

Abstract
Tuberculosis is caused by Mycobacterium tuberculosis (Mtb) and is a deadly disease resulting in the deaths of approximately 1.5 million people with 10 million infections reported in 2018. Recently, a key condensation step in the synthesis of mycolic acids was shown to require β-ketoacyl-ACP synthase (KasA). A crystal structure of KasA with the small molecule DG167 was recently described, which provided a starting point for using computational structure-based approaches to identify additional molecules binding to this protein. We now describe structure-based pharmacophores, docking and machine learning studies with Assay Central as a computational tool for the identification of small molecules targeting KasA. We then tested these compounds using nanoscale differential scanning fluorimetry and microscale thermophoresis. Of note, we identified several molecules including the Food and Drug Administration (FDA)-approved drugs sildenafil and flubendazole with Kd values between 30–40 μM. This may provide additional starting points for further optimization.
Introduction
Tuberculosis (TB) is a public health concern responsible for the deaths of around 1.5 million people with 10 million infections reported per year.1 Some estimates put 25% of the world’s population infected with the causative pathogen Mycobacterium tuberculosis (Mtb).1 Standard treatment of care for nondrug-resistant infections consists of a two-month regimen of four drugs, isoniazid, rifampin, pyrazinamide, and ethambutol, followed by 4 months of continued isoniazid and rifampin treatment.2 Of these first-line drugs, isoniazid is often given as a prophylactic in cases of exposure; however, resistance to this drug also occurs at a higher frequency than the other first-line drugs.3 In 2018, approximately half a million TB patients had either rifampin-resistant tuberculosis (RR-TB) or multidrug-resistant tuberculosis (MDR-TB), which describes resistance to at least isoniazid and rifampin.1 Cases of MDR-TB rely on the administration of second-line antibacterial drugs that are less effective and have more adverse effects than the first-line drugs, and the treatment time for these cases extends from 6 months to 18–24 months. Ten percent of MDR-TB cases are made up of extensively drug-resistant TB (XDR-TB), which is resistant to isoniazid, rifampin, a second-line injectable drug (consisting of amikacin, capreomycin, or kanamycin), and any fluoroquinolone.4 Based on the increasing proportion of drug-resistant cases of TB, development of new drugs has become imperative. Despite the increased focus on drug discovery in the past decade, finding new molecules that are active against TB has proven to be a difficult endeavor. These drugs must be able to overcome resistance of the bacterium to hydrophobic molecules, must target essential processes in the life cycle of the bacterium and must not overlap with the function of any of the drugs represented by MDR-TB. Recently, three drugs have come to the market for the treatment of MDR-TB: bedaquiline, delamanid, and pretomanid.5
The bacterial cell wall is historically one of the most successful targets for Mtb drug discovery. The most commonly used Mtb cell wall-active drugs are isoniazid (INH), ethambutol and ethionamide. The Mycobacterium genus has a unique cellular envelope that contains a layer of mycolic acids, the mycolic membrane or mycomembrane, which is covalently linked to the cell wall posing a permeability hurdle to most small molecules while also allowing the mycobacteria to survive inside the macrophages.6,7 Mycolic acids are structural elements of the cell wall and essential for Mtb in vivo pathogenesis.8−10 Mycolic acids are C60–C90 branched chain β-hydroxylated fatty acids.11 During mycolic acid biosynthesis, the type I fatty acid synthase (FAS-I) extends acyl-CoA primers to yield C16–C18 and C24–C26 fatty acids. The FAS-II system then completes chain elongation, producing meromycolic acids (C48–C64).10 The essentiality of mycolic acids to the Mtb cell wall has suggested that the different enzymatic steps involved in the mycolic acid biosynthesis would be good drug targets. INH is a critical component of front-line TB therapy. Its mechanism of action in vitro and in vivo is due primarily to its inhibition of the enoyl-acyl carrier protein reductase (InhA).12,13 InhA is a protein integral in the FAS-II cycle, a system that is distinct to the eukaryotic fatty acid biosynthesis.14 Other FAS-II enzymes are highly likely to be good Mtb drug targets for this same reason. Unfortunately, INH demonstrates biphasic kill kinetics in vitro, with resistance and persistence developing within one week of drug exposure, leading to decreased cidality.15 Drugs with a synergistic activity with INH should circumvent this issue, and those drugs with the ability to potentiate the activity of INH and eradicate INH-induced persisters would also likely provide substantial benefit in TB treatment.16
A recent drug target of focus is β-ketoacyl-ACP synthase (KasA), which performs the condensation step in the synthesis of mycolic acids.17,18 KasA is an essential protein in Mtb and a component of the fatty acid synthase II complex (FAS II), which unlike FAS I has no human equivalent.16,19−22 Small molecules have been identified that inhibit KasA, including derivatives of thiolactomycin23 and oxadiazolone.24 A screen of the GSK library of antitubercular hits25 for cell wall biosynthesis inhibitors was previously reported.16 GSK3011724A (renamed DG167; Figure S1) has a minimum inhibitor concentration (MIC) of 0.39 M, and the cocrystal structure with KasA was determined.16 Another group recently reported that this molecule targets KasA,22 but its original X-ray crystal structure bound to KasA was misinterpreted. This KasA–DG167 structure revealed that each monomer of KasA in the biological dimer binds to two molecules of DG167 instead of a single one. This X-ray crystal structure was recently leveraged by Inoyama et al. to afford a preclinical tuberculosis compound (JSF-3285).26 Previous computational studies have also proposed natural products27 and derivatives of thiolactomycin as KasA-targeting compounds.28 The current study aims to use an array of computational structure-based and ligand-based approaches to identify compounds binding to KasA as potential starting points for future optimization.
Results and Discussion
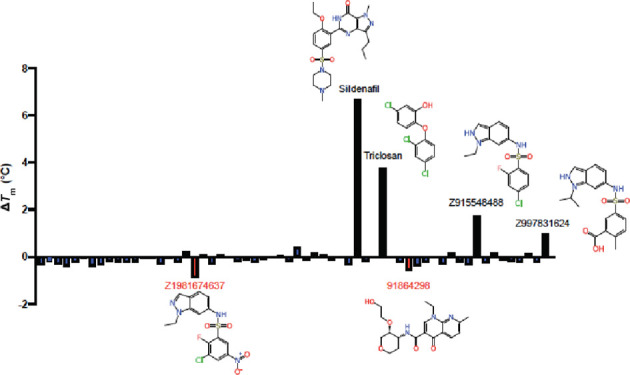
We used multiple approaches to identify additional molecules that potentially bind to KasA, including structure-based pharmacophores (Figure S2), shape-based pharmacophores (Figure S3), a whole cell Mtb machine learning model, docking, and a substructure similarity search using DG167. Using these approaches, 62 compounds were identified and tested using either nanoscale differential scanning fluorimetry (nanoDSF, Figures 1 and 2)29 and/or microscale thermophoresis (MST, Figure 3).30,31 Binding affinities using MST were assessed for eight compounds (Figure 3), and nanoDSF was performed for 60 compounds (Figure S4). Six of these showed notable changes in melting temperature (ΔTm) (Figure 1).
Figure 1.

Representation of the KasA nanoDSF results. ΔTm is calculated from a dimethyl sulfoxide (DMSO)-treated KasA control. Chemical structures are depicted for those compounds with the most pronounced ΔTm. Many of the identified compounds share a similar indazole core, suggesting that this may be an important structural element responsible for binding to KasA. Interestingly, two of these compounds with the indazole core seem to destabilize KasA as opposed to stabilizing it.
Figure 2.

NanoDSF traces for the compounds showing the most significant KasA stabilization. (A) Ratio of mean fluorescence (350/330 nm). (B) Ratio of mean fluorescence 350/330 nm (1st derivative). KasA (10 μM) was tested in the presence of 100 μM of compounds. Sildenafil (purple) and triclosan (green) have a ΔTm of 6.7 and 3.8 °C, respectively.
Figure 3.

Microscale thermophoresis data. Compounds chosen by various approaches were assessed for KasA binding. Each compound was run in triplicate, and ± represents the Kd confidence interval (68% CI). The compound 15407843 was also evaluated, but no binding could be determined via MST.
The molecules triclosan, metoclopramide, chlorpropamide, and flubendazole (Figure S2) were identified using a pharmacophore-based methodology. A structure-based pharmacophore resulted in two hydrophobic features: a hydrogen bond acceptor and a hydrogen bond donor as well as excluded volumes (Figure S2). These aforementioned molecules were shown to map well to this pharmacophore. The binding affinities were determined using MST for two of these, chlorpropamide (Kd = 90.60 ± 40.29 μM) and flubendazole (Kd = 38.52 ± 13.51 μM) (Figure 3). A second approach was employed using a shape-based pharmacophore including both DG167 molecules from the crystal structure PDB code 5W2O,16 potentially capturing more of the binding site volume. When this shape-based pharmacophore was used to search the Food and Drug Administration (FDA)-approved drugs, three drugs mapped well into this pocket: streptomycin, lovastatin, and sildenafil (Figure S3). Binding affinities were determined for lovastatin and sildenafil using MST (Kd = 224 ± 86 μM and Kd = 32.51 ± 14.36 μM, respectively) (Figure 3). As DG167 was previously shown to be a potent binder of KasA (EC50 of 130.9 ± 18.2 nM),16 additional potential KasA-targeting compounds were identified using a substructure search using DG167 in PubChem. We tested 16 compounds identified in this manner using nanoDSF, with Z915548488 and Z997831624 increasing and Z1981674637 decreasing Tm by almost 1 °C, respectively (Figure 1).
A very different approach utilized is reminiscent of a target-independent strategy, which is often used in Mtb screening due to the challenge of overcoming the low permeability of the mycomembrane. In this schema, we first used a previously published whole cell Mtb machine learning model developed with the Assay Central with a 100 nM MIC activity cutoff32 to screen ∼2.5 million commercially available molecules from ChemBridge, MCE, Enamine, Octava, and Selleck chemicals in an attempt to find compounds likely to have a whole cell activity. It should be noted that this process identified compounds that were predicted to be active against Mtb in general, and we were not selecting compounds specifically targeting KasA. The selected compounds were then docked in the KasA structure (PDB code 5W2O).16 Thirty-four compounds were selected from the top 50 that had a libdock score > 95 for purchase. The compounds 88983431, 4781032, 9278699, and 15407843 had libdock scores of 155.35, 145.32, 128.67, and 136.03, respectively, and were chosen to be assessed for KasA binding via MST (Figure 3). All of these compounds, with the exception of 15407843, showed quantifiable binding via MST, though lovastatin and chlorpropamide displayed weak binding (Kd > 90 μM). 15407843 showed an insufficient signal-to-noise ratio to bind at the concentrations tested using MST.
The majority of these compounds described earlier were assessed for KasA binding via nanoDSF (Figure S5 and Table S1), a technique more conducive to a higher throughput screening. In addition to these selected compounds, as a control, five random compounds were chosen from our library to be tested for binding to KasA using nanoDSF: naloxone, cisapride, N-acetyl procainamide, pyronaridine, and haloperidol.
In summary, in this study, we have used the previously generated crystal structure of DG167 in KasA16 to develop several computational approaches to screen drugs and commercial molecules and in the process have identified several FDA-approved drugs that bind to the KasA protein. The largest ΔTm values were identified for the compounds sildenafil and triclosan (Figure 2). Sildenafil was previously reported to inhibit Mtb via the host effect33,34 but we are not aware of any assessment of the direct activity against the bacteria in vitro or identification of potential targets in Mtb. In this study, it showed a large ΔTm (ΔTm 6.7 °C, Figure 1), indicative of binding to the protein. Triclosan has also been widely reported as an Mtb inhibitor (MIC 12.5 μM35,36), and others suggest that it inhibits InhA and not KasA in Mycobacterium smegmatis(37−39) Our data showed that triclosan can stabilize KasA with a ΔTm of 3.8 °C using nanoDSF (Figure 1); however, other experiments are necessary to verify if it can also inhibit the enzyme activity and to further identify the triclosan binding site (i.e., crystal structure).
In total, we tested 62 commercially available compounds identified through various computational methods. Seven molecules were assessed using MST (Figure 3) with the Kd values ranging from 20–224 μM. The highest affinity binding molecule was 92786999, identified through predictions based on the whole cell Mtb machine learning model, followed by sildenafil, identified through a shape-based pharmacophore (Figure 3), both of which represent different core structures (Figure S5) when compared to DG167 (Figure S1). Other compounds that showed binding to KasA are chlorpropamide and flubendazole, identified through a structure-based pharmacophore approach; lovastatin, identified through a shape-based pharmacophore; and compounds 47871032 and 89983431, identified through machine learning models. Using nanoDSF, in addition to sildenafil and triclosan (discussed above), Z915548488 and Z997831624 increased and Z1981674637 decreased Tm by almost 1 °C, respectively (Figure 1). These compounds were identified through a substructure similarity search with DG167. While being less pronounced, 91864296, identified through predictions based on the whole cell Mtb machine learning model, also appeared to show a decrease in Tm of note (Figure 1).
Differential scanning fluorimetry has been utilized primarily as a drug discovery method to identify promising lead compounds for a number of target proteins for decades.40 NanoDSF relies on the change of intrinsic tryptophan fluorescence at 330 and 350 nm, and Tm can be determined by measuring the ratio of the fluorescence at 330 and 350 nm against temperature. NanoDSF approaches are sensitive to the intrinsic fluorescence properties of the molecules present in the screen under examination, which can result in a wide variation in the background of thermal profiles—resulting in false negatives.44 Some compounds showed binding using the MST technique, but did not show significant changes in Tm, such as flubendazole, lovastatin, chlorpropamide, and 47871032. One potential explanation is that these compounds could bind to allosteric sites or not cause conformational changes that could increase or decrease the stability of the protein. Since nanoDSF is based on the intrinsic fluorescence of tryptophan, the binding of these compounds did not alter the exposure of tryptophan.
In conclusion, the combination of structure-based computational approaches combined with biophysical methods to validate binding to the proteins may provide additional information to build the structure–activity relationship for KasA and suggest additional molecules for in vitro and structural studies in the future.
Experimental Section
Materials
M. tuberculosis KasA was purified as previously described.16 Compounds 15407843, 25671189, 30253177, 30664114, 34490793, 40460913, 41386319, 47871032, 47939596, 54184949, 56835939, 63585074, 66085352, 66164428, 68068491, 85444047, 89807517, 89983431, 91864298, 92786999, and 86776267 were purchased from ChemBridge Corporation (San Diego, CA). Z1160866713, Z1914226114, Z1981674637, Z2027060424, Z2168541837, Z2185649167, Z2186204950, Z2186647917, Z2187106221, Z2187123225, Z2187298674, Z2191260537, Z2197830902, Z2198620308, Z2199688154, Z2297567622, Z872338402, Z915548396, Z915548454, Z915548488, Z915548528, Z915548646, Z915548688, Z915548714, Z997831398, Z997831408, Z997831442, and Z997831624 were purchased from Enamine Ltd. (Monmouth Jct., NJ). PB915625920 was purchased from UkrOrgSynthesis Ltd. (Kiev, Ukraine). Pyronaridine was purchased from BOC Sciences (Shirley, NY). Chlorpropamide, cisapride, flubendazole, haloperidol, lovastatin, metoclopramide, N-acetylprocainamide, sildenafil, streptomycin sulfate, and triclosan were purchased from MedChemExpress (MCE), and naloxone hydrochloride was purchased from TRC (Canada).
KasA Receptor-Based Pharmacophore
A receptor-based pharmacophore was generated for the DG167 crystal structure (PDB code 5W2O)16 using a method that has been previously described using Discovery Studio software (Biovia, San Diego, CA).41,42 The resulting pharmacophore was then used to search an FDA-approved drug library (MicroSource Spectrum). Several compounds (Figure S5, shown in orange) were selected for purchase and testing.
KasA Shape-Based Pharmacophore
Both DG167 molecules in the crystal structure PDB code 5W2O16 were used to build a combined 3D shape43 that was then used for shape-based searching in Discovery Studio software. This shape-based pharmacophore was also used to screen a library of >1000 FDA-approved drugs, and several molecules (Figure S5, shown in yellow) were selected that fit the shape-based pharmacophore volume well and were then selected for purchase.
Selection of Compounds with a Similar Substructure to DG167
As DG167 was previously shown to be a potent KasA binder, we utilized a substructure search of this compound in PubChem to identify other compounds that may target KasA in a similar manner. Sixteen compounds (Figure S5, shown in green) were found using this substructure search that was commercially available. These were purchased to determine their ability to bind to KasA.
Selection of Commercially Available Compounds Using a Combination of Machine Learning and Docking Studies
To identify additional compounds that may be active against Mtb, multiple vendor libraries from ChemBridge (Core, experimental, Lipinski), Enamine (advanced, diversity, HTS, screening), MCE (full), Octava (Drug-like green, In-house, lead-like), and Selleck (Bioactive, Bioactive II, Clinical, Drug repurposing, Express pick, FDA-approved) were scored for predicted activity in our previously published Assay Central Bayesian machine learning Mtb model32 with a 100 nM activity threshold cutoff (18,886 total, 645 active). A maximum model overlap threshold of 0.9 was applied to ensure diversity from the training set, but a minimum of 0.5 was used to enhance the model predictivity. In addition, a molecular weight (MW) cutoff of 500 Dal was used to remove large molecules. A chemical diversity filter selected the top 50 most diverse compounds out of the top 250 compounds predicted as active (≤ 0.5 prediction score). The “top” compounds were defined as those with the highest probability-like score to be “active” from each commercial library scored (14 libraries scored in all). As there were overlapping molecules in the commercial libraries, duplicates were removed and then the top 50 diverse molecules were selected from this concatenated data set. Each of these compounds were docked in the KasA crystal structure (PDB code 5W2O)16 using libdock in Discovery Studio. The 32 compounds (Figure S5, shown in red) chosen for purchase had a favorable libdock score of >95 (96.34–155.35), suggesting KasA may be a viable target for these molecules.
