

Abstract
We investigated seven children from six families to expand the phenotypic spectrum associated with an early infantile epileptic encephalopathy caused by biallelic pathogenic variants in the phosphatidylinositol glycan anchor biosynthesis class Q (PIGQ) gene. The affected children were all identified by clinical or research exome sequencing. Clinical data, including EEGs and MRIs, was comprehensively reviewed and flow cytometry and transfection experiments were performed to investigate PIGQ function. Pathogenic biallelic PIGQ variants were associated with increased mortality. Epileptic seizures, axial hypotonia, developmental delay and multiple congenital anomalies were consistently observed. Seizure onset occurred between 2.5 months and 7 months of age and varied from treatable seizures to recurrent episodes of status epilepticus. Gastrointestinal issues were common and severe, two affected individuals had midgut volvulus requiring surgical correction. Cardiac anomalies including arrythmias were observed. Flow cytometry using granulocytes and fibroblasts from affected individuals showed reduced expression of glycosylphosphatidylinositol (GPI)‐anchored proteins. Transfection of wildtype PIGQ cDNA into patient fibroblasts rescued this phenotype. We expand the phenotypic spectrum of PIGQ‐related disease and provide the first functional evidence in human cells of defective GPI‐anchoring due to pathogenic variants in PIGQ.
Keywords: epileptic encephalopathy, exome sequencing, GPI, IGD, PIGQ, rare diseases
Abbreviations
- ALP
alkaline phosphatase
- CHO
Chinese hamster ovary
- DWI
diffusion weighted imaging
- EEG
electroencephalogram
- EIMFS
epilepsy of infancy with migrating focal seizures
- FLAER
fluorescein‐labeled proaerolysin
- GDD
global developmental delay
- GPI
glycosylphosphatidylinositol
- GPI‐AP
GPI anchored protein
- IGD
inherited GPI disorder
- MRI
magnetic resonance imagining
- WT
wildtype
SYNOPSIS.
We expand the phenotypic spectrum of PIGQ‐related disease and provide the first functional evidence in human cells of defective GPI‐anchoring due to pathogenic variants in PIGQ.
1. INTRODUCTION
The GPI‐anchor anchors more than 150 proteins to the cell surface. 1 These GPI‐anchored proteins (GPI‐APs) play important roles in embryogenesis, cell signaling, immune response and neurogenesis. 2 , 3 , 4 , 5 Thirty‐one different genes are involved in the biosynthesis of the GPI‐anchor, 6 of which 22 genes have been associated with human disease. 7 Clinical features generally include global developmental delay (GDD), epileptic seizures, hypotonia, and congenital anomalies. 2 The clinical spectrum is likely related to the various cellular functions of GPI‐APs, which include adhesion molecules, complement regulatory proteins, hydrolytic enzymes, protease inhibitors, and receptors. 1 , 8
Phosphatidylinositol glycan anchor biosynthesis class Q (PIGQ) is a key co‐enzyme in the N‐acetylglucosamine transferase complex, the first step catalyzing the biosynthesis of the GPI‐anchor, 9 , 10 , 11 and is encoded by PIGQ (OMIM 605754). 12 To date, three patients with PIGQ variants have been described, 13 , 14 , 15 however, for the first two patients, limited clinical information was published. Martin et al showed that PIGQ‐deficient Chinese hamster ovary (CHO) cells transfected with human PIGQ cDNA lacking exon 3 did not restore surface expression of GPI‐APs as well as wildtype (WT), 13 however, functional data in human cells remained elusive. Recently, Starr et al 15 provided a detailed clinical description of a patient with compound heterozygous variants in PIGQ. Very little is known about the clinical and functional spectrum associated with PIGQ insufficiency.
Herein, we describe and expand the phenotypical and neurophysiological spectrum of PIGQ deficiency in seven affected individuals and show reduced expression of GPI‐APs in patient granulocytes and fibroblast cell lines, with reversibility using PIGQ cDNA.
2. METHODS
2.1. Patient recruitment
Ethics approvals were obtained from the local institutional review boards and informed consent was obtained from patients' legal guardians. Details of local ethics approvals are available in Supporting Information. Collaboration was made possible using Matchmaker Exchange. 16 Detailed clinical examinations, magnetic resonance imaging (MRIs) and electroencephalograms (EEGs) were collected for each subject. Here forth, we abbreviate each subject to “St” (St1, St2, and so on).
2.2. Exome sequencing
DNA was extracted from whole blood, purified, and analyzed using an exome sequencing trio approach when both parents were available. All exomes were aligned to the human reference genome GRCh37/hg19. Detailed protocols for exome capture, sequencing, pipeline and analysis for each patient/family are available in Supporting Information. Primers for Sanger sequencing confirmation of variants are found in Table S1.
2.3. Cell line establishment
For St3b, a skin biopsy was taken and sent to the Centre for Applied Genomics (Toronto, Canada), for establishment of a fibroblast cell line. A skin biopsy for St5 was similarly used for establishment of a fibroblast cell line at the Duke Cytogenetics Laboratory. A fibroblast cell line for St2 was established independently at the Children's Hospital of Philadelphia.
HyClone DMEM media (GE Healthcare Life Sciences) supplemented with 10% fetal bovine serum, penicillin‐streptomycin (SV30010, GE Healthcare Life Sciences) and 2 mM l‐glutamine (SH30033401, Thermo Scientific), was used for cell‐line maintenance.
