

Abstract
Porous shape‐persistent organic cages have become the object of interest in recent years because they are soluble and thus processable from solution. A variety of cages can be achieved by applying dynamic covalent chemistry (DCC), but they are less chemically stable. Here the transformation of a salicylimine cage into a quinoline cage by a twelve‐fold Povarov reaction as the key step is described. Besides the chemical stability of the cage over a broad pH regime, it shows a unique absorption and emission depending on acid concentration. Furthermore, thin films for the vapor detection of acids were investigated, showing color switches from pale‐yellow to red, and characteristic emission profiles.
Keywords: adsorption, imines, cage compounds, structure elucidation, thin films
Cage to cage: Soluble, porous, and chemically stable quinoline cages are synthesized from salicylimine cages in one post‐synthetic step. The cages show characteristic acidochromic behavior in bulk and solution, and as thin‐films.
Since the pioneering contributions of the research groups of Cooper as well as Atwood on permanent porosity of shape‐persistent organic cages,1 the field has developed tremendously fast.2 Shape‐persistent organic cages are synthesized in a variety of geometries and sizes and many of which were studied for gas sorption and separation.2e, 3 Among those systems there have been microporous organic cages reported with very high specific surface areas greater than 1500 m2 g−1,4 as well as even mesoporous cages with specific surface areas of 3758 m2 g−1,5 a value demonstrating that large cavities can be constructed in a manner so that they will not collapse upon evacuation.6 The vast majority of shape‐persistent organic cages was synthesized by exploiting the reversible bond breaking and bond making that dynamic covalent chemistry (DCC) reactions offer. Most typical DCC reactions used for cage synthesis are condensation reactions such as the formation of imines from aldehydes and amines,7 or boronic esters from boronic acids and diols.8 By far fewer examples have been reported to rely on the oxidative disulfide formation or alkyne metathesis.9, 10
Especially imine‐based cages offer the possibility for post‐synthetic transformation of the imine functional groups by different reactions. Among them, the most trivial one is the reduction of imines to amines. Unfortunately, because of the transformation of sp2‐ into sp3‐hybridized atoms, the overall scaffold becomes more flexible. As a consequence the cages lose their shape persistency and thus their porosity.4a, 11 Nevertheless, with a follow‐up reaction, porosity can be recovered. This has been exemplified, for example, for Cooper's smaller [4+6] amine cages where the reduced amine cage contains 1,2‐diamine units, which form aminals with aldehydes or ketones.12 Another similar approach is the fixation of the corresponding amines of [2+3] salicyl imine cages13 as carbamates.14 Recently, our group demonstrated that the twelve imine bonds of a rigid shape‐persistent [4+6] salicylimine cage can be transformed by Pinnick oxidations into chemically much more robust amide bonds with retention of the shape persistency.15 Indeed, this amide cage was stable in a slightly broader pH range (−1 to 14.5) than, for example, the carbamate cage (−1 to 14).14 This unprecedented stability opened the possibility for further chemical derivatization under harsh reaction conditions, such as selective twelve‐fold bromination or nitration, occurring in yields greater than 80 %. Unfortunately, the Pinnick reaction does not work in the presence of the phenolic hydroxy group, so that the six hydroxy groups of the salicylimine cage needed to be first transformed into methyl ethers.4c Despite the richness in imine chemistry,16 these are the only few examples where transformations have been exploited to post‐stabilize organic cage compounds under the conservation/retention of porosity.17, 18
