

Abstract
We describe a family severely affected by colorectal cancer (CRC) where whole‐exome sequencing identified the coinheritance of the germline variants encoding MSH6 p.Thr1100Met and MUTYH p.Tyr179Cys in, at least, three CRC patients diagnosed before 60 years of age. Digenic inheritance of monoallelic MSH6 variants of uncertain significance and MUTYH variants has been suggested to predispose to Lynch syndrome‐associated cancers; however, cosegregation with disease in the familial setting has not yet been established. The identification of individuals carrying multiple potential cancer risk variants is expected to rise with the increased application of whole‐genome sequencing and large multigene panel testing in clinical genetic counseling of familial cancer patients. Here we demonstrate the coinheritance of monoallelic variants in MSH6 and MUTYH consistent with cosegregation with CRC, further supporting a role for digenic inheritance in cancer predisposition.
Keywords: digenic inheritance, familial colorectal cancer, Lynch syndrome, MSH6, MUTYH, whole‐exome sequencing
1. INTRODUCTION
Approximately 25% of colorectal cancers (CRCs) are diagnosed in patients with a family history of CRC. However, the majority of familial CRC cannot be explained by clear‐cut genetic defects, which hampers appropriate genetic counselling. 1 The most frequent form of hereditary CRC is Lynch syndrome (OMIM#120435), which predisposes to cancers that develop in a context of DNA mismatch repair (MMR) deficiency, including CRC and endometrial cancer. It is caused by heterozygous, pathogenic variants affecting the DNA MMR genes, MLH1, MSH2, MSH6, or PMS2. MUTYH‐associated polyposis (MAP; OMIM#608456) is a recessively inherited CRC syndrome caused by biallelic variants in the base‐excision repair gene MUTYH. The potential of monoallelic, pathogenic MUTYH variants to predispose to CRC remains debatable. 1 Some MUTYH variants confer greater functional defects in vitro and are associated with more severe clinical phenotypes, such as the variant encoding p.Tyr179Cys compared to p.Gly396Asp. 2 , 3
Digenic inheritance of monoallelic MSH6 and MUTYH variants has been suggested to predispose to Lynch syndrome‐associated cancers; however, cosegregation of both variants within CRC families has not yet been demonstrated. 4 , 5 , 6 , 7 , 8 , 9 Here, we demonstrate, for the first time, the coinheritance of monoallelic variants in MSH6 and MUTYH consistent with the cosegregation with CRC, further supporting a role for digenic inheritance in cancer predisposition.
2. MATERIALS AND METHODS
2.1. Patients
Clinicopathological data of family members was obtained during consultations at the department of Clinical Genetics of the Amsterdam University Medical Centre, Vrije Universiteit Amsterdam. DNA was extracted from peripheral blood and formalin‐fixed paraffin‐imbedded tissues using standard techniques. All patients provided written informed consent. The study was approved by the Medical Ethical Committee of the Leiden University Medical Center, The Netherlands (protocol P01.019).
2.2. Whole‐exome sequencing
Whole‐exome sequencing was outsourced to BGI (BGI‐Shenzhen, Shenzhen, China); exome libraries were constructed with the BGI capture kit, followed by sequencing on the Complete Genomics' Sequencing Platform (Complete Genomics Inc., San Jose, California). Filtering and variant prioritization was performed as previously described. 10 All variants were selected based on a maximum population frequency <0.01 (in 1000 Genomes phase 3, ExAC 1.0, ESP6500SI‐V2 or GoNL release 5).
2.3. Variant screening
The MSH6 (p.Thr1100Met) and MUTYH (p.Tyr179Cys) variants were validated and investigated in additional family members by using Sanger sequencing of PCR products obtained under standard PCR conditions. The following M13‐tailed primer sets were used: 5′‐TGT AAA ACG ACG GCC AGT AAA ACC CCC AAA CGA TGA A‐3′ and 5′‐CAG GAA ACA GCT ATG ACC TGC TCC TCT TCC TCA CAG‐3′ for MSH6, and 5′‐GAC GTT GTA AAA CGA CGG CCA GTC CCT AGG GTA GGG GAA ATA GG‐3′ and 5′‐CAG GAA ACA GCT ATG ACC ATG AGT TCC TAC CCT CCT GCC ATC‐3′ for MUTYH (M13‐tails are underlined).