2.4. Flow cytometry
Fresh blood samples from St4 and St5, as well as from healthy controls were stained with the GPI‐AP markers: PE‐conjugated anti human CD16 (BioLegend), FITC‐conjugated mouse anti human CD55 and CD59 (BD Pharmingen), or Fluorescein‐labeled proaerolysin (FLAER)‐Alexa 448 (Cedarlane) for 1 hour on ice. Red blood cells were lysed in FACS Lysing Solution (BD Bioscience). Granulocytes from St1 as well as a control were stained with FLAER and analyzed by flow cytometry in a clinical lab.
For fibroblasts, cells were harvested at 80% to 90% confluency, stained with FLAER‐Alexa 448, FITC‐conjugated mouse anti human CD73 (BioLegend) or PE‐ conjugated mouse anti human CD109 (BioLegend) for 1 hour on ice in the incubation buffer containing 0.5% BSA, then fixed in 3.7% formaldehyde. For all assays, non‐specific binding was washed off before analyzing by a BD FACSCanto II system (BD Biosciences) followed by the FlowJo software analysis. We use at least three different controls at two different times for fibroblasts (before and after the rescue). Fibroblast characteristics may change with time in culture and with passages, we thus use for our analysis and figures the control sample which displayed similar cell population patterns (eg, size and granularity) as the test's fibroblasts. Fibroblasts established from St2 and a control were stained with FITC‐CD59 and analyzed by flow cytometry in an independent lab (see Supporting Information).
2.5. Rescue assays of GPI‐APs on fibroblasts
Lentiviruses carrying a wildtype PIGQ‐Lv105 (NM_004204.3) or an empty‐Lv105 construct (GeneCopoeia) with the presence of packaging plasmids pMD2.G and psPAX2 (AddGene) were produced in HEK293T cells. Fibroblasts (established from St3b and St5) were transduced with the lentiviruses (typically with transduction efficiency over 25%), and selected by Puromycin resistance, creating a stable cell line. Untransduced cells, rescued cells and control cells were subjected to flow cytometry analyses as described above for fibroblasts.
3. RESULTS
We identified seven affected individuals with biallelic PIGQ variants from six unrelated families. The cohort comprised of five females and two males, from various ancestries. Detailed clinical summaries can be found in Supporting Information as well as Table 1 and Table S2. EEG descriptions are found in Table S3, and MRI findings are summarized in Table S4.
TABLE 1.
Clinical features of seven new affected individuals from six families with biallelic variants in PIGQ, and review of the literature
| Subject ID/source | St1 | St2 | St3a | St3b | St4 | St5 | St6 |
|---|---|---|---|---|---|---|---|
| Variants & Inheritance (NM_148920.2) |
Homozygous: c.1611del p.R538Afs*24 |
Maternal: c.1199_1201del p.Y400del Paternal: c.942+1G>A IVS4+1G>A |
Maternal: c.1578_1579del p.Q527Afs*75 Paternal: c.1199_1201del p.Y400del |
Maternal: c.1578_1579del p.Q527Afs*75 Paternal: c.1199_1201del p.Y400del |
Maternal: c.1130_1168del p.A377_S389del Paternal: c.1345G>C p.G449R |
Maternal: c.49G>A p.G17R Paternal: c.942+1G>A IVS4+1G>A |
Homozygous: c.1670del p.G557Dfs*4 |
| Gender | Female | Female | Female | Female | Female | Male | Male |
| Ancestry | Turkish | European/Puerto Rican | British Isles/French Canadian | British Isles/French Canadian | Lebanese/Iraqi | Mexican | Afghani |
| Current age | 11 y | 6 y 6 mo | Deceased 2 d | Deceased 5 y | Deceased 9 m | 2 y 2 m | Deceased 3 y 9 m |
| Prenatal issues | − | Polyhydramnios | Prominent kidneys, premature rupture of membranes | − | Polyhydramnios, hepatomegaly, hydronephrosis | Dandy Walker malformation | − |
| Neonatal complications | − | Respiratory distress, hypoglycemia, failed newborn hearing screen in left ear. | Respiratory distress, renal and cardiac failure | Jaundice, secundum atrial ventricular defect detected after birth | Respiratory distress | Respiratory distress, feeding difficulties, jaundice, PDA, PFO | Feeding difficulties, hypertonia |
| Developmental delay | + | + | N/A | + | + | + | + |
| Seizure onset | 6 mo | 7 mo | N/A | Almost 4 mo | 7 mo | 6 mo | 2.5 mo |
| Hypotonia | + | + | N/A | + | + | + | + |
| Abnormal movements | + | + | N/A | + | + | + | + |
| Facial dysmorphism | − | + | + | + | + | + | + |
| Cranial shape anomalies | − | + | − | − | + | + | + |
| Teeth anomalies | − | + | N/A | + | Too young | + | − |
| Skeletal anomalies | + | + | − | + | + | + | − |
| Joint contractures | + | − | − | + | + | − | − |
| Other dysmorphic features | + | − | + | − | + | − | − |
| Deafness | − | − | − | − | Mild left conductive | − | − |
| Ophthalmological anomalies | + | + | N/A | + | + | + | +. |
| Cardiac anomalies | − | + | + | + | + | + | +. |
| Genitourinary abnormalities | + | + | + | No U/S done | + | + | + |
| Gastrointestinal issues | + | + | N/A | + | + | + | + |
| Serum alkaline phosphatase | Normal | Intermittently elevated | Not measured | Elevated | Elevated | Not measured | Normal |
| Subject ID/source | Martin et al 13 | Alazami et al 14 | Starr et al 15 |
|---|---|---|---|
| Variants & Inheritance (NM_148920.2) |
Homozygous: c.690‐2A>G |
Homozygous: c.619C>T p.R207* |
Maternal: c.968_969del p.L323Pfs*119 Paternal: c.1199_1201del p.Y400del |
| Gender | Male | N/A | Male |
| Ancestry | West African | N/A | N/A |
| Current age | Deceased 2 y 4 m | N/A | Deceased 10 mo |
| Prenatal issues | − | N/A | Severe polyhydramnios requiring multiple amniocentesis fluid reduction procedures |
| Neonatal complications | At 4 wk: cyanotic episodes with eye twitching, brief stiffening of upper body. | N/A | Feeding difficulties, trembling episodes |
| DD/ID | + | + | + |
| Seizure onset | 4 wk | N/A | 7 mo |
| Hypotonia | + | N/A | + |
| Abnormal movements | + | N/A | + |
| Facial dysmorphism | + | N/A | + |
| Cranial shape anomalies | − | N/A | + |
| Teeth anomalies | − | N/A | − |
| Skeletal anomalies | − | N/A | + |
| Joint contractures | − | N/A | N/A |
| Other dysmorphic features | + | N/A | + |
| Deafness | − | N/A | − |
| Ophthalmological anomalies | + | + | + |
| Cardiac anomalies | − | N/A | + |
| Genitourinary anomalies | − | N/A | + |
| Gastrointestinal issues | + | N/A | + |
| Serum alkaline phosphatase | N/A | N/A | Elevated |
Note: Further details are provided in Table S2.