Here we present our results on transforming a [4+6] salicylimine cage directly (without the necessity of protecting the phenolic hydroxy groups) into a quinoline cage by a twelve‐fold Povarov reaction19, 20 with subsequent oxidation (Scheme 1). The quinoline cage 2 was synthesized by treating the imine cage 1 7f in neat phenylacetylene as the solvent, with scandium triflate (3.6 equiv) as the Lewis acid and chloranil (12 equiv) as the oxidant. After heating at 100 °C for 20 hours, and after removal of by‐products by column chromatography the cage was isolated in 25 % yield. It is worth mentioning that this corresponds to 89 % per imine bond, which is even higher than we were able to realize for the bisquinoline model compound 3 (here the yield was 35 % and per imine bond 59 %. For structure see Figure 3. For details, see the Supporting Information). The reaction time plays a critical role. Before 20 hours, the conversion of all twelve imine units was not complete. After much more than 20 hours, further addition of phenylacetylene units to the cage is observed by mass spectrometry (see the Supporting Information). By HR‐MALDI mass spectrometry, a single peak for [M+H]+ at m/z=3418.387 (calc. m/z=3418.390) was detected for 2, having the expected isotope distribution pattern (Figure 1). The compound 2 is basic because of the quinoline nitrogen atoms and is easily protonated by traces of Brønsted acids, thus the NMR spectra needed to be recorded with 0.1 mol % d15‐triethylamine to guarantee the high molecular symmetry of the compound for a simple 1H NMR spectrum (Figure 2). Characteristic are the two triptycene bridgehead protons resonating at δ=5.54 and 6.31 ppm. Furthermore, the protons of the quinoline units are found as singlets at δ=6.62, 7.83, and 7.66 ppm besides a singlet at δ=7.67 ppm for the aromatic protons of the phenol units. The twelve peripheral phenyl units appear as a multiplet between δ=7.54 and 7.60 ppm. However, the integral of these phenyl protons is somewhat too high, suggesting that this has its origin in their rotational freedom and thus a different relaxation time than for all the other protons of the rigid scaffold.21 Indeed the number of aromatic carbon nuclei found by 13C NMR spectroscopy is 17, exactly as expected, excluding the presence of impurities (such as remaining phenylacetylene or other by‐products). By diffusion‐ordered NMR spectroscopy (DOSY) in CDCl3 at room temperature a diffusion coefficient of D=3.52 10−10 m2 s−1 was measured, corresponding to a solvodynamic radius of r S=1.14 nm, which is close to the maximum outer diameter (approx. 2.7 nm) found for this structure by X‐ray diffraction (Figure 4). In the IR two characteristic imine peaks are found 1550 cm−1 and 1625 cm−1, which are comparable to those of model compound 3 (1550 cm−1 and 1618 cm−1).
Scheme 1.
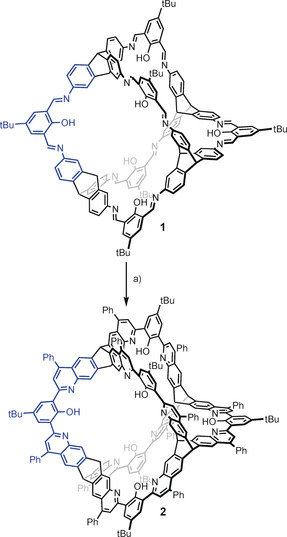
Twelve‐fold Povarov reaction of [4+6] imine cage 1 to the quinoline cage 2. a) Sc(OTf)3, chloranil, phenylacetylene (neat), 100 °C, 20 h, 25 % yield.
Figure 3.
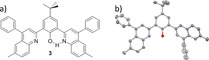
Molecular structure (a) and X‐ray structure30 (b) of the model compound 3. For details (synthesis and characterization) see the Supporting Information. Hydrogens are omitted in (b) for clarity. Ellipsoids are at a 50 % probability level.
Figure 1.
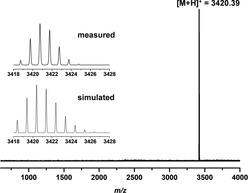
MALDI‐TOF mass spectrum of 2. The inset shows the isotopic distribution pattern of the molecular ion peak by HR‐MALDI MS.
Figure 2.
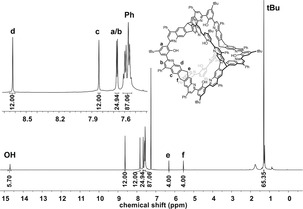
1H NMR spectrum (600 MHz, CDCl3 + 0.1 % d15‐NEt3) of 2.
Figure 4.
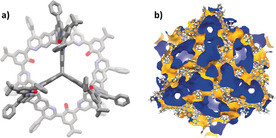
X‐ray crystal structure of 2.30 a) Molecular structure with hydrogen atoms omitted for clarity. b) Cubic unit cell with accessible surface area for a probe of radius 1.62 Å, view along cell diagonal.
