MDSCs, M‐MDSCs, and CD39 expression on CD8+ T cells were associated with clinical outcomes in patients with advanced NSCLC (stage IIIB‐IV) treated with anti‐PD‐1 immunotherapy. Low frequencies of each individual suppressive immune cell subsets were correlated with longer PFS and OS to anti‐PD‐1 therapy. When the analysis combined MDSCs and CD39+CD8+T cells, the differential effects of PFS, OS, and response rate were more prominent. Further, an independent cohort of patients with stage I‐IV disease showed that in advanced stage of disease, the amount of circulating suppressive immune cell types of PMN‐MDSCs, M‐MDSCs, and Treg cells were increased, whereas T cells expressed high CD39.
Previous studies have reported that clinical responders to ipilimumab therapy show significantly lower frequencies of M‐MDSCs in the peripheral blood when compared with nonresponders [11]. Moreover, a lower frequency of circulating MDSCs correlated with increased OS of patients with prostate cancer treated with a cancer vaccine in combination with ipilimumab [22], which is consistent with our findings. Further, combined analysis of MDSCs subtypes with CD39+CD8+ T cells revealed a significant difference in PFS and OS in patients treated with anti‐PD‐1 therapy. These findings indicate that circulating suppressive immune cells might offer a potential biomarker for selecting patients who will benefit from anti‐PD‐1 therapy.
MDSCs release various cytokines, including IL‐10, TGF‐β, and IL‐6 to suppress T cell, NKC, and DC function and activate TAMs and Treg cells at tumor sites [23, 24, 25]. We verified that IL‐10 possibly produced by suppressive immune cells, including MDSCs correlated with poor OS implying that IL‐10 release might be one of the mechanisms by which suppressive immune cells suppress tumor immunity.
Previously, our group reported that a higher fold change in the percentage of Ki‐67 7 days after anti‐PD‐1 therapy predicted a DCB and prolonged PFS in patients with thymic epithelial cell tumors and NSCLC [20]. In this study, only CD39+CD8+ T cells correlated with PFS, and PMN‐MDSCs, and M‐MDSCs were correlated with OS for discovery cohort. M‐MDSCs were correlated with PFS, and PMN‐MDSCs and CD39+CD8+ T cells were correlated with OS for validation cohort (Supporting information Fig. 3). The changes of MDSCs and CD39+CD8+ T cells after the therapy compared with baseline showed no differences in PFS and OS (Supporting information Fig. 4). Thus, we hypothesize that pre‐existing MDSCs at baseline might play more important role in predicting clinical outcomes for anti‐PD‐1 therapy.
Interestingly, PD‐L1 expression was not correlated with frequencies of suppressive immune cells, including PMN‐MDSCs and M‐MDSCs, or with clinical outcomes, including PFS, OS, and response rate (data not shown); suggesting that PD‐L1 expression itself alone is not a sufficient biomarker. Based on our results, circulating suppressive immune cells can be complementary predictive biomarkers along with PD‐L1 expression for anti‐PD‐1 immunotherapy.
Evaluation of tissue suppressive immune cells would be more representative than cells from the peripheral blood, but tissue biopsies are not always available. Recently, Yamauchi et al. reported that frequencies of MDSC subsets are higher in tumors than in the peripheral blood, but frequencies of MDSCs from the peripheral blood but not from tumor tissues predicted recurrence after surgery. Therefore, circulating suppressive immune cells might be used as an alternative resource to predict response to anti‐PD‐1 therapy [16].
Although our findings showed promising results for the use of circulating suppressive immune cells as predictive biomarkers, this study has limitations. The distinction between PMN‐MDSCs and neutrophils has been debated for many years for the similarity of their phenotype and morphology. Presently, the only method allowing for separation of neutrophils from PMN‐MDSC is standard Ficoll gradient, the method which we used in this study. PMN‐MDSCs are enriched in low‐density fraction, but still, low‐density fraction contains not only PMN‐MDSCs, but some activated neutrophils contaminate low‐density fraction of PMN‐MDSCs [26]. In recent study, lectin‐like oxidized low‐density lipoprotein receptor‐1 (Lox‐1) has been known to distinct PMN‐MDSCs from neutrophils [27, 28], therefore, we further analyzed PMN‐MDSCs with lox‐1 expression in NSCLC patients (n = 7) from PBMC isolated by standard Ficoll gradient and granulocytes obtained by RBC lysed whole blood. Lox‐1 expression varied depending on the patients which might suggest that PMN‐MDSCs and neutrophils are mixed together likewise each individual has different frequencies of PMN‐MDSCs and neutrophils (Supporting information Fig. 6). However, as neutrophils also express 5‐15% of Lox‐1, more studies are needed to potentially serve it as a marker for identification of PMN‐MDSC in cancer patients [27]. PMN‐MDSCs could be considered as pathologically activated neutrophils, or these cells might take a different role in different situation for example of tumor microenvironment or in different diseases. Therefore, more investigation is needed until we could either separate or unify PMN‐MDSCs from neutrophils [27, 29].
In conclusion, baseline circulating PMN‐MDSCs and M‐MDSCs, and CD39+CD8+ T cells are correlated with clinical outcomes including PFS and OS to anti‐PD‐1 therapy in NSCLC patients. Combined analysis of MDSC subsets along with CD39+CD8+ T cells showed more prominent differential effects. Also, IL‐10 possibly released by suppressive immune cells including MDSCs correlated with MDSCs frequencies, and IL‐10 alone or together with suppressive immune cells correlated with clinical outcomes. These results indicate that circulating suppressive immune cells, including PMN‐MDSCs and M‐MDSCs, might be used as potential biomarkers to predict clinical outcomes of anti‐PD‐1 immunotherapy. Combinatorial strategies targeting MDSCs or IL‐10 should be investigated to further improve outcomes of immune checkpoint inhibitors in advanced NSCLC.
Material and methods
Study samples
Baseline blood samples from stage I to IV lung cancer patients (n = 59), and baseline and 1 week after the therapy paired blood samples from stage IIIB to IV NSCLC patients (discovery cohort, n = 83; validation cohort, n = 49) undergoing anti‐PD‐1 immunotherapy either pembrolizumab (200 mg every 3 weeks) or nivolumab (2 mg/kg every 2 weeks) were collected as a part of a phase II clinical trial (NCT02607631) at Samsung Medical Center (Korea) from March 2017 to February 2018 for discovery cohort, and March 2018 to March 2019 for validation cohort. All the patients had no previous treatment with immune checkpoint inhibitors and the baseline peripheral blood was collected prior to the first line of the immunotherapy. All protocols were approved by the Institutional Review Board and all the participants signed informed consent. All studies in patients were conducted in accordance with the ethical guidelines stated by Samsung Medical Center. Blood donor characteristics are shown in Table 1 and Supporting information Table 1. Patients who benefited from anti‐PD‐1 longer than 6 months defined as durable clinical benefiters (DCB) and less than 6 months for nondurable benefiters (NDB).
Blood preparation
PBMCs from whole blood mixed 1:1 with PBS was isolated by density centrifugation using Ficoll Paque (GE healthcare, Chicago, IL, USA), at 400 × g for 25 min with brakeoff at room temperature. Isolated PBMCs were washed with RPMI (Gibco, Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 400 × g for 10 min at 4°C. PMN‐MDSCs and M‐MDSCs were analyzed on the same day of PBMC isolation and for other immune cell population analysis, PBMCs were cryopreserved for the later use.
Flow cytometry analysis
For MDSCs, isolated PBMCs were stained with anti‐CD3‐BV421 (UCHT1/562426), CD19‐BV421 (HIB19/562440), CD56‐BV421 (NCAM16.2/562751), CD20‐BV421 (2H7/562873), CD11b‐BB515 (ICRF44/564517), CD15‐PerCP‐Cy 5.5 (HI98/560828), CD14‐APC (M5E2/555399), and HLA‐DR‐PE (G46‐6/555812) antibodies (BD Biosciences, San Jose, CA, USA) for 45 min. For lectin‐type oxidized LDL receptor 1 (Lox‐1) expression on PMN‐MDSCs, lox‐1‐BV421 (15C4/358610) (Biolegend, San Diego, CA, USA) was used. For Treg cells, isolated PBMCs were stained with anti‐CD4‐FITC (RPA‐T4/555346), CD25‐APC (M‐A251/555434), CD45RA‐PerCP‐Cy 5.5 (HI100/563429), and Foxp3‐PE (259D/C7/560046) antibodies (BD Biosciences) for 45 min. For CD8+ T cells, isolated PBMCs were stained with anti‐CD8‐APC (RPA‐T8/555369) (BD Biosciences), CD39‐PE‐Cy7 (A1/328212) (Biolegend), and PD‐1‐PerCP‐Cy5.5 (EH12.1 /561273) (BD Biosciences) antibodies. Samples were washed twice and then read on a BD FACSVerse (BD Biosciences) flow cytometer. Dead cells were excluded using 7‐Amino‐Actinomycin D (7AAD) (Biolegend). Gating strategies for PMN‐MDSCs, M‐MDSCs, and CD8+ T cells are shown in Supporting information Fig. 1A‐C. All the process of T‐cell assays and flow cytometry analysis (linear axis) adhered to the guidelines of MIATA compliant and Cossarizza [30].
Intracellular staining
For intracellular staining, isolated and washed PBMCs were stained with appropriate antibodies for 30 min and washed. Cells were fixed and permeabilized with Cytofix/Cytoperm and TF fix/perm (BD Biosciences) for 20 and 40 min respectively, and then washed with Perm Wash Buffer (BD Biosciences). Cells were then stained with Foxp3‐PE (259D/C7/560046) and Ki‐67‐PerCP‐Cy 5.5 (B56/561284) (BD Biosciences) according to the manufacturer's instructions. Samples were washed twice and then read on a BD FACSVerse (BD Biosciences).
Suppressive assays
In order to test suppressive functions of MDSC subtypes, PMN‐MDSCs and T cells were isolated from PBMCs obtained from the same patient using a MACS Separator (Miltenyi Biotec, Bergisch Gladbach, Germany). First, PBMCs were isolated from whole blood as described above. To isolate PMN‐MDSCs from PBMCs, CD15 microbeads were used. The purity of isolated PMN‐MDSCs was 87% or higher as measured on a FACSVerse (BD Biosciences) with PMN‐MDSCs cell surface markers. Isolated PMN‐MDSCs were then cocultured with T cells isolated from the same patient by CD3 microbeads (Miltenyi Biotec). T cells were preactivated with 2 μg/mL anti‐CD3 (OKT‐3/ BE0001‐2) and anti‐CD28 (9.3/BE0248) (Bio X Cell, West Lebanon, NH, USA) antibodies as effector cells and co‐cultured with or without PMN‐MDSCs at different ratios in AIM V Medium (Fisher Scientific, Hampton, NH, USA) with 2 μmol/L of ATP (Sigma, St. Louis, MO, USA) for 5 days. After coculture, cells were harvested and stained with anti‐CD3 (UCHT1/562426) antibody (BD Biosciences) for 30 min, washed twice, and permeabilized in 100 μL of Perm/Fix (BD Biosciences) solution for 40 min. The cells were then washed twice, stained with anti‐Ki‐67 (B56/561284) antibody (BD Bioscience) for 30 min, washed twice, and then Ki‐67 expression on CD3+ T cells was analyzed. For CD39+CD8+ T cells, after 5 days of coculture, cells were harvested and stained with antibodies against CD39 and CD8 for 30 min, washed twice, and then CD39 expression on CD8+ T cells was analyzed on a FACSVerse (BD Biosciences).
Protein expression — ELISA
To measure IL‐10, TGF‐β, and IL‐6 protein expression, we collected plasma from NSCLC patients. The Human IL‐10 Quantikine ELISA Kit, Human TGF‐β 1 Quantikine ELISA Kit, and Human IL‐6 Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA) were used to detect plasma levels of each cytokine according to the manufacturer's instructions. Cytokine levels were read using a SPECTRA max plus microplate reader set to 450 nm (Molecular Devices, San Jose, CA, USA) and analyzed by GraphPad Prism 5 (GraphPad, La Jolla, CA).
mRNA expression — real‐time quantitative PCR
To measure IL‐10, TGF‐β, and IL‐6 suppressive cytokine gene expression, we isolated total RNA from PBMCs of patients using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. cDNA was then constructed from isolated total RNA using the Superscript III first strand synthesis system (Invitrogen, Carlsbad, CA, USA). RT PCR was performed using IL‐10, TGF‐β 1, IL‐6, and β‐actin TaqMan TM Gene Expression Assays and TaqMan TM Gene Expression Master Mix (Thermo Fisher Scientific). Gene expression was measured with an Applied Biosystem PRISM 7900HT (384 well mode) (Applied Biosystem, Foster City, CA, USA) and analyzed by SDS2.4 software.
Statistical analyses
Data were analyzed by independent two‐tailed Student's t tests, One‐way ANOVA (and nonparametric tests), and Mann‐Whitney U test with 95% confidence intervals. Survival curves were obtained using the Kaplan–Meier method and comparisons were made using the log‐rank (Mantel‐Cox) test. All statistical analyses were performed with GraphPad Prism 5 (GraphPad, La Jolla, CA) and R statistical software (version 3.2.2, The R Foundation for Statistical Computing, Vienna, Austria). Two‐tailed p‐values < 0.05 were considered significant.
Conflict of Interest
The authors have declared that no commercial or financial conflict of interest exists.
Abbreviations
- M‐MDSC
monocytic‐myeloid derived suppressor cell
- PMN‐MDSC
polymorphonuclear‐myeloid derived suppressor cell
- TGF‐β
transforming growth factor‐beta
- IL
interleukin
Supporting information
Supporting Information.
Acknowledgments
This research was supported by NRF (National Research Foundation of Korea) funded by the Korean Government (NRF‐2017H1A2A1044327 Global Ph.D. Fellowship Program), and the Bio & Medical Technology Development Program of the NRF (National Research Foundation) funded by the Korean Government (MSIT) (NRF‐2017M3A9G5060259).
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202048534
References
- 1. Herbst, R. S. , Morgensztern, D. and Boshoff, C. , The biology and management of non‐small cell lung cancer. Nature 2018. 553: 446–454. [DOI] [PubMed] [Google Scholar]
- 2. Zappa, C. and Mousa, S. A. , Non‐small cell lung cancer: current treatment and future advances. Transl. Lung Cancer Res. 2016. 5: 288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Garon, E. B. , Rizvi, N. A. , Hui, R. , Leighl, N. , Balmanoukian, A. S. , Eder, J. P. , Patnaik, A . et al., Pembrolizumab for the treatment of non‐small‐cell lung cancer. N. Engl. J. Med. 2015. 372: 2018–2028. [DOI] [PubMed] [Google Scholar]
- 4. Borghaei, H. , Paz‐Ares, L. , Horn, L. , Spigel, D. R. , Steins, M. , Ready, N. E. , Chow, L. Q . et al., Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N. Engl. J. Med. 2015. 373: 1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rittmeyer, A. , Barlesi, F. , Waterkamp, D. , Park, K. , Ciardiello, F. , von Pawel, J. , Gadgeel, S. M. et al., Atezolizumab versus docetaxel in patients with previously treated non‐small‐cell lung cancer (OAK): a phase 3, open‐label, multicentre randomised controlled trial. Lancet (London, England) 2017. 389: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Creasy, C. A. , Forget, M. A. , Singh, G. , Tapia, C. , Xu, M. , Stephen, B. , Sabir, S . et al., Exposure to anti‐PD‐1 causes functional differences in tumor‐infiltrating lymphocytes in rare solid tumors. Eur. J. Immunol. 2019. 49: 2245–2251. [DOI] [PubMed] [Google Scholar]
- 7. Raju, S. , Joseph, R. and Sehgal, S. , Review of checkpoint immunotherapy for the management of non‐small cell lung cancer. ImmunoTargets Ther. 2018. 7: 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Webb, E. S. , Liu, P. , Baleeiro, R. , Lemoine, N. R. , Yuan, M. and Wang, Y. H. , Immune checkpoint inhibitors in cancer therapy. J. Biomed. Res. 2018. 32: 317–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wei, S. C. , Duffy, C. R. and Allison, J. P. , Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018. 8: 1069–1086. [DOI] [PubMed] [Google Scholar]
- 10. Cho, J. H. , Immunotherapy for non‐small‐cell lung cancer: current status and future obstacles. Immune Netw. 2017. 17: 378–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meyer, C. , Cagnon, L. , Costa‐Nunes, C. M. , Baumgaertner, P. , Montandon, N. , Leyvraz, L. , Michielin, O. et al., Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunotherap: CII 2014. 63: 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beury, D. W. , Parker, K. H. , Nyandjo, M. , Sinha, P. , Carter, K. A. and Ostrand‐Rosenberg, S. , Cross‐talk among myeloid‐derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J. Leukocyte Biol. 2014. 96: 1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gabrilovich, D. I. , Myeloid‐derived suppressor cells. Cancer Immunol Res. 2017. 5: 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Condamine, T. and Gabrilovich, D. I. , Molecular mechanisms regulating myeloid‐derived suppressor cell differentiation and function. Trends Immunol. 2011. 32: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lindau, D. , Gielen, P. , Kroesen, M. , Wesseling, P. and Adema, G. J. , The immunosuppressive tumour network: myeloid‐derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013. 138: 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yamauchi, Y. , Safi, S. , Blattner, C. , Rathinasamy, A. , Umansky, L. , Juenger, S. , Warth, A. et al., Circulating and tumor myeloid‐derived suppressor cells in resectable non‐small cell lung cancer. Am J. Respir. Crit. Care Med. 2018. 198: 777–787. [DOI] [PubMed] [Google Scholar]
- 17. Chanmee, T. , Ontong, P. , Konno, K. and Itano, N. , Tumor‐associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 2014. 6: 1670–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hope, H. C. and Salmond, R. J. , Targeting the tumor microenvironment and T cell metabolism for effective cancer immunotherapy. Eur. J. Immunol. 2019. 49: 1147–1152. [DOI] [PubMed] [Google Scholar]
- 19. Canale, F. P. , Ramello, M. C. , Nunez, N. , Bossio, S. N. , Piaggio, E. , Gruppi, A. , Rodriguez, E. V. A. and Montes, C. L. , CD39 expression defines cell exhaustion in tumor‐infiltrating CD8(+) T cells‐response. Cancer Res. 2018. 78: 5175. [DOI] [PubMed] [Google Scholar]
- 20. Kim, K. H. , Cho, J. , Ku, B. M. , Koh, J. , Sun, J. M. , Lee, S. H. , Ahn, J. S . et al., The first‐week proliferative response of peripheral blood PD‐1+CD8+ T cells predicts the response to anti‐PD‐1 therapy in solid tumors. Clin. Cancer Res. An Off. J. Am. Assoc. Cancer Res. 2019. 25: 2144–2154. [DOI] [PubMed] [Google Scholar]
- 21. Gabrilovich, D. I. and Nagaraj, S. , Myeloid‐derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009. 9: 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Santegoets, S. J. , Stam, A. G. , Lougheed, S. M. , Gall, H. , Jooss, K. , Sacks, N. , Hege, K . et al., Myeloid derived suppressor and dendritic cell subsets are related to clinical outcome in prostate cancer patients treated with prostate GVAX and ipilimumab. J. Immunother. Cancer 2014. 2: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao, Y. , Wu, T. , Shao, S. , Shi, B. and Zhao, Y. , Phenotype, development, and biological function of myeloid‐derived suppressor cells. Oncoimmunology 2016. 5: e1004983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Umansky, V. , Blattner, C. , Gebhardt, C. and Utikal, J. , The role of myeloid‐derived suppressor cells (MDSC) in cancer progression. Vaccines 20164: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park, M. J. , Lee, S. H. , Kim, E. K. , Lee, E. J. , Baek, J. A. , Park, S. H. , Kwok, S. K. and Cho, M. L. , Interleukin‐10 produced by myeloid‐derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci. Rep. 2018. 8: 3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bronte, V. , Brandau, S. , Chen, S. H. , Colombo, M. P. , Frey, A. B. , Greten, T. F. , Mandruzzato, S . et al., Recommendations for myeloid‐derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016. 7: 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Condamine, T. , Dominguez, G. A. , Youn, J. I. , Kossenkov, A. V. , Mony, S. , Alicea‐Torres, K. , Tcyganov, E. et al., Lectin‐type oxidized LDL receptor‐1 distinguishes population of human polymorphonuclear myeloid‐derived suppressor cells in cancer patients. Sci. Immunol. 20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim, H. R. , Park, S. M. , Seo, S. U. , Jung, I. , Yoon, H. I. , Gabrilovich, D. I. , Cho, B. C . et al., The ratio of peripheral regulatory T cells to Lox‐1(+) polymorphonuclear myeloid‐derived suppressor cells predicts the early response to anti‐PD‐1 therapy in patients with non‐small cell lung cancer. Am. J. Respir. Crit. Care. Med. 2019. 199: 243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou, J. , Nefedova, Y. , Lei, A. , and Gabrilovich, D. , Neutrophils and PMN‐MDSC: Their biological role and interaction with stromal cells. Semin. Immunol. 2018. 35: 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cossarizza, A. , Chang, H. D. , Radbruch, A. , Acs, A. , Adam, D. , Adam‐Klages, S. , Agace, W. W . et al., Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur. J. Immunol. 2019. 49: 1457–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.