Abbreviations: N/A, not applicable; PDA, patent ductus arteriosus; PFO, patent foramen ovale; U/S, ultrasound.
3.1. Variants identified
We identified seven previously unreported PIGQ (NM_148920.2) variants predicted to be damaging and one previously identified variant (p.Y400del). 15 All were either in a homozygous or compound heterozygous state. The novel variants included two missense variants (p.G17R; p.G449R), a canonical splice site substitution (c.942+1G>A), an in‐frame deletion (p.A377_S389del) and three frameshifts (p.Q527Afs*75, p.R538Afs*24 and p.G557Dfs*4) (Figure 1A). Variants identified in other genes are listed in the detailed clinical descriptions in Supporting Information but were excluded as disease‐causing due to phenotype incompatibility, predicted benign pathogenicity, or mode of inheritance.
FIGURE 1.
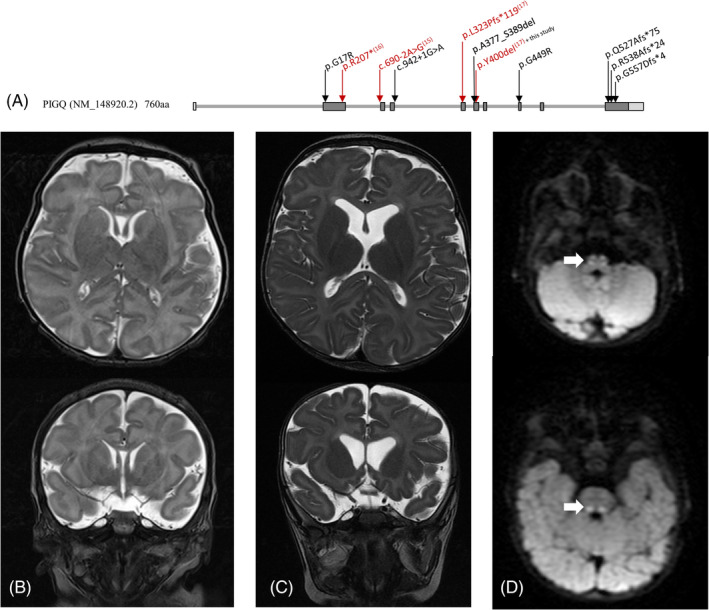
A, Variants in PIGQ identified in this study localized on isoform 1 (NM_148920.2) of the gene. Variants reported in previous studies are in red with corresponding reference numbers in superscript. Brain MRI of subject 4 was performed at age 9 days, B, and 7 months, C. At each age, T2‐weighted image sequences were performed with axial (top) and coronal (bottom) images shown. Brain MRI at 7 months showed progressive cortical volume loss with increased prominence of the lateral ventricles, consistent with loss of subcortical white matter volume compared to the prior study. Mild delay in myelination was apparent. D, Diffusion weighted axial images identified restricted diffusion in the medial lemniscus tracts, bilaterally
3.2. Phenotypic analysis
There was significant morbidity and mortality associated with biallelic PIGQ variants. Four of the seven patients died prematurely (St3a at 2 days of age secondary to cardiac and renal failure, St3b at 5 years of age due to asystole, St4 at 9 months of age under palliation, and St6 at 3 years and 9 months of age due to recurrent pneumonias). A presumed affected older brother of St5 died a few hours after birth, but DNA was not collected so the relationship to the familial variants cannot be confirmed.
Abnormal prenatal findings included polyhydramnios (St2 and St4), prominent kidneys and premature rupture of membranes at 28 weeks (St3a), severe fetal hepatomegaly and hydronephrosis (St4) and enlarged lateral ventricles and hypoplastic cerebellar vermis suggestive of a Dandy Walker anomaly (St5). Neonatal complications were present in six patients, including respiratory distress (St2, St3a, St4, and St5), jaundice (St3b and St5), heart failure (St3a), atrial ventricular defect (St3b), patent ductus arteriosus and patent foramen ovale (St5), hypoglycemia (St2) and feeding difficulties with irritability and limb hypertonia of the extremities (St2).
