

Abstract
By secreting insulin and glucagon, the β‐ and α‐cells of the pancreatic islets play a central role in the regulation of systemic metabolism. Both cells are equipped with ATP‐regulated potassium (KATP) channels that are regulated by the intracellular ATP/ADP ratio. In β‐cells, KATP channels are active at low (non‐insulin‐releasing) glucose concentrations. An increase in glucose leads to KATP channel closure, membrane depolarization and electrical activity that culminates in elevation of [Ca2+]i and initiation of exocytosis of the insulin‐containing secretory granules. The α‐cells are also equipped with KATP channels but they are under strong tonic inhibition at low glucose, explaining why α‐cells are electrically active under hypoglycaemic conditions and generate large Na+‐ and Ca2+‐dependent action potentials. Closure of residual KATP channel activity leads to membrane depolarization and an increase in action potential firing but this stimulation of electrical activity is associated with inhibition rather than acceleration of glucagon secretion. This paradox arises because membrane depolarization reduces the amplitude of the action potentials by voltage‐dependent inactivation of the Na+ channels involved in action potential generation. Exocytosis in α‐cells is tightly linked to the opening of voltage‐gated P/Q‐type Ca2+ channels, the activation of which is steeply voltage‐dependent. Accordingly, the inhibitory effect of the reduced action potential amplitude exceeds the stimulatory effect resulting from the increased action potential frequency. These observations highlight a previously unrecognised role of the action potential amplitude as a key regulator of pancreatic islet hormone secretion.

Keywords: diabetes, glucagon, insulin, KATP channels, membrane potential
Abstract figure legend The confocal images of a mouse pancreatic islet with β‐cells in red (top left), α‐cells in green (middle left) and merged image (bottom left). The images were taken at the top of an islet using an upright confocal microscope, which is why a clear separation of the β‐ and α‐cells is not as evident as in the centre of the islet. Panels to the right show the effects of KATP channel closure on electrical activity in β‐ (top right) and α‐cells (middle right). The schematic (lower right) summarizes the relationship between KATP channel closure/electrical activity on insulin and glucagon secretion. A decrease in KATP channel activity leads to a biphasic stimulation of insulin secretion (from a low basal value) but inhibition of glucagon secretion (from a high basal rate) despite similar effects on electrical activity in α‐ and β‐cells. This Topical Review discusses this paradox.
Introduction
In most cells, potassium (K+) channel activity constitutes the principal background membrane conductance, which explains why the resting membrane potential usually approximates the K+ equilibrium potential (EK; ∼‐70 mV). In general, an increase in K+ channel activity lowers electrical excitability by clamping the membrane potential at EK and reducing the input resistance; both factors making it more difficult for the cell to generate action potentials. Conversely, a decrease in K+ channel activity increases electrical excitability by producing membrane depolarization and increasing the membrane resistance. Under the latter conditions, the opening of individual Na+ or Ca2+ channels may suffice for the cell to depolarize to the threshold for action potential firing (Fenwick et al. 1982).
ATP‐sensitive potassium (KATP) channels are expressed in a number of cells including cardiomyocytes, skeletal muscle, smooth muscle cells and certain neurones (Huang et al. 2019). However, their role is particularly evident in the β‐cells of the pancreatic islets (Ashcroft, 2007). The pancreatic islets represent the endocrine part of the pancreas and make up about 1% of its mass, corresponding to ∼1 g in humans (Rorsman & Ashcroft, 2018). The islets are micro‐organs consisting of (on average) ∼200 endocrine cells: insulin‐producing β‐cells (75% of islet cells) and glucagon‐producing α‐cells (15–20%). In addition, there are a small number (5%) of somatostatin‐releasing δ‐cells. Insulin and glucagon are the body's principal glucose‐regulating hormones (Frayn & Evans, 2019). They are released in response to increases (hyperglycaemia) and decreases (hypoglycaemia) in plasma glucose concentrations, respectively. Collectively they ensure that plasma glucose is maintained at ∼5 mm in humans.
In β‐cells, an increase in plasma glucose (for example, following ingestion of a carbohydrate‐rich meal) stimulates insulin secretion by closing KATP channels (Rorsman & Ashcroft, 2018). Intriguingly, KATP channels are also found in the glucagon‐secreting α‐cells (Bokvist et al. 1999). Given the reciprocal regulation of insulin and glucagon secretion by glucose, the question arises as to how KATP channel closure stimulates secretion in β‐cells but inhibits it in α‐cells. In this Topical Review we consider this conundrum.
KATP channels
KATP channels are octameric heterocomplexes of four KIR6.2 (encoded by Kcnj11) and four SUR1 (encoded by Abcc8) subunits (Seino, 1999). They are regulated by variations of the intracellular ATP concentrations: they are active in the absence of ATP and are inhibited by increasing concentrations of ATP with an IC50 of ∼10μm in both α‐ and β‐cells (Cook & Hales, 1984; Bokvist et al. 1999). KATP channels closed by ATP can be reactivated by Mg‐ADP (Dunne & Petersen, 1986; Kakei et al. 1986) and in the intact cell the channel activity is therefore believed to be regulated by the submembrane ATP/ADP ratio.
The KATP channel is the molecular target of the hypoglycaemic sulphonylureas (SUs) (Trube et al. 1986), compounds that have been used to treat type‐2 diabetes (T2D) since the early 1950s. Tolbutamide is an example of the first generation SUs and blocks the KATP channels with an IC50 of 7 μm; second generation SUs (like glibenclamide) inhibit channel activity at nanomolar concentrations. Conversely, the KATP channels are activated by K+ channel openers such as diazoxide, which activates the channel with an EC50 of 20–30 μm in β‐ and α‐cells (Zunkler et al. 1988; Zhang et al. 2013).
Because of its central role in the control of systemic metabolism, the β‐cell can be regarded as the archetypal metabolically regulated cell. We will therefore start by summarizing the role of the KATP channel in insulin secretion. We will then consider the more controversial role of KATP channels in the glucagon‐secreting α‐cell against the backdrop of the β‐cell. There are several excellent reviews on the structure and function of the KATP channels and the respective roles of the two subunits in its regulation (Seino, 1999; Nichols, 2006; McTaggart et al. 2010). These aspects will therefore not be reviewed here.
KATP channels in metabolic sensing: the β‐cell
β‐cell electrical activity is tightly correlated with insulin secretion (Rorsman & Ashcroft, 2018). At low (1 mm) glucose (when insulin secretion is suppressed), the β‐cell is hyperpolarised (‐70 mV) and electrically silent. Increasing glucose to concentrations ≥6 mm (i.e. close to the normal plasma glucose levels) induces membrane depolarization. When the membrane potential reaches a threshold of ∼‐50 mV, the β‐cells start generating oscillatory electrical activity consisting of bursts of action potentials that originate from depolarized plateaux separated by electrically silent repolarized intervals. The bursts of action potentials leads to increases in [Ca2+]i that trigger exocytosis of the insulin‐containing secretory granules by mechanisms similar to those involved in neurotransmitter release (Fig. 1A and C ) (Rorsman & Ashcroft, 2018). As in neurones, exocytosis of the insulin‐containing granules appears to be determined by the local [Ca2+]i in close proximity to the inner mouth of the Ca2+ channels (Pertusa et al. 1999; Barg et al. 2001).
Figure 1. KATP channels in β‐cells.
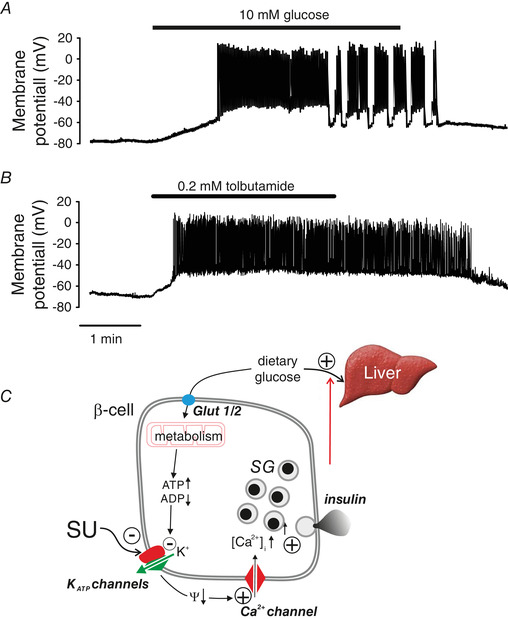
A, electrical activity in β‐cells evoked by an increase in plasma glucose from 1 to 10 mm. B, as in A but tolbutamide (0.2 mm) applied instead of glucose. C, schematic of consensus model of glucose‐induced insulin secretion in β‐cells. Glut1/2, glucose transporters 1 and 2 (human and mouse, respectively); KATP channels, ATP‐sensitive K+ channels; Ψ, membrane potential; SG, secretory granules. The + and ‐ signs denote stimulation and inhibition, respectively, whereas the arrows (↑,↓) indicate an increase or decrease of the indicated parameter. The red arrow indicates feedback regulation of insulin secretion via changes in plasma glucose due to (for example) hepatic glucose disposition.
In β‐cells, the glucose‐induced membrane depolarization and associated initiation of action potential firing are mediated by changes in KATP channel activity. Under hypoglycaemic conditions, KATP channel activity is high because of a low cytoplasmic ATP/ADP ratio and the combination of a negative membrane potential and low membrane resistance prevents electrical activity. When plasma glucose rises, accelerated glucose uptake into the β‐cells and the associated stimulation of metabolism increase the cytoplasmic ATP/ADP ratio and thereby causes KATP channel closure.