2.4. Tumor analysis
MMR deficiency in tumor samples was assessed by microsatellite instability analysis and immunohistochemical detection of the four MMR proteins (MLH1, MSH2, MSH6, and PMS2). 11 KRAS codon 12/13 mutations were screened with Sanger sequencing. 12
2.5. Functional MMR assay
In vitro MMR activity assay was performed as previously described. 13
3. RESULTS
We performed germline whole‐exome sequencing on three CRC patients diagnosed before 60 years of age (III‐1, III‐7, III‐8, Figure 1A) and who belonged to a CRC family comprising of seven cancer patients divided over two generations. Twenty‐two rare variants were shared by the three patients (Tables 1 and S1), including variants in the MSH6 (NM_000179.2: c.3299C > T, p.Thr1100Met) and MUTYH (NM_001128425.1: c.536A > G, p.Tyr179Cys) genes, while the other 20 genes could not be clearly linked to cancer predisposition. The identified MSH6 variant was classified as a variant of uncertain significance (VUS) in the Leiden Open Variant Database and the InSiGHT DNA Variant Database. 14 , 15 The MUTYH variant is the most common pathogenic variant found in the Netherlands. 2
FIGURE 1.
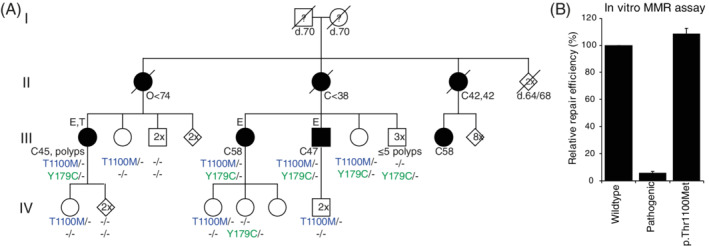
The digenic inheritance of MSH6 and MUTYH variants. A, The pedigree shows the coinheritance of the monoallelic variants which encode MSH6 p.Thr1100Met and MUTYH p.Tyr179Cys in a family affected by colorectal cancer. All spouses were unrelated and unaffected by cancer. Genotypes: MSH6 p.Thr1100Met (T1100M; blue); MUTYH p.Tyr179Cys (Y179C; green); ‐, wild type. E, whole‐exome sequencing analysis; T, tumor analysis; ?, unknown phenotype; numbers in symbols, number of unaffected relatives merged for clarity; filled symbols, cancer patients; C, colorectal cancer; E, endometrial cancer; O, ovarian cancer; d., age at death; followed by the age at diagnosis or death. B, in vitro mismatch repair (MMR) activity assay shows wild‐type MMR activity of MSH6 p.Thr1100Met, compared to wild‐type MSH6 (p.Gly529Gly) and a pathogenic MSH6 mutant (p.Gly1139Ser). Data are shown as mean ± SEM of three independent experiments [Color figure can be viewed at wileyonlinelibrary.com]
TABLE 1.
All rare variants shared by the three individuals from whole‐exome sequencing data
| Chr | Gene | RefSeq accession number | mRNA change | Protein change | Population frequency a | ClinVar classification b | Franklin classification c | Cancer gene census |
|---|---|---|---|---|---|---|---|---|
| 1 | EBNA1BP2 | NM_001159936 | c.1034A > T | p.Asn345Ile | 0.006009 | — | Benign | — |
| 1 | MUTYH | NM_001128425 | c.536A > G | p.Tyr179Cys | 0.001538 | Pathogenic | Pathogenic | Yes |
| 1 | TESK2 | NM_007170 | c.983A > G | p.Gln328Arg | 0.0006052 | — | VUS | — |
| 1 | CAPN9 | NM_006615 | c.55G > T | p.Ala19Ser | 0.00006365 | — | VUS | — |
| 2 | MSH6 | NM_000179 | c.3299C > T | p.Thr1100Met | 0.00004243 | Uncertain | VUS | Yes |
| 3 | C3orf20 | NM_032137 | c.1746C > G | p.Phe582Leu | 0.005847 | — | Likely benign | — |
| 5 | DNAH5 | NM_001369 | c.1781A > G | p.Glu594Gly | — | — | VUS | — |
| 7 | KIAA1324L | NM_001142749 | c.2369 T > C | p.Val790Ala | 0.0006585 | — | VUS | — |
| 7 | TRIP6 | NM_003302 | c.822G > C | p.Glu274Asp | 0.0009893 | — | VUS | — |
| 7 | CUX1 | NM_001202543 | c.1438A > G | p.Ser480Gly | 0.001128 | — | Likely benign | Yes |
| 7 | ZNF783 | NM_001195220 | c.46A > G | p.Thr16Ala | 0.001083 | — | VUS | — |
| 8 | PDP1 | NM_018444 | c.283A > C | p.Ser95Arg | — | — | VUS | — |
| 9 | NMRK1 | NM_017881 | c.304C > G | p.Leu102Val | 0.001419 | — | VUS | — |
| 9 | GAPVD1 | NM_015635 | c.850G > A | p.Val284Met | 0.003596 | — | Benign | — |
| 11 | INTS5 | NM_030628 | c.1436A > G | p.Asn479Ser | 0.00004607 | — | VUS | — |
| 11 | GAL3ST3 | NM_033036 | c.326G > A | p.Arg109His | 0.00004731 | — | VUS | — |
| 11 | SORL1 | NM_003105 | c.3346A > G | p.Ile1116Val | 0.005308 | — | VUS | — |
| 14 | LTBP2 | NM_000428 | c.1226G > A | p.Arg409His | 0.0000203 | — | VUS | — |
| 15 | RYR3 | NM_001036 | c.7812C > G | p.Asn2604Lys | 0.002144 | Likely benign | Likely benign | — |
| 15 | DAPK2 | NM_014326 | c.179G > A | p.Arg60Gln | 0.003725 | — | Likely benign | — |
| 16 | NLRC5 | NM_032206 | c.1219G > A | p.Ala407Thr | 0.000003542 | — | VUS | — |
| 20 | C20orf85 | NM_178456 | c.101G > A | p.Arg34Gln | 0.00192 | — | Likely benign | — |
Abbreviations: Chr, chromosome; VUS, variant of uncertain significance.