By diffusion of an ammonia/ethyl acetate mixture into a solution of 2 in dichloromethane at room temperature single‐crystals of 2 were obtained, which were analyzed by X‐ray diffraction (Figure 4). The compound crystallizes in the cubic space group with eight molecules in the unit cell. Electron density of highly disordered solvate molecules needed to be removed by the SQUEEZE routine function of PLATON to allow the structure to be solved and refined.22 By the Povarov cyclization the flexibility of the bonds within 2 is substantially reduced in comparison to those in 1. As a consequence, all nitrogen electron lone‐pairs are conformationally fixed endo to the cavities interior. Therefore, two nitrogen atoms have to strongly interact with the phenolic hydroxy groups, leading to a conformationally restricted repulsion of lone pairs, which is reflected in larger thermal ellipsoids of these quinoline and phenol atoms, which most likely rapidly slide back and forth even in the crystalline state. The distance between the inner triptycene bridgeheads form an almost regular tetrahedron with edge length of 11.1 Å. The cages are loosely packed by intermolecular forces, such as week CH–π or π–π stacking and London dispersion interactions.
The as‐synthesized material of 2 has an amorphous but porous structure (see SEM images and PXRD in the Supporting Information). The pores were activated by immersing the material in Et2O (2×) and n‐pentane (5×) before thermal treatment at 250 °C and vacuum (10−3 mbar) overnight. By nitrogen sorption at 77 K a specific surface area of 698 m2 g−1 was measured (Figure 5). The isotherms are best described as a combination of type I (microporous) and type II (macroporous or nonporous).23 By QS‐DFT24 the most intensive peak is found for a pore‐width at 12.6 Å besides less intensive broad peaks at 22.6 and 40 Å. The first one is in good agreement to the minimum inner diameter of the cage model (15 Å). The uptake of other gases was investigated too. At 273 K and 1 bar, 2.72 mmol g−1 (12.0 wt %) CO2, 1.15 mmol g−1 (1.85 wt %) CH4, and 5.17 mmol g−1 (1.0 wt %) H2 were adsorbed.
Figure 5.
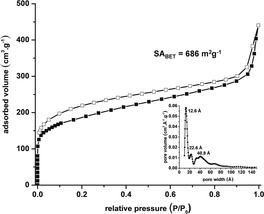
Nitrogen sorption isotherm at 77 K and QS‐DFT pore‐size distribution (inset) of 2. Filled symbols: adsorption, open symbols: desorption.
The chemical and thermal stability of the compound was tested. A sample was heated to 350 °C and showed, after cooling to room temperature, no difference in the 1H NMR spectrum. Still, the MS showed only the peak of the quinoline cage at m/z=3418 with its characteristic isotopic distribution pattern. Neither under very harsh acidic conditions (concentrated sulfuric acid, pH −1.9), nor under harsh basic conditions (concentrated NaOH, pH 15.2) does the compound decompose. The nearly quantitative recovered material was neutralized and the recorded 1H NMR spectra again showed no difference to the compound before treatment. This range is to the best of our knowledge, the largest pH window in which a shape‐persistent organic cage is stable (for comparison the [4+6] amide cage is pH‐stable from −1 to 14.5).15 It is worth mentioning that 1 decomposes at pH 3 or beyond pH 11 (see the Supporting Information).
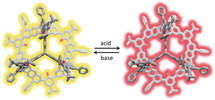
In contrast to the previously reported [4+6] amide cage, the extended fused aromatic quinoline units allowed us to study the acidochromism of this cage. By the naked eye, a clear color change is detected both in the solid state and in solution. Under basic or pH neutral conditions the compound is pale yellow, whereas when acidic it is bright red to orange (Figure 7). Because 2 is well soluble in CHCl3 and CH2Cl2, (>130 mg mL−1) and also in THF or toluene (approx. 30 mg mL−1), the reversible acidochromism was studied in more detail. By UV/Vis titration of 2 with trifluoroacetic acid in dichloromethane two pK b values of 9.2 and 13.2 were determined. These values are comparable but slightly different to those of the model compound 3 (pKb1=9.1 and pKb2 13.5).
Figure 7.
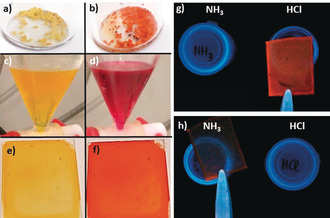
Acidochromism of 2 in the bulk (a,b), solution (c,d), and as thin film (e–h). a,c,e) pH neutral or basic. b,d,f) pH acidic. g) Film under UV (254 nm) held over an open vial containing HClaq. h) Film under UV (254 nm) held over an open vial containing NH3,aq.