Age at onset of epileptic seizures in the remaining six patients ranged from 2.5 to 7 months of age. Seizure types included focal tonic seizures (St1, St2, and St6, evolving to focal status epilepticus for St2 and St6), bilateral tonic‐clonic (St2, St4, St5, and St6) evolving to status epilepticus (St4 and St6), myoclonic jerks (St3b and St5), epileptic spasms (St4), absence seizures (St1) and migrating focal seizures (St6) (Table S2). Seizure outcome ranged from partially controlled with occasional breakthroughs with illness (St1 and St2), to recurrent status epilepticus (St4, St6), daily focal seizures (St6) or daily clustered myoclonic jerks (St5). Seizures often clustered (St1, St3b, and St5). Notably, St3b was seizure‐free from 4 months until age 2 years, but thereafter had seizures in cycles that would progressively worsen and taper off, with seizure‐free periods of 7 to 12 days (Table S2).
All six patients had a severe to profound GDD. Axial hypotonia was observed in all patients (Table 1, Table S2). Appendicular hypotonia was observed in St2 and St5, whereas appendicular hypertonia was observed in the remaining patients. Severe head lag was observed in St2, St5, and St6. Stereotypic movements, opisthotonos, hyperkinetic movements, or random non‐epileptic jerks were present in all patients (Table S2).
Visual problems were diagnosed in all patients that survived the neonatal period. St3b, St4, St5, and St6 were diagnosed with cortical visual impairment. St1 had short fixation/tracking abilities, whereas an electroretinogram for St2 suggested impaired photoreceptor transmission in the retina. St4 had a mild unilateral conductive hearing deficit, whereas the other subjects had normal hearing (Table S2).
Six of the seven patients had facial dysmorphisms including coarse facies and macroglossia. Four (St2, St4, St5, and St6) had cranial shape anomalies and three (St1, St2, and St3b) had pectus excavatum. Other findings included inverted nipples (St1), scoliosis (St2), delayed dentition (St2, St3b, and St5), bilateral pulmonary interstitial emphysema (St3a), an accessory spleen (St3a), increased nuchal redundancy, short stature, hyperextension of the interphalangeal joints and deep palmar and plantar creases (St4), and an enlarged pinna (St5) (Table S2).
Cardiac anomalies were observed in six patients, including prolapsed mitral valve (St2), pulmonary stenosis (St5), pulmonary hypertension (St6), heart block and arrythmia (St2, St3b, and St4), and right ventricular hypertrophy with poor heart function (St3a, though hypertrophy was not found on autopsy) (Table S2). Genitourinary issues included bilateral hydronephrosis (St4, St5), renal stones with slightly enlarged kidneys (St4) and vesicoureteral reflux (St5). Autopsy of St3a revealed dilated/tortuous ureters with hypoplastic renal pelvis and calyces. Most patients had significant gastrointestinal issues including G‐tube dependency (St2, St3b, St4, St5, and St6), constipation (St1, St2, and St6), idiopathic volvulus (St4, who also had superior mesenteric artery and vein reversal, and St5 who had a markedly dilated colon; both had Ladd's procedure), and duodenal web (St5) (Table S2).
EEG findings were in keeping with clinical presentations and ranged from normal findings early in the disease course, to burst‐suppression patterns, background slowing with interictal multifocal sharp waves, hypsarrhythmia and ictal focal spike and slow wave complexes (Table S3).
MRI findings showed no initial abnormalities in the cerebral cortex, but subsequent imaging showed progressive cortical volume loss (St4, Figure 1B/C; St5). There was poor or incomplete myelination or demyelination in four of the six subjects with available imaging study results (St1, St3b, St4, and St5; Table S4). While St3a did not have brain imaging studies, an autopsy showed pyknotic nuclei of the cerebral cortex, as well as ischemic white matter changes with necrosis and karyorrhexis. There was a strongly positive glial fibrillary acidic protein signal and no striking anoxic changes, giving an overall impression of periventricular leukomalacia. Additional MRI findings included broad periventricular spaces (St1 and St3b), prominent frontal horns of the lateral ventricles (St2), volume loss of the vermis (St3b and St5), dangling choroids (St5) and pituitary hypoplasia with preservation of the stalk (St5; Table S4). MRS showed borderline lactate peaks (St2) and diffusion‐weighted imaging (DWI) showed increased intensity in the bilateral medial lemniscus tracts (St4, Figure 1D; Table S4).
Serum alkaline phosphatase levels were slightly elevated in St3b, and intermittently slightly elevated in St2 and St4 (Table S2). There were no obvious bone abnormalities associated.
3.3. Flow cytometry
In blood, both St4 and St5 had very low levels of FLAER and CD16 cell surface localization. In brief, granulocytes from St4 had only 6% the level of FLAER compared to normal while this marker in St5 was 35% (mean fluorescence intensity). For CD16, expression was 28% compared to normal for St4, and 20% in St5. CD55 and CD59 in St5 were normal whereas CD55 was decreased to 68% in St4. However, this patient has an increase in CD59 (Figure 2). Analysis of granulocytes by a clinical lab for St1 showed that the level of FLAER was 64.7% relative to the control.
FIGURE 2.
