

Abstract
Aim
To report an analysis of ~1 year of setmelanotide treatment for obesity and hunger, as well as metabolic and cardiac outcomes, in individuals with Bardet‐Biedl syndrome (BBS).
Materials and methods
Individuals aged 12 years and older with BBS received once‐daily setmelanotide. The dose was titrated every 2 weeks to establish the individual therapeutic dose (≤3 mg); treatment continued for an additional 10 weeks. Participants who lost 5 kg or more (or ≥5% of body weight if <100 kg at baseline) continued into the 52‐week extension phase. The primary outcome was mean percent change from baseline in body weight at 3 months. Hunger scores and safety were secondary outcomes.
Results
From February 2017 and February 2018, 10 individuals were screened; eight completed the 3‐month treatment phase and seven completed the extension phase. Mean percent change in body weight from baseline to 3 months was −5.5% (90% CI, −9.3% to −1.6%; n = 8); change from baseline was −11.3% (90% CI, −15.5% to −7.0%; n = 8) at 6 months and −16.3% (90% CI, −19.9% to −12.8%; n = 7) at 12 months. All participants reported at least one treatment‐emergent adverse event (AE), most commonly injection‐site reaction. No AEs led to study withdrawal or death. Most, morning, and average hunger scores were reduced across time points.
Conclusions
Setmelanotide reduced body weight and hunger in individuals with BBS and had a safety profile consistent with previous reports. Setmelanotide may be a treatment option in individuals with BBS‐associated obesity and hyperphagia.
Keywords: antiobesity drug, appetite control, obesity therapy, phase I‐II study
1. INTRODUCTION
Bardet‐Biedl syndrome (BBS) is a rare, genetically heterogeneous syndrome associated with function‐altering variants in 24 or more possible causative genes, including BBS1‐21, NPHP1, FBN3 and CEP19, each of which plays a role in primary cilia function. 1 , 2 , 3 , 4 In Europe and the USA, function‐altering variants in BBS1 (23.2%) and BBS10 (20.0%) are most commonly found among individuals with BBS. 2 In addition to early‐onset obesity and hyperphagia, 5 BBS is also characterized by retinal degeneration, cognitive disability, polydactyly, renal abnormalities and hypogonadism. 1 , 2 , 6
The hypothalamic melanocortin‐4 receptor (MC4R) neuronal pathway regulates energy balance and body weight. 1 , 7 Rare function‐diminishing variants in genes involved in this pathway have been associated with hyperphagia, or insatiable hunger, which leads to increased food intake, and development of obesity in early childhood. 7 , 8 , 9 No specific pharmacotherapies exist for obesity and hyperphagia in most rare genetic disorders of obesity. Although the pathophysiology of BBS is not completely established, it is hypothesized that the associated obesity is at least in part a result of hypothalamic dysfunction. 1 Data from rodents show that a dysfunctional BBS protein can impair the trafficking of leptin receptor (LEPR) in hypothalamic proopiomelanocortin (POMC) neurons, 10 , 11 thus reducing the activation of MC4R. Further, serum leptin concentrations in people with BBS are higher than expected when compared with body mass index (BMI)‐matched controls. 12 This finding suggests that, in BBS, leptin resistance could be associated with diminished leptin signalling in the hypothalamus, 12 which, in turn, would reduce downstream activation of MC4R and contribute to the development of severe obesity and hyperphagia. 1 Indeed, targeted deletion of BBS1 from LEPR‐expressing cells in mice causes hyperphagia and obesity, but, when BBS1 is ablated from adipocytes, obesity does not occur. 13 Finally, administration of the MC4R agonist melanotan II reduces food intake and weight in BBS knockout mice, implying that the downstream appetite‐regulating melanocortin signalling pathway is intact. 10
Setmelanotide is an eight‐amino‐acid cyclic peptide that preferentially binds to MC4R and acts as a substitute for melanocyte‐stimulating hormone for MC4R‐expressing neurons. 14 , 15 The unique mechanism of action of setmelanotide activates MC4R and can overcome many of the effects of genetic deficiencies that occur upstream in the pathway in some individuals with rare genetic disorders of obesity. In previous phase 2 studies, setmelanotide treatment led to reductions in body weight and hunger in individuals with POMC and LEPR deficiency obesities. 14 , 15 Setmelanotide was well tolerated, and participants did not experience clinically substantial increases in blood pressure or heart rate, which had been observed with first‐generation MC4R agonists. 16 However, it remains unclear whether activation of MC4R by setmelanotide can reduce hunger and improve weight loss in individuals with syndromic forms of obesity that may be associated with impaired activity in the MC4R pathway, including BBS. We report an analysis of ~1 year of setmelanotide treatment for severe obesity and hunger, as well as metabolic and cardiac outcomes, in individuals with BBS.
2. MATERIALS AND METHODS
2.1. Study design and participants
The data were collected in an ongoing phase 2, open‐label, single‐arm, basket‐design pilot study composed of several distinct cohorts of individuals aged 12 years or older diagnosed with one of several rare genetic disorders of obesity, including BBS, POMC deficiency, LEPR deficiency, Alström syndrome, Smith‐Magenis syndrome, SRC1 deficiency and SH2B1 deficiency (ClinicalTrials.gov identifier: NCT03013543). This analysis reports data from participants with a genetic or clinical diagnosis of BBS, according to the criteria of Beales et al. 6 All participants (or guardians/legal representatives) signed written informed consent forms, and child participants provided assent before any study‐specific procedures were performed. This study was conducted in accordance with the International Council on Harmonisation for Good Clinical Practice, Declaration of Helsinki, and the appropriate regulatory requirements. To safeguard the rights, safety and well‐being of all participants, all study documentation was reviewed and approved by the institutional review boards of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health (Bethesda, MD, USA) and Marshfield Clinic Health System (Marshfield, WI, USA).
Eligible participants for the current analysis were adults (aged ≥18 years) with a BMI of 30 kg/m2 or higher, and adolescents (aged 12‐17 years) with a body weight greater than in the 97th percentile (adjusted for age and sex) who had a diagnosis of BBS. Participants could not have achieved a greater than 2% weight loss from intensive diet or exercise regimens within 2 months of enrolment or greater than 10% weight loss durably maintained after gastric bypass surgery. Other exclusion criteria included: diagnosis of a mental disorder that could substantially interfere with study adherence; any suicidal ideation or history of suicide attempt; clinically significant pulmonary, cardiac or oncologic disease (including dermatologic findings related to melanoma); history of liver disease other than non‐alcoholic fatty liver disease; impaired glomerular filtration rate (≤30 mL/min/1.73 m2); family history of skin cancer, melanoma or oculocutaneous albinism; or an inability to adhere to a once‐daily injection.
2.2. Procedures
Adults and adolescents received subcutaneous injections of 1.0 and 0.5 mg/day, respectively, with a dose titration of 0.5 mg increments every 2 weeks up to a maximum of 3.0 mg. Individualized therapeutic doses were determined primarily on weight loss (~2‐3 kg lost per week for adults; ~1‐2 kg lost per week for adolescents) as well as hunger (target score of 0‐2 for adults; score reduction that remained >0 for adolescents). The dose‐titration phase varied in duration from 2 to 12 weeks depending on the number of titrations needed to establish the therapeutic dose. The last 2 weeks of the dose‐titration phase that established the therapeutic dose were considered the first 2 weeks of the treatment phase. Participants received an additional 10 weeks of treatment for a total of 12 weeks (3 months) of the treatment phase at the therapeutic dose (Figure S1). A 52‐week treatment extension phase followed the treatment phase to evaluate the efficacy and safety of setmelanotide at 6 and 12 months in participants who achieved weight reduction from baseline of 5 kg or higher (or ≥5% if baseline body weight was <100 kg) without evidence of severe or clinically relevant adverse events (AEs) or changes in vital signs, safety laboratory measurements or electrocardiographic findings. Because of the variable length of the titration phase, each participant's 3‐, 6‐ and 12‐month time point occurred at different weeks of the overall follow‐up.
Body weight and safety assessments, including blood pressure and heart rate, were recorded at each visit. Body composition assessments (Tanita Scale SC‐240 [TANITA Corporation, Tokyo, Japan] at the Marshfield Clinic or iDXA [Lunar Corporation, Madison, WI, USA] at the National Institutes of Health) and physical examinations, plus metabolic, endocrine, haematologic and pharmacokinetic testing, were also conducted at regular intervals. Depression and suicidality were assessed using the Columbia Suicide Severity Rating Scale and Patient Health Questionnaire‐9 and were monitored over the entire course of the trial.
Participants self‐assessed their hunger levels daily by answering the following questions: ‘In the last 24 hours, how hungry did you feel when you were the most hungry?’; ‘This morning when you woke up for the day, how hungry did you feel?’; and ‘In the last 24 hours, on average, how hungry did you feel?’. The assessments used a Likert‐type scale, where 0 = no hunger at all and 10 = most hunger, to generate a hunger score.
Exploratory observer‐related questionnaires included a food problem diary (FPD) and significant event questionnaire (SEQ), which were completed by caregivers of individuals with cognitive impairment, and were intended to capture common and rare food‐related behaviours, respectively. 17 , 18 The FPD is a 10‐item, observer‐reported outcome measure derived from an instrument used for individuals with Prader‐Willi syndrome designed to capture common food‐related behaviours as recorded daily by caregivers. 19 Total scores range from 0 to 30, with higher scores suggestive of more severe hyperphagia/food‐related behaviours. The SEQ is a novel instrument composed of an eight‐item, observer‐reported outcome measure designed to capture rare food‐related behaviours (i.e. behaviours expected to occur only with reduction in hyperphagia in response to treatment) as recorded weekly by caregivers. 17 , 18 Total scores range from 0 to 24, with higher scores suggestive of more significant appetite suppression.
2.3. Outcomes
The primary outcome was percent change in body weight after 3 months of treatment at the therapeutic dose in participants completing the treatment phase. Percent change in body weight was also assessed after 6 and 12 months of treatment at the therapeutic dose as additional exploratory efficacy outcomes. Other key secondary or exploratory outcomes included daily hunger scores, BMI, body fat mass, glucose‐related variables, waist circumference, safety and tolerability (as assessed by frequency and severity of AEs and serious AEs), changes in physical examinations, electrocardiography, vital signs (including resting blood pressure and heart rate) and clinical laboratory evaluations, and injection‐site reactions (ISRs) over 12 months.
2.4. Statistical analysis
Given the exploratory nature of the early efficacy signals in the phase 2, proof‐of‐concept study in individuals with rare genetic disorders of obesity, 14 , 15 as well as the rarity of this disorder, the sample size for each genetic disorder is not primarily driven by statistical testing considerations, but rather by clinical considerations. For all outcomes, the statistical analysis considered baseline as the last value obtained before the first dose of active treatment. Some patients may have completed their clinic visit outside of the protocol‐specified window. To include as many data points as possible in the present analysis, a ±1 month window was applied on the 3‐month time point, and a ±2 month window was applied on the 6‐ and 12‐month time points. Outcomes of participants completing each study phase (per‐protocol population) were summarized using descriptive statistics. Weight outcomes were also summarized for the intent‐to‐treat (ITT) population using a last observation carried forward analysis. Unless otherwise stated, P values (via a one‐sample t test at a one‐sided .05 significance level) and corresponding 90% two‐sided confidence intervals (CIs) were given as appropriate; however, the P values and associated CIs should be considered exploratory for clinical scrutiny and estimation purposes rather than for formal statistical hypothesis testing. No adjustments for multiplicity were conducted.
3. RESULTS
Ten participants with BBS were screened for eligibility from February 2017 and February 2018, and all were enrolled. Eight participants had genetic confirmation of BBS with variants in a single BBS gene prior to enrolment; two were screened and enrolled in the study with clinical diagnoses of BBS. Regarding the latter two participants, genetic tests confirmed one participant to be composite heterozygous for BBS1/BBS10; the other participant was clinically diagnosed with BBS as per Beales et al. 6 criteria without genetic confirmation. In each of the latter two cases, symptoms and progression of the disorder were consistent with BBS. Study investigators enrolled these participants because known disease‐causing biallelic variants are identified in only 80% of individuals with BBS. 20 , 21 Eight participants completed the 3‐month treatment period and entered the 52‐week extension phase; of those, seven completed the 52‐week extension phase (Figure 1). Demographics and baseline characteristics of the ITT population are shown in Table 1. The average age of the participants was 22.5 years. Mean baseline weight and BMI were 128.1 kg and 44.8 kg/m2, respectively. Baseline hunger scores had mean values of 8 for most hunger, 6 for average hunger and 5 for morning hunger.
FIGURE 1.
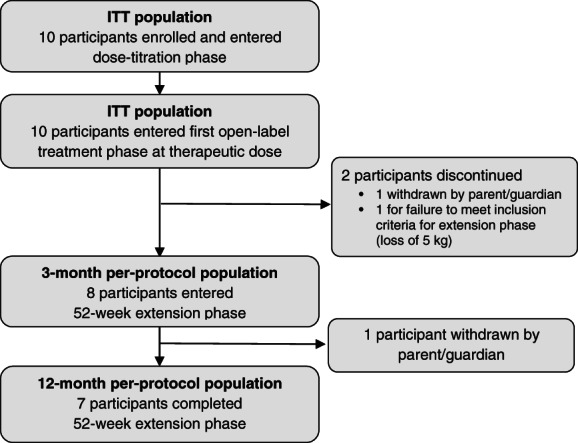
Participant enrolment and disposition. ITT, intent to treat
TABLE 1.
Baseline participant and disease characteristics (ITT) a
| Participants | |
|---|---|
| Age, y | 22.5 (14.7) |
| 12‐18 | 6 (60) |
| >18 | 4 (40) |
| Sex | |
| Female | 6 (60) |
| Male | 4 (40) |
| Race | |
| Black or African American | 1 (10) |
| White | 9 (90) |
| Ethnicity | |
| Hispanic or Latino | 1 (10) |
| Not Hispanic or Latino | 8 (80) |
| Unknown | 1 (10) |
| Weight, kg | 128.1 (28.6) |
| BMI, kg/m2 | 44.8 (4.1) |
| Hunger scores b | |
| Most hunger score c | 7 (1.4) |
| Average hunger score | 6 (1.7) |
| Morning hunger score | 5 (2.7) |
| Total fat mass, g d | 59 776.3 (15 080.7) |
| Total body mass, g d | 122 536.3 (25 316.8) |
| Waist circumference, cm | 126.2 (19.3) |
| Glucose concentration, mg/dL | 103.7 (32.5) |
| HbA1c, % | 5.8 (1.5) |
Abbreviations: BMI, body mass index; ITT, intent to treat; SD, standard deviation.
Note: Data are mean (SD) or n (%).
N = 10 unless otherwise noted.
n = 6.
Most hunger score was determined on a 0 to 10 Likert scale based on the question, ‘In the last 24 hours, how hungry did you feel when you were the most hungry?’
n = 9.
Participants completing the initial treatment phase achieved a significant mean percent change in body weight from baseline at 3 months (−5.5% [90% CI, −9.3% to −1.6%]; n = 8; P = .02). In the long‐term extension phase, mean percent change in body weight was also significant at 6 months (−11.3% [90% CI, −15.5% to −7.0%]; n = 8; P <.001) and 12 months (−16.3% [90% CI, −19.9% to −12.8%]; n = 7; P <.0001; Figure 2A,B). A last observation carried forward analysis also showed significant mean percent change in body weight from baseline in the ITT population at 3 months (−4.6% [90% CI, −8.3% to −1.0%]; n = 10; P = .02), 6 months (−8.1% [90% CI, −13.2% to −3.1%]; n = 10; P = .01) and 12 months (−10.5% [90% CI, −16.4% to −4.5%]; n = 10; P = .01; Table S1). Participants also experienced a significant mean percent change in BMI from baseline at 3 months (−5.5%; n = 8; P = .01), 6 months (−11.1%; n = 8; P <.001) and 12 months (−16.2%; n = 7; P <.0001; Table 2).
FIGURE 2.
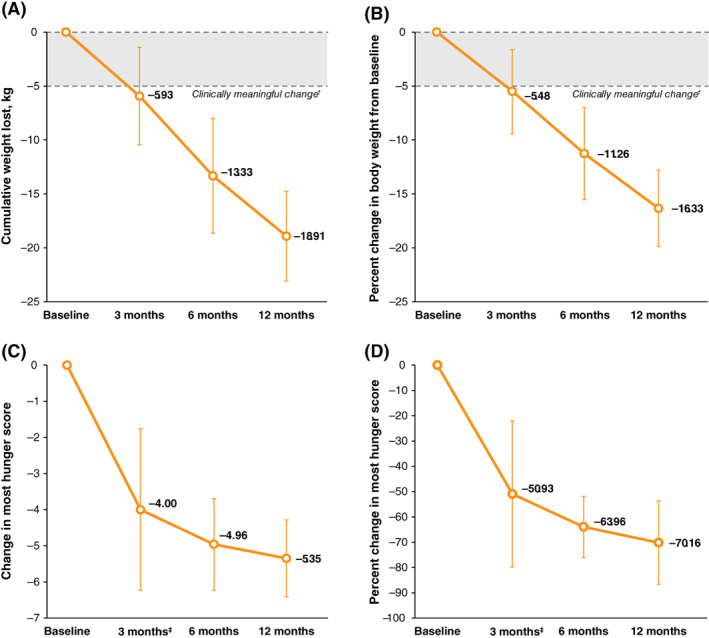
Mean weight and hunger changes with setmelanotide treatment in the per‐protocol population. (A) Mean change from baseline in body weight at each visit over 1 year of setmelanotide treatment. (B) Percent change from baseline in mean body weight over 1 year of setmelanotide treatment. (C) Mean change from baseline in most hunger score at each visit over 1 year of setmelanotide treatment. (D) Percent change in mean most hunger score over 1 year of setmelanotide treatment. (A) and (B): N = 8; (C) and (D): N = 5. Error bars are the 90% confidence interval. †Clinically meaningful change defined as weight loss of ≥5 kg (or ≥5% in patients with baseline body weight <100 kg). ‡n = 6
TABLE 2.
Changes in body mass index (BMI) and hunger scores from baseline through 12 months of treatment (per‐protocol population)
| BMI, kg/m2 | Most hunger score a | Average hunger score a | Morning hunger score a | |||||
|---|---|---|---|---|---|---|---|---|
| Mean (SD) | Percent change (SD) | Mean (SD) | Percent change (SD) | Mean (SD) | Percent change (SD) | Mean (SD) | Percent change (SD) | |
| Baseline | 44.8 (4.1) | — | 7.5 (1.4) | — | 6.4 (1.7) | — | 5.3 (2.7) | — |
| n = 10 | ||||||||
| 3 mo | 41.6 (5.5) | −5.5 (5.6) | 3.7 (2.0) b | −50.9 (35.1) | 3.3 (1.8) b | −46.5 (36.8) | 2.2 (2.4) b | −61.0 (52.1) |
| n = 8 | n = 8 | P = .01 | P = .01 | P = .02 | ||||
| P = .01 | ||||||||
| 6 mo | 38.9 (5.2) | −11.1 (6.3) | 3.2 (1.5) | −64.0 (12.7) c | 2.6 (0.8) | −61.2 (13.3) c | 1.3 (1.2) | −81.6 (18.0) c |
| n = 8 | n = 8 | P < .001 | P < .001 | P < .001 | ||||
| P < .001 | ||||||||
| 12 mo | 36.0 (4.0) | −16.2 (5.3) | 3.0 (2.1) | −70.2 (17.2) c | 2.7 (1.4) | −63.9 (16.5) c | 1.3 (1.2) | −81.3 (18.1) c |
| n = 7 | n = 7 | P < .001 | P < .001 | P < .001 | ||||
| P < .0001 | ||||||||
Abbreviation: SD, standard deviation.
n = 6 unless otherwise noted; three participants with cognitive impairment were given different assessments, and further fluctuation of n was caused by missing values at given time points.
n = 7.
n = 5.
In addition, participants reported significant reductions in the most hunger score at 3, 6 and 12 months (P <.05; Figure 2C,D; Table 2). Participants also reported significant reductions in morning and average hunger scores at 3, 6 and 12 months (P <.05; Table 2). Three participants were unable to complete the self‐reported hunger questionnaire because of cognitive impairment characterized by neuropsychologists as an extremely low level of intelligence (n = 2) and autism with mild cognitive impairment (n = 1). For these participants, hunger was evaluated by caregivers using a FPD and SEQ. Among these participants, FPD scores improved from 23 to 6 at 12 weeks, 6 to 0 at 70 weeks, and 18 to 12 at 19 weeks. Two participants experienced improvements in SEQ scores at the last evaluated time point from baseline, increasing from 3 to 24 at 12 weeks, and from 3 to 24 at 58 weeks. The third participant experienced no change from baseline to week 4.
At 3 months, participants experienced significant percent change from baseline in body fat mass (−9.3%; n = 9; P = .01) and total body mass (−5.3%; n = 9; P = .01). Similarly, at 12 months, participants experienced significant percent change from baseline in body fat mass (−24.0%; n = 7; P <.01) and total body mass (−15.8%; n = 7; P <.0001). A non‐significant change at 3 months from baseline was observed in waist circumference (−4.9%; n = 8; P = .06). Participants achieved significant percent change from baseline in waist circumference at both 6 (−10.8%; n = 8; P <.01) and 12 months (−17.0%; n = 7; P <.001).
At baseline, individuals had normal to mild elevation in mean (standard deviation [SD]) levels of high‐density lipoprotein cholesterol (HDL‐C) and low‐density lipoprotein cholesterol (LDL‐C) (44.2 [10.3] and 106.6 [18.8] mg/dL, respectively). Slight improvements but no meaningful changes in lipid profiles were observed with setmelanotide. At 3, 6 and 12 months, mean percent changes in HDL‐C from baseline were −5.8% (n = 9; 90% CI: −14.3% to 2.7%), −1.1% (n = 8; 90% CI: −13.3% to 11.2%) and 13.0% (n = 6; 90% CI: −2.5% to 28.5%), respectively. Mean percent changes in LDL‐C from baseline at 3, 6 and 12 months were −10.1% (n = 9; 90% CI: −20.8% to 0.7%), −9.0% (n = 8; 90% CI: −24.6% to 6.6%) and −1.9% (n = 7; 90% CI: −17.6% to 13.8%), respectively. The mean (SD) baseline triglyceride value was 166.6 (85.8) mg/dL; mean percent changes in triglyceride values from baseline were −15.0% (n = 9; 90% CI: −31.1% to 1.1%), −30.5% (n = 8; 90% CI: −46.7% to −14.3%) and −28.8% (n = 7; 90% CI: −40.1% to −17.5%), respectively.
Baseline mean (SD) glucose concentrations were normal to mildly elevated (103.7 [32.5] mg/dL). Setmelanotide was not associated with meaningful changes in glucose variables. Mean percent change in fasting glucose from baseline at 3, 6 and 12 months was −0.6% (n = 9; 90% CI: −9.7% to 8.5%), −4.8% (n = 8; 90% CI: −14.8% to 5.2%) and −0.4% (n = 7; 90% CI: −7.4% to 6.5%), respectively. The baseline mean (SD) HbA1c value was 5.8% (1.5%); setmelanotide was not associated with a significant percent change from baseline in HbA1c at any time point.
At baseline, mean (SD) diastolic and systolic blood pressure values were normal (75.7 [14.5] and 109.5 [13.9] mmHg, respectively). Mean percent change in diastolic blood pressure values from baseline at 3 and 12 months were −3.2% (n = 8; 90% CI: −12.5% to 6.0%) and −5.3% (n = 7; 90% CI: −20.9% to 10.4%), respectively. Mean percent change in systolic blood pressure values from baseline at 3 and 12 months were 8.9% (n = 8; 90% CI:−0.2% to 17.9%) and 8.9% (n = 7; 90% CI: −1.0% to 18.8%), respectively. The baseline mean (SD) heart rate was 68.0 (12.3) beats per minute; mean percent changes in heart rate from baseline at 3 and 12 months were −1.4% (n = 8; 90% CI: −14.7% to 12.0%) and 5.2% (n = 7; 90% CI: −5.7% to 14.4%), respectively. None of the changes in blood pressure or heart rate were statistically significant.
Setmelanotide was generally well tolerated. All participants reported at least one treatment‐emergent AE and at least one drug‐related AE (Table 3). The most common treatment‐emergent AEs were ISRs (100%) and hyperpigmentation (80%). The one serious treatment‐emergent AE was a case of rotavirus in one participant, which was not related to setmelanotide treatment. There were no treatment‐emergent AEs leading to study withdrawal or death.
TABLE 3.
Treatment‐emergent AEs (ITT)
| n (%) | |
|---|---|
| Treatment‐emergent AE | 10 (100) |
| Drug‐related | 10 (100) |
| Leading to permanent discontinuation | 0 |
| Serious treatment‐emergent AE | 1 (10) |
| Drug‐related | 0 |
| Treatment‐emergent AE of special interest | |
| Injection‐site reaction | 10 (100) |
| Hyperpigmentation | 8 (80) |
| Nausea | 3 (30) |
| Vomiting | 2 (20) |
| Deaths | 0 |
Abbreviations: AE, adverse event; ITT, intent to treat.
4. DISCUSSION
We report the first clinical study to investigate the efficacy of an MC4R agonist for treatment of BBS, a condition that reduces LEPR signalling and therefore leads to insufficient melanocortin action at hypothalamic centres involved in appetite. 1 , 10 , 11 This open‐label, phase 2 trial investigated the efficacy of setmelanotide for BBS‐associated obesity and hyperphagia as well as laboratory variables and safety. Setmelanotide was associated with substantial reduction in body weight after 3 months of treatment. Significant weight loss was observed at all time points both in the per‐protocol population as well as the ITT population. Because of the proportion of study withdrawals, the per‐protocol population analysis may have overestimated the efficacy of setmelanotide. Conversely, the ITT population analysis may have underestimated weight loss because weight outcomes were assessed using a last observation carried forward analysis. Together, these analyses provide support for setmelanotide efficacy for weight loss in individuals with BBS.
Setmelanotide had previously showed long‐term efficacy in weight loss in individuals with other rare genetic disorders of obesity due to variants in the MC4R pathway, including POMC and LEPR deficiency obesities. 14 , 15 The efficacy of setmelanotide in the current study suggests that BBS‐related obesity may also be mediated by the MC4R pathway. Although the mechanism of obesity in BBS is not fully understood, evidence from rodent models suggests that variants in BBS affect the MC4R signalling pathway. 10 , 11 , 22 , 23 BBS proteins appear to be involved in primary cilia generation and maintenance, and rodent models lacking cilia on POMC neurons had significant increases in weight and hyperphagia. 9 , 11 Mice with BBS variants have leptin resistance due to attenuated LEPR signalling, leading to improper cellular response by POMC neurons. 10 In addition, depletion of BBS protein leads to LEPR mistrafficking. 10 Selective disruption of the BBS protein complex in POMC neurons leads to weight gain and increased fat mass in mice driven by hyperphagia. 23 The clinical efficacy of MC4R agonism in individuals with BBS provides evidence that, despite the clinical complexity of BBS, the observed hyperphagia and obesity are at least in part caused by insufficient MC4R action.
The safety profile of setmelanotide was consistent with that observed in previous clinical studies in individuals with POMC or LEPR deficiency obesity. 14 , 15 In participants with BBS, setmelanotide was well tolerated with no discontinuations because of AEs, and the most common treatment‐emergent AE was ISR. In the current study, setmelanotide was not associated with statistically significant changes in blood pressure or heart rate. In previous studies of setmelanotide, blood pressure and heart rate did not substantially change or were decreased. 14 , 15 Because baseline blood pressure levels were normal, the effect of setmelanotide in individuals with high baseline blood pressure could not be evaluated. The impact of setmelanotide on blood pressure and heart rate should be evaluated in future trials and closely monitored in individuals receiving setmelanotide.
Currently, no approved targeted pharmacotherapies exist for the treatment of obesity and hyperphagia in individuals with BBS. Current treatments focus on management of symptoms associated with the disorder and may include bariatric surgery, antiobesity medications developed for the general population, and lifestyle or dietary management for treatment of obesity. 1 , 4 , 24 However, effective and durable treatments, specifically those addressing hyperphagia in individuals with rare genetic disorders of obesity, are lacking. 24 , 25 The efficacy of setmelanotide in reducing body weight and hunger scores suggests it is a useful treatment option for severe obesity and hyperphagia in individuals with BBS. Uncontrolled obesity can lead to co‐morbidities, including cardiovascular issues, metabolic syndrome, diabetes and reduced quality of life. 26 , 27 , 28 Because obesity management can control these co‐morbidities, weight reduction through hyperphagia management with setmelanotide could potentially effectively manage or alleviate obesity‐related co‐morbidities in individuals with BBS. 29 , 30
The current study has some limitations. The study design is open‐label, uncontrolled and non‐comparative. In addition, the participants did not undergo follow‐up after a study medication withdrawal period. Therefore, it is possible that a placebo effect of setmelanotide could have affected outcomes. The open‐label, non‐randomized design of the current study is supported by the substantial clinical response of setmelanotide shown in previous open‐label studies. 14 , 15 However, randomized trials to evaluate setmelanotide compared with placebo are needed. Finally, the minimum age for enrolled participants was 12 years, which may have limited the generalizability of these findings for younger individuals. An ongoing phase 3 study of setmelanotide in individuals with BBS includes a double‐blind randomized treatment period and will enrol participants aged 6 years or older, thereby addressing several of these limitations (NCT03746522).
Setmelanotide reduced body weight and hunger in participants with BBS during 1 year of treatment. Setmelanotide was well tolerated, and no new safety signals emerged over the course of treatment. The results of this study suggest that setmelanotide may be a useful pharmacotherapy for treatment of obesity and hyperphagia in individuals with BBS.
CONFLICT OF INTEREST
RH is a consultant for Rhythm Pharmaceuticals, Inc., and Trinity Life Sciences. He receives grant funding from the Bardet‐Biedl Syndrome Foundation. JY receives grant support for clinical investigations from the NICHD, NIH, Soleno Pharmaceuticals Inc., and Rhythm Pharmaceuticals, Inc. GY, GG and MS are employed by and may own stock in Rhythm Pharmaceuticals, Inc. KF, ED and SB have no conflicts of interest to disclose.
AUTHOR CONTRIBUTIONS
The study sponsor, RH and JY were responsible for conception and design of the study. RH, SB, ED, KF and JY acquired data for the study. RH, GY and MS were responsible for analysis of the data. RH, GY, GG, MS and JY were responsible for interpretation of the data. All authors were involved in drafting and critically revising the manuscript for important intellectual content or specific study site information and gave final approval of the manuscript for publication. All authors had full access to the data and are responsible for the accuracy and completeness of this report. The corresponding author had final responsibility for the decision to submit the report for publication.
DATA SHARING
Anonymized individual participant data and study documents can be requested for further research.
Supporting information
Figure S1. Phase 2 basket study design and treatment duration. QD, once daily. †The last 2 weeks of the open‐label dose‐titration phase in which the therapeutic dose for a participant is established is considered the first 2 weeks of the open‐label treatment phase. Participants then receive an additional 10 weeks of active treatment in the open‐label treatment phase for a total of 12 weeks of treatment at the therapeutic dose.
Table S1. Change in Weight From Baseline Through 12 Months of Treatment (ITT)†
ACKNOWLEDGMENTS
Editorial assistance was provided under the direction of the authors by Rhyomi Sellnow, PhD, Jennifer Rossi, MA, ELS, and Sherri Damlo, ELS, MedThink SciCom, and was funded by Rhythm Pharmaceuticals, Inc. We thank Kong Y. Chen, PhD, Peter Pinto, MD, PhD, and Heidi Kong, MD for their assistance with this protocol at the NIH Clinical Center. This study was sponsored by Rhythm Pharmaceuticals, Inc. JY, ED and SB were supported by the Intramural Research Program of the National Institutes of Health (Z1AHD00641). The funder of the study had a role in the study design, data collection, data analysis, data interpretation and writing of the report.
Haws R, Brady S, Davis E, et al. Effect of setmelanotide, a melanocortin‐4 receptor agonist, on obesity in Bardet‐Biedl syndrome. Diabetes Obes Metab. 2020;22:2133–2140. 10.1111/dom.14133
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1111/dom.14133.
Funding information This study was sponsored by Rhythm Pharmaceuticals, Inc. JY, ED and SB were supported by the Intramural Research Program of the National Institutes of Health (Z1AHD00641).