As mentioned above, 2 shows a pronounced acidochromism. It is pale‐yellow in neutral or basic media and red in acidic media. Furthermore, 2 shows in the protonated form a characteristic emission dependency on the acid concentration (Figure 6). For instance, if trifluoracetic acid is added, the emission increases up to the point of three equivalents of acid to show a local maximum at 565 nm. After that, the emission decreases upon further addition of acid, before getting much stronger with a slightly bathochromically shifted peak at 575 nm, having an emission maximum at 3×104 equivalents of TFA. In the presence of sulfuric acid, a very similar behavior can be found, with the same two emission maxima. In contrast, the much weaker acetic acid (pK a=4.76) is not able to protonate each bisquinoline unit twice and thus a high concentration of 104 equivalents is needed to show the first emission maximum at 565 nm. At higher concentration the second emission maximum at 575 nm is not observed, clearly underlining the prior determined pK a values. Those phenol quinoline units should also be able to act as a ligand for transition metals or “BF2 ” units to generate boroquinoles.25 Therefore, we tested the emission dependency upon the addition of BF3 etherate. Here we see three emissions: 1) 566 nm at low concentration; 2) 580 nm at higher concentration, comparable to the event with TFA; and 3) at larger BF3⋅Et2O concentration the emission is hypsochromically shifted towards 483 nm, which is different from the events observed for Brønsted acids.
Figure 6.
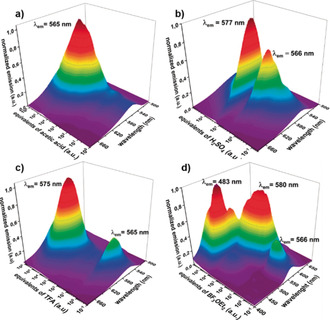
Acid concentration dependent emission profiles of a solution of 2 (c=2.76×10−6 mol L−1) in dichloromethane. a) Acetic acid; b) Sulfuric acid, c) TFA, and d) BF3⋅Et2O.
Recently, covalent organic frameworks (COFs) were described for humidity sensing26 and acid sensing,27 showing detectable color changes by the naked eye. As‐synthesized COFs are usually insoluble solids or powders and to generate thin‐films of those, they need to be grown on the substrate by distinct protocols. However, COFs are made by reversible reactions, such as imine condensations, and are therefore, with a few exceptions, chemically less robust against aqueous basic or acidic conditions. This lack of robustness may explain why only pure water, pure organic solvents, or pure acids have been used for sensing these by COFs. As has been demonstrated previously by us and others, porous organic cages have the advantage to be solution processable.28 Therefore, thin films of 2 were made by drop‐casting a dioxane solution of 2 onto glass (Figure 7). This thin‐film could be switched in color from pale yellow to bright (fluorescent) red by exposing it to (diluted) hydrochloric acid vapors or aqueous ammonia vapors, and it is highly reversible (see the Supporting Information). After the adsorption of acidic vapors (here HCl), the film can also be regenerated by heat. Furthermore, the film‐coated glass can be dipped into water, aqueous solutions of hydrochloride acid, or ammonia and stays fully intact. MALDI‐TOF MS analysis afterwards showed that 2 does not decompose. It is worth mentioning that the imine cage 1 could also be used to make a thin‐film on glass, showing a comparable switch in color and fluorescence, if treated with acidic or basic vapors and is thus comparable to the aforementioned recently reported COF‐films.27a But as soon as this film is dipped into acidic or basic aqueous solutions, the film, as well as 1, are rapidly decomposed by hydrolysis—something that most likely will also happen to thin films of imine linked COFs.
To summarize, a chemically and thermally very robust shape‐persistent quinoline cage has been synthesized in 25 % yield by a twelve‐fold Povarov reaction of a [4+6] salicylimine cage with phenylacetylene. In contrast to prior post functionalizations of this and other imine cages,14, 15, 29 this conversion was achieved in one step. The quinoline cage shows a characteristic acidochromism, which was used in combination with its solubility to generate thin‐films for sensing of either acid or base vapors.
Experimental Section
For Experimental Details, see the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank the European Research Council (ERC) for funding this project in the frame of the consolidators grant CaTs n DOCs (grant agreement no. 725765). Open access funding enabled and organized by Projekt DEAL.