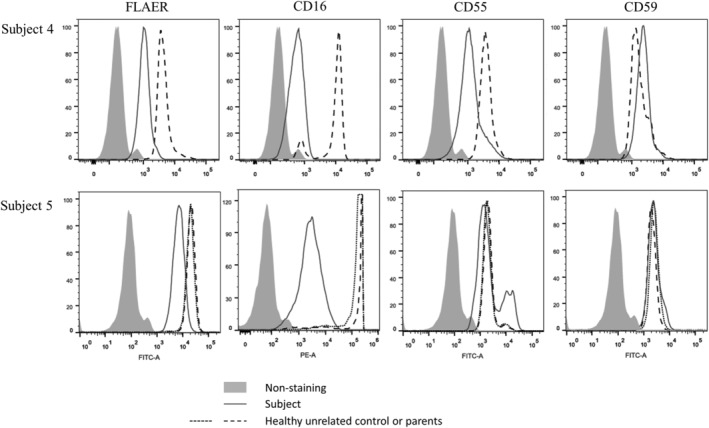
Impact of the PIGQ variants on the expression of GPI‐APs. Flow cytometry analysis of granulocytes from fresh blood from subject 4 (top row) and subject 5 (bottom row) and compared to healthy controls and parents, respectively. Fluorescently labeled proaerolysin “FLAER” directly binds the GPI‐anchor, whereas CD16, CD55, and CD59 all stain for GPI‐APs
For fibroblasts, levels of all markers were decreased in St3b and St5. St3b showed decreases in FLAER, CD73 and CD109 to 45%, 56%, and 20%, respectively relative to control, and these markers in St5 were reduced to 45%, 28%, and 20%, respectively (Figure 3). Analysis of CD59 expression in an independent lab showed a mean fluorescence intensity of 65.2% in St2‐derived fibroblasts relative to the control (Figure S2). While transduction with an empty lentivirus did not affect GPI‐AP levels, a lentivirus expressing wildtype PIGQ completely restored GPI‐AP expression in St3b, and there was partial rescue in St5 (FLAER, CD73, and CD109 were increased to 56%, 70%, and 46% vs control; Figure 3).
FIGURE 3.
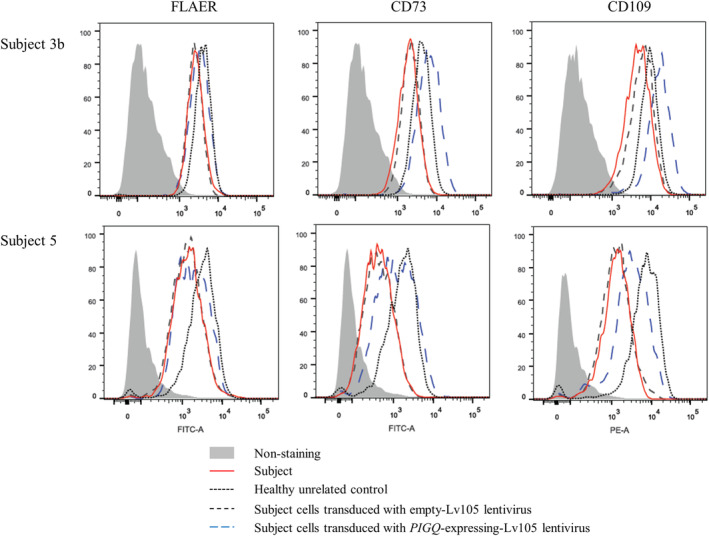
Impact of the PIGQ variants on the expression of GPI‐APs. Flow cytometry analysis of fibroblasts derived from subject 3b (top row) and subject 5 (bottom row) and compared to healthy controls and parental derived cell‐lines, respectively. Subject cell lines were further transfected with empty lentivirus or lentivirus expressing WT PIGQ cDNA. Fluorescently labeled proaerolysin “FLAER” directly binds the GPI‐anchor, whereas CD73 and CD109 stain for GPI‐APs
4. DISCUSSION
This cohort of seven new individuals affected by biallelic pathogenic variants in PIGQ enables us to broaden the phenotypic spectrum of this rare condition (Tables 1 and S2) and to demonstrate the impact of PIGQ on GPI‐anchored proteins.
Patients with biallelic PIGQ variants show a broad phenotypic spectrum including epileptic seizures, GDD, hypotonia, feeding difficulties, and multiple congenital anomalies (Tables 1 and S2), clinical features also observed in other inherited GPI‐deficiencies (IGDs). 2 Most affected individuals with biallelic PIGQ variants have delayed myelination, but generally lack gross structural lesions on MRI. Of note, while the affected individual reported by Starr et al 15 had significant left‐sided ventriculomegaly, we did not observe this in our cohort. A limiting factor could be that our subjects generally underwent MRI before the age of 10 months and that a long‐term follow‐up of MRI findings was not available. Individuals with pathogenic variants affecting other genes in the pathway have shown development of both cerebral and cerebellar loss of tissue over time. 17 , 18
While we also did not observe long bone radiolucent lesions as previously described, 15 skeletal anomalies and delayed dentition were common. Of note, St4 and St5 had midgut volvulus requiring surgery, and St5 had a duodenal web requiring resection. Furthermore, our cohort showed a variety of cardiac issues, including arrhythmia and fatal heart block.
Seizure type and control were highly variable (Tables S2 and S3). Martin et al reported a burst‐suppression pattern on EEG and Ohtahara syndrome, 13 which was not observed in subsequent patients, but has been reported in other IGDs, 19 Two patients developed epileptic spasms and St3b showed a recurrent pattern of seizure activity. Interestingly, St6 was diagnosed with epilepsy of infancy with migrating focal seizures (EIMFS), which is a very severe epileptic syndrome with poor outcome, previously known as malignant migrating partial seizures of infancy or Coppola‐Dulac syndrome. 20 The most common genetic cause of this syndrome is gain‐of‐function mutations in a gene encoding a potassium channel (KCNT1), but recently, in a cohort of 135 patients with EIMFS, two were found with de novo heterozygous pathogenic variants in PIGA, an X‐linked gene involved in GPI‐anchor biosynthesis, implicating this pathway in this particular epileptic syndrome. 21 Presently, the downstream GPI‐AP target(s) involved in the pathogenesis of IGDs remain unidentified.
St1, who was homozygous for the p.(R538Afs*24) variant, seems to have the mildest phenotype in our cohort. She is currently 11 years old and although she has chewing difficulties, she remains the only living patient who is not G‐tube dependent. While she has limited fixing/tracking abilities, she is also the only patient without a cortical visual impairment. While she is homozygous for a frameshift in the last exon, it is unclear if this can explain her milder phenotype, as St6, with a more severe phenotype and who eventually died, had an even later frameshift p.(G557Dfs*4). This may be in part because St6 was born from consanguineous parents thus other recessive genes may be contributing, or there may not be a clear genotype‐phenotype correlation. Indeed, even the two siblings in our cohort (St3a and St3b) had different clinical presentations and survived to 2 days and 5 years, respectively. Analysis of further affected individuals may reveal a genotype‐phenotype correlation, and given that PIGQ is part of the N‐acetylglucosamine complex, the protein location of the disease‐causing variants may result in differential impact for the binding of other enzymes in the complex. Compared to other IGDs, there are more often bi‐allelic nonsense and/or frameshift variants in PIGQ, but there is also increased mortality. 22 This might be because bi‐allelic loss‐of‐function mutations in other GPI‐biosynthesis genes lead to embryonic lethality, while it does not for PIGQ.
Previous studies have correlated variants in PIGT, another key component to GPI‐biosynthesis, to preferential atrophy of the cerebellar vermis, 18 , 23 , 24 , 25 a finding found inconsistently in a subsequent cohort. 17 Similarly, only two of our patients (St3b and St5) had MRI results showing atrophy affecting the cerebellar vermis, however, interestingly, an MRI of St5 at age 2 years found diffuse cerebral atrophy, but cerebellar atrophy was not appreciated. Given that variants in other genes in this pathway have been linked to cerebral and cerebellar loss of tissue over time, it is worth considering further imaging later in childhood.
Although serum alkaline phosphatase (ALP) has been linked with IGDs, evidence suggests that elevated ALP levels are most often associated with variants in later steps of GPI biosynthesis, whereas earlier steps are associated with ER‐associated degradation of the anchor. 26 , 27 Thus, it is not surprising that elevated serum ALP is not common among subjects with PIGQ pathogenic variants.
Treatments remain limited, though butyrate helped treat intractable epilepsy due to variants in the PIGM promoter, 28 and IGDs due to variants in PIGV, PIGO, and PIGS have been partially treated with vitamin B6 supplementation. 29 , 30 , 31 In St6, there was phenotypic overlap with cerebral folate deficiency due to FOLR1‐mutations, 32 in which the GPI‐anchored folate receptor alpha fails to provide folate to the brain. This can be by‐passed by providing folinic acid supplementation, however, unfortunately, St6 did not respond to this therapy, though it should be noted it was initiated late in his disease course.
Given the function of the proteins involved in GPI‐anchor biosynthesis, the gold standard for evaluation of functional defects in this pathway is using flow cytometry to assess the effect of the variants on GPI‐anchored proteins. 29 In granulocytes from fresh blood and fibroblasts, we show reduced expression of GPI‐anchored proteins in five unrelated affected individuals, a phenotype that was partially or fully reversible in fibroblasts by transfecting WT cDNA. Taken together with previous work on CHO cells, we show that biallelic PIGQ variants impact GPI‐AP expression.
In summary, we expand the phenotypic spectrum of IGDs related to biallelic PIGQ pathogenic variants and show that the impaired expression of GPI‐APs in human cells can be improved in vitro by the transduction of WT PIGQ cDNA. These variants lead to impaired GPI‐biosynthesis and subsequently reduced localization of GPI‐APs, resulting in a broad phenotypic spectrum. While the phenotypic spectrum is broad, the clinical overlap of IGDs is significant, and collectively they may represent as much as 0.15% of all developmental disorders. 33 Thus, there is a need for future work to develop therapies that reduce the impact of these devastating diseases. 34
CONFLICT OF INTEREST
Devon L. Johnstone, Thi Tuyet Mai Nguyen, Jessica Zambonin, Kristin Kernohan, Anik St‐Denis, Nissan V. Baratang, Taila Hartley, Michael T. Geraghty, Julie Richer, Jacek Majewski, Eric Bareke, Andrea Guerin, Manuela Pendziwiat, Loren D.M. Pena, Hilde M. H. Braakman, Karen W. Gripp, Andrew C. Edmondson, Miao He, Rebecca C. Spillmann, Erik A. Eklund, Allan Bayat, Hugh J.McMillan, Kym M. Boycott, and Philippe M. Campeau declare that they have no conflict of interest.
ETHICS STATEMENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. 35 Informed consent was obtained from all patients for being included in the study.