Population frequency (gnomAD 2.1.1).
ClinVar clinical significance (ClinVar database version August 5, 2019).
Franklin by Genoox (accessed on May 20, 2020).
Fourteen relatives, all unaffected by cancer or polyposis, were genotyped for these MSH6 and MUTYH variants, identifying one additional carrier of both variants, five MSH6‐only carriers and four MUTYH‐only carriers. In all probability, the mothers of the sequenced patients, II‐1 and II‐2, who were affected by ovarian cancer bellow age 74 and CRC at 38 years old respectively, were obligate carriers of both variants; however, DNA was unavailable for testing and, formally, inheritance through the fathers to the sequenced individuals (III‐1, III‐7, III‐8) cannot be excluded. MMR deficiency was not detected in the colorectal carcinoma of patient III‐1, which also lacked the KRAS mutation typical for MAP tumors (c.34C > T; Table S2). Functional analysis of the MSH6 p.Thr1100Met variant showed retained MMR function in vitro (Figure 1B).
4. DISCUSSION
Digenic inheritance of monoallelic MSH6 and MUTYH variants has been suggested to predispose to Lynch syndrome‐associated cancers. The involvement of both MSH6 and MUTYH in oxidative DNA damage repair and their physical interaction enhancing MUTYH's repair activity, substantiates the association of variants in these genes. 16 From earlier studies, the inheritance of monoallelic MUTYH variants seemed primarily relevant in patients carrying MSH6 VUSs, which are less strongly associated with MMR deficiency than pathogenic MSH6 variants (Table S2). 4 , 5 , 6 , 7 , 8 , 9 Furthermore, a digenic inheritance model was proposed once before for CRC predisposition in a carrier of variants in the oxidative DNA damage repair genes MUTYH and OGG1. 17 Although the functional evidence of combined defects in oxidative DNA damage repair genes is still lacking, the coinheritance of MSH6 and MUTYH variants in at least three, but likely five cancer cases within one family warrants further mechanistic and clinical studies. The absence of cancer and numerous polyps in nondigenic carriers further substantiates this association. Tumor analysis of the tumor of one of the digenic carriers and the in vitro MMR activity assay indicated retention of MMR function of MSH6 p.Thr1100Met protein. In addition, the genetic marker for MAP‐tumors (KRAS c.34G > T) was absent in this tumor, which points toward retained MUTYH repair activity. The combined inheritance of both genetic variants could still result in impaired repair of oxidative DNA damage. More extensive somatic mutation analysis to assess this was, however, not possible, because of low quality of the DNA sample and the unavailability of additional tumor material.
Next to MSH6 and MUTYH, CUX1 has been described as a cancer‐driving gene. 18 CUX1 is implicated in inflammatory bowel disease and various cancer types, although primarily due to loss‐of‐function somatic mutations. 18 , 19 This gene codes for several isoforms, including the ubiquitously expressed p200 CUX1, which, among other functions, has been shown to stimulate the repair of oxidized DNA bases by OGG1. 20 The identified CUX1 (NM_001202543: c.1438A > G, p.Ser480Gly) variant, however, was classified as likely benign by the Franklin variant classification tool. 21 Additional gene reportedly linked to tumorigenesis include RYR3, 22 EBNA1BP2, 23 TRIP6, 24 and CAPN9. 25 The RYR3 (NM_001036: c.7812C > G, p.Asn2604Lys) and EBNA1BP2 (NM_001159936: c.1034A > T, p.Asn345Ile) variants were classified as likely benign and benign, respectively, while the TRIP6 (NM_003302: c.822G > C, p.Glu274Asp) and the CAPN9 (NM_006615: c.55G > T, p.Ala19Ser) variants were classified as VUS. 21 TRIP6 promotes cell migration and invasion through Wnt/β‐catenin signaling and was shown to be upregulated in colorectal tumors. 24 Therefore, TRIP6 variants that increase protein stability or expression could potentially stimulate colorectal tumorigenesis. In addition, lost‐of‐function variants in CAPN9 might promote tumor formation, as Calpain‐9 induces cell cycle arrest and apoptosis, and low expression predicts a poorer prognosis in gastric cancer patients. 25 The contribution of the genetic variants, other than MSH6 and MUTYH, to cancer risk cannot be completely excluded. However, none of these variants have been functionally investigated and especially the variants predicted as benign or likely benign are less likely to contribute to an increased cancer risk. Besides, none of these genes have, to date, been associated with a genetic predisposition to any types of cancer.
In conclusion, with the increased application of whole‐genome sequencing or large multigene panel testing in clinical genetic counseling, the number of identified individuals carrying multiple potential risk variants is expected to rise. Here, we demonstrate the coinheritance of MSH6 and MUTYH variants consistent with the cosegregation with cancer, further supporting a role for digenic inheritance in CRC predisposition. Our results reiterate that digenic inheritance should be considered as cause of genetic diseases.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Tom van Wezel, Noel F. C. C. de Miranda, and Hans Morreau conceived and designed the study. Dina Ruano performed next‐generation sequencing analyses. Noel F. C. C. de Miranda and Stephanie A. Schubert performed analysis and interpretation of whole‐exome sequencing data. Mark Drost and Yvonne Tiersma performed functional analysis. Maartje Nielsen and Liselotte P. van Hest performed patient counseling and clinical data acquisition. Hans Morreau performed the pathology review of the samples. Tom van Wezel, Noel F. C. C. de Miranda, Mark Drost, and Niels de Wind supervised the work. Stephanie A. Schubert, Noel F. C. C. de Miranda, and Tom van Wezel wrote the manuscript. All authors read and approved the manuscript.
Supporting information
Supplementary Table S1 . All rare variants shared by the three individuals from whole‐exome sequencing data
Supplementary Table S2: Digenic inheritance of MSH6 and MUTYH variants.
ACKNOWLEDGMENTS
The authors thank Juul T. Wijnen for the collection of clinicopathological data and samples. The authors also thank Julia van Hees and Anniek van Veen for their technical support. The authors are thankful to the Leiden University Fund/Nypels‐van der Zee Fonds. This project was funded by research grants from the Dutch Digestive Foundation (MLDS FP13‐13) and Stichting Sacha Swarttouw‐Hijmans awarded to T.v.W. N.F.C.C.d.M. is supported by the KWF Bas Mulder Award UL (2015‐7664) and the ZonMw Veni grant (016.176.l44). This article is based upon work from COST Action CA17118, supported by European Cooperation in Science and Technology (COST).
Schubert SA, Ruano D, Tiersma Y, et al. Digenic inheritance of MSH6 and MUTYH variants in familial colorectal cancer. Genes Chromosomes Cancer. 2020;59:697–701. 10.1002/gcc.22883
Funding information KWF Kankerbestrijding, Grant/Award Number: 2015‐7664; Maag Lever Darm Stichting, Grant/Award Number: MLDS FP13‐13; Stichting Sacha Swarttouw‐Hijmans; ZonMw Veni, Grant/Award Number: 016.176.l44; European Cooperation in Science and Technology (COST); Leiden University Fund/Nypels‐van der Zee Fonds
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. The data are not publicly available due to privacy restrictions.