P.-E. Alexandre, W.-S. Zhang, F. Rominger, S. M. Elbert, R. R. Schröder, M. Mastalerz, Angew. Chem. Int. Ed. 2020, 59, 19675.
References
- 1.
- 1a. Tozawa T., Jones J. T. A., Swamy S. I., Jiang S., Adams D. J., Shakespeare S., Clowes R., Bradshaw D., Hasell T., Chong S. Y., Tang C., Thompson S., Parker J., Trewin A., Bacsa J., Slawin A. M. Z., Steiner A., Cooper A. I., Nat. Mater. 2009, 8, 973–978; [DOI] [PubMed] [Google Scholar]
- 1b. Tian J., Thallapally P. K., Dalgarno S. J., McGrail P. B., Atwood J. L., Angew. Chem. Int. Ed. 2009, 48, 5492–5495; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5600–5603. [Google Scholar]
- 2.
- 2a. Mastalerz M., Angew. Chem. Int. Ed. 2010, 49, 5042–5053; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5164–5175; [Google Scholar]
- 2b. Mastalerz M., Synlett 2013, 24, 781–786; [Google Scholar]
- 2c. Zhang G., Mastalerz M., Chem. Soc. Rev. 2014, 43, 1934–1947; [DOI] [PubMed] [Google Scholar]
- 2d. Mastalerz M., Chem. Eur. J. 2012, 18, 10082–10091; [DOI] [PubMed] [Google Scholar]
- 2e. Mastalerz M., Acc. Chem. Res. 2018, 51, 2411–2422; [DOI] [PubMed] [Google Scholar]
- 2f. Holst J. R., Trewin A., Cooper A. I., Nat. Chem. 2010, 2, 915; [DOI] [PubMed] [Google Scholar]
- 2g. Hasell T., Cooper A. I., Nat. Rev. Mater. 2016, 1, 16053; [Google Scholar]
- 2h. Beuerle F., Gole B., Angew. Chem. Int. Ed. 2018, 57, 4850–4878; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4942–4972; [Google Scholar]
- 2i. Acharyya K., Mukherjee P. S., Angew. Chem. Int. Ed. 2019, 58, 8640–8653; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8732–8745; [Google Scholar]
- 2j. Mukhopadhyay R. D., Kim Y., Koo J., Kim K., Acc. Chem. Res. 2018, 51, 2730–2738; [DOI] [PubMed] [Google Scholar]
- 2k. Little M. A., Cooper A. I., Adv. Funct. Mater. 2020, 30, 1909842. [Google Scholar]
- 3. Santolini V., Miklitz M., Berardo E., Jelfs K. E., Nanoscale 2017, 9, 5280–5298. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Mastalerz M., Schneider M. W., Oppel I. M., Presly O., Angew. Chem. Int. Ed. 2011, 50, 1046–1051; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1078–1083; [Google Scholar]
- 4b. Schneider M. W., Oppel I. M., Ott H., Lechner L. G., Hauswald H.-J. S., Stoll R., Mastalerz M., Chem. Eur. J. 2012, 18, 836–847; [DOI] [PubMed] [Google Scholar]
- 4c. Schneider M. W., Oppel I. M., Griffin A., Mastalerz M., Angew. Chem. Int. Ed. 2013, 52, 3611–3615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3699–3703; [Google Scholar]
- 4d. Zhang G., Presly O., White F., Oppel I. M., Mastalerz M., Angew. Chem. Int. Ed. 2014, 53, 5126–5130; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5226–5230. [Google Scholar]
- 5. Zhang G., Presly O., White F., Oppel I. M., Mastalerz M., Angew. Chem. Int. Ed. 2014, 53, 1516–1520; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1542–1546. [Google Scholar]
- 6.
- 6a. Jelfs K. E., Wu X., Schmidtmann M., Jones J. T. A., Warren J. E., Adams D. J., Cooper A. I., Angew. Chem. Int. Ed. 2011, 50, 10653–10656; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10841–10844; [Google Scholar]
- 6b. Skowronek P., Warzajtis B., Rychlewska U., Gawronski J., Chem. Commun. 2013, 49, 2524–2526; [DOI] [PubMed] [Google Scholar]
- 6c. Briggs M. E., Jelfs K. E., Chong S. Y., Lester C., Schmidtmann M., Adams D. J., Cooper A. I., Cryst. Growth Des. 2013, 13, 4993–5000. [Google Scholar]
- 7.
