

Abstract
Prior pharmacokinetic (PK) analyses of the antibody‐drug conjugate (ADC) brentuximab vedotin (1.8 mg/kg every 3 weeks) in pediatric patients with relapsed/refractory hematologic malignancies found that patients aged <12 years exhibited decreased ADC area under the curve (AUC) compared with those aged ≥12 years. This population PK (POPPK) analysis used data from pediatric (NCT01492088) and adult (NCT00430846) studies of brentuximab vedotin to quantify body size effects on ADC exposure. Data were collected from 84 patients with a median age of 25.7 years (range, 7.7‐87.3 years), 34 of whom (40.5%) were aged <18 years; median patient weight was 67 kg (range, 21‐154 kg), and median body surface area was 1.8 m2 (range, 0.87‐2.81 m2). ADC PK was described by a linear 3‐compartment model with zero‐order input and first‐order elimination. POPPK modeling indicated that dosing brentuximab vedotin at 1.8 mg/kg every 3 weeks or 1.2 mg/kg every 2 weeks resulted in lower ADC AUC values in small/moderate‐sized pediatric patients (<28 kg and 28‐49 kg, respectively) compared with large pediatric/adult patients (50‐100 kg). Dosing at 71.5 mg/m2 every 3 weeks and 47.7 mg/m2 every 2 weeks was predicted to achieve comparable AUC values across all body weight ranges and a similar AUC to that in the 50‐ to 100‐kg group at the standard doses of 1.8 mg/kg every 3 weeks and 1.2 mg/kg every 2 weeks, respectively. These results have generated a hypothesis to support evaluation of brentuximab vedotin at 48 mg/m2 every 2 weeks in combination with adriamycin, vinblastine, and dacarbazine chemotherapy in an ongoing pediatric trial in frontline Hodgkin lymphoma (NCT02979522).
Keywords: oncology, pediatrics, population pharmacokinetics
Brentuximab vedotin is a CD30‐directed antibody‐drug conjugate (ADC) that comprises a monoclonal human/murine antibody linked to the microtubule‐disrupting agent monomethyl auristatin E (MMAE) via a protease‐cleavable linker. 1 Brentuximab vedotin binds to cell‐surface CD30, following which the ADC‐CD30 complex is internalized and processed into lysosomal vesicles. MMAE is then released into the cytoplasm by proteolytic cleavage, inhibiting microtubule polymerization and inducing cell‐cycle arrest and apoptosis (Figure 1). 1 , 2 , 3 Brentuximab vedotin specifically targets cells that overexpress cell‐surface CD30, such as classical Hodgkin lymphoma (cHL) and systemic anaplastic large‐cell lymphoma (sALCL). 2 , 4
Figure 1.
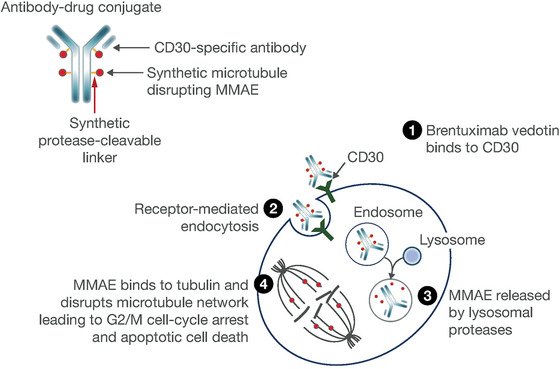
Brentuximab vedotin mechanism of action. Figure reproduced from Suri A, Mould DR, Liu Y, Jang G, Venkatakrishnan K. Population PK and exposure‐response relationships for the antibody‐drug conjugate brentuximab vedotin in CTCL patients in the phase III ALCANZA study. Clin Pharmacol Ther. 2018;104(5)989‐999. ©2018 The Authors. Clinical Pharmacology & Therapeutics published by Wiley Periodicals, Inc. on behalf of the American Society for Clinical Pharmacology and Therapeutics. MMAE, monomethyl auristatin E.
In a phase 2 study (NCT00848926) of patients with relapsed/refractory (R/R) cHL, patients treated with 1.8 mg/kg brentuximab vedotin every 3 weeks 5 achieved an overall response rate of 75% and a median progression‐free survival of 5.6 months, with a manageable safety profile. 5 Based on these results, the United States Food and Drug Administration approved brentuximab vedotin for the treatment of adult patients with R/R cHL after failure of autologous hematopoietic stem‐cell transplant (auto‐HSCT) or at least 2 prior multiagent therapies in adult patients who are ineligible for auto‐HSCT. 6 Based on pivotal studies, brentuximab vedotin has since been approved by the Food and Drug Administration to treat adult patients with 6 cHL who are at high risk of relapse or progression post‐auto‐HSCT as consolidation therapy, 7 primary cutaneous anaplastic large‐cell lymphoma or CD30‐expressing mycosis fungoides following previous systemic therapy, 8 sALCL after failure of at least 1 prior multiagent chemotherapy regimen, 9 previously untreated sALCL or other CD30‐expressing peripheral T‐cell lymphomas in combination with cyclophosphamide, doxorubicin, and prednisone, 10 and previously untreated stage III/IV cHL in combination with doxorubicin, vinblastine, and dacarbazine (AVD). 11
Brentuximab vedotin is not currently approved for use in pediatric patients; however, the potential use in the pediatric setting is supported by data from a phase 1/2 open‐label, single‐agent multicenter dose‐escalation study (NCT01492088), which assessed patients aged 7‐18 years who had R/R cHL or sALCL and for whom standard treatment was not available or was no longer effective. 12 In pediatric patients who received the adult dosage (1.8 mg/kg every 3 weeks), brentuximab vedotin had a manageable safety profile with clinically meaningful responses (overall response rate of 47% in patients with cHL and 53% for those with sALCL). 12
In anticancer drug development, population pharmacokinetics (POPPK) analyses play a crucial part in characterizing the mechanistically and clinically relevant determinants of systemic drug exposure in identifying covariates that influence response and toxicity and in optimizing posology that maximizes benefit versus risk. 13 , 14 , 15 , 16 , 17
Brentuximab vedotin pharmacokinetics (PK) were previously assessed in adult patients with HL, sALCL, or cutaneous T‐cell lymphoma 3 , 8 , 18 , 19 , 20 , 21 , 22 , 23 and in pediatric patients with R/R sALCL or HL (NCT01492088). 12 , 24 ADC concentrations are typically dose‐proportional, with peak concentrations reached at the end of infusion, and with a mean steady‐state volume of distribution of approximately 6‐10 L. 25 MMAE is a substrate of cytochrome P450 3A, 26 and after the initial dose of ADC, brentuximab vedotin‐derived MMAE exposure decreases to 50%‐80% of the original exposure for subsequent cycles. 6 In the pediatric study (NCT01492088), PK evaluation suggested a trend toward lower systemic exposure of ADC in younger pediatric patients compared with older pediatric patients; median area under the concentration‐time curve from time 0 to infinity (AUC0‐∞) of ADC following body weight‐normalized dosing was approximately 14% lower in patients aged <12 years and 3% lower in those aged ≥12 years than the ADC AUC0‐∞ observed in adults. 12
Here we report POPPK analyses using data from 2 clinical studies of brentuximab vedotin: 1 in pediatric patients (NCT01492088) and 1 in adult patients (NCT00430846). Key objectives of the POPPK analyses were to develop a POPPK model to describe ADC and MMAE concentration‐time data in both pediatric and adult patients using data from 2 clinical studies of brentuximab vedotin, to simulate ADC and MMAE pharmacokinetics in pediatric patients aged 6‐9 years from the currently available pediatric data/model, to compare ADC and MMAE in pediatric patients with those in adults, and to evaluate the performance characteristics of body surface area (BSA)‐based dosing scenarios in pediatric patients with respect to their ability to achieve exposures similar to those using adult body weight‐based dosing.
Methods
Clinical Studies
This POPPK analysis used data from 2 studies of brentuximab vedotin: a phase 1/2 study (NCT01492088) in pediatric patients with R/R sALCL or cHL 12 and a phase 1 dose‐escalation study (NCT00430846) in adult patients with R/R CD30‐positive hematologic malignancies. 21
In the pediatric study, patients received 1.4 or 1.8 mg/kg brentuximab vedotin every 3 weeks; the maximum tolerated dose was not reached, and the recommended phase 2 dose was identified as 1.8 mg/kg. 12 In the adult study, brentuximab vedotin was administered intravenously every 3 weeks at doses between 0.1 and 3.6 mg/kg; the maximum tolerated dose was found to be 1.8 mg/kg. 21
Details of sampling schedules in each study are shown in Supplementary Table S1. ADC concentrations were measured using a validated enzyme‐linked immunosorbent assay, and unconjugated MMAE concentration was established using a liquid chromatography‐tandem mass spectrometry assay as described previously. 26
Both studies were approved by the institutional review board or independent ethics committee at each study site and were conducted in accordance with the Declaration of Helsinki and International Conference of Harmonization Guideline for Good Clinical Practice. Written informed consent was provided by all adult patients and by the parents or legal guardians of pediatric patients.
POPPK Model Development
Patients who had at least 1 adequately documented ADC or MMAE concentration were evaluable for analysis.
The structural PK models for ADC and MMAE employed in this analysis were based on previously reported models 27 and consisted of 3 basic components: first, the structural PK model component, defining PK parameters and describing concentration‐time profiles of ADC and MMAE; second, the interindividual error model component, describing interindividual variation in PK parameters after correction for fixed effects; and third, the residual error model component, describing the underlying distribution of the error in the measured PK observation (ie, intraindividual variability). Individual ADC PK parameter estimates were required for MMAE PK to be evaluated. The concentration‐time data collected in the studies were analyzed using mixed‐effects modeling methods as implemented by the computer program NONMEM (version 7.4; Icon Development Solutions, Dublin, Ireland; with Intel Visual Fortran Intel 64 Compiler XE, version 12.0.0.104 Build 20101006 [Santa Clara, California]). 28 Missing data were excluded from the analyses. A log‐transform both‐sides approach was used with an additive residual error (proportional on back‐transform). Modeling was performed using the first‐order conditional estimation method. The final basic structural model was selected on the basis of goodness of fit; the chi‐square test (P < .005) for the log‐likelihood difference in objective function (OBJ) between nested models, with degrees of freedom equal to the difference in number of parameters between models, was used to declare superiority of 1 model over another.
Covariate Analysis
Prespecified covariates for potential inclusion in the final model were age, weight, sex, race, ethnicity, BSA, disease type, treatment cycle, antidrug antibody (ADA) status, creatinine clearance, and concentrations of albumin, alanine aminotransferase, aspartate aminotransferase, bilirubin, and creatinine. Covariates were tested for statistical significance as single covariate models, further supplemented by visual inspection of graphical trends. Covariates identified to individually reduce OBJ by >7.9 were pooled into a full model, which was subjected to backward elimination, with the final model only retaining covariates whose removal increased OBJ by ≥10.8 (P < .001). Prior to the inclusion of covariates in the model, interindividual parameter shrinkage was evaluated. The magnitude of the impact of the covariates was also considered; if the magnitude of the impact was small (<20% change over the range of covariate values in the database) or the covariate effect was poorly estimated (eg, standard error > 45%), then the covariate was evaluated for reparameterization or discarded.
For the visual predictive check evaluation, the 2.5th and 97.5th prediction intervals were constructed by simulating replicates of the data set from which the model was developed. The observed data were then overlaid and compared with the prediction intervals. Separate plots for pediatric versus adult patients were created. For the MMAE model, in which the M3 method for handling below‐the‐limit‐of‐quantitation (BLQ) samples was implemented, an additional plot of the actual proportion of BLQ samples over time compared with the 95% prediction interval of this proportion was created. 29
Model Evaluation
Model stability was tested through the evaluation of the condition number, which was calculated as the square root of the ratio of the largest to the smallest eigenvalue of the correlation matrix. A condition number <20 suggested that the degree of collinearity of the parameter estimates was acceptable. A condition number >100 indicated that the model may be unstable because of high collinearity. 25 In such cases the model was simplified, the condition number recomputed, and the model reevaluated.
Bootstrapping was used to evaluate parameter precision. Five hundred bootstrap data sets were generated and run using the ADC final model, and 200 bootstrap data sets were generated and run using the MMAE final model; a lower number of data sets were used for the MMAE final model because of longer run times. The data sets were stratified by study. The percentile bootstrap confidence intervals were constructed by taking the lower 5% and the upper 95% values of each parameter estimate from runs that converged successfully, as this interval should cover the true value of the parameter estimate approximately 90% of the time without imposing an assumption of symmetry on the distribution.
Model‐Based Simulations
The final PK models for ADC and MMAE were used to simulate the dosing regimens shown in Table 1. A dose was administered as a 30‐minute infusion every 14 or 21 days for ≥10 cycles. The baseline covariate values from 72 patients in the data set who weighed ≤100 kg were used in the simulations. One hundred replicates were simulated, resulting in 7200 simulated patients for each regimen. ADA‐negative concentration profiles were simulated. Rich sampling schemes were used to capture the peak and trough concentrations.
Table 1.
Simulated Dose Scenarios
| Dose Frequency | Dose Metric | Dose |
|---|---|---|
| Once every 3 weeks | mg/kg | 1.8 |
| Once every 3 weeks | mg/m2 | 71.5 |
| Once every 2 weeks | mg/kg | 1.2 |
| Once every 2 weeks | mg/m2 | 47.7 |
AUC (0‐21 days) and maximum serum concentration (Cmax) values were calculated from the simulated concentrations, and the results were compared based on body weight ranges expected for small pediatric patients (<28 kg; approximately 6‐9 years of age), moderate‐size pediatric patients (28‐49 kg or approximately 9‐14 years of age), and large pediatric/adult patients (50‐100 kg). 30
Results
POPPK Analysis Data Set
PK data were collected from 84 patients (34 pediatric and 50 adult patients) from studies NCT01492088 (pediatric n = 34, adult n = 2) and NCT00430846 (adult n = 48). Overall, 63 patients had HL, 17 had sALCL, and 4 had other diagnoses. Thirteen patients (15.5%) were ADA positive, 10 (27.8%) from study NCT01492088 and 3 (6.3%) from study NCT00430846. The majority of patients were white (86.9%) and male (64.3%). Median age was 25.65 years (range, 7.65‐87.24 years), and median creatinine clearance was 70.72 mL/min (range, 25.00‐123.76 mL/min). Additional details on patient baseline characteristics and demographics are shown in Table 2.
Table 2.
Demographics and Disease Characteristics for Pharmacokinetic‐Evaluable Patients Summarized for Categorical and Continuous Covariates
| Covariate | Value |
|---|---|
| Patients in each study, n (%) | |
| NCT01492088 | 36 (42.9) |
| ADA positive | 10 (27.8) |
| NCT00430846 | 48 (57.1) |
| ADA positive | 3 (6.3) |
| Age (years) | |
| Median (range) | 25.65 (7.65‐87.24) |
| Mean (SD) | 27.59 (16.76) |
| Adult age group, n (%) | 50 (59.5) |
| Pediatric age group, n (%) | 34 (40.5) |
| Sex, n (%) | |
| Male | 54 (64.3) |
| Female | 30 (35.7) |
| Race, n (%) | |
| White | 73 (86.9) |
| Black | 5 (6.0) |
| Asian | 4 (4.8) |
| Not reported | 2 (2.4) |
| Ethnicity, n (%) | |
| Not Hispanic | 71 (84.5) |
| Hispanic | 4 (4.8) |
| Not reported | 9 (10.7) |
| Height (cm) | |
| Median (range) | 170.00 (123.7‐194.0) |
| Mean (SD) | 166.66 (15.96) |
| Weight (kg) | |
| Overall median (range) | 66.70 (21.2‐153.8) |
| Overall mean (SD) | 68.41 (25.83) |
| Pediatric median (range) | 49.45 (21.20‐87.00) |
| Pediatric mean (SD) | 49.51 (17.60) |
| Adult median (range) | 77.90 (44.90‐153.80) |
| Adult mean (SD) | 81.26 (22.47) |
| BMI, kg/m2 | |
| Overall median (range) | 22.66 (12.94‐44.94) |
| Overall mean (SD) | 23.95 (6.85) |
| Pediatric median (range) | 18.36 (12.74‐32.41) |
| Pediatric mean (SD) | 19.59 (4.88) |
| Adult median (range) | 26.13 (16.90‐44.94) |
| Adult mean (SD) | 26.91 (6.43) |
| BSA (m2) | |
| Overall median (range) | 1.80 (0.87‐2.81) |
| Overall mean (SD) | 1.76 (0.40) |
| Pediatric median (range) | 1.49 (0.87‐2.02) |
| Pediatric mean (SD) | 1.46 (0.32) |
| Adult median (range) | 1.94 (1.43‐2.81) |
| Adult mean (SD) | 1.96 (0.30) |
| Disease, n (%) | |
| HL | 63 (75.0) |
| sALCL | 17 (20.2) |
| Other | 4 (4.8) |
| Albumin, g/L | |
| Median (range) | 4.77 (0.82‐19.08) |
| Mean (SD) | 5.94 (3.89) |
| ALT (U/L) | |
| Median (range) | 39.00 (22‐51) |
| Mean (SD) | 38.04 (6.46) |
| AST (U/L) | |
| Median (range) | 18.00 (5‐140) |
| Mean (SD) | 26.12 (24.22) |
| Bilirubin (µmol/L) | |
| Median (range) | 22.00 (8‐163) |
| Mean (SD) | 26.35 (19.46) |
| Creatinine (µmol/L) | |
| Median (range) | 6.00 (2‐20) |
| Mean (SD) | 7.44 (4.19) |
| CrCl (mL/min) | |
| Median (range) | 70.72 (25.00‐123.76) |
| Mean (SD) | 67.66 (24.69) |
ADA, antidrug antibody; ALT, alanine aminotransferase; AST, aspartate transaminase; BMI, body mass index; BSA, body surface area; CrCl, creatinine clearance; HL, Hodgkin lymphoma; sALCL, systemic anaplastic large‐cell lymphoma; SD, standard deviation.
From 84 patients, 8234 records were collected for analysis, 2664 from study NCT01492088 and 5570 from study NCT00430846. The ADC PK data set included 3020 records: 524 dosing records (pediatric study NCT01492088, n = 269; adult study NCT00430846, n = 255) and 2496 concentration records (pediatric study, n = 799, adult study, n = 1697). The MMAE PK data set included 3164 records: 524 dosing records (pediatric study, n = 269, adult study, n = 255) and 2640 concentration records (pediatric study, n = 797, adult study, n = 1843).
ADC and MMAE PK Models
ADC PK was described by a linear 3‐compartment model with zero‐order input and first‐order elimination based on previous POPPK models 31 , 32 , 33 , 34 , 35 , 36 for single‐agent brentuximab vedotin (Figure 2). The concentration of ADC decreased in a multiexponential manner, indicating a multicompartment model was appropriate. The final model parameters are shown in Table 3. Body size had a strong influence on clearance and central volume of distribution (V1), and BSA was found to be the best predictor of body size for V1. Weight was a better predictor for clearance than BSA, as determined by the reduction in objective function, but BSA was also a statistically significant predictor of clearance and thus was selected for both parameters for consistency.
Figure 2.
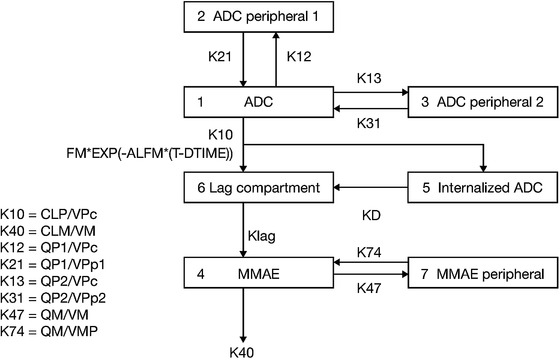
Brentuximab vedotin final ADC and MMAE pharmacokinetic model. ADC, antibody‐drug conjugate; ALFM, rate constant to describe the decline in direct conversion of ADC to MMAE following TAD; CLM, apparent MMAE clearance; CLP, ADC clearance; EXP, exponential; FM, fraction metabolized; KD, binding rate constant; Klag, rate constant for lag compartment; MMAE, monomethyl auristatin E; QM, apparent MMAE intercompartmental clearance; QP1 and QP2, ADC intercompartmental clearance from central to first and second peripheral compartments; T‐DTIME, time since last dose; VM and VMP, apparent volume of MMAE central and peripheral compartments; VPc, volume of ADC central compartment; VPp1 and VPp2, volume of ADC first and second peripheral compartments.
Table 3.
ADC and MMAE Final Model Parameters
| ADC | MMAE | |||
|---|---|---|---|---|
| Parameter | Population Mean (SE%) | %CV IIV (Shrinkage) | Population Mean (SE%) | %CV IIV (Shrinkage) |
| Clearance, L/h | 0.05 (10.3%) | 53.8 (4.2%) | 0.604 (8.7%) | 53.7 (11.2%) |
| Central volume (V1), L | 3.71 (3.2%) | 15.1 (10.9%) | 10.5 (7.1%) | 75.2 (16.3%) |
| Binding rate constant (Kd 1/h) | ― | ― | 0.00374 (12.3%) | 172.6 (13.6%) |
| Intercompartmental clearance (Q2), L/h | 0.02 (17.3%) | 63.7 (24.1%) | ― | ― |
| ― | ― | 13.1 (9.8%) | ― | |
| Peripheral volume (V2), L | 206 (16.6%) | ― | ― | ― |
| ― | ― | 22.2 (10.4%) | 63.6 (15.5%) | |
| Fraction metabolized | ― | ― | 1 FIX | ― |
| Intercompartmental clearance (Q3), L/h | 0.08 (5.0%) | ― | ― | ― |
| ADC to MMAE conversion rate (ALFM 1/h) | ― | ― | 2.68 (1.9%) | ― |
| Peripheral volume (V3), L | 5.0 (10.9%) | 48.1 (10.1%) | ― | ― |
| Lag compartment rate constant (Klag 1/h) | ― | ― | 15.3 (33.1%) | ― |
| BSA on central volume (V1) | 1.36 (8.6%) | ― | ― | ― |
| ― | ― | 1.22 (35.4%) | ― | |
| BSA on clearance | 1.66 (17.8%) | ― | ― | ― |
| ADA positive on clearance | 1.82 (1.7%) | ― | ― | ― |
| Albumin concentration on clearance | −0.90 (10.4%) | ― | ― | ― |
| Bilirubin on clearance | 0.15 (13.1%) | ― | ― | ― |
| AST on Kd | ― | ― | −0.672 (11.3%) | ― |
| BSA on Kd | ― | ― | −3.35 (26.8%) | ― |
| BSA on peripheral volume (V2), L | ― | ― | 1.81 (22.2%) | ― |
| BSA on peripheral volume (V3) | 1.15 (24.6%) | ― | ― | ― |
| Residual variability | 28.6%CV (0.9%) | ― | 35.7%CV (0.8%CV) | ― |
ADA, antidrug antibody; ADC, antibody‐drug conjugate; ALFM, rate constant to describe the decline in direct conversion of ADC to MMAE following time after dose; AST, aspartate transaminase; BSA, body surface area; CV, coefficient of variation; IIV, interindividual variability; Kd, binding rate constant; Klag, lag compartment rate constant; MMAE, monomethyl auristatin E; Q2, intercompartmental clearance from compartment 1 to compartment 2; Q3, intercompartmental clearance from compartment 2 to 3; SE, standard error; V1, central volume of distribution; V2, first peripheral volume of distribution; V3, second peripheral volume of distribution; 1 FIX, value fixed to 1.
MMAE PK was described by a 2‐compartment model with first‐order elimination and formation of MMAE both directly from ADC and through binding of ADC to a hypothetical target (Figure 2). The final model parameters are shown in Table 3.
Visual Predictive Check
Visual predictive check plots by age group versus time since first and last doses (Figure 3) show the observed concentrations over time. Ideally, the observed 95% and 50% intervals would be within the shaded areas, indicating that the model predicts the observed values adequately. For the most part this is the case, and when the observed intervals are outside the shaded areas, it is only marginally so. Therefore, the model adequately predicts the concentrations observed in each of the studies and for each age group.
Figure 3.
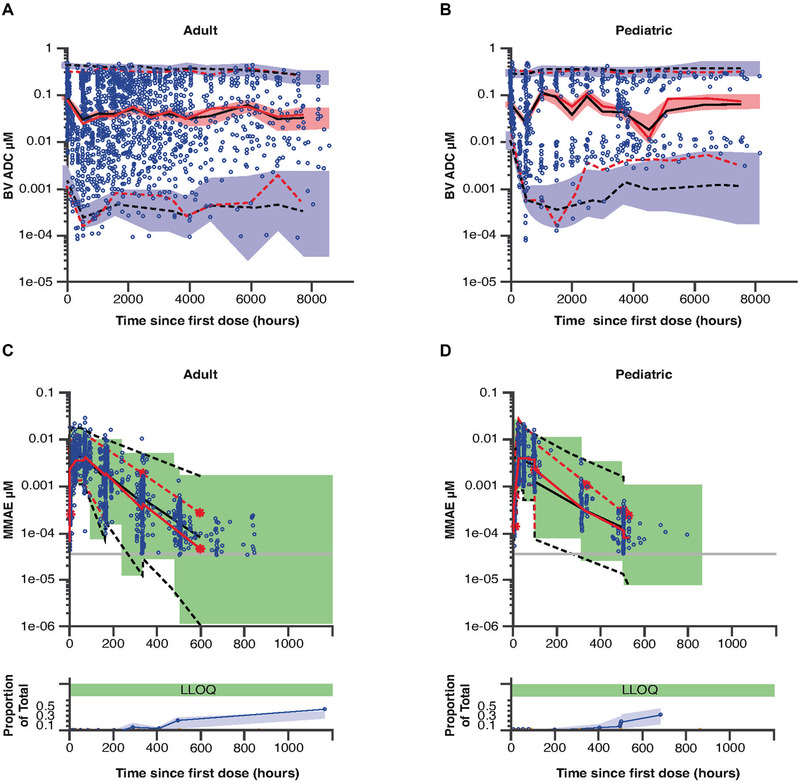
Final pharmacokinetic model visual predictive checks for ADC (A, B) and MMAE (C, D) by age group (adult [A, C] or pediatric [B, D]), by time since first dose. For ADC (A, B), observed concentrations are represented by open blue circles. The 95% prediction interval is shown by the dashed black lines, and the 50th percentile is shown as the solid black line. The 95% interval of the observed data is shown by the dashed red lines and the median is shown as the solid red line. The purple‐shaded area is the 95% confidence interval of the 95% prediction interval, and the pink‐shaded area is the 95% confidence interval of the median. For MMAE (C, D), the top panels show the model‐predicted 95% prediction interval (black dashed lines and shaded green area), the observed concentrations (blue circles), and the 95% interval of the observed values (red dashed lines). The lower panels show the observed proportion of BLQ samples (blue lines with blue circles), and the 90% prediction interval of this proportion. ADC, antibody‐drug conjugate; BLQ, below the limit of quantitation; BV, brentuximab vedotin; LLOQ, lower limit of quantitation; MMAE, monomethyl auristatin E.
Simulations of Dosing Regimens
With the final models for ADC and MMAE, simulations were performed using the patients in the analysis data sets at the typical dose of 1.8 mg/kg brentuximab vedotin and an alternate BSA‐based dose of 71.5 mg/m2. A rich sampling scheme over ten 21‐day single‐dose cycles was employed. A second set of simulations was performed to identify an equivalent BSA‐based dose for each of the approved body weight‐based doses (1.2 and 1.8 mg/kg), in which a lower dose was administered more frequently (every 2 weeks): doses were based on body weight (1.2 mg/kg) or BSA (47.7 mg/m2).
ADC Simulations at 1.8 mg/kg and 71.5 mg/m2 Every‐3‐Week Dosing
The first set of simulations used the approved starting dose of 1.8 mg/kg brentuximab vedotin monotherapy administered every 21 days (Table 1). This dosing regimen resulted in patients with smaller body weight having a 30% lower median AUC than patients with larger body weight (Figure 4A), with minimal effects on accumulation (Figure 4B). Therefore, pediatric patients would need a higher dose than 1.8 mg/kg to achieve AUC values similar to those observed in adults. The difference in median Cmax (Supplementary Figure S1A) between the body weight groups was 17%, lower than the difference observed for AUC.
Figure 4.
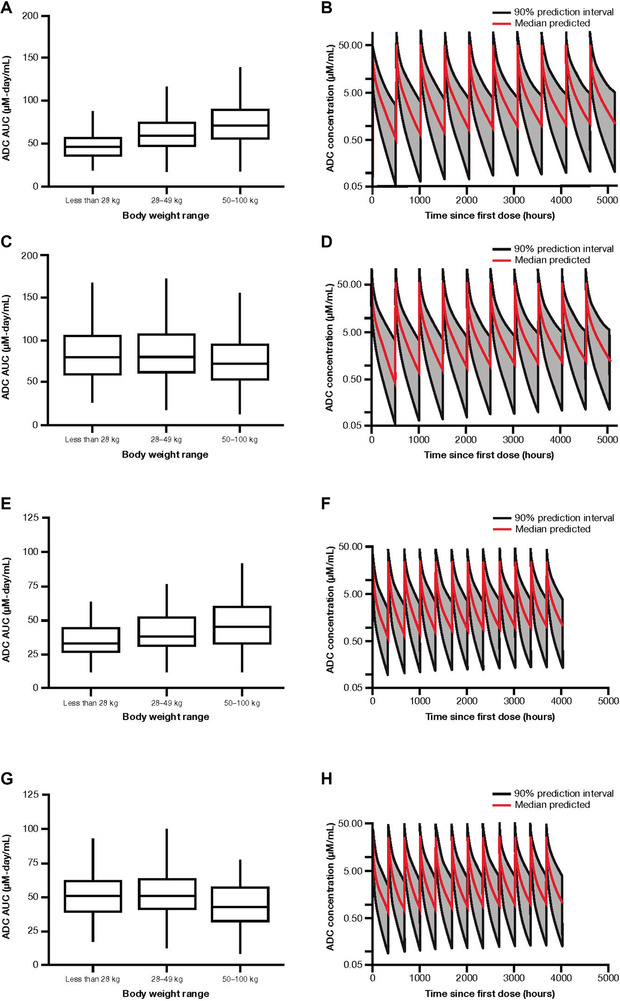
Simulated ADC AUC by body weight range (A, C, E, G) and concentration‐to‐time profiles (B, D, F, H) following 1.8 mg/kg every‐3‐week dosing (A, B), 71.5 mg/m2 every‐3‐week dosing (C, D), 1.2 mg/kg every‐2‐week dosing (E, F), and 47.7 mg/m2 every‐2‐week dosing (G, H). Each box plot illustrates: the median by the horizontal line in the center, the 25th percentile by the lower end of the box, the 75th percentile by the upper end of the box, the 5th percentile by the lower whisker, and the 95th percentile by the upper whisker. ADC, antibody‐drug conjugate; AUC, area under the concentration‐to‐time curve.
BSA‐based dosing simulations using 71.5 mg/m2 brentuximab vedotin every‐3‐week dosing showed AUC in smaller patients similar to that achieved with body weight‐based dosing in adults (Figure 4C). As with body weight‐based dosing, BSA‐based dosing had minimal effects on accumulation over time (Figure 4D). The geometric mean Cmax values for small‐ and moderate‐sized pediatric patients administered 71.5 mg/m2 were approximately 33% and 11% higher, respectively, than those in large pediatric/adult patients administered the BSA‐based dose or the 1.8 mg/kg dose (Supplementary Figure S1A,C); however, the magnitude of this difference is relatively small compared with the observed overall variability in Cmax (34%‐36% coefficient of variation) with 1.8 mg/kg every‐3‐week dosing.
MMAE Simulations at 1.8 mg/kg and 71.5 mg/m2 Every‐3‐Week Dosing
Infusion of 1.8 mg/kg brentuximab vedotin over 30 minutes resulted in a 37% lower AUC in the small‐ and moderate‐sized pediatric patient weight groups compared with the large pediatric/adult weight group (Figure 5A); the concentration‐time curve showed a noticeably higher exposure for the first dose compared with subsequent doses (Figure 5B), consistent with prior observations. 6 A dose of 71.5 mg/m2 resulted in overall AUC values that were more similar across the 3 body size groups than was seen with the body weight‐based dosing approach (Figure 5C). The overall Cmax values for pediatric patients who received a dose of 71.5 mg/m2 every 3 weeks were similar to those in large pediatric/adult patients dosed at 1.8 mg/kg every 3 weeks (Supplementary Figure S1B,D).
Figure 5.
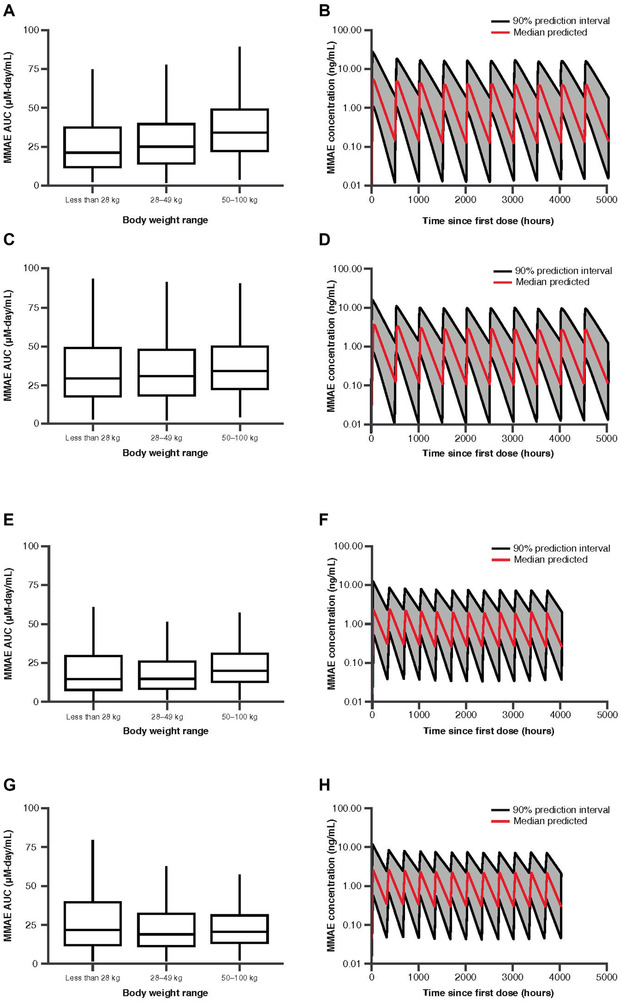
Simulated MMAE AUC by body weight range (A, C, E, G) and concentration‐to‐time profiles (B, D, F, H) following 1.8 mg/kg every‐3‐week dosing (A, B), 71.5 mg/m2 every‐3‐week dosing (C, D), 1.2 mg/kg every‐2‐week dosing (E, F), and 47.7 mg/m2 every‐2‐week dosing (G, H). Each box plot illustrates: the median by the horizontal line in the center, the 25th percentile by the lower end of the box, the 75th percentile by the upper end of the box, the 5th percentile by the lower whisker, and the 95th percentile by the upper whisker. AUC, area under the concentration‐to‐time curve; MMAE, monomethyl auristatin E.
Simulations at 1.2 mg/kg and 47.7 mg/m2 Every‐2‐Week Dosing
Results of simulations using 1.2 mg/kg and 47.7 mg/m2 brentuximab vedotin administered every 2 weeks paralleled those for 1.8 mg/kg and 71.5 mg/m2 brentuximab vedotin given every 3 weeks. The lowest AUC overall was observed in the <28‐kg body weight group treated with 1.2 mg/kg brentuximab vedotin (Figure 4E), whereas AUCs achieved following 47.7 mg/m2 dosing were more consistent across each body weight range (Figure 4G), suggesting that BSA‐based dosing provides more consistent exposure than body weight‐based dosing in smaller patients. Both every‐2‐week regimens showed minimal accumulation of ADC throughout the treatment period (Figure 4F,H). Every‐2‐week body weight‐based dosing resulted in smaller patients having lower Cmax values than larger patients (Supplementary Figure S1E), whereas BSA‐based dosing resulted in higher Cmax in lower‐body‐weight patients (Supplementary Figure S1G).
Every‐2‐week dosing resulted in MMAE AUC values that were comparable to every‐3‐week dosing for both body weight‐based dosing (Figure 5E) and BSA‐based dosing (Figure 5G). There was little difference between every 3 weeks (Supplementary Figure S1B,D) and every‐2‐week dosing (Supplementary Figure S1F,H) on MMAE Cmax.
Discussion
Prior noncompartmental PK analysis of pediatric patients (NCT01492088) highlighted a trend for reduced ADC and MMAE exposures in patients aged <12 years. 12 The current study aimed to identify the sources of ADC and MMAE exposure variability, in particular, the impact of body weight on exposure. The purpose of the current POPPK analysis was to generate hypotheses for alternate dosing regimens that may normalize ADC and MMAE exposures across all body weights. BSA‐based dosing of brentuximab vedotin was identified as having the potential to normalize ADC and MMAE exposures across all body weights.
The initial structural models for ADC and MMAE were based on the previously developed models. 26 The model for ADC PK was a linear 3‐compartment model with zero‐order input and first‐order elimination. BSA, over body weight and age, was found to be the best predictor of body size in the model based on the lowest OBJ. ADA status was modeled as a categorical covariate (positive/negative). Once a positive value was detected, the patient's ADA status was categorized as positive for all subsequent records. ADA positivity was a predictor of ADC clearance; patients who were ADA positive had an approximately 82% higher mean clearance than patients who were ADA negative (Table 3). The precision of parameter estimates and the residual variability for the final model were acceptable. The shrinkage for the final model parameters was low to moderate and was considered acceptable for simulation of ADC concentrations and subsequent estimation of exposure through AUC and Cmax.
ADC concentrations in the MMAE model were estimated based on the individual parameter estimates from the ADC model. The PK of MMAE was described by a 2‐compartment model with first‐order elimination and formation of MMAE both directly from ADC and through binding of ADC to a hypothetical target. The model had a lag compartment to describe the delay in formation of MMAE, both directly from ADC and through binding of ADC to the target. The fraction of MMAE formed directly from ADC decreased following ADC administration, relative to time after dose. BLQ handling was included in the model and implemented with the M3 method, with which the likelihood that a concentration is less than the lower limit of quantitation was modeled. Body size was found to be a predictor of MMAE V1, V2, and binding rate constant, and aspartate transaminase concentration was a predictor of binding rate constant. This final model showed acceptable residual variability, precision of parameter estimates, and shrinkage. The model was well conditioned based on the condition number.
With these final models for ADC and MMAE, simulations were performed using the patients in the analysis data sets at the approved 1.8 mg/kg brentuximab vedotin monotherapy dose and a 71.5 mg/m2 test dose. A rich sampling scheme over ten 21‐day single‐dose cycles was employed. AUC (0‐21 days) and Cmax values were calculated from the simulated concentrations, and the results were compared based on body weight ranges expected for small pediatric patients (<28 kg, or approximately 6‐9 years of age), moderate‐sized pediatric patients (28‐49 kg, or approximately 9‐14 years of age), and large pediatric/adult patients (50‐100 kg). The ADC simulation results showed that a 1.8 mg/kg dose infused over 30 minutes resulted in lower AUC and Cmax values in small‐/moderate‐sized pediatric patients than in large pediatric/adult patients. Using a BSA‐based dose of 71.5 mg/m2 resulted in similar AUC values for both pediatric and adult patients that were similar to the AUC in adult patients administered the 1.8 mg/kg dose. The geometric mean ADC Cmax values for small‐ and moderate‐size pediatric patients who received 71.5 mg/m2 were approximately 33% and 11% higher, respectively, than those in large pediatric/adult patients administered the BSA‐based dose or the 1.8 mg/kg dose. These differences were less than the overall variability in MMAE Cmax (coefficient of variation, 34%‐36% following dosing at 1.8 mg/kg).
The MMAE simulation results for these doses showed that a 1.8 mg/kg brentuximab vedotin dose infused over 30 minutes resulted in a lower AUC and Cmax in small‐ and moderate‐sized pediatric patients compared with the large pediatric/adult size weight group. A dose of 71.5 mg/m2 resulted in AUC values that were more similar across the 3 body size groups than those seen with the mg/kg dosing approach. The Cmax values for pediatric patients administered a dose of 71.5 mg/m2 were comparable with the values for adult patients administered the approved dose of 1.8 mg/kg.
A second set of simulations was performed in which a lower brentuximab vedotin dose was administered more frequently (every 2 weeks). Doses were based on body weight (1.2 mg/kg) and BSA (47.7 mg/m2). Results similar to the every‐3‐week simulations were observed with the every‐2‐week simulations, with BSA‐based dosing resulting in more consistent exposures across body sizes than body weight‐based dosing.
Limitations of this investigation include the relatively small number of patients overall (n = 84) and the relatively larger number of adult patients compared with pediatric patients (50 and 34, respectively). Further studies using this model may overcome these limitations by including data from a larger number of trials to increase the sample size and aiming to better balance the numbers of adult and pediatric patients.
Guided by the hypotheses generated from this analysis, BSA‐based dosing of brentuximab vedotin (47.7 mg/m2 every 2 weeks) in combination with AVD is currently being assessed as an alternative dosing option in a phase 1/2 study to evaluate the efficacy and safety of frontline brentuximab vedotin, doxorubicin, vinblastine, and dacarbazine in pediatric patients with cHL (NCT02979522). 37 Emerging data indicate exposure, safety, and efficacy results that are generally consistent with those previously reported from body weight‐based dosing in adult patients with cHL. 24
Conclusion
In summary, POPPK modeling suggests that body size is an important predictor of the variability in brentuximab vedotin PK. The investigational 47.7 mg/m2 pediatric dosing schedule is expected to provide AUC values matching those achieved with the adult brentuximab vedotin 1.2 mg/kg every‐2‐week dosing schedule across a range of pediatric body weights, and this hypothesis is currently being evaluated in an ongoing trial in pediatric patients with cHL in the frontline treatment setting (NCT02979522). Our findings show that model‐informed approaches to pediatric oncology drug development enable dose selection aimed at minimizing interpatient variability during clinical evaluation of safety and efficacy.
Conflicts of Interest
A.S., G.S., and J.K. disclose employment by and D.R.M. is a paid consultant for Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. K.V. discloses previous employment by Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, and current employment by EMD Serono, Inc. Writing assistance for this article was provided by Rebecca Vickers and Hedley Coppock of FireKite, an Ashfield company, part of UDG Healthcare plc, and was funded by Millennium Pharmaceuticals, Inc. All editorial procedures complied with Good Publication Practice 3 guidelines (Battisti WP, et al. Ann Intern Med. 2015;163:461‐464).
Funding
All clinical data were generated in studies sponsored by Seattle Genetics, Inc. and Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Data Sharing
Takeda makes patient‐level deidentified data sets and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met as set forth in Takeda's data‐sharing policy (see https://www.takedaclinicaltrials.com for details). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requester's qualifications and conflicts of interest that can result in potential bias. Once approved, qualified researchers who sign a data‐sharing agreement are provided access to these data in a secure research environment.
Supporting information
Supporting information
Figure S1. Simulated ADC and MMAE Cmax by body weight range, following 1.8 mg/kg every‐3‐week dosing (A, B), 71.5 mg/m2 every‐3‐week dosing (C, D), 1.2 mg/kg every‐2‐week dosing (E, F), and 47.7 mg/m2 every‐2‐week dosing (G, H). Each box plot illustrates: the median by the horizontal line in the center, the 25th percentile by the lower end of the box, the 75th percentile by the upper end of the box, the 5th percentile by the lower whisker, and the 95th percentile by the upper whisker. ADC, antibody‐drug conjugate; Cmax, maximum serum concentration; MMAE, monomethyl auristatin E.
Table S1. Planned Pharmacokinetic Sampling Times
Acknowledgments
The authors acknowledge all patients who participated in the studies and their families, as well as all the investigators and site staff who made this study possible. All clinical data were generated in studies sponsored by Seattle Genetics, Inc. and Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
References
- 1. Berger GK, McBride A, Lawson S, et al. Brentuximab vedotin for treatment of non‐Hodgkin lymphomas: a systematic review. Crit Rev Oncol Hematol. 2017;109:42‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alperovich A, Younes A. Targeting CD30 using brentuximab vedotin in the treatment of Hodgkin lymphoma. Cancer J. 2016;22(1):23‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Suri A, Mould DR, Liu Y, Jang G, Venkatakrishnan K. Population PK and exposure‐response relationships for the antibody‐drug conjugate brentuximab vedotin in CTCL patients in the phase III ALCANZA study. Clin Pharmacol Ther. 2018;104(5):989‐999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van der Weyden CA, Pileri SA, Feldman AL, Whisstock J, Prince HM. Understanding CD30 biology and therapeutic targeting: a historical perspective providing insight into future directions. Blood Cancer J. 2017;7(9):e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol. 2012;30(18):2183‐2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. US FDA . ADCETRIS (brentuximab vedotin) Highlights of Prescribing Information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125388s099lbl.pdf. Published 2018. Accessed June 15, 2020.
- 7. Moskowitz CH, Nademanee A, Masszi T, et al. Brentuximab vedotin as consolidation therapy after autologous stem‐cell transplantation in patients with Hodgkin's lymphoma at risk of relapse or progression (AETHERA): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2015;385(9980):1853‐1862. [DOI] [PubMed] [Google Scholar]
- 8. Prince HM, Kim YH, Horwitz SM, et al. Brentuximab vedotin or physician's choice in CD30‐positive cutaneous T‐cell lymphoma (ALCANZA): an international, open‐label, randomised, phase 3, multicentre trial. Lancet. 2017;390(10094):555‐566. [DOI] [PubMed] [Google Scholar]
- 9. Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN‐35) in patients with relapsed or refractory systemic anaplastic large‐cell lymphoma: results of a phase II study. J Clin Oncol. 2012;30(18):2190‐2196. [DOI] [PubMed] [Google Scholar]
- 10. Horwitz S, O'Connor OA, Pro B, et al. Brentuximab vedotin with chemotherapy for CD30‐positive peripheral T‐cell lymphoma (ECHELON‐2): a global, double‐blind, randomised, phase 3 trial. Lancet. 2019;393(10168):229‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Connors JM, Jurczak W, Straus DJ, et al. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin's lymphoma. N Engl J Med. 2018;378(4):331‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Locatelli F, Mauz‐Koerholz C, Neville K, et al. Brentuximab vedotin for paediatric relapsed or refractory Hodgkin's lymphoma and anaplastic large‐cell lymphoma: a multicentre, open‐label, phase 1/2 study. Lancet Haematol. 2018;5(10):e450‐e461. [DOI] [PubMed] [Google Scholar]
- 13. Faucette S, Wagh S, Trivedi A, Venkatakrishnan K, Gupta N. Reverse translation of US Food and Drug Administration reviews of oncology new molecular entities approved in 2011‐2017: lessons learned for anticancer drug development. Clin Transl Sci. 2018;11(2):123‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ji Y, Jin JY, Hyman DM, Kim G, Suri A. Challenges and opportunities in dose finding in oncology and immuno‐oncology. Clin Transl Sci. 2018;11(4):345‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Venkatakrishnan K, Friberg LE, Ouellet D, et al. Optimizing oncology therapeutics through quantitative translational and clinical pharmacology: challenges and opportunities. Clin Pharmacol Ther. 2015;97(1):37‐54. [DOI] [PubMed] [Google Scholar]
- 16. Gupta N, Zhao Y, Hui AM, Esseltine DL, Venkatakrishnan K. Switching from body surface area‐based to fixed dosing for the investigational proteasome inhibitor ixazomib: a population pharmacokinetic analysis. Br J Clin Pharmacol. 2015;79(5):789‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bullock JM, Lin T, Bilic S. Clinical pharmacology tools and evaluations to facilitate comprehensive dose finding in oncology: a continuous risk‐benefit approach. J Clin Pharmacol. 2017;57(suppl 10):S105‐S115. [DOI] [PubMed] [Google Scholar]
- 18. Fanale MA, Forero‐Torres A, Rosenblatt JD, et al. A phase I weekly dosing study of brentuximab vedotin in patients with relapsed/refractory CD30‐positive hematologic malignancies. Clin Cancer Res. 2012;18(1):248‐255. [DOI] [PubMed] [Google Scholar]
- 19. Li H, Han TH, Hunder NN, Jang G, Zhao B. Population pharmacokinetics of brentuximab vedotin in patients with CD30‐expressing hematologic malignancies. J Clin Pharmacol. 2017;57(9):1148‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ogura M, Tobinai K, Hatake K, et al. Phase I/II study of brentuximab vedotin in Japanese patients with relapsed or refractory CD30‐positive Hodgkin's lymphoma or systemic anaplastic large‐cell lymphoma. Cancer Sci. 2014;105(7):840‐846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN‐35) for relapsed CD30‐positive lymphomas. N Engl J Med. 2010;363(19):1812‐1821. [DOI] [PubMed] [Google Scholar]
- 22. Zhao B, Chen R, O'Connor OA, et al. Brentuximab vedotin, an antibody‐drug conjugate, in patients with CD30‐positive haematologic malignancies and hepatic or renal impairment. Br J Clin Pharmacol. 2016;82(3):696‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suri A, Mould DR, Song G, et al. Population pharmacokinetic modeling and exposure‐response assessment for the antibody‐drug conjugate brentuximab vedotin in Hodgkin's lymphoma in the phase III ECHELON‐1 study. Clin Pharmacol Ther. 2019;106(6):1268‐1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sekimizu M, Iguchi A, Mori T, et al. Phase I clinical study of brentuximab vedotin (SGN‐35) involving children with recurrent or refractory CD30‐positive Hodgkin's lymphoma or systemic anaplastic large cell lymphoma: rationale, design and methods of BV‐HLALCL study: study protocol. BMC Cancer. 2018;18(1):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Glantz SA, Slinker BA. Primer of Applied Regression and Analysis of Variance. New York, NY: McGraw‐Hill; 1990. [Google Scholar]
- 26. Han TH, Gopal AK, Ramchandren R, et al. CYP3A‐mediated drug‐drug interaction potential and excretion of brentuximab vedotin, an antibody‐drug conjugate, in patients with CD30‐positive hematologic malignancies. J Clin Pharmacol. 2013;53(8):866‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Han TH, Kennedy D, Hayes S, Lynch CM. The pharmacokinetics of brentuximab vedotin (SGN‐35), an antibody‐drug conjugate (ADC) [abstract]. Clin Pharmacol Ther. 2012;91(suppl 1):PII‐1. [Google Scholar]
- 28. Boeckmann AJ, Beal SL, Sheiner LB. Parts I through VIII, NONMEM Installation Guide. NONMEM Project Group, University of California; 1998. [Google Scholar]
- 29. Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28(5):481‐504. [DOI] [PubMed] [Google Scholar]
- 30. Centers for Disease Control and Prevention (CDC) . Clinical growth charts. https://www.cdc.gov/growthcharts/clinical_charts.htm. Accessed May 27, 2020.
- 31. Schmitz N, Pfistner B, Sextro M, et al. Aggressive conventional chemotherapy compared with high‐dose chemotherapy with autologous haemopoietic stem‐cell transplantation for relapsed chemosensitive Hodgkin's disease: a randomised trial. Lancet. 2002;359(9323):2065‐2071. [DOI] [PubMed] [Google Scholar]
- 32. Santoro A, Bredenfeld H, Devizzi L, et al. Gemcitabine in the treatment of refractory Hodgkin's disease: results of a multicenter phase II study. J Clin Oncol. 2000;18(13):2615‐2619. [DOI] [PubMed] [Google Scholar]
- 33. Zinzani PL, Bendandi M, Stefoni V, et al. Value of gemcitabine treatment in heavily pretreated Hodgkin's disease patients. Haematologica. 2000;85(9):926‐929. [PubMed] [Google Scholar]
- 34. Venkatesh H, Di Bella N, Flynn TP, Vellek MJ, Boehm KA, Asmar L. Results of a phase II multicenter trial of single‐agent gemcitabine in patients with relapsed or chemotherapy‐refractory Hodgkin's lymphoma. Clin Lymphoma. 2004;5(2):110‐115. [DOI] [PubMed] [Google Scholar]
- 35. Oki Y, Pro B, Fayad LE, et al. Phase 2 study of gemcitabine in combination with rituximab in patients with recurrent or refractory Hodgkin lymphoma. Cancer. 2008;112(4):831‐836. [DOI] [PubMed] [Google Scholar]
- 36. Linch DC, Winfield D, Goldstone AH, et al. Dose intensification with autologous bone‐marrow transplantation in relapsed and resistant Hodgkin's disease: results of a BNLI randomised trial. Lancet. 1993;341(8852):1051‐1054. [DOI] [PubMed] [Google Scholar]
- 37. Franklin A, Zecca M, Fagioli F, et al. Phase 1 results from a phase 1/2 study to assess the safety, tolerability and recommended phase 2 dose (RP2D) of brentuximab vedotin plus doxorubicin, vinblastine and dacarbazine (A+AVD) in pediatric patients (pts) with advanced stage newly diagnosed classical Hodgkin Lymphoma (cHL). Blood. 2018;132(1):1644. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Figure S1. Simulated ADC and MMAE Cmax by body weight range, following 1.8 mg/kg every‐3‐week dosing (A, B), 71.5 mg/m2 every‐3‐week dosing (C, D), 1.2 mg/kg every‐2‐week dosing (E, F), and 47.7 mg/m2 every‐2‐week dosing (G, H). Each box plot illustrates: the median by the horizontal line in the center, the 25th percentile by the lower end of the box, the 75th percentile by the upper end of the box, the 5th percentile by the lower whisker, and the 95th percentile by the upper whisker. ADC, antibody‐drug conjugate; Cmax, maximum serum concentration; MMAE, monomethyl auristatin E.
Table S1. Planned Pharmacokinetic Sampling Times