When KATP channel activity is reduced, the β‐cells depolarize because the residual K+ permeability is no longer sufficient to counteract the depolarizing influence of other membrane conductances that were previously too small to affect the membrane potential. The identity of the depolarizing background membrane conductance remains to be unequivocally identified; candidates include TRPM2 or Piezo1 (Yosida et al. 2014; Deivasikamani et al. 2019) and/or Cl−‐permeable LRRC8 channels (also known as SWELL1 or VRAC) (Stuhlmann et al. 2018).
As reviewed previously (Rorsman & Ashcroft, 2018), β‐cell electrical activity reflects the activation of voltage‐gated Ca2+ and K+ channels, which underlie the up‐ and downstroke of the action potential, respectively. In mouse β‐cells, the Ca2+ channels are L‐ and R‐type whereas in human β‐cells, L‐ and P/Q‐type Ca2+ channels predominate. Voltage‐gated Na+ channels also contribute but mainly at glucose concentrations just above the threshold for insulin secretion (Braun et al. 2008). However, in mouse β‐cells the Na+ channels exhibit an unusual voltage dependence of inactivation and in most cells they are completely inactivated at the normal resting potential of ‐70 mV (Zhang et al. 2014; Godazgar et al. 2018).
The repolarization of the action potential results from the opening of Kv2.1 (mouse) or Kv2.2 (human) and large‐conductance Ca2+‐activated K+ channels (Jacobson et al. 2007; Braun et al. 2008; Houamed et al. 2010). Pharmacological inhibition of the voltage‐gated K+ channels amplifies glucose‐induced insulin secretion by increasing the duration and amplitude of the action potentials (Rorsman et al. 2011).
The oscillatory electrical activity seen at the intermediate glucose concentration and its conversion into uninterrupted action potential reflects the complex cross‐talk between Ca2+ entry, intracellular Ca2+ ATPase activity and Ca2+‐induced (via changes in the ATP/ADP ratio) of KATP channel activity (Kanno et al. 2002).
Like glucose, SUs promptly and reversibly depolarize the β‐cell and initiate action potential firing in β‐cells (Fig. 1B ), which accounts for their insulin‐releasing capacity. When applied at intermediate glucose concentrations (10 mm), they also convert oscillatory electrical activity into continuous action potential firing (Henquin, 1988; Kanno et al. 2002), in keeping with the idea that electrical activity transiently reactivates KATP channels until the intracellular ATP/ADP ratio has been restored (Tarasov et al. 2012). Conversely, addition of the KATP channel activator diazoxide suppresses glucose‐induced electrical activity in β‐cells (Henquin & Meissner, 1982). Two important conclusions can be drawn from these observations: First, KATP channel closure is involved in both the initiation and modulation of β‐cell electrical activity. Second, there must be sufficient depolarizing background in β‐cells even at low glucose to account for the rapid initiation of electrical activity upon addition of SUs.
Following its release into the bloodstream, secreted insulin acts on the target organs (chiefly the liver) where it promotes glucose storage until normal blood glucose is restored. When this has occurred, insulin secretion stops by the reversal of the process described above. Thus, insulin secretion is under feedback control via changes in plasma glucose (Fig. 1C ).
Variations on the KATP channel theme: the α‐cell
As mentioned above, the glucagon‐producing α‐cells are also equipped with KATP channels. At the molecular level, they are identical to those in β‐cells and they are similarly influenced by ATP and ADP (Bokvist et al. 1999). Unlike β‐cells, α‐cells are electrically active at low (1 mm), or even the complete absence of, glucose (Gopel et al. 2000). A role for the KATP channels in α‐cells may seem counterintuitive given that glucose reciprocally regulates insulin and glucagon secretion. However, studies in KATP channel knockout mice firmly establish their role in glucagon secretion. In islets from such mice, glucagon secretion is strongly inhibited at low glucose, and high glucose and tolbutamide have (unlike what is seen in control islets) little or no further glucagonostatic effect (Gromada et al. 2004; Munoz et al. 2005; Shiota et al. 2005; MacDonald et al. 2007; Cheng‐Xue et al. 2013). We acknowledge that glucagon secretion may also be under paracrine regulation by insulin (Unger & Orci, 2010) or somatostatin (Hauge‐Evans et al. 2009). Indeed, it is possible that the small inhibition that persists in KATP channel‐deficient islets (10% of that in control islets) (Cheng‐Xue et al. 2013) reflects such paracrine actions. However, whilst somatostatin may contribute to the glucagonostatic effect at high glucose concentrations that are associated with stimulation of somatostatin secretion, its role at low glucose (when somatostatin release is low) must be limited (Zhang et al. 2007; Briant et al. 2017; Lai et al. 2018; Kellard et al. 2020). Here we will therefore focus on the alternative possibility that the glucagonostatic actions of glucose and tolbutamide reflect direct effects within the α‐cell itself and that they are mediated by inhibition of KATP channels.
KATP channels in α‐cells vs. β−cells: quantitative aspects
Glucagon secretion is traditionally measured in isolated intact pancreatic islets. Many of the earlier studies that suggest little role for the KATP channels in α‐cells were conducted on isolated cells maintained in tissue culture (Barg et al. 2000; Quoix et al. 2009), which may not be fully representative of the situation in vivo. When KATP channel activity is instead measured in α‐cells in acutely isolated intact islets, the resting membrane conductance was found to be 270 pS at 1 mm glucose and reduced to ∼200 pS after application of tolbutamide or increasing glucose to 6 mm (the concentration producing maximal inhibition of glucagon secretion) (Zhang et al. 2013). The corresponding values in β‐cells are 4 nS and 1 nS (Gopel et al. 1999). Thus, the net whole‐cell KATP conductance in α‐cells is only 70 pS at 1 mm glucose (∼2% of the 3 nS in β‐cells). It is likely that most of the residual KATP‐independent (tolbutamide‐resistant) membrane conductance largely reflects ‘leak’ across the lipid membrane itself and around the recording electrode as it is not affected by any experimental manipulation and the giga‐seal typically has a resistance of 5–10 GΩ (i.e. 100–200 pS) (Hamill et al. 1981).
Strong tonic inhibition of KATP channels in α‐cells at low glucose
The low resting conductance of α‐cells explains why they are electrically active at low glucose. Importantly, it is not simply a consequence of a low expression of the KATP channel in α‐cells. The KATP channel density can be estimated from the increase in K+ conductance following the wash‐out of intracellular ATP. Such experiments reveal that the KATP channel density (normalised to membrane area) is, if anything, considerably higher in α‐than in β‐cells (Bokvist et al. 1999), in agreement with gene expression data (DiGruccio et al. 2016). This suggests that the KATP channels must be under strong tonic inhibition even in α‐cells exposed to low glucose. Exactly how this occurs remains to be elucidated but it may reflect effective metabolism of fatty acids in α‐cells under hypoglycaemic conditions (Briant et al. 2018). Carnitine palmitoyltransferase 1 (CPT1) is a mitochondrial transmembrane enzyme responsible for the formation of acyl‐carnitine from long‐chain acyl‐coenzyme A. This enzyme is considered rate‐limiting for β‐oxidation of long‐chain fatty acids. In mice that lack CPT1 in the α‐cells, glucagon secretion at 1 mm glucose is reduced by 40%. Thus, there is a redundancy of ATP‐producing mechanisms in α‐cells. It is therefore of interest that α‐cells (unlike β‐ and δ‐cells) also express the low‐Km hexokinase‐1 (Hk1) in addition to glucokinase (Gck; a high‐Km hexokinase). Genetic ablation of glucokinase in α‐cells has no effect on α‐cell electrical activity and glucagon secretion at low (1 mm) glucose but abolishes the glucagonostatic effect of high (6 and 20 mm) glucose (Basco et al. 2018). However, the impact of genetically ablating hexokinase‐1 in α‐cells has not yet been investigated.
Although α‐cell KATP channel activity in the absence of glucose is very low, it is greater than zero; the observed net membrane conductance of 70 pS is equivalent to an average of five KATP channels being active in the entire cell, suggesting that the open probability of the α‐cell's >1000 KATP channels (Bokvist et al. 1999) is <0.5%. However, even a KATP channel activity as low as this is sufficient to keep the α‐cell membrane potential partially repolarized (∼‐55 mV) as witnessed by the 10–15 mV depolarization observed when the channels are inhibited by tolbutamide (Fig. 2A ) or high glucose (Zhang et al. 2013; Babinsky et al. 2017). As in β‐cells, this depolarization increases action potential firing in α‐cells but this is associated with a suppression rather than stimulation of glucagon secretion. Below we discuss how this paradox arises.
Figure 2. KATP channels in α‐cells.