- 7a. MacDowell D., Nelson J., Tetrahedron Lett. 1988, 29, 385–386; [Google Scholar]
- 7b. Quan M. L. C., Cram D. J., J. Am. Chem. Soc. 1991, 113, 2754–2755; [Google Scholar]
- 7c. Ro S., Rowan S. J., Pease A. R., Cram D. J., Stoddart J. F., Org. Lett. 2000, 2, 2411–2414; [DOI] [PubMed] [Google Scholar]
- 7d. Rue N. M., Sun J., Warmuth R., Isr. J. Chem. 2011, 51, 743–768; [Google Scholar]
- 7e. Skowronek P., Gawronski J., Org. Lett. 2008, 10, 4755–4758; [DOI] [PubMed] [Google Scholar]
- 7f. Mastalerz M., Chem. Commun. 2008, 4756–4758. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Christinat N., Scopelliti R., Severin K., Angew. Chem. 2008, 120, 1874–1878; [DOI] [PubMed] [Google Scholar]
- 8b. Hutin M., Bernardinelli G., Nitschke J. R., Chem. Eur. J. 2008, 14, 4585–4593; [DOI] [PubMed] [Google Scholar]
- 8c. Nishimura N., Kobayashi K., Angew. Chem. Int. Ed. 2008, 47, 6255–6258; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6351–6354; [Google Scholar]
- 8d. Klotzbach S., Scherpf T., Beuerle F., Chem. Commun. 2014, 50, 12454–12457; [DOI] [PubMed] [Google Scholar]
- 8e. Dhara A., Beuerle F., Chem. Eur. J. 2015, 21, 17391–17396; [DOI] [PubMed] [Google Scholar]
- 8f. Klotzbach S., Beuerle F., Angew. Chem. Int. Ed. 2015, 54, 10356–10360; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10497–10502; [Google Scholar]
- 8g. Ono K., Johmoto K., Yasuda N., Uekusa H., Fujii S., Kiguchi M., Iwasawa N., J. Am. Chem. Soc. 2015, 137, 7015–7018; [DOI] [PubMed] [Google Scholar]
- 8h. Elbert S. M., Regenauer N. I., Schindler D., Zhang W.-S., Rominger F., Schröder R. R., Mastalerz M., Chem. Eur. J. 2018, 24, 11438–11443. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Tam-Chang S.-W., Stehouwer J. S., Hao J., J. Org. Chem. 1999, 64, 334–335; [Google Scholar]
- 9b. Naumann C., Place S., Sherman J. C., J. Am. Chem. Soc. 2002, 124, 16–17. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Zhang C., Wang Q., Long H., Zhang W., J. Am. Chem. Soc. 2011, 133, 20995–21001; [DOI] [PubMed] [Google Scholar]
- 10b. Wang Q., Zhang C., Noll B. C., Long H., Jin Y., Zhang W., Angew. Chem. Int. Ed. 2014, 53, 10663–10667; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10839–10843; [Google Scholar]
- 10c. Lee S., Yang A., Moneypenny T. P., Moore J. S., J. Am. Chem. Soc. 2016, 138, 2182–2185; [DOI] [PubMed] [Google Scholar]
- 10d. Moneypenny T. P., Yang A., Walter N. P., Woods T. J., Gray D. L., Zhang Y., Moore J. S., J. Am. Chem. Soc. 2018, 140, 5825–5833. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Jin Y., Voss B. A., Noble R. D., Zhang W., Angew. Chem. Int. Ed. 2010, 49, 6348–6351; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6492–6495; [Google Scholar]
- 11b. Jin Y., Voss B. A., Jin A., Long H., Noble R. D., Zhang W., J. Am. Chem. Soc. 2011, 133, 6650–6658. [DOI] [PubMed] [Google Scholar]
- 12. Liu M., Little M. A., Jelfs K. E., Jones J. T. A., Schmidtmann M., Chong S. Y., Hasell T., Cooper A. I., J. Am. Chem. Soc. 2014, 136, 7583–7586. [DOI] [PubMed] [Google Scholar]
- 13. Schneider M. W., Oppel I. M., Mastalerz M., Chem. Eur. J. 2012, 18, 4156–4160. [DOI] [PubMed] [Google Scholar]
- 14. Hu X.-Y., Zhang W.-S., Rominger F., Wacker I., Schröder R. R., Mastalerz M., Chem. Commun. 2017, 53, 8616–8619. [DOI] [PubMed] [Google Scholar]