AUTHOR CONTRIBUTIONS
Devon L. Johnstone, exome analysis, Sanger sequencing, collection and analysis of clinical data, functional work, drafting of manuscript. Thi Tuyet Mai Nguyen, flow cytometry, experiment planning, drafting of manuscript; Jessica Zambonin, collection and analysis of patient data, revision of supplemental clinical data summaries; Kristin Kernohan, exome analysis, experiment planning, editing of manuscript; Anik St‐Denis, flow cytometry, editing of manuscript; Nissan V. Baratang, flow cytometry, editing of manuscript; Taila Hartley, exome analysis, editing of manuscript; Michael T. Geraghty, collection and analysis of patient data, editing of manuscript; Julie Richer, collection and analysis of patient data, editing of manuscript; Jacek Majewski, exome analysis, editing of manuscript; Eric Bareke, exome analysis, editing of manuscript; Andrea Guerin, collection and analysis of patient data, editing of manuscript; Manuela Pendziwiat, collection and analysis of patient data, editing of manuscript; Loren D.M. Pena, collection and analysis of patient data, editing of manuscript; Hilde M. H. Braakman, collection and analysis of patient data, editing of manuscript; Karen W. Gripp, collection and analysis of patient data, editing of manuscript; Andrew C. Edmondson, collection and analysis of patient data, functional data, editing of manuscript; Miao He, cell culture and flow cytometry, editing of manuscript; Rebecca C. Spillmann, collection and analysis of patient data, editing of manuscript; Erik A. Eklund, collection and analysis of patient data, editing of manuscript; Allan Bayat, collection and analysis of patient data, editing of manuscript; Hugh J. McMillan, collection and analysis of patient data, MRI analysis, editing of manuscript; Kym M. Boycott, co‐senior author; Philippe M. Campeau, co‐senior author.
Supporting information
APPENDIX S1: Supporting Information
ACKNOWLEDGMENTS
We wish to thank the patients and their families for participating in this study, without whom none of this would be possible. This work was partially supported by the Care4Rare Canada Consortium funded by Genome Canada and the Ontario Genomics Institute (OGI‐147), the Canadian Institutes of Health Research, Ontario Research Fund, Genome Alberta, Genome British Columbia, Genome Quebec, Fonds de la recherche en santé du Quebec and Children's Hospital of Eastern Ontario Foundation. Research reported in this manuscript was also supported by the NIH Common Fund through the Office of Strategic Coordination/Office of the NIH Director under Award Number(s) [U01HG007672 (PI Dr. Vandana Shashi, Duke University)] and NIH grant T32GM008638 (Andrew C. Edmondson). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Devon L. Johnstone was supported by a Vanier Canada Graduate Scholarship. Kym M. Boycott was supported by a CIHR Foundation Grant (FDN‐154279) and a Tier 1 Canada Research Chair in Rare Disease Precision Health.
Johnstone DL, Nguyen TTM, Zambonin J, et al. Early infantile epileptic encephalopathy due to biallelic pathogenic variants in PIGQ: Report of seven new subjects and review of the literature. J Inherit Metab Dis. 2020;43:1321–1332. 10.1002/jimd.12278
Communicating Editor: Eva Morava
Funding information Canadian Institutes of Health Research, Grant/Award Number: FDN‐154279; National Institutes of Health, Grant/Award Number: T32GM008638; Office of Strategic Coordination, Grant/Award Number: U01HG007672; Ontario Genomics Institute, Grant/Award Number: OGI‐147
Contributor Information
Kym M. Boycott, Email: kboycott@cheo.on.ca.
Philippe M. Campeau, Email: p.campeau@umontreal.ca.
REFERENCES
- 1. Kinoshita T. Biosynthesis and deficiencies of glycosylphosphatidylinositol. Proc Jpn Acad Ser B Phys Biol Sci. 2014;90(4):130‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bellai‐Dussault K, Nguyen TTM, Baratang NV, Jimenez‐Cruz DA, Campeau PM. Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clin Genet. 2019;95(1):112‐121. [DOI] [PubMed] [Google Scholar]
- 3. McKean DM, Niswander L. Defects in GPI biosynthesis perturb Cripto signaling during forebrain development in two new mouse models of holoprosencephaly. Biol Open. 2012;1(9):874‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nozaki M, Ohishi K, Yamada N, Kinoshita T, Nagy A, Takeda J. Developmental abnormalities of glycosylphosphatidylinositol‐anchor‐deficient embryos revealed by Cre/loxP system. Lab Invest. 1999;79(3):293‐299. [PubMed] [Google Scholar]
- 5. Park S, Lee C, Sabharwal P, Zhang M, Meyers CL, Sockanathan S. GDE2 promotes neurogenesis by glycosylphosphatidylinositol‐anchor cleavage of RECK. Science. 2013;339(6117):324‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Knaus A, Kortum F, Kleefstra T, et al. Mutations in PIGU impair the function of the GPI transamidase complex, causing severe intellectual disability, epilepsy, and brain anomalies. Am J Hum Genet. 2019;105:395‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carmody LC, Blau H, Danis D, et al. Significantly different clinical phenotypes associated with mutations in synthesis and transamidase+remodeling glycosylphosphatidylinositol (GPI)‐anchor biosynthesis genes. Orphanet J Rare Dis. 2020;15(1):40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kinoshita T, Fujita M, Maeda Y. Biosynthesis, remodelling and functions of mammalian GPI‐anchored proteins: recent progress. J Biochem. 2008;144(3):287‐294. [DOI] [PubMed] [Google Scholar]
- 9. Hong Y, Ohishi K, Watanabe R, Endo Y, Maeda Y, Kinoshita T. GPI1 stabilizes an enzyme essential in the first step of glycosylphosphatidylinositol biosynthesis. J Biol Chem. 1999;274(26):18582‐18588. [DOI] [PubMed] [Google Scholar]