REFERENCES
- 1. Schubert SA, Morreau H, de Miranda NFCC, van Wezel T. The missing heritability of familial colorectal cancer. Mutagenesis. 2020;35(3):221‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nielsen M, Joerink‐van de Beld MC, Jones N, et al. Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH‐associated polyposis. Gastroenterology. 2009;136(2):471‐476. [DOI] [PubMed] [Google Scholar]
- 3. Komine K, Shimodaira H, Takao M, et al. Functional complementation assay for 47 MUTYH variants in a MutY‐disrupted Escherichia coli strain. Hum Mutat. 2015;36(7):704‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Niessen RC, Sijmons RH, Ou J, et al. MUTYH and the mismatch repair system: partners in crime? Hum Genet. 2006;119(1–2):206‐211. [DOI] [PubMed] [Google Scholar]
- 5. Steinke V, Rahner N, Morak M, et al. No association between MUTYH and MSH6 germline mutations in 64 HNPCC patients. Eur J Human Genet. 2008;16(5):587‐592. [DOI] [PubMed] [Google Scholar]
- 6. Giraldez MD, Balaguer F, Caldes T, et al. Association of MUTYH and MSH6 germline mutations in colorectal cancer patients. Fam Cancer. 2009;8(4):525‐531. [DOI] [PubMed] [Google Scholar]
- 7. Giraldez MD, Balaguer F, Bujanda L, et al. MSH6 and MUTYH deficiency is a frequent event in early‐onset colorectal cancer. Clin Cancer Res. 2010;16(22):5402‐5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Puijenbroek M, Nielsen M, Reinards THCM, et al. The natural history of a combined defect in MSH6 and MUTYH in a HNPCC family. Fam Cancer. 2007;6(1):43‐51. [DOI] [PubMed] [Google Scholar]
- 9. Win AK, Reece JC, Buchanan DD, et al. Risk of colorectal cancer for people with a mutation in both a MUTYH and a DNA mismatch repair gene. Fam Cancer. 2015;14(4):575‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schubert SA, Ruano D, Elsayed FA, et al. Evidence for genetic association between chromosome 1q loci and predisposition to colorectal neoplasia. Br J Cancer. 2017;117(6):1215‐1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Jong AE, van Puijenbroek M, Hendriks Y, et al. Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10(3):972‐980. [DOI] [PubMed] [Google Scholar]
- 12. Nielsen M, Poley JW, Verhoef S, et al. Duodenal carcinoma in MUTYH‐associated polyposis. J Clin Pathol. 2006;59(11):1212‐1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Drost M, Tiersma Y, Glubb D, et al. Two integrated and highly predictive functional analysis‐based procedures for the classification of MSH6 variants in Lynch syndrome. Genet Med. 2020;22(5):847‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. LOVD . Leiden Open Variant Database v.3.0 Build 22 https://www.lovd.nl. Accessed May 11, 2020.
- 15. InSiGHT . DNA Variant Database http://insight-database.org/. Accessed May 11, 2020.
- 16. Gu Y, Parker A, Wilson TM, Bai H, Chang DY, Lu AL. Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J Biol Chem. 2002;277(13):11135‐11142. [DOI] [PubMed] [Google Scholar]
- 17. Morak M, Massdorf T, Sykora H, Kerscher M, Holinski‐Feder E. First evidence for digenic inheritance in hereditary colorectal cancer by mutations in the base excision repair genes. Eur J Cancer. 2011;47(7):1046‐1055. [DOI] [PubMed] [Google Scholar]
- 18. Wong CC, Martincorena I, Rust AG, et al. Inactivating CUX1 mutations promote tumorigenesis. Nat Genet. 2014;46(1):33‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Darsigny M, St‐Jean S, Boudreau F. Cux1 transcription factor is induced in inflammatory bowel disease and protects against experimental colitis. Inflamm Bowel Dis. 2010;16(10):1739‐1750. [DOI] [PubMed] [Google Scholar]
- 20. Ramdzan ZM, Vadnais C, Pal R, et al. RAS transformation requires CUX1‐dependent repair of oxidative DNA damage. PLoS Biol. 2014;12(3):e1001807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Franklin . franklin.genoox.com. Accessed May 20, 2020.
- 22. Chae YS, Kim JG, Kang BW, et al. Functional polymorphism in the MicroRNA‐367 binding site as a prognostic factor for colonic cancer. Anticancer Res. 2013;33(2):513‐519. [PubMed] [Google Scholar]
- 23. Liao P, Wang W, Shen M, et al. A positive feedback loop between EBP2 and c‐Myc regulates rDNA transcription, cell proliferation, and tumorigenesis. Cell Death Dis. 2014;5(1):e1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chastre E, Abdessamad M, Kruglov A, et al. TRIP6, a novel molecular partner of the MAGI‐1 scaffolding molecule, promotes invasiveness. FASEB J. 2009;23(3):916‐928. [DOI] [PubMed] [Google Scholar]
- 25. Peng P, Wu W, Zhao J, et al. Decreased expression of Calpain‐9 predicts unfavorable prognosis in patients with gastric cancer. Sci Rep. 2016;6:29604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1 . All rare variants shared by the three individuals from whole‐exome sequencing data
Supplementary Table S2: Digenic inheritance of MSH6 and MUTYH variants.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. The data are not publicly available due to privacy restrictions.