- 15. Bhat A. S., Elbert S. M., Zhang W.-S., Rominger F., Dieckmann M., Schröder R. R., Mastalerz M., Angew. Chem. Int. Ed. 2019, 58, 8819–8823; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8911–8915. [Google Scholar]
- 16. Layer R. W., Chem. Rev. 1963, 63, 489–510. [Google Scholar]
- 17. Culshaw J. L., Cheng G., Schmidtmann M., Hasell T., Liu M., Adams D. J., Cooper A. I., J. Am. Chem. Soc. 2013, 135, 10007–10010. [DOI] [PubMed] [Google Scholar]
- 18. Acharyya K., Mukherjee P. S., Chem. Eur. J. 2015, 21, 6823–6831. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Povarov L. S., Russ. Chem. Rev. 1967, 36, 656–670; [Google Scholar]
- 19b. Kouznetsov V. V., Tetrahedron 2009, 65, 2721–2750; [Google Scholar]
- 19c. Park Y. S., Dibble D. J., Kim J., Lopez R. C., Vargas E., Gorodetsky A. A., Angew. Chem. Int. Ed. 2016, 55, 3352–3355; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3413–3416. [Google Scholar]
- 20. Li X., Zhang C., Cai S., Lei X., Altoe V., Hong F., Urban J. J., Ciston J., Chan E. M., Liu Y., Nat. Commun. 2018, 9, 2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Claridge T. D. W. in High-Resolution NMR Techniques in Organic Chemistry (Third Edition) (Ed.: T. D. W. Claridge), Elsevier, Boston, 2016, pp. 61–132. [Google Scholar]
- 22. Spek A., Acta Crystallogr. Sect. D 2009, 65, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thommes M., Kaneko K., Neimark Alexander V., Olivier James P., Rodriguez-Reinoso F., Rouquerol J., Sing Kenneth S. W., Pure Appl. Chem. 2015, 87, 1051. [Google Scholar]
- 24. Neimark A. V., Lin Y., Ravikovitch P. I., Thommes M., Carbon 2009, 47, 1617–1628. [Google Scholar]
- 25. Elbert S. M., Wagner P., Kanagasundaram T., Rominger F., Mastalerz M., Chem. Eur. J. 2017, 23, 935–945. [DOI] [PubMed] [Google Scholar]
- 26. Jhulki S., Evans A. M., Hao X.-L., Cooper M. W., Feriante C. H., Leisen J., Li H., Lam D., Hersam M. C., Barlow S., Brédas J.-L., Dichtel W. R., Marder S. R., J. Am. Chem. Soc. 2020, 142, 783–791. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Ascherl L., Evans E. W., Gorman J., Orsborne S., Bessinger D., Bein T., Friend R. H., Auras F., J. Am. Chem. Soc. 2019, 141, 15693–15699; [DOI] [PubMed] [Google Scholar]
- 27b. Kulkarni R., Noda Y., Kumar Barange D., Kochergin Y. S., Lyu P., Balcarova B., Nachtigall P., Bojdys M. J., Nat. Commun. 2019, 10, 3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Brutschy M., Schneider M. W., Mastalerz M., Waldvogel S. R., Adv. Mater. 2012, 24, 6049–6052; [DOI] [PubMed] [Google Scholar]
- 28b. Brutschy M., Schneider M. W., Mastalerz M., Waldvogel S. R., Chem. Commun. 2013, 49, 8398–8400; [DOI] [PubMed] [Google Scholar]
- 28c. Hasell T., Zhang H., Cooper A. I., Adv. Mater. 2012, 24, 5732–5737; [DOI] [PubMed] [Google Scholar]
- 28d. Jiang S., Song Q., Massey A., Chong S. Y., Chen L., Sun S., Hasell T., Raval R., Sivaniah E., Cheetham A. K., Cooper A. I., Angew. Chem. Int. Ed. 2017, 56, 9391–9395; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9519–9523. [Google Scholar]
- 29. Schick T. H. G., Lauer J. C., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2019, 58, 1768–1773; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1782–1787. [Google Scholar]
- 30.CCDC 2002767 (2) and 2002766 (3) contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary