

Abstract
Cyclodextrins are widely used pharmaceutical excipients, particularly for insoluble compounds dosed orally, such as the oral solution of itraconazole, which is frequently used in clinical drug–drug interaction studies to inhibit cytochrome P450 3A. Since cyclodextrins act by forming inclusion complexes with their coformulated drug, they could have an unintended consequence of affecting absorption if they form a strong complex with the potential victim drug in an itraconazole drug–drug interaction study. This observation was made in a drug–drug interaction study with the Bruton’s tyrosine kinase (BTK) inhibitor fenebrutinib and itraconazole, in which, relative to the control group, the expected increase in fenebrutinib maximum plasma concentration (Cmax) was not observed in the itraconazole group, and a delay in time to reach maximum plasma concentration (Tmax) was observed in the itraconazole group. The in vitro binding constant between fenebrutinib and hydroxypropyl‐β‐cyclodextrin was determined to be 2 × 105 M−1, and the apparent permeability of fenebrutinib across a Madin‐Darby canine kidney cell monolayer decreased in a cyclodextrin concentration‐dependent manner. This observation was confirmed in vivo, in a pentagastrin‐pretreated dog model, in which fenebrutinib was administered with or without cyclodextrin; a reduction in Cmax, a prolonged Tmax, and increased fenebrutinib recovery in feces replicated the previous observation in healthy volunteers and supported the hypothesis that complexation with cyclodextrin decreased rate and extent of fenebrutinib absorption. Physiologically‐based pharmacokinetic modeling was used to translate the in vitro effect of cyclodextrin on fenebrutinib apparent permeability to the in vivo effect on absorption, which was then confirmed using the in vivo dog pharmacokinetic data.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Cyclodextrins are commonly used excipients, but their ability to produce meaningful clinical changes in pharmacokinetics of coadministered drugs was heretofore unknown.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study validated the hypothesis that hydroxypropyl‐β‐cyclodextrin (HP‐β‐CD) in the itraconazole formulation reduced the absorption of fenebrutinib, confounding the DDI study.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This study identified a potential confounding factor in a commonly used drug–drug interaction (DDI) study protocol, since itraconazole oral solution is frequently used to determine the contribution of cytochrome P450 3A (CYP3A) to the metabolism of a drug and potential for CYP3A‐mediated DDIs. This study demonstrated that excipient–drug interactions should not be overlooked.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Many new molecular entities (NMEs) undergo a clinical DDI study to determine label recommendations, frequently carried out with itraconazole solution. This study suggests that additional conditions be met prior to carrying out such a DDI study: that the binding constant with HP‐β‐CD be determined prior to using itraconazole solution in a DDI study with NMEs, and if complexation with HP‐β‐CD is high to consider using a different formulation of itraconazole that does not contain HP‐β‐CD.
Cyclodextrins are a common food additive and pharmaceutical excipient. They are macrocycles comprised of glucose subunits, with α‐cyclodextrin, β‐cyclodextrin, and γ‐cyclodextrin denoting a 6‐subunit, 7‐subunit, or 8‐subunit oligosaccharide, respectively. Cyclodextrins are frequently used to improve the apparent solubility of highly insoluble compounds. Their hydrophobic interior allows them to form a complex with the insoluble, hydrophobic compound, while their hydrophilic exterior allows them to form hydrogen bonds with aqueous solvents and form a solution. Attaining a solution is essential for intravenous dosing and can also be beneficial to increase absorption of an oral or parenteral dose of a hydrophobic compound. 1
One example of a formulation in which hydroxypropyl‐β‐cyclodextrin (HP‐β‐CD) is used as an excipient is the itraconazole solution for oral dosing (Sporanox). While itraconazole was originally marketed as an antifungal agent, at present, it is primarily used as a strong cytochrome P450 3A (CYP3A) inhibitor in drug–drug interaction (DDI) studies to determine the effect of CYP3A inhibition on the pharmacokinetics of potential CYP3A substrates. While itraconazole is available in both solution and solid dosage formulations, the solution formulation is more commonly used because it has higher systemic exposure and less variability than the capsule formulation. 2
Fenebrutinib, also known as GDC‐0853, is an orally administered, reversible Bruton’s tyrosine kinase (BTK) inhibitor, under development for several autoimmune disease indications. 3 , 4 Like most drugs entering drug development, an in vitro assessment of DDI potential was conducted for fenebrutinib, and it was determined that fenebrutinib is a substrate of CYP3A ( Supplemental Data ). Since fenebrutinib is to be administered chronically in patients who may be receiving concomitant therapies, we also conducted an in vivo DDI assessment in healthy subjects using itraconazole as a strong CYP3A inhibitor, in which the expected increase in fenebrutinib maximum plasma concentration (Cmax) was not observed in the itraconazole group relative to control, and furthermore, a delay in time to reach maximum plasma concentration (Tmax) was observed in the itraconazole group relative to control. The extent of this expected change depends on the fraction of fenebrutinib metabolized by CYP3A, which is currently unknown. We hypothesized that an interaction between fenebrutinib and the HP‐β‐CD in the itraconazole formulation was responsible for these unexpected results. The present report outlines the clinical DDI study and its unusual results, and also outlines multiple studies conducted to test the hypothesis that these unusual results were caused by a fenebrutinib–cyclodextrin interaction, including (i) determination of binding constant between fenebrutinib and HP‐β‐CD, (ii) characterization of the effect of HP‐β‐CD on in vitro apparent permeability (P app) of fenebrutinib, (iii) in vivo confirmation of cyclodextrin–fenebrutinib interaction in dogs in vivo, and (iv) modeling using GastroPlus software (Lancaster, CA) to simulate HP‐β‐CD concentration in different parts of the intestine, which were in turn used to predict the changes in apparent permeability in those intestinal regions. These changes in permeability were then used to estimate the pharmacokinetic (PK) profile relative to the observed data in dog and human. This study presents, for the first time (to the authors’ knowledge), an example of cyclodextrin–drug interaction that confounded results of a DDI study.
MATERIALS AND METHODS
Itraconazole clinical DDI study
This evaluation consisted of an open‐label fixed‐sequence three‐period drug–drug interaction study. In the first period, subjects received a single oral dose of 100 mg fenebrutinib on Day 1, consisting of two 50 mg tablets (tablet composition provided in Supplemental Data ). After a washout period of 3 days, subjects initiated the second period in which they received six doses of 200 mg itraconazole orally once daily as an oral solution on Days 4 through 9. In the third period, a single oral dose of 100 mg fenebrutinib was coadministered with 200 mg itraconazole on Day 10, followed by one oral dose of 200 mg itraconazole on Day 11. Blood samples for PK analysis of fenebrutinib were collected predose and up to 48 hours postdose on Days 1 and 10. Subjects were confined at the study site from the time of check‐in (Day −1) until clinic discharge on Day 13. All doses of study drug were administered with 240 mL of room temperature water following an overnight fast, food was restricted for 4 hours following dosing, and water was restricted for an hour before and after oral dosing.
Subjects were healthy males and females (not of childbearing potential) between 18 and 60 years of age, with body mass index between 18 and 31 kg/m2, and in good health based on the results of medical history, physical examination, 12‐lead electrocardiogram, and laboratory results. Subjects were ineligible if there was any prior history of immunodeficiency (or current infection), if they had used nicotine or nicotine‐containing products during the last 6 months, if they could not refrain from use of any medication or supplements starting 2 weeks prior to the study through the duration of the study, or if they could not refrain from using grapefruit or its products starting 72 hours prior to the study through the duration of the study. This study was conducted in accordance with Good Clinical Practice and the ethical principles of the Declaration of Helsinki. All subjects provided written informed consent before beginning any study procedures. The protocols, amendments, and subject‐informed consents received appropriate Institutional Review Board approval prior to initiation.
Determination of the binding constant
The binding constant (k complex) was measured by adding an excess amount of fenebrutinib into individual glass vials containing an aqueous solution of different concentrations of either HP‐β‐CD or α‐cyclodextrin (α‐CD) and sealed. The mixtures were placed on a shaker and shaken for a period of 48 hours at ambient conditions to allow for equilibration. After equilibration, each mixture was centrifuge filtered to remove excess solids. The concentration of the fenebrutinib in each filtrate was determined by using high‐performance liquid chromatography with diode array detector. The k complex was calculated using Eq. 1, in which [FEN‐CD] is the fenebrutinib–cyclodextrin complex in the filtrate, [FEN] is the intrinsic solubility of fenebrutinib and [CD] is the concentration of free HP‐β‐CD.
| (1) |
Permeability across an MDCK cell monolayer
Madin‐Darby canine kidney cells (MDCK) were obtained from ATCC, (Manassas, VA). CRISPR (clustered regularly interspaced short palindromic repeats) Cas9 (CRISPR associated protein 9) was used to knock out the endogenous canine Mdr1 gene as previously reported. 5 Cells were maintained in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum, 1% pen‐strep and 5 µg/mL plasmocin (InvivoGen, San Diego, CA) before seeding on Millipore Millicell‐24 well plates with polyester membrane and 1 µm pores (EMD Millipore, Darmstadt, Germany) at 2.5 × 105 cells/mL and allowed to grow for 5 days. Prior to the permeability experiment cell monolayers were equilibrated in transport buffer (Hank’s Balanced Salt Solution with 10 mM Hepes, pH 7.4) for 20 minutes at 37°C with 5% carbon dioxide and 95% relative humidity. Fenebrutinib dose solutions were prepared at 20 µM concentration in transport buffer containing the monolayer integrity marker lucifer yellow (50 µM) and various percentages (weight/volume, w/v) of HP‐β‐CD (0–20%). Dose solutions were added to the donor chambers and transport buffer was added to all receiver chambers. The permeability was examined in the apical to basolateral (A:B) direction (n = 4). The receiver chambers were sampled at 30, 60, and 90 minutes and were replenished with fresh transport buffer. At the end of the experiment lucifer yellow was measured using a SpectraMax i3 fluorescence plate reader (Molecular Devices, San Jose, CA) with an excitation wavelength of 425 nm and an emission of 530 nm. Samples were diluted fivefold with analytical internal standard (20 nM propranolol in 100% acetonitrile containing 0.1% formic acid) before being analyzed in Multiple Reaction Monitoring mode on a Sciex 6500Qtrap (Foster City, CA) with Shimadzu Nextera‐series high performance liquid chromatography pumps and autosampler (Kyoto, Japan). P app was calculated using Eq. 2, in which dQ/dt is the rate of compound appearance in the receiver compartment; A = surface area of the insert; and C 0 = initial substrate concentration at time 0 minute.
| (2) |
In Vivo studies in dogs
Eight nonnaïve male beagle dogs were fasted overnight prior to the study. Approximately 30 minutes prior to dosing fenebrutinib, animals were administered a 6 µg/kg intramuscular dose of pentagastrin. The dogs were divided into two groups with four animals per group. Animals in group 1 were administered 20 mL water (H2O) (pH 2) plus 230 mL H2O by oral gavage and then were given two 50 mg fenebrutinib tablets for a total dose of 100 mg. Animals in group 2 were administered 20 mL of 400 mg/mL HP‐β‐CD in H2O (pH 2) plus 230 mL H2O by oral gavage and then were given two 50 mg fenebrutinib tablets for a total dose of 100 mg. Blood samples were collected from the cephalic or saphenous vein at 0 (predose), 5, 15, and 30 minutes postdose and 1, 3, 6, 9, and 24 hours postdose. The concentrations of fenebrutinib in plasma were quantified using liquid chromatography with tandem mass spectrometry (lower limit of quantification = 1.02 ng/mL).
Modeling with GastroPlus
GastroPlus 9.6 was used to fit a model to the human and dog PK data and then simulate the effect of HP‐β‐CD on the fenebrutinib PK profile by altering the intestinal permeability as a function of HP‐β‐CD concentration in each segment. The effect of HP‐β‐CD on permeability was calculated from the MDCK data, and not from the in vitro complexation data. The physiochemical properties used for fenebrutinib are LogD at pH 7.4 of 1.6, pKas of 5 and 3.7, particle size distribution median diameter (D50) of 12 µm, and solubility values of 36, 6.5, 0.036, 0.0001, 0.009, and 0.018 mg/mL in buffer at pH 2.3, pH 2.6, pH 3.8, pH 7.8, FaSSIF at pH 6.5, and FeSSIF at pH 5.0, respectively. HP‐β‐CD, was administered as a solution, and a solubility of 1,000 mg/mL was used in all conditions, since HP‐β‐CD is highly soluble in aqueous solvents.
Statistics
In the clinical study, PK parameters were log transformed and analyzed using a mixed model that included treatment as a fixed effect and subject as a random effect. Least square means were calculated for test (100 mg fenebrutinib coadministered with 200 mg itraconazole) and reference (100 mg fenebrutinib alone) treatments, and the mean differences between the treatments were calculated. The residual variance from the mixed model was used to calculate 90% confidence interval for the difference between the test and reference treatments. These values were back‐transformed to give geometric least square means, a point estimate, and 90% confidence interval for the ratio of the test treatment relative to the reference treatment. Differences between groups in the clinical DDI study were assessed using Student’s two‐tailed t‐test, with P < 0.05 set as being statistically significant. Differences between groups in the dog study were assessed using one‐way analysis of variance, with P < 0.05 set as being statistically significant.
RESULTS
Fenebrutinib DDI study in healthy subjects
The PK profiles of fenebrutinib in the presence and absence of steady‐state itraconazole were compared (Figure 1 ). Typically, the perpetrator of a CYP3A‐mediated DDI causes an increase in both Cmax and area under the concentration‐time curve (AUC) of a victim drug, usually without a change in Tmax. 6 However, in the itraconazole–fenebrutinib DDI study, fenebrutinib AUC0–24h increased significantly from 1.11 ± 0.359 to 2.45 ± 0.588 µg/hour/mL in the presence of itraconazole, but Cmax decreased from 0.221 ± 0.0825 to 0.192 ± 0.0647 µg/mL and this change was not found to be statistically significant. In addition, there was a delay in fenebrutinib absorption with a statistically significant delay in Tmax from 1.56 ± 0.629 to 3.46 ± 2.58 hours. As expected, the terminal half‐life increased from 8.26 ± 2.33 to 15.9 ± 3.02 hours and this was statistically significant, indicating that itraconazole was still inhibiting CYP3A.
Figure 1.
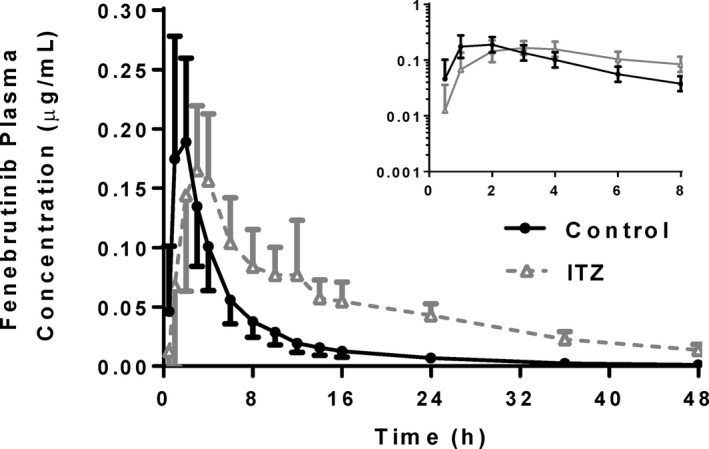
Fenebrutinib plasma concentration‐time profiles in healthy volunteers receiving 100 mg fenebrutinib, or a 200 mg itraconazole pretreatment, once daily × 3 days followed by 100 mg fenebrutinib. Itraconazole did not produce the expected increase in Cmax, but still decreased the slope of the elimination phase. Tmax was also delayed in the itraconazole arm. (n = 14–16 healthy volunteers per arm). Cmax, maximum plasma concentration; ITZ, itraconazole; Tmax, time to reach maximum plasma concentration.
Fenerutinib forms a strong complex with HP‐β‐CD and not α‐CD
The relationship between HP‐β‐CD concentration and fenebrutinib solubility was found to be linear between the concentrations of 0.1 and 130 µM and exhibited a 1:1 stoichiometry (Figure 2a ). The k complex was estimated to be ~ 2 × 105 M−1. Fenebrutinib was found to form a much weaker complex with α‐CD (Figure 2b ); k complex with α‐CD was estimated to be 4,000 M−1. A comparison of the complexation data is shown in Figure 2c .
Figure 2.
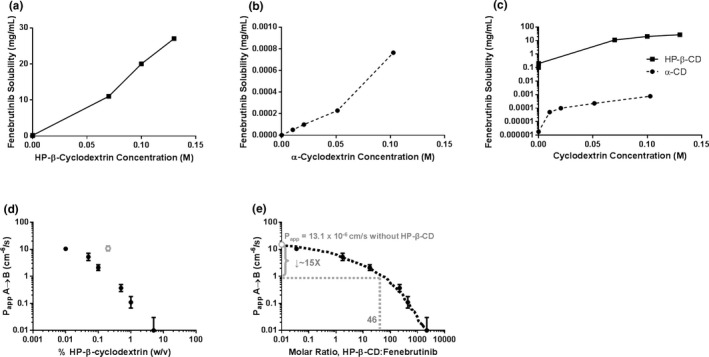
The (a) k complex of fenebrutinib and HP‐β‐CD or (b) α‐CD was estimated by determining the solubility of fenebrutinib at different concentrations of cyclodextrin. (c) k complex of fenebrutinib with HP‐β‐CD was 45‐fold higher than that with α‐CD. (d) HP‐β‐CD reduces apparent apical‐to‐basolateral permeability of fenebrutinib across an MDCK cell monolayer in a concentration‐dependent manner. (e) Expressing HP‐β‐CD concentration as a molar ratio of HP‐β‐CD to fenebrutinib allows for the estimation of the change in permeability in the itraconazole DDI study. At an HP‐β‐CD:fenebrutinib ratio of 46, P app was expected to drop about 16‐fold. (n = 3. An outlier was removed, for analysis, shown in gray). α‐CD, α‐cyclodextrin; DDI, drug–drug interaction; HPβCD, hydroxypropyl‐β‐cyclodextrin; k complex, binding constant; MDCK, Madin‐Darby canine kidney cells; P app, apparent permeability; w/v, weight/volume.
HP‐β‐CD decreases apparent permeability of fenebrutinib across an MDCK cell monolayer
The P app of fenebrutinib was determined in Transwell experiments, across an MDCK cell monolayer, from a donor (apical) compartment to a receiver (basolateral) compartment. As the (w/v) percentage of HP‐β‐CD increased, the apparent permeability decreased (Figure 2d ). When HP‐β‐CD amount was expressed as a molar ratio to fenebrutinib (Figure 2e ), the expected drop in permeability could be compared with the molar ratio of HP‐β‐CD to fenebrutinib that occurred in the DDI study, in which 100 mg of fenebrutinib was administered with 8 g of HP‐β‐CD, resulting in a molar ratio of 46. Interpolation from Figure 2e shows that an approximate 16‐fold drop in apparent permeability of fenebrutinib is expected between the control arm and the itraconazole arm in the DDI study.
HP‐β‐CD decreases absorption of fenebrutinib in pentagastrin‐pretreated dogs
A pentagastrin‐pretreated dog model 7 was used to establish the in vitro–in vivo translation of the fenebrutinib–cyclodextrin interaction, and to test the effect of HP‐β‐CD on fenebrutinib without the confounding factor of itraconazole. Pentagastrin‐pretreated dogs were administered two 50 mg tablets of fenebrutinib, plus 250 mL of solution of placebo or the amount of HP‐β‐CD contained in the itraconazole formulation at full strength, quarter‐strength, and 10% strength HP‐β‐CD concentration, for a total HP‐β‐CD dose of 8 g, 2 g, or 0.8 g, respectively. The plasma concentration‐time profiles are compared in Figure 3a . Cmax decreased about 9‐fold, 3.5‐fold, and 2‐fold, and AUC about 7‐fold, 3.5 fold, and 1.5‐fold at HP‐β‐CD doses of 8 g, 2 g, or 0.8 g, respectively (Table 1 ). Similar to what was observed in the DDI study, Tmax was prolonged from 0.88 to 3.25 hours postdose in the dogs receiving 8 g of HP‐β‐CD (Table 1 ). Variability in AUC appeared to be greater in the control and low‐dose HP‐β‐CD groups than in the mid‐dose and high‐dose HP‐β‐CD groups. Recovery of fenebrutinib in feces increased about 6.5‐fold, 4.5 fold, and 3‐fold at HP‐β‐CD doses of 8 g, 2 g, or 0.8 g, respectively (Figure 3b ; Table 1 ).
Figure 3.
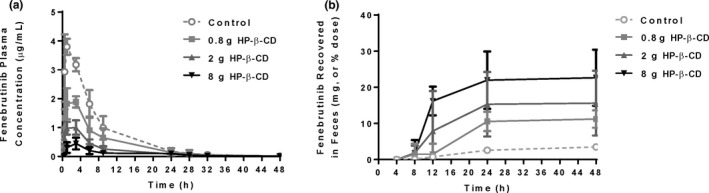
Fenebrutinib (a) plasma concentration‐time profiles and (b) cumulative recovery in feces in pentagastrin‐pretreated dogs receiving a 100 mg fenebrutinib tablet, or a 100 mg fenebrutinib tablet with 0.8 g, 2 g, or 8 g of HP‐β‐CD. The 8 g dose of HP‐β‐CD was the same amount that was administered in the itraconazole DDI study. Cmax and AUC decreased, recovery in feces increased, and Tmax was prolonged, all in an HP‐β‐CD dose‐dependent manner (n = 4 dogs per group). AUC, area under the concentration‐time curve; Cmax, maximum plasma concentration; DDI, drug–drug interaction; HP‐β‐CD, hydroxypropyl‐β‐cyclodextrin; Tmax, time to reach maximum plasma concentration.
Table 1.
Pharmacokinetic parameters compared between the control and HP‐β‐CD arms from the pharmacokinetic study in pentagastrin‐pretreated dogs
| Fenebrutinib Dose (mg) | HP‐β‐CD Administered (g) | AUC 0‐∞ (µg/hour/mL) | Tmax (hour) | Cmax (µg/mL) | Fenebrutinib recovered in feces (mg) |
|---|---|---|---|---|---|
| 100 | 0 | 28.9 ± 6.98 | 0.875 ± 0.250 | 3.94 ± 0.225 | 3.48 ± 0.681 |
| 100 | 0.8 | 17.4 ± 6.92* | 1.38 ± 1.11 | 2.09 ± 0.243* | 11.2 ± 2.39 |
| 100 | 2 | 8.38 ± 1.94* | 2.00 ± 1.15 | 1.11 ± 0.142* | 15.6 ± 8.99* |
| 100 | 8 | 4.46 ± 2.07* | 3.00 ± 0.00* | 0.447 ± 0.204* | 22.7 ± 7.76* |
AUC, area under the concentration‐time curve; Cmax, maximum plasma concentration; HP‐β‐CD; hydroxypropyl‐β‐cyclodextrin; Tmax, time to reach maximum plasma concentration.
Denotes P < 0.05 between control and treatment arm using one‐way ANOVA (analysis of variance) with Bonferroni’s multiple comparisons test.
Modeling to relate the decrease in apparent permeability to the plasma concentration‐time profiles
GastroPlus 9.6 was used to fit a model to the human and dog PK data and then simulate the effect of HP‐β‐CD on the PK profile. A two‐compartment model was used to fit human PK under fasted conditions and opt logD model SA/V6.1 (for the absorption scaling factor) was selected. First the PK data from the control group were used to establish the model, and all PK parameters were fit based on clinical data from the control group; this fit is shown in Figure 4b relative to the observed data in the control group. In this model, the systemic clearance was 2.1 L/hour/kg, volume of distribution in the central compartment was 4.3 L/kg, transfer rate constant from the central to peripheral compartment was 0.5 hour−1, transfer rate constant from the peripheral to central compartment was 0.19 hour−1, and fraction absorbed (Fa) was 1. Fenebrutinib systemic clearance was estimated to be reduced by approximately 45% based on the use of the residual method on the elimination phases of the concentration‐time curves for fenebrutinib alone or in the presence of itraconazole. 8 Assuming the volume of distribution does not change as a result of itraconazole administration, the systemic clearance in the itraconazole group was reduced from 2.1 L/hour/kg to 1.15 L/hour/kg.
Figure 4.
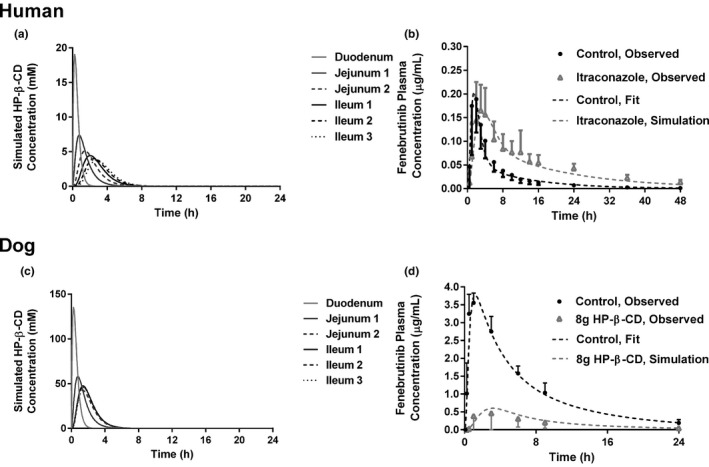
Simulated (a) HP‐β‐CD concentrations in regions of the human small intestine following an 8 g dose. (b) The model, fitted to the control arm data from the human DDI is shown next to the observed data for the control arm. The predicted effect of 8 g HP‐β‐CD and itraconazole is shown relative to the observed data for the itraconazole arm. (c) Simulated HP‐β‐CD concentrations in regions of the dog small intestine following an 8 g dose. (d) The model, fitted to the intravenous bolus data and control arm data from the dog PK study is shown next to the observed data for the control arm. The predicted effect of 8 g HP‐β‐CD is shown relative to the observed data for the 8 g HP‐β‐CD arm. DDI, drug–drug interaction; HP‐β‐CD, hydroxypropyl‐β‐cyclodextrin; PK, pharmacokinetics.
This model was used to simulate the impact on fenebrutinib absorption in the itraconazole group. The concentration of HP‐β‐CD was simulated in each segment of the intestine following an oral dose of 8 g, in solution, with permeability assigned to a value of 1 × 10‐10 cm/s, to reflect minimum oral bioavailability 9 with solubility assigned at 1,000 mg/mL, since HP‐β‐CD is highly soluble in water. The concentration‐time profiles of HP‐β‐CD in each intestinal segment is shown in Figure 4a . The logarithmic average concentration of HP‐β‐CD in each segment was calculated using the formula in which AUCt 1− t 2 is the AUC between t1 and t2, t1 and t2 represent the transit window between HP‐β‐CD entering the segment (t1) and the HP‐β‐CD concentration dropping to 1% of the maximum concentration in that segment (t2) (i.e., for the duodenum, t1 = 0.08 hour and t2 = 2.00 hours).
| (3) |
The average HP‐β‐CD concentration in each small intestinal segment was calculated to be 6.50, 3.31, 2.47, 2.42, 2.24, and 2.12 mM for the duodenum, jejunum 1, jejunum 2, ileum 1, ileum 2, and ileum 3, respectively. The P app of the fenebrutinib as a function of HP‐β‐CD concentration was calculated using the in vitro MDCK permeability data (Figure 3 ) and the corresponding human P app value of each data point from the in vitro MDCK data was calculated by using the GastroPlus user setting and the MDCK calibration curve. Then, the regional effective permeability (P eff) of fenebrutinib in each small intestine segment following an 8 g dose of HP‐β‐CD was determined with the Absorption Systems Caco‐2 calibration option ( Supplemental Data ). These values were used as regional P eff values for fenebrutinib in any dosing group receiving HP‐β‐CD and the model was used to simulate the fenebrutinib plasma concentration‐time profile in the itraconazole group. This refined model predicted that in the presence of HP‐β‐CD, the Fa of fenebrutinib was reduced from 1.0 to 0.91 with a slower absorption rate. The human model predicted that fenebrutinib Cmax would drop approximately 16% from 0.19 µg/mL (control) to 0.16 µg/mL (itraconazole) and Tmax will increase from 1.5 hours (control) to 3.5 hours (itraconazole). The human model accurately predicted the PK curve of the itraconazole group (Figure 4b ) using only the control PK data, the difference in terminal half‐life between the control and IT data, and the in vitro permeability data for fenebrutinib at different concentrations of HP‐β‐CD.
To test this modeling paradigm in the absence of itraconazole, a similar approach was used, utilizing the data from groups 1 and 4 of the dog PK study. Intravenous PK data were used to obtain PK parameters for the dog model ( Supplemental Data ); the systemic clearance of the fenebrutinib was determined to be 0.36 L/hour/kg, volume of distribution in the central compartment was 2 L/kg, transfer rate constant from the central to peripheral compartment was 0.08 hour−1, and transfer rate constant from the peripheral to central compartment was 0.2 hour−1. Fa was 1. Following the same approach used for the human model, the average HP‐β‐CD concentrations for dog in each small intestine segment were calculated to be 48.5, 27.0, 24.5, 22.5, 20.9, and 20.5 mM for the duodenum, jejunum 1, jejunum 2, ileum 1, ileum 2, and ileum 3, respectively (Figure 4c ). P app and dog P eff were calculated as described previously and are shown in the Supplemental Data . These values were used in the GastroPlus dog model to simulate the fenebrutinib plasma concentration‐time profile in the group receiving 8 g of HP‐β‐CD. The model predicted a reduction in Fa from 1 (control) to 0.24 (HP‐β‐CD), a reduction in Cmax from 3.8 µg/mL to 0.6 µg/mL, and an increase in Tmax from 1.2 to 3.1 hours. The predicted values are in good agreement with the observed in vivo data for the group receiving 8 g of HP‐β‐CD (Figure 4d ).
DISCUSSION
In order to rationalize the unexpected decrease in fenebrutinib Cmax and delay in fenebrutinib Tmax observed in the presence of itraconazole in the clinical DDI study, we hypothesized that the HP‐β‐CD in the itraconazole formulation complexed with fenebrutinib, decreasing the absorption of fenebrutinib in that arm, and adding a confounding factor to the study. This hypothesis was tested with in vitro and in vivo studies, and the results were used to simulate the impact on fenebrutinib PK in both dog and human.
When compared with other drugs that form a 1:1 complex with HP‐β‐CD, fenebrutinib had the highest k complex of all drugs for which information was available (Table 2 ). This high k complex value is likely due to the size of fenebrutinib relative to the cavity size of HP‐β‐CD, since the interaction with α‐CD was about 45‐fold weaker. This is an important consideration, since α‐CD is a commonly used food additive and is present at high concentrations in many processed foods, with the top 90th percentile of average daily consumption in the United States reaching 19.6 g. 10 Dividing this number by three meals per day yields a value of 6.5 g of α‐CD; slightly less than the amount of HP‐β‐CD administered in the itraconazole DDI study. Overall, the risk of α‐CD having a meaningful impact of fenebrutinib absorption is considered to be low. Similarly, although γ‐CD is present in pharmaceuticals and foods, the risk of meaningful drug interaction with fenebrutinib was expected to be low due to the larger cavity size and the greater ease of digestion. 9
Table 2.
k complex for other drugs known to form a 1:1 complex with HP‐β‐CD
| Drug | k complex (M−1) | Source, Ref. No. |
|---|---|---|
| Fenebrutinib | 200,000 | Internal data |
| Diethylstilbestrol | 59,200 | 11 |
| Estradiol | 38,600 | 11 |
| Ibprofen | 10,000 | Internal data |
| Digoxin | 6,800 | 12 |
| Celecoxib | 3,000 | Internal data |
| Hydrocortisone | 1,900 | 11 |
| Diflunisal | 1,600 | Internal data |
| Flunitrazepam | 1,100 | 12 |
| Prednisolone | 958 | 11 |
| Naproxen | 780 | 12 |
| Sulfathiazole | 450 | Internal data |
| Sulfamethoxazole | 360 | 12 |
| Miconazole | 260 | 12 |
| Alprazolam | 250 | 12 |
| Triazolam | 200 | 12 |
| Rofecoxib | 190 | Internal data |
| Camptothecin | 184 | 13 . |
| Diazepam | 184 | 11 |
| Econazole | 180 | 12 |
| Acetazolamide | 85 | 12 |
| Norfloxacin | 5 | Internal data |
HP‐β‐CD, hydroxypropyl‐β‐cyclodextrin;k complex, binding constant.
It should be noted that the in vitro complexation experiment was not performed in the presence of bile salts, which would be present at mM concentrations in the in vivo setting. Since fenebrutinib does exhibit pH‐dependent changes in solubility, bile salts may increase the baseline solubility of fenebrutnib, but this difference is unlikely to have a pronounced effect in vivo, owing to the very strong complexation between fenebrutnib and HP‐β‐CD.
It is also important to understand if an excipient–drug interaction could occur if fenebrutinib were to be given concomitantly with another drug that is formulated with HP‐β‐CD. To address this concern, an important factor for consideration is the concentration of cyclodextrin in other drug products compared with itraconazole, summarized in Table 3 . The oral formulation of itraconazole appears to have the highest amount of cyclodextrin by a factor of 10 or greater. Of all the drugs for which information was available, the median amount of cyclodextrin in the theoretical maximum daily dose was 266 mg. This was 30‐fold lower than the dose of cyclodextrin administered in the present itraconazole DDI study. In the case that fenebrutinib was administered with 266 mg of HP‐β‐CD, the molar ratio of HP‐β‐CD to fenebrutinib would be 1.56, resulting in an estimated 2‐fold reduction in P app, instead of the 16‐fold reduction in P app that was observed with the 8 g of HP‐β‐CD that was given with itraconazole. In the dog study, only a twofold reduction in fenebrutinib AUC and Cmax was observed in the group in which fenebrutinib was co‐dosed with 800 mg of HP‐β‐CD, a dose which is threefold higher than the median quantity of HP‐β‐CD present in the marketed drugs in Table 3 . In addition to this, the PBPK model showed that the HP‐β‐CD effect on fenebrutinib absorption was greater in dog than in human, thus the HP‐β‐CD present in potential concomitant medications with which fenebrutinib may be administered would not be expected to have a meaningful clinical impact on fenebrutnib PK, with the exception of some oral solution or suspension formulations that contain HP‐β‐CD. In the event that fenebrutinib needs to be administered with an oral suspension containing HP‐β‐CD, simulations showed that administration of the oral suspension after the majority of fenebrutinib absorption has occurred (at or after Tmax) could circumvent a potential complexation interaction.
Table 3.
β‐Cyclodextrin is present in numerous marketed drugs
| Generic name | Formulation | Cyclodextrin | Highest amount of cyclodextrin per unit dose | Amount of cyclodextrin with maximum daily dose | Reference(s) |
|---|---|---|---|---|---|
| Aceclofenac | Capsule | βCD | 100 mg | 200 mg | 14 |
| Cetirizine | Tablet | βCD | 133 mg a | 266 mg a | 14 |
| Cisapride | Suppository | HP‐β‐CD | Not administered orally | 9 | |
| Chlordiazepoxide | Tablet | βCD | 133 mg a | 1596 mg a | 15 |
| Dextromethorphan | Tablet | βCD | 133 mg a | 1596 mg a | 16 |
| Diphenhydramine HCl | Tablet | βCD | 133 mg a | 1596 mg a | 15 |
| Ethinyl Estradiol | Tablet | βCD | 160 mg | 160 mg | 17, 18 |
| Hydrocortisone | Mouth wash | HP‐β‐CD | If used as directed, 0 mg. | 19 | |
| Itraconazole | Solution | HP‐β‐CD | 4,000 mg | 16,000 mg | 20 |
| Nicotine | Sublingual tablet | βCD | 30.4 mg | 608 mg | 21 |
| Nimesulide | Powder | βCD | 300 mg | 1200 mg | 22 |
| Omeprazole | Film Coated Tablet | βCD | 82.5 mg a | 165 mg a | 15 |
| Perindopril | Tablet | HP‐β‐CD | 26.7 mg | 107 mg | 23 |
| Piroxicam | Tablet; granule | βCD | 171 mg | 171 mg | 24 |
| Risperidone | Solution | HP‐β‐CD | 400 mg a | 3,200 mg a | 25 |
| Tiaprofenic acid | Tablet | Diethyl‐βCD | 133 mg a | 266 mg a | 26 |
Maximum amounts of cyclodextrin derived either from drug labels or formulation patents directly or from FDA Inactive Ingredient Database maximums when compound name but not dose was listed.
βCD, β‐cyclodextrin; FDA, US Food and Drug Administration; HP‐β‐CD, hydroxypropyl‐β‐cyclodextrin.
Indicates when FDA Inactive Ingredient maximum assumed.
This study underscores the importance of not overlooking the possibility of drug–excipient interactions. While fenebrutinib was shown to have an unusually high k complex, and the oral formulation of itraconazole contained an unusually high amount of HP‐β‐CD relative to other marketed drugs, the oral formulation of itraconazole is widely used to determine the extent to which a compound is metabolized by CYP3A (fm,CYP3A), a parameter which is crucial in predicting DDIs with CYP3A inhibitors or inducers. If a new molecular entity should form as strong a complex with HP‐β‐CD as fenebrutinib did, a DDI study with itraconazole could easily be confounded, as was the case for fenebrutinib. It may be prudent for development programs to determine the k complex of their new molecular entity with HP‐β‐CD prior to embarking on a DDI study with itraconazole as an oral formulation, and if strong complexation is observed, alternate formulations of itraconazole that do not contain HP‐β‐CD is advisable.
Translation of preclinical data to predict in vivo outcomes in humans is most often a very daunting task. The present study demonstrates that hypothesis testing and careful attention to the scientific method can and should be used to explain unusual results in the clinic. It also demonstrates that more attention should be paid not just to drugs that can cause DDIs, but also excipients which may do the same.
Funding
This study was funded by Genentech Inc.
Conflict of Interest
All authors are employed by Genentech.
Author Contributions
M.R.D., N.S.J., and P.‐C.C. wrote the manuscript. M.R.D., Y.C., C.M., P.‐C.C., E.G.P., K.N., and L.W.C. designed the research. E.G.P., P.‐C.C., J.L., and S.W. performed the research. M.R.D., J.Z.C., E.G.P., and P.‐C.C. analyzed the data. P.‐C.C. and C.M. contributed new reagents/analytical tools.
Supporting information
Supplementary Material
Acknowledgments
The authors wish to thank the US Food and Drug Administration's (FDA’s) Office of Clinical Pharmacology for the robust discussion of the topic and deeply appreciate their input with respect to study design.