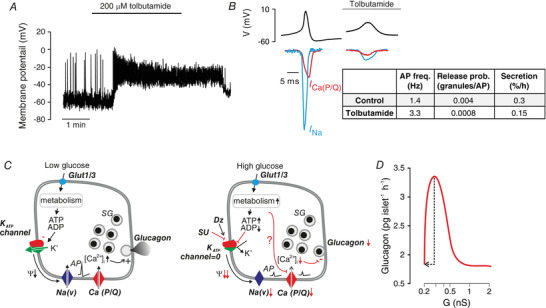
A, effects of tolbutamide (200 μm) on action potential firing in an α‐cell within a mouse islet exposed to 1 mm glucose. Note that the α‐cell is electrically active and that tolbutamide leads to membrane depolarization and increases action potential firing. B, schematic of α‐cell action potentials at 1 mm glucose in the absence and presence of tolbutamide; membrane potential (V) shown in top trace (black) and membrane currents flowing through voltage‐gated Na+ channels (I Na; blue) and P/Q‐type Ca2+ channels (I Ca(P/Q); red) are indicated. The table (inset, right) summarizes the action potential (AP) frequency (freq.; taken from Zhang et al. (2013)), release probability (prob.) and fractional glucagon release (% of content/h; estimated from reported glucagon secretion rates and islet glucagon contents (Knudsen et al. 2019)) and converted to number of granules released based on each α‐cell containing 7000 granules (Barg et al. 2000). C, schematic of effects of glucose and tolbutamide on electrical activity and glucagon secretion in α‐cells at low glucose (left) and at high glucose or in the presence of a high dose of SU (right). At low glucose, KATP channel activity is low but >0 and this keeps the membrane potential (Ψ) of the α‐cell sufficiently depolarized to allow AP firing whilst preventing voltage‐dependent inactivation of the voltage‐gated Na+ channels (Na[V]). The large‐amplitude action potentials activate the P/Q‐type Ca2+ channels (Ca[P/Q]). The associated increase in [Ca2+]i triggers exocytosis of glucagon‐containing secretory granules (SGs). When glucose is elevated, increased glucose uptake via Glut1 or 3 and the associated increase in ATP/ADPi, KATP channel activity fall to zeros, resulting in strong membrane depolarization, inactivation of the Na(V) channels, reduced AP amplitude, less Ca2+ entry via Ca(P/Q) activation and a fall in [Ca2+]i, culminating in suppression of glucagon secretion. It is also possible that a glucose metabolite directly modulates Ca(P/Q) and thereby reduces Ca2+ entry and glucagon secretion (indicted by ‘?’). Diazoxide may reverse the glucagonostatic effect of high glucose by activation of KATP channels (black arrow) when used at an appropriate concentration leading to an increase in channel activity comparable to the decrease produced by glucose. D, relationship between whole‐cell KATP channel conductance (G) and glucagon secretion. The black arrow indicates changes in KATP channel activity and the resulting change in glucagon secretion when glucose is increased from 1 to 6 mm (arrow). The red curve was estimated by parallel measurements of glucagon secretion at 6 mm glucose in the presence of increasing concentrations of diazoxide (1–100 μm). Data in panel D from Zhang et al. (2013) (redrawn).
Functional consequences of KATP channel closure: role of voltage‐gated Na+ and Ca2+ channels
Glucagon secretion in α‐cells is the product of action potential frequency and the amount of glucagon secreted for each action potential. Normally, each action potential evokes glucagon granule exocytosis, albeit with a probability as low as 0.004 granules/action potential (Fig. 2B ). Following the application of tolbutamide, the release probability is reduced by 80% (to 0.0008 granules/action potential) and although there is a 150% increase in action potential frequency, the combination of these effects produces a net 50% decrease in glucagon secretion (Zhang et al. 2013).
Why is the release probability reduced? The action potentials in α‐cells reflect the activation of voltage‐gated Na+ channels and P/Q‐type Ca2+ channels. Na+ channels (including those in α‐cells) (Zhang et al. 2014) have a dual dependence on membrane potential: depolarization both activates and inactivates the channels. Unlike what is observed in β‐cells where most (if not all) Na+ channels are locked in the inactivated state, Na+ channels in α‐cells exist in an ‘activatable’ state (Zhang et al. 2014). The α‐cells express Nav1.3 Na+ channels whereas Nav1.7 predominates in β‐cells but this difference is only part of the reason why the Na+ currents in the α‐cells inactivate at 50 mV more depolarized membrane potentials than in β‐cells (midpoint of inactivation ‐50 mV rather than ‐100 mV); most of the discrepancy is instead attributable to differences in the Na+ channels’ lipid environment in the α‐ and β‐cell plasma membranes (Godazgar et al. 2018). The central role of Na+ channels in the glucagon secretion at low glucose is underscored by the strong inhibition of glucagon secretion produced by the Na+ channel blocker tetrodotoxin (TTX) (Zhang et al. 2013, 2014).
Because of Na+ channel inactivation, the peak voltage of the action potentials declines from +10 mV when the membrane potential is ‐55 mV (or more negative) to ‐10 mV when the membrane potential is ‐45 mV (or more positive). This change in peak voltage is highly significant because exocytosis in α‐cell is steeply voltage‐dependent, largely reflecting the voltage dependence of the P/Q‐type Ca2+ channels that mediate the Ca2+ entry for glucagon secretion at low glucose (Fig. 2B ). The change in action potential amplitude produced by application of tolbutamide can be estimated to reduce glucagon exocytosis by 80% (Zhang et al. 2013), similar to the estimated reduction of glucagon granules’ release probability (see above).
It is implicit from the model that exocytosis of the glucagon granules is regulated by [Ca2+]i close to the inner mouth of the P/Q‐type Ca2+ channels. This is supported by the findings that exocytosis is resistant to intracellular application of millimolar concentrations of the Ca2+ chelator EGTA (Zhang et al. 2013). It remains an open question whether hyperglycaemia inhibits glucagon secretion exclusively by the same (membrane potential‐dependent) mechanism or whether glucose metabolism additionally modulates P/Q‐type Ca2+ channel activity by a more direct inhibitory effect. The latter possibility is suggested by studies on the glucagonostatic effects of the incretin hormone GLP‐1 (De Marinis et al. 2010). Such a direct modulatory effect of glucose would be consistent with the observation that glucose inhibits glucagon secretion evoked by high K+ (70 mm) stimulation in human islets (Ramracheya et al. 2018) (Fig. 2C ), an effect that clearly cannot be accounted for by modulation of action potential height. Whether this also applies to mouse α‐cells and the underlying mechanism have not been elucidated. However, it is clear that the glucose‐induced changes in action potential height are sufficient to account for most of the glucagonostatic effect of high glucose but this does not exclude the contribution of additional mechanisms.
Voltage‐gated K+ channels
Action potential repolarization in α‐cells is mediated by the activation of voltage‐gated K+ currents. Gene expression and electrophysiological data suggest evidence for the presence of Kv2.1/2.2 and Kv11.1/11.2 delayed rectifying and Kv4.1 (mouse) and Kv4.3 (human) A‐type K+ currents. Unlike what is observed in β‐cells, pharmacological inhibition of these channels in α‐cells leads to suppression of glucagon secretion at 1 mm glucose (Gromada et al. 2004; Ramracheya et al. 2010; Spigelman et al. 2010), possibly by resulting in a more depolarized interspike voltage that prevents complete Na+ channel reactivation between the action potentials (Braun & Rorsman, 2010).
Bell‐shaped relationship between KATP channel activity and glucagon secretion: impact of pharmacological activation of KATP channels
If tolbutamide inhibits glucagon secretion, the KATP channel opener diazoxide should have the opposite effect. Indeed, when tested at low concentrations, diazoxide reverses the glucagonostatic effect of high glucose by reversal of the process shown in Fig. 2C . Notably, the concentration of diazoxide that stimulates glucagon secretion must be carefully titrated and depends on the experimental conditions such as the concentration of bovine serum albumin (commonly included in the extracellular medium in glucagon secretion studies). At high concentrations, the repolarizing effect may become so strong that any stimulatory effect on glucagon secretion due to the increase in action potential height might be cancelled out by the inhibition (partial or complete) of action potential firing. Collectively, these considerations suggest that there is a bell‐shaped relationship between KATP channel activity and glucagon secretion (Fig. 2D ). This concept may – as we will elaborate below – have important implications for the understanding of the dysregulation of glucagon secretion associated with diabetes.
Role of [Ca2+]i in glucagon secretion
As discussed above, changes in KATP channel activity modulate action potential firing in α‐cells. Because electrical activity involves the opening of voltage‐gated Ca2+ channels, electrical activity in α‐cells is associated with oscillations in [Ca2+]i (Kellard et al. 2020). Despite the robust inhibitory effects of high glucose on glucagon secretion this associates with surprisingly subtle effects on [Ca2+]i (Nadal et al. 1999; Quoix et al. 2009; Le Marchand & Piston, 2012; Zhang et al. 2013; Li et al. 2015) and although a glucose‐induced reduction of [Ca2+]i is observed in some cells, it continues to oscillate in other cells (Fig. 3A ). A similar variability is observed in the responses to tolbutamide when the compound is tested in intact islets (Fig. 3B ) (Quesada et al. 1999). This has led to the suggestions that glucagon is regulated distal to the elevation of [Ca2+]i by a direct effect on exocytosis (Hughes et al. 2018). Recently, based on imaging of several hundreds of α‐cells, we were able to demonstrate that (despite a great cell‐to‐cell variability) increasing glucose from 1 to 6 mm produced a statistically significant reduction of [Ca2+]i manifested as a reduction of both frequency and amplitude of [Ca2+]i oscillations (Kellard et al. 2020). The effect on the amplitude of the [Ca2+]i oscillations resembles that produced by blocking the subset of Ca2+ channels (P/Q‐type) linked to glucagon secretion at low glucose (De Marinis et al. 2010), in keeping with the idea that the effect of glucose culminates in reduced Ca2+ entry.
Figure 3. Effects of glucose and tolbutamide on [Ca2+]i in α‐cells.