REFERENCES
- 1. Huvenne H, Dubern B, Clement K, Poitou C. Rare genetic forms of obesity: clinical approach and current treatments in 2016. Obes Facts. 2016;9(3):158‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Geets E, Meuwissen MEC, Van Hul W. Clinical, molecular genetics and therapeutic aspects of syndromic obesity. Clin Genet. 2019;95(1):23‐40. [DOI] [PubMed] [Google Scholar]
- 3. Weihbrecht K, Goar WA, Pak T, et al. Keeping an eye on Bardet‐Biedl syndrome: a comprehensive review of the role of Bardet‐Biedl syndrome genes in the eye. Med Res Arch. 2017;5(9). Epub ahead of print. 10.18103/mra.v5i9.1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Forsythe E, Kenny J, Bacchelli C, Beales PL. Managing Bardet‐Biedl syndrome ‐ now and in the future. Front Pediatr. 2018;6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sherafat‐Kazemzadeh R, Ivey L, Kahn SR, et al. Hyperphagia among patients with Bardet‐Biedl syndrome. Pediatr Obes. 2013;8(5):e64‐e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet‐Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437‐446. [PMC free article] [PubMed] [Google Scholar]
- 7. Yazdi FT, Clee SM, Meyre D. Obesity genetics in mouse and human: back and forth, and back again. Peer J. 2015;3:e856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dayton K, Miller J. Finding treatable genetic obesity: strategies for success. Curr Opin Pediatr. 2018;30(4):526‐531. [DOI] [PubMed] [Google Scholar]
- 9. Heymsfield SB, Avena NM, Baier L, et al. Hyperphagia: current concepts and future directions proceedings of the 2nd international conference on hyperphagia. Obesity. 2014;22(suppl 1):S1‐S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC. Requirement of Bardet‐Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet. 2009;18(7):1323‐1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davenport JR, Watts AJ, Roper VC, et al. Disruption of intraflagellar transport in adult mice leads to obesity and slow‐onset cystic kidney disease. Curr Biol. 2007;17(18):1586‐1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feuillan PP, Ng D, Han JC, et al. Patients with Bardet‐Biedl syndrome have hyperleptinemia suggestive of leptin resistance. J Clin Endocrinol Metab. 2011;96(3):E528‐E535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo DF, Cui H, Zhang Q, et al. The BBSome controls energy homeostasis by mediating the transport of the leptin receptor to the plasma membrane. PLoS Genet. 2016;12(2):e1005890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clement K, Biebermann H, Farooqi IS, et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat Med. 2018;24(5):551‐555. [DOI] [PubMed] [Google Scholar]
- 15. Kühnen P, Clément K, Wiegand S, et al. Proopiomelanocortin deficiency treated with a melanocortin‐4 receptor agonist. N Engl J Med. 2016;375(3):240‐246. [DOI] [PubMed] [Google Scholar]
- 16. Greenfield JR, Miller JW, Keogh JM, et al. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360(1):44‐52. [DOI] [PubMed] [Google Scholar]
- 17. Haws RM, Fletty KL, McIntee TJ, et al. Effect of the mealnocortin‐4 receptor agonist, setmelanotide, on obesity and hyperphagia in individuals affected by Bardet‐Biedl syndrome. Poster presented at: 57th Annual Meeting of the European Society of Paediatric Endocrinology; September 27‐29, 2018; Athens, Greece.
- 18. Haws RM, Fletty KL, McIntee TJ, et al. Obesity and hyperphagia therapy in Bardet‐Biedl syndrome with a melanocortin‐4 receptor agonist. Poster presented at: Obesity Week 2017; October 29‐November 2, 2017; Washington, DC.
- 19. Dykens EM, Maxwell MA, Pantino E, Kossler R, Roof E. Assessment of hyperphagia in Prader‐Willi syndrome. Obesity. 2007;15(7):1816‐1826. [DOI] [PubMed] [Google Scholar]
- 20. Forsythe E, Beales PL. Bardet‐Biedl syndrome. Eur J Hum Genet. 2013;21(1):8‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suspitsin EN, Imyanitov EN. Bardet‐Biedl syndrome. Mol Syndromol. 2016;7(2):62‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo D‐F, Rahmouni K. Molecular basis of the obesity associated with Bardet‐Biedl syndrome. Trends Endocrinol Metab. 2011;22(7):286‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo DF, Lin Z, Wu Y, et al. The BBSome in POMC and AgRP neurons is necessary for body weight regulation and sorting of metabolic receptors. Diabetes. 2019;68(8):1591‐1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kenny J, Forsythe E, Beales P, Bacchelli C. Toward personalized medicine in Bardet‐Biedl syndrome. Per Med. 2017;14(5):447‐456. [DOI] [PubMed] [Google Scholar]
- 25. Crinò A, Fintini D, Bocchini S, Grugni G. Obesity management in Prader‐Willi syndrome: current perspectives. Diabetes Metab Syndr Obes. 2018;11:579‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pulgaron ER. Childhood obesity: a review of increased risk for physical and psychological comorbidities. Clinical Ther. 2013;35(1):A18‐A32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Andolfi C, Fisichella PM. Epidemiology of obesity and associated comorbidities. J Laparoendosc Adv Surg Tech A. 2018;28(8):919‐924. [DOI] [PubMed] [Google Scholar]
- 28. Kushner RF, Foster GD. Obesity and quality of life. Nutrition. 2000;16(10):947‐952. [DOI] [PubMed] [Google Scholar]
- 29. Boscolo M, Féry F, Cnop M. Beneficial outcomes of sleeve gastrectomy in a morbidly obese patient with Bardet‐Biedl syndrome. J Endocr Soc. 2017;1(4):317‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ryan DH, Yockey SR. Weight loss and improvement in comorbidity: differences at 5%, 10%, 15%, and over. Curr Obes Rep. 2017;6(2):187‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Phase 2 basket study design and treatment duration. QD, once daily. †The last 2 weeks of the open‐label dose‐titration phase in which the therapeutic dose for a participant is established is considered the first 2 weeks of the open‐label treatment phase. Participants then receive an additional 10 weeks of active treatment in the open‐label treatment phase for a total of 12 weeks of treatment at the therapeutic dose.
Table S1. Change in Weight From Baseline Through 12 Months of Treatment (ITT)†