References
- 1. Uekama, K. , Hirayama, F. & Irie, T. Cyclodextrin drug carrier systems. Chem. Rev. 98, 2045–2076 (1998). [DOI] [PubMed] [Google Scholar]
- 2. Liu, L. et al Best practices for the use of itraconazole as a replacement for ketoconazole in drug–drug interaction studies. J. Clin. Pharmacol. 56, 143–151 (2016). [DOI] [PubMed] [Google Scholar]
- 3. Crawford, J.J. et al Discovery of GDC‐0853: a potent, selective, and noncovalent Bruton's tyrosine kinase inhibitor in early clinical development. J. Med. Chem. 61, 2227–2245 (2018). [DOI] [PubMed] [Google Scholar]
- 4. Herman, A.E. et al Safety, pharmacokinetics, and pharmacodynamics in healthy volunteers treated with GDC‐0853, a selective reversible Bruton's tyrosine kinase inhibitor. Clin. Pharmacol. Ther. 103, 1020–1028 (2018). [DOI] [PubMed] [Google Scholar]
- 5. Chen, E.C. et al Evaluating the utility of canine Mdr1 knockout Madin‐Darby canine kidney I cells in permeability screening and efflux substrate determination. Mol. Pharm. 15, 5103–5113 (2018). [DOI] [PubMed] [Google Scholar]
- 6. Ahonen, J. , Olkkola, K.T. & Neuvonen, P.J. Effect of itraconazole and terbinafine on the pharmacokinetics and pharmacodynamics of midazolam in healthy volunteers. Br. J. Clin. Pharmacol. 40, 270–272 (1995). [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou, R. et al pH‐dependent dissolution in vitro and absorption in vivo of weakly basic drugs: development of a canine model. Pharm. Res. 22, 188–192 (2005). [DOI] [PubMed] [Google Scholar]
- 8. Gibaldi, M. & Perrier, D. Pharmacokinetics, 2nd edn. (Marcel Dekker, New York, 1983). [Google Scholar]
- 9. European Medicines Agency . Cyclodextrins Used as Excipients European Medical Agency https://www.ema.europa.eu/en/documents/scientific-guideline/questions-answers-cyclodextrins-used-excipients-medicinal-products-human-use_en.pdf (October 2017).
- 10. Reuscher, H. GRAS notice for alpha‐cyclodextrin wacker chemical corporation. US Food and Drug Administration https://www.fda.gov/media/101653/download (November 2016).
- 11. Másson, M. , Sigurdardóttir, B.V. , Matthíasson, K. & Loftsson, T. Investigation of drug–cyclodextrin complexes by a phase‐distribution method: some theoretical and practical considerations. Chem. Pharm. Bull. (Tokyo) 53, 958–964 (2005). [DOI] [PubMed] [Google Scholar]
- 12. Loftsson, T. & Brewster, M.E. Cyclodextrins as functional excipients: methods to enhance complexation efficiency. J. Pharm. Sci. 101, 3019–3032 (2012). [DOI] [PubMed] [Google Scholar]
- 13. Saetern, A.M. , Nguyen, N.B. , Bauer‐Brandl, A. & Brandl, M. Effect of hydroxypropyl‐beta‐cyclodextrin‐complexation and pH on solubility of camptothecin. Int. J. Pharm. 284, 61–68 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Bextrin Cap, MEDPLUSMART <https://www.medplusmart.com/product/BEXTRIN‐CAP/BEXT0002>. Accessed August 7, 2019.
- 15. El‐Kattan, E. Oral Bioavailability Assessment: Basics and Strategies for Drug Discovery and Development (John Wiley & Sons, Hoboken, NJ, 2017). [Google Scholar]
- 16. Loftsson, T. & Brewster, M.E. Pharmaceutical applications of cyclodextrins: basic science and product development. J. Pharm. Pharmacol. 62, 1607–1621 (2010). [DOI] [PubMed] [Google Scholar]
- 17. Backensfeld, T. , Heil, W. & Lipp, R. Compositions comprising drospirenone and a complex between ethinyl‐estradiol and a cyclodextrin. EP1632237B1. European Patent Office 2001.
- 18. Center for Drug Evaluation and Research, US Food and Drug Administration . Application 21‐676 <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021676s000_CLINPHARMR.pdf> (2004). Accessed August 7, 2019.
- 19. Holbrook, W.P. , Kristmundsdóttir, T. & Loftsson, T. Aqueous hydrocortisone mouthwash solution: clinical evaluation. Acta. Odontol. Scand. 56, 157–160 (1998). [DOI] [PubMed] [Google Scholar]
- 20. Sporanox [package insert] Janssen Pharmaceutica N.V. (2010).
- 21. Carlsson, T. & Andersson, S. Smoking substitute. US5512306A. United States Patent Office 1996. <https://patentimages.storage.googleapis.com/ee/ac/3b/6ea67303dc39ad/US5512306.pdf>. Accessed August 7, 2019
- 22. Bocanegra, M. , Seijas, A. & Yibirín, M.G. Efficacy and tolerability of conventional nimesulide versus beta‐cyclodextrin nimesulide in patients with pain after surgical dental extraction: a multicenter, prospective, randomized, double‐blind, double‐dummy study. Curr. Ther. Res. Clin. Exp. 64, 279–289 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kovacic, M. & Husu‐Kovacevic, B. A pharmaceutical composition comprising perindopril. EP1815857A1. European Patent Office 2007.
- 24. Scarpignato, C. Piroxicam‐β‐cyclodextrin: a GI safer piroxicam. Curr. Med. Chem. 20, 2415–2437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Public Assessment Report . Decentralised procedure. Risperidone 1mg/mL Oral Solution <https://www.medicines.org.uk/emc/files/pil.3048.pdf>; <https://www.fda.gov/drugs/drug‐approvals‐and‐databases/inactive‐ingredients‐database‐download>. Accessed August 7, 2019.
- 26. Vakily, M. , Pasutto, F.M. , Daneshtalab, M. & Jamali, F. Inclusion complexation of heptakis (2,6‐di‐O‐ethyl)‐beta‐cyclodextrin with tiaprofenic acid: pharmacokinetic consequences of a pH‐dependent release and stereoselective dissolution. J. Pharm. Sci. 84, 1014–1019 (1995). [DOI] [PubMed] [Google Scholar]
- 27. Bilensoy, E. Cyclodextrins in Pharmaceutics, Cosmetics, and Biomedicine: Current and Future Industrial Applications (John Wiley & Sons, Hoboken, NJ, 2011). [Google Scholar]
- 28. Cyclodextrin News. February 2013, Volume 27, No. 2, ISSN 0951‐256X. [Google Scholar]
- 29. CERSI Center of Excellence in Regulatory Science and Innovation . Excipients Browser <http://excipients.ucsf.bkslab.org/index>. Accessed August 7, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material