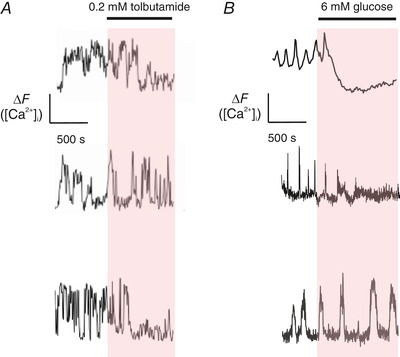
A, effects of application of 0.2 mm tolbutamide in three different α‐cells in intact islets exposed to 1 mm glucose. B, as in A but effects of increasing glucose from 1 to 6 mm. Measurements were made using fluo‐3 in A and the lower trace in B. The other measurements were performed in α‐cells expressing the Ca2+ sensor GCaMP3 under the proglucagon promoter.
In addition to the P/Q‐type Ca2+ channels, α‐cells are also equipped with L‐type Ca2+ channels. Although these channels contribute to action potential firing in α‐cells, blocking them with isradipine or nifedipine does not affect glucagon secretion at 1 mm glucose (MacDonald et al. 2007; De Marinis et al. 2010). However, L‐type Ca2+ channels play a key role in glucagon secretion evoked by adrenaline (and presumably other agents increasing intracellular cAMP) (De Marinis et al. 2010) by promoting intracellular Ca2+ release (Hamilton et al. 2018).
Consistent with a bell‐shaped relationship between KATP channel activity and α‐cell [Ca2+]i/glucagon secretion, diazoxide abolished [Ca2+]i oscillations at low glucose (Le Marchand & Piston, 2012) but was without consistent inhibitory effect or even increased their frequency at high glucose in some cells (Li et al. 2015; Quesada et al. 1999; MacDonald et al. 2007), whereas complete inhibition of the β‐cells was observed. The latter difference may be related to the fact that diazoxide's capacity to activate the KATP channels is diminished by high intracellular ATP concentration (Zunkler et al. 1988), a concept that would be consistent with the strong tonic inhibition of KATP channels in α‐cells.
Comparison of secretory responses in α‐ and β‐cells
Clearly, the mechanism we propose for glucose‐ and tolbutamide‐induced inhibition of glucagon secretion bears a strong resemblance to that which we outline for biphasic insulin secretion (see Abstract illustration). During sustained glucose or SU stimulation of β‐cells, there is a time‐dependent reduction in action potential amplitude as well as frequency because of voltage‐dependent and Ca2+‐dependent inactivation of Ca2+ and Na+ channels (partial or complete, respectively). As in α‐cells, exocytosis is β‐cells is voltage‐dependent (Gopel et al. 2004) and depends on the local Ca2+ concentration at the release sites (Barg et al. 2001). This will reduce the rate of insulin because of two synergistic effects: fewer Ca2+ channels remain to be activated (inactivation) and there is less voltage‐dependent activation of these (reduced action potential amplitude). It has been argued that this (in combination with the depletion of a pool of readily releasable secretory granules) contributes to the termination of first phase insulin secretion and that second phase secretion results from the generation of smaller action potentials (Rorsman & Ashcroft, 2018). Importantly, the rate of insulin release during the second phase remains much higher than the very low rate of release under basal conditions prior to the initiation of electrical activity. As discussed below, this is highly significant for an understanding of why glucose differentially regulates insulin and glucagon secretion.
In α‐cells, the application of tolbutamide and high glucose also leads to a reduction of action potential height. As in β‐cells, this leads to a decrease in exocytosis. However, because α‐cells (unlike β‐cells) are electrically active at low glucose, ‘basal’ glucagon secretion is high and the reduction of action potential height therefore leads to a net inhibition of glucagon secretion (as discussed above) (see Abstract illustration). In conclusion, time‐dependent changes in action potential frequency and amplitude shape the secretory responses in both β‐ and α‐cells.
KATP channels also control glucagon secretion in humans
The model outlined above for the regulation of glucagon secretion is largely based on studies of α‐cells in isolated mouse islets. However, key elements of the model have been confirmed in isolated human pancreatic islets; whereas tolbutamide depolarizes human α‐cells and inhibits glucagon secretion (Ramracheya et al. 2010), low concentrations of diazoxide reverse the glucagonostatic effect of high glucose (MacDonald et al. 2007). Importantly, there are also clinical data to support the model. In healthy individuals, glucagon secretion increases when plasma glucose is lowered to ≤3.6 mm (by administration of exogenous insulin; Banarer et al. (2002)). Parenthetically, the fact that insulin is commonly used to produce hypoglycaemia and stimulate glucagon secretion militates against the prevailing dogma that insulin is a major glucagonostatic factor (Unger & Orci, 2010). This stimulation is abolished in the presence of tolbutamide, suggesting that counter‐regulatory glucagon secretion is mediated by an increase in KATP channel activity. Notably, tolbutamide only affects glucagon secretion at low glucose and it is ineffective under normoglycaemic conditions (Fig. 4A ), presumably because the KATP channels are already (nearly) maximally inhibited under the latter condition. Application of tolbutamide leads to stimulation of the insulin secretion rate (estimated from the increase in plasma C‐peptide, which reflects endogenous insulin release; exogenous insulin will not contain C‐peptide); this effect was strongest under normoglycaemic conditions and in terms of absolute magnitude it was reduced by nearly 50% following induction of hypoglycaemia (Fig. 4B ). Although the data were interpreted in terms of intra‐islet insulin exerting a glucagonostatic effect (Banarer et al. 2002), the findings that KATP channels are present in human α‐cells and control glucagon secretion rather suggest that it reflects a direct effect of the SU on the α‐cells. In agreement with this idea, the KATP channel activator diazoxide also inhibits glucagon secretion evoked by hypoglycaemia without affecting intra‐islet insulin secretion (again estimated from C‐peptide measurements) (Fig. 4C–D ) (Raju & Cryer, 2005). Diazoxide reduced C‐peptide by 50% whilst not affecting glucagon secretion when applied under normoglycaemic conditions. By contrast, when the KATP channel opener was tested after induction of hypoglycaemia there was no effect on endogenous insulin release and yet counter‐regulatory glucagon secretion was reduced. The finding that both KATP channel activators and inhibitors inhibit glucagon secretion becomes understandable from the bell‐shaped relationship documented in vitro (Zhang et al. 2013).
Figure 4. Effects of tolbutamide and diazoxide on glucagon secretion in clinical studies.
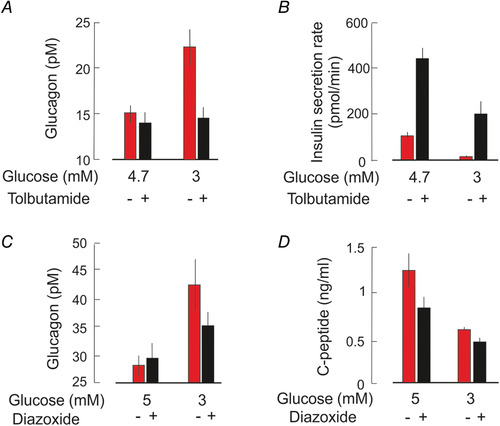
A–B, glucagon (A) and insulin secretion rate (B; estimated from the release of C‐peptide, a proxy for endogenous insulin secretion) measured in the absence and presence of tolbutamide (1 g/h intravenously) under essentially normo‐ (4.7 mm) and hypoglycaemic (3 mm) conditions. Hypoglycaemia induced by administering insulin intravenously (1.5 or 6 pmol kg‐1 min‐1). Data from Banarer et al. (2002) (redrawn). C–D, as in A–B but measured in the absence and presence of diazoxide (6 mg kg−1, orally). Data from Raju & Cryer (2005) (redrawn).
Islet KATP channels and diabetes: pathophysiological and therapeutic implications
There are – broadly speaking – two forms of diabetes: type 1 diabetes (T1D) that has been attributed to the autoimmune destruction of the β‐cells (resulting in a dramatically reduced insulin content) (Brissova et al. 2018) and type‐2 diabetes (T2D) that is caused by insufficient insulin secretion (often but not always combined with insulin resistance) (Ahlqvist et al. 2018). In T2D, the capacity of glucose to evoke insulin secretion is selectively reduced whereas the insulinotropic capacity of other secretagogues like GLP‐1 and arginine are maintained (Ward et al. 1984; Deacon & Holst, 2013). These compounds stimulate insulin secretion by mechanisms that are (partially or wholly) independent of KATP channel closure (Shigeto et al. 2015). This raises the interesting possibility that the impairment of glucose‐induced insulin secretion in T2D is caused by the failure of glucose to inhibit the KATP channel (possibly reflecting impaired mitochondrial ATP production) (Del Guerra et al. 2005). This would account for the therapeutic capacity of the KATP channel‐blocking SUs; by closing the KATP channel they restore action potential firing and insulin secretion in T2D patients. A problem with the SUs is that they, because of their mode of action (inhibiting the KATP channels by a direct/non‐metabolic effect), disable the normal feedback control of β‐cell electrical activity and insulin secretion. Consequently, insulin secretion remains inappropriately stimulated even after normal plasma glucose has been restored or even fallen below the normal range (see Fig. 4B ).
Although it has attracted much less attention, both T1D and T2D are also associated with defective glucagon secretion. The dysregulation is dual: too much glucagon is secreted under hyperglycaemic conditions (when it is not needed) and too little is released in response to hypoglycaemia (when it is needed) (Muller et al. 1970). A similar dysregulation is seen in 50% of human islet preparations from donors diagnosed with T2D (Zhang et al. 2013): in these preparations glucagon secretion at 1 mm glucose is lower than in islets from non‐diabetic donors and increasing glucose stimulates rather than inhibits glucagon release (Fig. 5A ). These defects can be recapitulated in islets from non‐diabetic donors by exposure to low concentrations of diazoxide (Fig. 5B ) or by metabolical poisoning (Zhang et al. 2013), suggesting that the glucagon secretion defects in T2D result from an increase in KATP channel activity due to metabolic derangement in the α‐cell interfering with ATP production (Knudsen et al. 2019). In patients with T1D, glucagon secretion is completely refractory to changes in plasma glucose (Gerich et al. 1973).This may be also due to an increase in α‐cell KATP channel activity. This would explain the observation that a high dose of the SU glimepiride stimulates glucagon secretion in T1D patients whereas it slightly inhibits the release of the hormone in healthy individuals (Cooperberg & Cryer, 2009) (Fig. 5C ). We attribute the weak effect of glimepiride in the healthy controls to the fact that this experiment was performed under normoglycaemic conditions (5 mm).
Figure 5. KATP channels and dysregulation of glucagon secretion in diabetes.
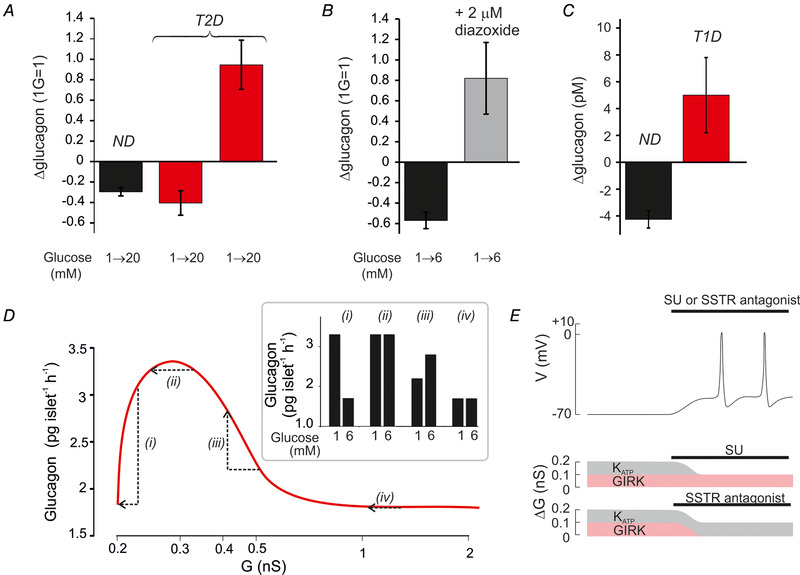
A, relative change in glucagon secretion measured in islets from healthy donors (n = 40) and donors diagnosed with T2D (n = 10) in response to an elevation of glucose from 1 to 20 mm. Responses in T2D islets fell into two groups; one with normal regulation (n = 5) and one with inverted regulation (n = 5). B, glucagon secretion in islets from two healthy organ donors at 1 and 6 mm glucose in the absence and presence of 2 μm diazoxide. C, net change of plasma glucagon measured in healthy individuals and in patients with T1D 4 h after administration of the SU glimepiride (4 mg, orally). Data from Cooperberg & Cryer (2009) (redrawn). D, relationship between total whole‐cell K channel conductance (G) and glucagon secretion (continuous curve) in T2D patients with normal KATP channel regulation (i) and with small (ii), moderate (iii) or large (iv) increases in KATP channel activity. The arrows indicate the changes in glucagon secretion produced by an elevation of glucose from 1 to 6 mm. The bar graphs (Inset) show the predicted effects of increasing glucose with variable degrees of basal KATP channel hyperactivity. E, schematic illustrating the effects of SU or an SSTR antagonist. The α‐cell is initially repolarized (top trace) because resting whole‐cell K+ conductance in excess of background (ΔG) is increased from the optimal 0.1 nS to 0.2 nS (see Fig. 2). This increase may result from either an increase in KATP (grey; due to a metabolic defect in the α‐cells) and/or GIRK (red; due to increased somatostatin release from δ‐cells) channel activity. Inhibition of KATP channels with SU (middle) or GIRK channels with an SSTR antagonist reduces ΔG to the 0.1 nS, which is optimal for α‐cell electrical activity and glucagon secretion.
Based on the bell‐shaped relationship between KATP channel activity and glucagon secretion, we propose that glucagon secretion is suppressed under hypoglycaemic conditions in diabetic patients because KATP channel activity has increased beyond the optimal range (Fig. 5D ). The severity of the metabolic defect will determine the clinical phenotype and range (depending on the increase in KATP channel activity) between nearly normal (i), high rate of glucagon secretion but no effect of glucose (ii), inverted glucose responses (iii) and low rate of glucagon secretion with no effect of glucose at all (iv) as illustrated schematically in Fig. 5D (inset).
These considerations suggest that it may be possible to pharmacologically restore normal glucose regulation of glucagon secretion using SUs. There is evidence from studies in isolated islets from donors diagnosed with T2D that this works in vitro (Zhang et al. 2013). It will be interesting to establish whether this is also the case in T1D islets. However, for SUs to be effective in patients with diabetes and impaired glucagon secretion, they must be carefully dosed to reduce KATP channel activity into the range where glucagon secretion is maximal; the high concentrations normally used for stimulating insulin secretion will probably inhibit glucagon secretion by producing complete inhibition of the KATP channels. In fact, such an effect may contribute to the (unacceptably) high risk of hypoglycaemia associated with SU therapy (Ruan et al. 2020), which is one of the reasons these compounds are no longer favoured in T2D therapy (Khunti et al. 2018). This is because not only do they stimulate the release of insulin under hypoglycaemic conditions (see above), they also abolish the body's counter‐regulatory response.
We point out that the bell‐shaped relationship between K+ channel activity and glucagon secretion is agnostic with regard to the type of K+ channels involved. We have previously proposed that the resting K+ conductance in α‐cells may be a mosaic of different K+ channel activities (Briant et al. 2016). In healthy α‐cells, KATP channels predominate but the situation might be different after the onset of diabetes (Fig. 5E ). Indeed, T2D is associated with hypersecretion of somatostatin at low glucose (Vergari et al. 2020). Somatostatin inhibits glucagon secretion by several mechanisms including the activation of G protein‐coupled inward‐rectifying K+ channels (GIRK) and a direct effect on exocytosis (Rorsman & Huising, 2018). Interestingly, the somatostatin receptor (SSTR) antagonist CYN154806 restores glucagon secretion at 1 mm glucose in diabetic mice and in some islet preparations from donors with T2D (Vergari et al. 2020). In such patients, blocking the KATP channel component, low‐dose SUs might lower the resting K+ conductance sufficiently to stimulate secretion at 1 mm glucose but it would not correct the suppression due to inhibition of exocytosis. This would account for the fact that SUs, whilst restoring normal glucose regulation, fail to normalise glucagon secretion at 1 mm glucose to the level seen in islets from healthy mice/organ donors without T2D (Knudsen et al. 2019). Conversely, an SSTR antagonist would prevent activation of the GIRK channels and reverse the effect on exocytosis but not the metabolic defect leading to increased KATP channel activity. Thus, a combination of SSTR antagonists and SUs may be required to fully restore normal metabolic regulation of glucagon secretion in patients with T1D and T2D.
Yin and Yang of K+ channel activity
In this Topical Review we show – based on observations in pancreatic α‐ and β‐cells – that K+ channel closure has a dual effect on electrical excitability and hormone/neurotransmitter release. Normally, a reduction of K+ channel activity increases electrical excitability by producing membrane depolarization and increasing the membrane resistance (as exemplified by the β‐cell). However, K+ channel activity must not be reduced too much; excessive and maintained membrane depolarization may lead to voltage‐dependent inactivation of the membrane conductances involved in action potential firing, resulting in a paradoxical decrease in electrical excitability (as exemplified by the α‐cell). Under these conditions, (high membrane) resistance is indeed futile. Thus, there is a yin and yang of K+ channel activity and electrical excitability. Although our argument here focuses on the role of KATP channels and the pancreatic islet α‐ and β‐cells, the concept might be equally applicable to other types of K+ channel and other cell types/tissues. For example, in the brain's supra‐chiasmatic nucleus, closure of small conductance Ca2+‐activated K+‐channels causes membrane depolarization and yet suppresses action potential firing (Belle et al. 2009). Our data suggest that the glucagon‐secreting α‐cells are regulated similarly and do not obey conventional electrophysiological expectations.
The way forward
We acknowledge that this Topical Review has a slant towards electrophysiological explanations. However, this does not mean that events beneath the plasma membrane are irrelevant or uninteresting! As we have tried to discuss above, there must be metabolic differences between the β‐ and α‐cells that ensure that ATP is maintained at sufficiently high levels to keep the KATP channels almost fully closed even during protracted hypoglycaemia to ensure continuous release of glucagon but how this occurs is not known. If this mechanism becomes defective in diabetes, it might explain the dysregulation of glucagon secretion associated with the disease and the loss of appropriate counter‐regulatory glucagon secretion in some of the patients.