- 10. Watanabe R, Inoue N, Westfall B, et al. The first step of glycosylphosphatidylinositol biosynthesis is mediated by a complex of PIG‐A, PIG‐H, PIG‐C and GPI1. EMBO J. 1998;17(4):877‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tiede A, Daniels RJ, Higgs DR, Mehrein Y, Schmidt RE, Schubert J. The human GPI1 gene is required for efficient glycosylphosphatidylinositol biosynthesis. Gene. 2001;271(2):247‐254. [DOI] [PubMed] [Google Scholar]
- 12. Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33(Database issue:D514‐D517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martin HC, Kim GE, Pagnamenta AT, et al. Clinical whole‐genome sequencing in severe early‐onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet. 2014;23(12):3200‐3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alazami AM, Patel N, Shamseldin HE, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole‐exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10(2):148‐161. [DOI] [PubMed] [Google Scholar]
- 15. Starr LJ, Spranger JW, Rao VK, Lutz R, Yetman AT. PIGQ glycosylphosphatidylinositol‐anchored protein deficiency: characterizing the phenotype. Am J Med Genet A. 2019;179(7):1270‐1275. [DOI] [PubMed] [Google Scholar]
- 16. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Human Mut. 2015;36(10):928‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bayat A, Knaus A, Juul AW, et al. PIGT‐CDG, a disorder of the glycosylphosphatidylinositol anchor: description of 13 novel patients and expansion of the clinical characteristics. Genet Med. 2019;21:2216‐2223. [DOI] [PubMed] [Google Scholar]
- 18. Lam C, Golas GA, Davids M, et al. Expanding the clinical and molecular characteristics of PIGT‐CDG, a disorder of glycosylphosphatidylinositol anchors. Mol Genet Metab. 2015;115(2–3):128‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnstone DL, Nguyen TT, Murakami Y, et al. Compound heterozygous mutations in the gene PIGP are associated with early infantile epileptic encephalopathy. Hum Mol Genet. 2017;26(9):1706‐1715. [DOI] [PubMed] [Google Scholar]
- 20. Coppola G, Plouin P, Chiron C, Robain O, Dulac O. Migrating partial seizures in infancy: a malignant disorder with developmental arrest. Epilepsia. 1995;36(10):1017‐1024. [DOI] [PubMed] [Google Scholar]
- 21. Burgess R, Wang S, McTague A, et al. The genetic landscape of epilepsy of infancy with migrating focal seizures. Ann Neurol. 2019;86(6):821‐831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baratang NV, Jimenez Cruz DA, Ajeawung NF, Nguyen TTM, Pacheco‐Cuellar G, Campeau PM. Inherited glycophosphatidylinositol deficiency variant database and analysis of pathogenic variants. Mol Genet Genom Med. 2019;7(7):e00743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakashima M, Kashii H, Murakami Y, et al. Novel compound heterozygous PIGT mutations caused multiple congenital anomalies‐hypotonia‐seizures syndrome 3. Neurogenetics. 2014;15(3):193‐200. [DOI] [PubMed] [Google Scholar]
- 24. Skauli N, Wallace S, Chiang SC, et al. Novel PIGT variant in two brothers: expansion of the multiple congenital anomalies‐Hypotonia seizures syndrome 3 phenotype. Genes (Basel). 2016;7(12):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang L, Peng J, Yin XM, et al. Homozygous PIGT mutation Lead to multiple congenital anomalies‐Hypotonia seizures syndrome 3. Front Genet. 2018;9:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murakami Y, Tawamie H, Maeda Y, et al. Null mutation in PGAP1 impairing Gpi‐anchor maturation in patients with intellectual disability and encephalopathy. PLoS Genet. 2014;10(5):e1004320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murakami Y, Kanzawa N, Saito K, et al. Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J Biol Chem. 2012;287(9):6318‐6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Almeida AM, Murakami Y, Baker A, et al. Targeted therapy for inherited GPI deficiency. N Engl J Med. 2007;356(16):1641‐1647. [DOI] [PubMed] [Google Scholar]
- 29. Kuki I, Takahashi Y, Okazaki S, et al. Vitamin B6‐responsive epilepsy due to inherited GPI deficiency. Neurology. 2013;81(16):1467‐1469. [DOI] [PubMed] [Google Scholar]
- 30. Thompson MD, Killoran A, Percy ME, Nezarati M, Cole DE, Hwang PA. Hyperphosphatasia with neurologic deficit: a pyridoxine‐responsive seizure disorder? Pediatr Neurol. 2006;34(4):303‐307. [DOI] [PubMed] [Google Scholar]
- 31. Nguyen TTM, Murakami Y, Wigby KM, et al. Mutations in PIGS, encoding a GPI transamidase, cause a neurological syndrome ranging from fetal Akinesia to epileptic encephalopathy. Am J Hum Genet. 2018;103(4):602‐611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steinfeld R, Grapp M, Kraetzner R, et al. Folate receptor alpha defect causes cerebral folate transport deficiency: a treatable neurodegenerative disorder associated with disturbed myelin metabolism. Am J Hum Genet. 2009;85(3):354‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pagnamenta AT, Murakami Y, Taylor JM, et al. Analysis of exome data for 4293 trios suggests GPI‐anchor biogenesis defects are a rare cause of developmental disorders. Eur J Hum Genet. 2017;25(6):669‐679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ng BG, Freeze HH. Human genetic disorders involving glycosylphosphatidylinositol (GPI) anchors and glycosphingolipids (GSL). J Inherit Metab Dis. 2015;38(1):171‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. World medical association declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2000;284(23):3043‐3045. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1: Supporting Information