It is currently unknown whether the glucagon secretion defects associated with diabetes develop in α‐cells independently of the disruption of β‐cell function or whether they are a consequence of diabetes. Data from mouse models of diabetes suggest that the dysregulation of glucagon secretion is secondary to hyperglycaemia‐induced intracellular acidification of the α‐cell that interferes with mitochondrial ATP production that in turn results in KATP channel activation and suppression of glucagon secretion (Knudsen et al. 2019). It will be important to establish whether this also applies to human α‐cells and whether these defects can be reversed by pharmacological interventions. The finding that low concentrations of SUs partially correct the glucagon secretion in diabetic islets in vitro suggests that this is feasible (Zhang et al. 2013) but whether these findings can be translated into improved clinical management remains to be established in clinical trials. Importantly, the SUs must be carefully dosed to reduce KATP channel activity by ∼50% (IC50) as higher concentrations inhibit glucagon secretion (Zhang et al. 2013). A stable plasma concentration around the IC50 is difficult to achieve therapeutically with currently available SUs and small changes in the concentration will have large effects on KATP channel activity/glucagon secretion. Successful translation into the clinic may therefore require a KATP channel blocker with a mode of action distinct from that of the currently used lipophilic SUs that reach the binding site following solvation in the plasma membrane (Zunkler et al. 1989).
Finally, impaired counter‐regulatory glucagon secretion with the risk of severe (potentially fatal) hypoglycaemia represents a barrier towards optimal glycaemic control. The only effective treatment available for recurrent life‐threatening hypoglycaemia is islet or pancreas transplantation but the need for lifelong immunosuppression makes this a last resort (Harlan, 2016). Importantly, not all patients with diabetes experience severe hypoglycaemia and 80% of patients with T2D and 60% of patients with T1D seem protected even after 5 years of insulin therapy (UK Hypoglycaemia Study(Group, 2007). Why hypoglycaemia only affects some patients is not known but it is tempting to speculate that this subgroup is genetically predisposed. Whilst there has been much progress in the understanding of the genetic basis of the insulin secretion defects in T2D (Mahajan et al. 2018), hardly anything is known about the genetics of the glucagon secretion defects. With the advent of human iPSC‐derived glucose‐responsive α‐cells with electrophysiological properties that closely resemble those of primary human α‐cells (Peterson et al. 2020), it might be possible to compare glucagon secretion in response to hypoglycaemia and its relationship to electrical activity in α‐cells derived from diabetes patients with good glycaemic control and those with an elevated risk of hypoglycaemia. This represents an exciting – but challenging – area of future research into the dysregulation of glucagon secretion in diabetes.
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
QZ and HD researched the data. PR drafted the manuscript, which was edited and approved by all authors.
Funding
Supported by Diabetes UK (QZ), the Swedish Research Council (PR) and the Helmsley Trust (PR). Concepts presented in this Topical Review are based on studies supported by the Wellcome Trust.
Biographies
Quan Zhang obtained his BSc at Shandong University and his PhD at the University of Lund. He currently holds a university research lectureship at the University of Oxford (funded by an RD Lawrence Fellowship from Diabetes UK).

Haiqiang Dou obtained his BSc and PhD at Peking University. He currently holds a tenured research position at the University of Gothenburg in Sweden.
Patrik Rorsman is Professor of Diabetic Medicine at the University of Oxford and a Distinguished Investigator at the University of Gothenburg.
Edited by: Ian Forsythe & Ruth Murrell‐Lagnado
This is an Editor's Choice article from the 1 November 2020 issue.
References
- Ahlqvist E, Storm P, Karajamaki A, Martinell M, Dorkhan M, Carlsson A, Vikman P, Prasad RB, Aly DM, Almgren P, Wessman Y, Shaat N, Spegel P, Mulder H, Lindholm E, Melander O, Hansson O, Malmqvist U, Lernmark A, Lahti K, Forsen T, Tuomi T, Rosengren AH & Groop L (2018). Novel subgroups of adult‐onset diabetes and their association with outcomes: a data‐driven cluster analysis of six variables. Lancet Diabetes Endocrinol 6, 361–369. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM (2007). The Walter B. Cannon physiology in perspective lecture, 2007. ATP‐sensitive K+ channels and disease: from molecule to malady. Am J Physiol Endocrinol Metab 293, E880‐889. [DOI] [PubMed] [Google Scholar]
- Babinsky VN, Hannan FM, Ramracheya RD, Zhang Q, Nesbit MA, Hugill A, Bentley L, Hough TA, Joynson E, Stewart M, Aggarwal A, Prinz‐Wohlgenannt M, Gorvin CM, Kallay E, Wells S, Cox RD, Richards D, Rorsman P & Thakker RV (2017). Mutant Mice With Calcium‐Sensing Receptor Activation Have Hyperglycemia That Is Rectified by Calcilytic Therapy. Endocrinology 158, 2486–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banarer S, McGregor VP & Cryer PE (2002). Intraislet hyperinsulinemia prevents the glucagon response to hypoglycemia despite an intact autonomic response. Diabetes 51, 958–965. [DOI] [PubMed] [Google Scholar]
- Barg S, Galvanovskis J, Gopel SO, Rorsman P & Eliasson L (2000). Tight coupling between electrical activity and exocytosis in mouse glucagon‐secreting alpha‐cells. Diabetes 49, 1500–1510. [DOI] [PubMed] [Google Scholar]
- Barg S, Ma X, Eliasson L, Galvanovskis J, Gopel SO, Obermuller S, Platzer J, Renstrom E, Trus M, Atlas D, Striessnig J & Rorsman P (2001). Fast exocytosis with few Ca(2+) channels in insulin‐secreting mouse pancreatic B cells. Biophys J 81, 3308–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basco D, Zhang Q, Salehi A, Tarasov A, Dolci W, Herrera P, Spiliotis I, Berney X, Tarussio D, Rorsman P & Thorens B (2018). alpha‐cell glucokinase suppresses glucose‐regulated glucagon secretion. Nat Commun 9, 546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belle MD, Diekman CO, Forger DB & Piggins HD (2009). Daily electrical silencing in the mammalian circadian clock. Science 326, 281–284. [DOI] [PubMed] [Google Scholar]
- Bokvist K, Olsen HL, Hoy M, Gotfredsen CF, Holmes WF, Buschard K, Rorsman P & Gromada J (1999). Characterisation of sulphonylurea and ATP‐regulated K+ channels in rat pancreatic A‐cells. Pflugers Arch 438, 428–436. [DOI] [PubMed] [Google Scholar]
- Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR & Rorsman P (2008). Voltage‐gated ion channels in human pancreatic beta‐cells: electrophysiological characterization and role in insulin secretion. Diabetes 57, 1618–1628. [DOI] [PubMed] [Google Scholar]
- Braun M & Rorsman P (2010). The glucagon‐producing alpha cell: an electrophysiologically exceptional cell. Diabetologia 53, 1827–1830. [DOI] [PubMed] [Google Scholar]
- Briant L, Salehi A, Vergari E, Zhang Q & Rorsman P (2016). Glucagon secretion from pancreatic alpha‐cells. Ups J Med Sci 121, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briant LJ, Zhang Q, Vergari E, Kellard JA, Rodriguez B, Ashcroft FM & Rorsman P (2017). Functional identification of islet cell types by electrophysiological fingerprinting. J R Soc Interface 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briant LJB, Dodd MS, Chibalina MV, Rorsman NJG, Johnson PRV, Carmeliet P, Rorsman P & Knudsen JG (2018). CPT1a‐dependent long‐chain fatty acid oxidation contributes to maintaining glucagon secretion from pancreatic islets. Cell Rep 23, 3300–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissova M, Haliyur R, Saunders D, Shrestha S, Dai C, Blodgett DM, Bottino R, Campbell‐Thompson M, Aramandla R, Poffenberger G, Lindner J, Pan FC, von Herrath MG, Greiner DL, Shultz LD, Sanyoura M, Philipson LH, Atkinson M, Harlan DM, Levy SE, Prasad N, Stein R & Powers AC (2018). alpha cell function and gene expression are compromised in type 1 diabetes. Cell Rep 22, 2667–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng‐Xue R, Gomez‐Ruiz A, Antoine N, Noel LA, Chae HY, Ravier MA, Chimienti F, Schuit FC & Gilon P (2013). Tolbutamide controls glucagon release from mouse islets differently than glucose: involvement of K(ATP) channels from both alpha‐cells and delta‐cells. Diabetes 62, 1612–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DL & Hales CN (1984). Intracellular ATP directly blocks K+ channels in pancreatic B‐cells. Nature 311, 271–273. [DOI] [PubMed] [Google Scholar]
- Cooperberg BA & Cryer PE (2009). Beta‐cell‐mediated signaling predominates over direct alpha‐cell signaling in the regulation of glucagon secretion in humans. Diabetes Care 32, 2275–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marinis YZ, Salehi A, Ward CE, Zhang Q, Abdulkader F, Bengtsson M, Braha O, Braun M, Ramracheya R, Amisten S, Habib AM, Moritoh Y, Zhang E, Reimann F, Rosengren A, Shibasaki T, Gribble F, Renstrom E, Seino S, Eliasson L & Rorsman P (2010). GLP‐1 inhibits and adrenaline stimulates glucagon release by differential modulation of N‐ and L‐type Ca2+ channel‐dependent exocytosis. Cell Metab 11, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon CF & Holst JJ (2013). Dipeptidyl peptidase‐4 inhibitors for the treatment of type 2 diabetes: comparison, efficacy and safety. Expert Opin Pharmacother 14, 2047–2058. [DOI] [PubMed] [Google Scholar]
- Deivasikamani V, Dhayalan S, Abudushalamu Y, Mughal R, Visnagri A, Cuthbertson K, Scragg JL, Munsey TS, Viswambharan H, Muraki K, Foster R, Sivaprasadarao A, Kearney MT, Beech DJ & Sukumar P (2019). Piezo1 channel activation mimics high glucose as a stimulator of insulin release. Sci Rep 9, 16876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, Torri S, Pollera M, Boggi U, Mosca F, Del Prato S & Marchetti P (2005). Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 54, 727–735. [DOI] [PubMed] [Google Scholar]
- DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing‐Zitron C, van der Meulen T & Huising MO (2016). Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol Metab 5, 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne MJ & Petersen OH (1986). Intracellular ADP activates K+ channels that are inhibited by ATP in an insulin‐secreting cell line. FEBS Lett 208, 59–62. [DOI] [PubMed] [Google Scholar]
- Fenwick EM, Marty A & Neher E (1982). A patch‐clamp study of bovine chromaffin cells and of their sensitivity to acetylcholine. J Physiol 331, 577–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frayn KN & Evans R (2019). Human Metabolism: A Regulatory Perspective. Wiley Blackwell's, Oxford. [Google Scholar]
- Gerich JE, Langlois M, Noacco C, Karam JH & Forsham PH (1973). Lack of glucagon response to hypoglycemia in diabetes: evidence for an intrinsic pancreatic alpha cell defect. Science 182, 171–173. [DOI] [PubMed] [Google Scholar]
- Godazgar M, Zhang Q, Chibalina MV & Rorsman P (2018). Biphasic voltage‐dependent inactivation of human NaV 1.3, 1.6 and 1.7 Na(+) channels expressed in rodent insulin‐secreting cells. J Physiol 596, 1601–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopel S, Kanno T, Barg S, Galvanovskis J & Rorsman P (1999). Voltage‐gated and resting membrane currents recorded from B‐cells in intact mouse pancreatic islets. J Physiol 521 Pt 3, 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopel S, Zhang Q, Eliasson L, Ma XS, Galvanovskis J, Kanno T, Salehi A & Rorsman P (2004). Capacitance measurements of exocytosis in mouse pancreatic alpha‐, beta‐ and delta‐cells within intact islets of Langerhans. J Physiol 556, 711–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopel SO, Kanno T, Barg S & Rorsman P (2000). Patch‐clamp characterisation of somatostatin‐secreting ‐cells in intact mouse pancreatic islets. J Physiol 528, 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromada J, Ma XS, Hoy M, Bokvist K, Salehi A, Berggren PO & Rorsman P (2004). ATP‐sensitive K+ channel‐dependent regulation of glucagon release and electrical actiflty by glucose in wild‐type and SUR1(‐/‐) mouse alpha‐cells. Diabetes 53, S181‐S189. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B & Sigworth FJ (1981). Improved patch‐clamp techniques for high‐resolution current recording from cells and cell‐free membrane patches. Pflugers Arch 391, 85–100. [DOI] [PubMed] [Google Scholar]
- Hamilton A, Zhang Q, Salehi A, Willems M, Knudsen JG, Ringgaard AK, Chapman CE, Gonzalez‐Alvarez A, Surdo NC, Zaccolo M, Basco D, Johnson PRV, Ramracheya R, Rutter GA, Galione A, Rorsman P & Tarasov AI (2018). Adrenaline Stimulates Glucagon Secretion by Tpc2‐Dependent Ca(2+) Mobilization From Acidic Stores in Pancreatic alpha‐Cells. Diabetes 67, 1128–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlan DM (2016). Islet Transplantation for Hypoglycemia Unawareness/Severe Hypoglycemia: Caveat Emptor. Diabetes Care 39, 1072–1074. [DOI] [PubMed] [Google Scholar]
- Hauge‐Evans AC, King AJ, Carmignac D, Richardson CC, Robinson IC, Low MJ, Christie MR, Persaud SJ & Jones PM (2009). Somatostatin secreted by islet delta‐cells fulfills multiple roles as a paracrine regulator of islet function. Diabetes 58, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC (1988). ATP‐sensitive K+ channels may control glucose‐induced electrical activity in pancreatic B‐cells. Biochem Biophys Res Commun 156, 769–775. [DOI] [PubMed] [Google Scholar]
- Henquin JC & Meissner HP (1982). Opposite effects of tolbutamide and diazoxide on 86Rb+ fluxes and membrane potential in pancreatic B cells. Biochem Pharmacol 31, 1407–1415. [DOI] [PubMed] [Google Scholar]
- Houamed KM, Sweet IR & Satin LS (2010). BK channels mediate a novel ionic mechanism that regulates glucose‐dependent electrical activity and insulin secretion in mouse pancreatic beta‐cells. J Physiol 588, 3511–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Hu D, Huang C & Nichols CG (2019). Genetic Discovery of ATP‐Sensitive K(+) Channels in Cardiovascular Diseases. Circ Arrhythm Electrophysiol 12, e007322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JW, Ustione A, Lavagnino Z & Piston DW (2018). Regulation of islet glucagon secretion: Beyond calcium. Diabetes Obes Metab 20 Suppl 2, 127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson DA, Kuznetsov A, Lopez JP, Kash S, Ammala CE & Philipson LH (2007). Kv2.1 ablation alters glucose‐induced islet electrical activity, enhancing insulin secretion. Cell Metab 6, 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakei M, Kelly RP, Ashcroft SJ & Ashcroft FM (1986). The ATP‐sensitivity of K+ channels in rat pancreatic B‐cells is modulated by ADP. FEBS Lett 208, 63–66. [DOI] [PubMed] [Google Scholar]
- Kanno T, Rorsman P & Gopel SO (2002). Glucose‐dependent regulation of rhythmic action potential firing in pancreatic beta‐cells by K(ATP)‐channel modulation. J Physiol 545, 501–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellard JA, Rorsman NJG, Hill TG, Armour SL, van der Bunt M, Rorsman P, Knudsen JG & Briant LJB (2020). Reduced somatostatin signalling leads to hypersecretion of glucagon in mice fed a high‐fat diet. Molecular metabolism, 101021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khunti K, Chatterjee S, Gerstein HC, Zoungas S & Davies MJ (2018). Do sulphonylureas still have a place in clinical practice? Lancet Diabetes Endocrinol 6, 821–832. [DOI] [PubMed] [Google Scholar]
- Knudsen JG, Hamilton A, Ramracheya R, Tarasov A, Brereton M, e Haythorne, Chibalina MV, Spegel P, Mulder H, Zhang Q, Ashcroft FM, Adam J & Rorsman P (2019). Dysregulation of glucagon secretion by hyperglycemia‐induced sodium‐dependent reduction of ATP production. Cell Metab 29, 430–442.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai BK, Chae H, Gomez‐Ruiz A, Cheng P, Gallo P, Antoine N, Beauloye C, Jonas JC, Seghers V, Seino S & Gilon P (2018). Somatostatin Is Only Partly Required for the Glucagonostatic Effect of Glucose but Is Necessary for the Glucagonostatic Effect of KATP Channel Blockers. Diabetes 67, 2239–2253. [DOI] [PubMed] [Google Scholar]
- Le Marchand SJ & Piston DW (2012). Glucose decouples intracellular Ca2+ activity from glucagon secretion in mouse pancreatic islet alpha‐cells. PLoS One 7, e47084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yu Q, Ahooghalandari P, Gribble FM, Reimann F, Tengholm A & Gylfe E (2015). Submembrane ATP and Ca2+ kinetics in alpha‐cells: unexpected signaling for glucagon secretion. FASEB J 29, 3379–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald PE, De Marinis YZ, Ramracheya R, Salehi A, Ma X, Johnson PR, Cox R, Eliasson L & Rorsman P (2007). A K ATP channel‐dependent pathway within alpha cells regulates glucagon release from both rodent and human islets of Langerhans. PLoS Biol 5, e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, Cook JP, Schmidt EM, Wuttke M, Sarnowski C, Mägi R, Nano J, Gieger C, Trompet S, Lecoeur C, Preuss MH, Prins BP, Guo X, Bielak LF, Below JE, Bowden DW, Chambers JC, Kim YJ, Ng MCY, Petty LE, Sim X, Zhang W, Bennett AJ, Bork‐Jensen J, Brummett CM, Canouil M, Ec kardt K‐U, Fischer K, Kardia SLR, Kronenberg F, Läll K, Liu C‐T, Locke AE, Luan JA, Ntalla I, Nylander V, Schönherr S, Schurmann C, Yengo L, Bottinger EP, Brandslund I, Christensen C, Dedoussis G, Florez JC, Ford I, Franco OH, Frayling TM, Giedraitis V, Hackinger S, Hattersley AT, Herder C, Ikram MA, Ingelsson M, Jørgensen ME, Jørgensen T, Kriebel J, Kuusisto J, Ligthart S, Lindgren CM, Linneberg A, Lyssenko V, Mamakou V, Meitinger T, Mohlke KL, Morris AD, Nadkarni G, Pankow JS, Peters A, Sattar N, Stančáková A, Strauch K, Taylor KD, Thorand B, Thorleifsson G, Thorsteinsdottir U, Tuomilehto J, Witte DR, Dupuis J, Peyser PA, Zeggini E, Loos RJF, Froguel P, Ingelsson E, Lind L, Groop L, Laakso M, Collins FS, Jukema JW, Palmer CNA, Grallert H, Metspalu A, Dehghan A, Köttgen A, Abecasis GR, Meigs JB, Rotter JI, Marchini J, Pedersen O, Hansen T, Langenberg C, Wareham NJ, Stefansson K, Gloyn AL, Morris AP, Boehnke M & McCarthy MI (2018). Fine‐mapping type 2 diabetes loci to single‐variant resolution using high‐density imputation and islet‐specific epigenome maps. Nat Genet 50, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTaggart JS, Clark RH & Ashcroft FM (2010). The role of the KATP channel in glucose homeostasis in health and disease: more than meets the islet. J Physiol 588, 3201–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA, Faloona GR, Aguilar‐Parada E & Unger RH (1970). Abnormal alpha‐cell function in diabetes. Response to carbohydrate and protein ingestion. N Engl J Med 283, 109–115. [DOI] [PubMed] [Google Scholar]
- Munoz A, Hu M, Hussain K, Bryan J, Aguilar‐Bryan L & Rajan AS (2005). Regulation of glucagon secretion at low glucose concentrations: evidence for adenosine triphosphate‐sensitive potassium channel involvement. Endocrinology 146, 5514–5521. [DOI] [PubMed] [Google Scholar]
- Nadal A, Quesada I & Soria B (1999). Homologous and heterologous asynchronicity between identified alpha‐, beta‐ and delta‐cells within intact islets of Langerhans in the mouse. J Physiol 517(Pt 1), 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG (2006). KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476. [DOI] [PubMed] [Google Scholar]
- Pertusa JA, Sanchez‐Andres JV, Martin F & Soria B (1999). Effects of calcium buffering on glucose‐induced insulin release in mouse pancreatic islets: an approximation to the calcium sensor. J Physiol 520(Pt 2), 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson QP, Veres A, Chen L, Slama MQ, Kenty JHR, Hassoun S, Brown MR, Dou H, Duffy CD, Zhou Q, Matveyenko AV, Tyrberg B, Sorhede‐Winzell M, Rorsman P & Melton DA (2020). A method for the generation of human stem cell‐derived alpha cells. Nat Commun 11, 2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada I, Nadal A & Soria B (1999). Different effects of tolbutamide and diazoxide in alpha, beta‐, and delta‐cells within intact islets of Langerhans. Diabetes 48, 2390–2397. [DOI] [PubMed] [Google Scholar]
- Quoix N, Cheng‐Xue R, Mattart L, Zeinoun Z, Guiot Y, Beauvois MC, Henquin JC & Gilon P (2009). Glucose and pharmacological modulators of ATP‐sensitive K+ channels control [Ca2+]c by different mechanisms in isolated mouse alpha‐cells. Diabetes 58, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju B & Cryer PE (2005). Loss of the decrement in intraislet insulin plausibly explains loss of the glucagon response to hypoglycemia in insulin‐deficient diabetes: documentation of the intraislet insulin hypothesis in humans. Diabetes 54, 757–764. [DOI] [PubMed] [Google Scholar]
- Ramracheya R, Chapman C, Chibalina M, Dou H, Miranda C, Gonzalez A, Moritoh Y, Shigeto M, Zhang Q, Braun M, Clark A, Johnson PR, Rorsman P & Briant LJB (2018). GLP‐1 suppresses glucagon secretion in human pancreatic alpha‐cells by inhibition of P/Q‐type Ca(2+) channels. Physiol Rep 6, e13852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramracheya R, Ward C, Shigeto M, Walker JN, Amisten S, Zhang Q, Johnson PR, Rorsman P & Braun M (2010). Membrane potential‐dependent inactivation of voltage‐gated ion channels in alpha‐cells inhibits glucagon secretion from human islets. Diabetes 59, 2198–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P & Ashcroft FM (2018). Pancreatic beta‐cell electrical activity and insulin secretion: of mice and men. Physiol Rev 98, 117–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, Eliasson L, Kanno T, Zhang Q & Gopel S (2011). Electrophysiology of pancreatic beta‐cells in intact mouse islets of Langerhans. Prog Biophys Mol Biol 107, 224–235. [DOI] [PubMed] [Google Scholar]
- Rorsman P & Huising MO (2018). The somatostatin‐secreting pancreatic delta‐cell in health and disease. Nature Reviews Endocrinology 14, 404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Y, Moysova Z, Tan GD, Lumb A, Davies J & Rea RD (2020). Inpatient hypoglycaemia: understanding who is at risk. Diabetologia 63, 1299–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S (1999). ATP‐sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annu Rev Physiol 61, 337–362. [DOI] [PubMed] [Google Scholar]
- Shigeto M, Ramracheya R, Tarasov AI, Cha CY, Chibalina MV, Hastoy B, Philippaert K, Reinbothe T, Rorsman N, Salehi A, Sones WR, Vergari E, Weston C, Gorelik J, Katsura M, Nikolaev VO, Vennekens R, Zaccolo M, Galione A, Johnson PR, Kaku K, Ladds G & Rorsman P (2015). GLP‐1 stimulates insulin secretion by PKC‐dependent TRPM4 and TRPM5 activation. J Clin Invest 125, 4714–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota C, Rocheleau JV, Shiota M, Piston DW & Magnuson MA (2005). Impaired glucagon secretory responses in mice lacking the type 1 sulfonylurea receptor. Am J Physiol Endocrinol Metab 289, E570‐577. [DOI] [PubMed] [Google Scholar]
- Spigelman AF, Dai X & MacDonald PE (2010). Voltage‐dependent K(+) channels are positive regulators of alpha cell action potential generation and glucagon secretion in mice and humans. Diabetologia 53, 1917–1926. [DOI] [PubMed] [Google Scholar]
- Stuhlmann T, Planells‐Cases R & Jentsch TJ (2018). LRRC8/VRAC anion channels enhance beta‐cell glucose sensing and insulin secretion. Nat Commun 9, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, Sekler I, Rizzuto R & Rutter GA (2012). The mitochondrial Ca2+ uniporter MCU is essential for glucose‐induced ATP increases in pancreatic beta‐cells. PLoS One 7, e39722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trube G, Rorsman P & Ohno‐Shosaku T (1986). Opposite effects of tolbutamide and diazoxide on the ATP‐dependent K+ channel in mouse pancreatic beta‐cells. Pflugers Arch 407, 493–499. [DOI] [PubMed] [Google Scholar]
- UK Hypoglycaemia Study Group (2007). Risk of hypoglycaemia in types 1 and 2 diabetes: effects of treatment modalities and their duration. Diabetologia 50, 1140–1147. [DOI] [PubMed] [Google Scholar]
- Unger RH & Orci L (2010). Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci U S A 107, 16009–16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward WK, Bolgiano DC, McKnight B, Halter JB & Porte D, Jr (1984). Diminished B cell secretory capacity in patients with noninsulin‐dependent diabetes mellitus. J Clin Invest 74, 1318–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergari E, Denwood G, Salehi A, Zhang Q, Adam J, Alrifaiy A, Wernstedt Asterholm I, Benrick A, Chibalina MV, Eliasson L, Guida C, Hill TG, Hamilton A, Ramracheya R, Reimann F, Rorsman NJG, Spilliotis I, Tarasov AI, Walker JN, Rorsman P & Briant LJB (2020). Somatostatin secretion by Na(+)‐dependent Ca(2+)‐induced Ca(2+) release in pancreatic delta‐cells. Nature metabolism 2, 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosida M, Dezaki K, Uchida K, Kodera S, Lam NV, Ito K, Rita RS, Yamada H, Shimomura K, Ishikawa SE, Sugawara H, Kawakami M, Tominaga M, Yada T & Kakei M (2014). Involvement of cAMP/EPAC/TRPM2 activation in glucose‐ and incretin‐induced insulin secretion. Diabetes 63, 3394–3403. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Bengtsson M, Partridge C, Salehi A, Braun M, Cox R, Eliasson L, Johnson PR, Renstrom E, Schneider T, Berggren PO, Gopel S, Ashcroft FM & Rorsman P (2007). R‐type Ca(2+)‐channel‐evoked CICR regulates glucose‐induced somatostatin secretion. Nat Cell Biol 9, 453–460. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Chibalina MV, Bengtsson M, Groschner LN, Ramracheya R, Rorsman NJ, Leiss V, Nassar MA, Welling A, Gribble FM, Reimann F, Hofmann F, Wood JN, Ashcroft FM & Rorsman P (2014). Na+ current properties in islet alpha‐ and beta‐cells reflect cell‐specific Scn3a and Scn9a expression. J Physiol 592, 4677–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Ramracheya R, Lahmann C, Tarasov A, Bengtsson M, Braha O, Braun M, Brereton M, Collins S, Galvanovskis J, Gonzalez A, Groschner LN, Rorsman NJ, Salehi A, Travers ME, Walker JN, Gloyn AL, Gribble F, Johnson PR, Reimann F, Ashcroft FM & Rorsman P (2013). Role of KATP channels in glucose‐regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab 18, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunkler BJ, Lenzen S, Manner K, Panten U & Trube G (1988). Concentration‐dependent effects of tolbutamide, meglitinide, glipizide, glibenclamide and diazoxide on ATP‐regulated K+ currents in pancreatic B‐cells. Naunyn Schmiedebergs Arch Pharmacol 337, 225–230. [DOI] [PubMed] [Google Scholar]
- Zunkler BJ, Trube G & Panten U (1989). How do sulfonylureas approach their receptor in the B‐cell plasma membrane? Naunyn Schmiedebergs Arch Pharmacol 340, 328–332. [DOI] [PubMed] [Google Scholar]