Thermal Shift Experiments Using NanoDSF
The compounds lovastatin, flubendazole, chlorpropamide, sildenafil, metoclopramide, triclosan, and streptomycin, identified through the shape-based pharmacophore analysis, and additional compounds identified through docking and machine learning studies described above, were tested using nanoDSF Prometheus (NanoTemper). A total of 60 compounds were assessed for KasA binding using this experimental technique. NanoDSF is a modified differential scanning fluorimetry method to determine the protein stability by employing intrinsic tryptophan or tyrosine fluorescence. This technique was used to verify if the molecules identified through shape-based pharmacophore searching and docking could bind to KasA and stabilize the protein by increasing the Tm. For the experiment, 10 μM of KasA in MST buffer (150 mM NaCl, 10 mM HEPES pH 7.4, 1 mM β-mercaptoethanol) was incubated with 100 μM of compounds and analyzed using Prometheus (NanoTemper).
Binding Studies Using MST
We also conducted MST to determine Kd for eight compounds using Monolith (NanoTemper). MST is a sensitive method that can be used to assess biomolecular interactions30 and has been utilized to study interactions between a variety of binding partners of various molecular sizes: protein–protein, antibody–antigen, protein–DNA, and protein–RNA interactions and the binding of ligand to ternary complexes.30,31
For KasA labeling, a Monolith His-Tag Labeling Kit RED-tris-NTA 2nd Generation kit (NanoTemper) was used according to the manufacturer’s protocol. In short, once the initial Kd of the his-tagged KasA protein was calculated to be ∼14 nM in PBS-T, the KasA protein stock was diluted to a concentration of 20× the Kd in the supplied PBS-T buffer. The protein and the dye were incubated with the dye for 30 min at room temperature, which was followed by a centrifugation of 15,000 g for 10 min (4 °C). The concentration after labeling was 200 nM. The labeled protein was kept on ice during the MST experiments. KasA binding affinity measurements were performed in the supplied PBS-T buffer. The final concentration of KasA in each assay was 10 nM. Each compound had an initial concentration of 10 mM and was serially diluted 15 times in 100% DMSO creating 16 independent stocks. For each experiment, 0.5 μL of the compound was transferred to 19.5 μL of labeled KasA. The final concentration of DMSO in the experiment was 2.5%. KasA was incubated in the presence of compounds for 20 min at room temperature in a nonbinding 384-well plate.
Acknowledgments
Dr. Alex M. Clark is acknowledged for assistance with the Assay Central, and Dr. Dinorah Leyva and Perry Ripa from NanoTemper are acknowledged for discussions and help with initial MST assays.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c04271.
Structure of DG167, nanoDSF results, a summary of approaches used to find compounds that bind to KasA, a complete list of molecules tested in this study, and ΔTm of compounds tested using nanoDSF (PDF)
Author Contributions
# A.C.P. and T.R.L. are joint first authors. A.C.P. and T.R.L. generated and analyzed the nanoDSF and MST data. T.R.L. performed the docking study. S.E. performed all pharmacophore approaches. T.R.L. and K.M.Z. performed the machine learning study. G.C.C., M.B.N., and J.S.F. provided the protein. S.E. drafted the manuscript. S.E. and J.S.F. obtained funding for the project. All authors were involved in contributing and editing the manuscript.
We kindly acknowledge the NIH funding NIAID R41AI13456 and NIGMS R44GM122196-02A1.
The authors declare the following competing financial interest(s): S.E. is the CEO of Collaborations Pharmaceuticals, Inc. A.C.P., T.R.L., P.A.V., and K.M.Z. are employees at Collaborations Pharmaceuticals, Inc. G.C.C., M.B.N., and J.S.F. do not have conflicts of interest.
Supplementary Material
References
- WHO Global tuberculosis report 2018. https://www.who.int/tb/publications/global_report/en/ (accessed March 3).
- Nagarajan S.; Whitaker P. Management of adverse reactions to first-line tuberculosis antibiotics. Curr. Opin. Allergy Clin. Immunol. 2018, 18, 333–341. 10.1097/ACI.0000000000000462. [DOI] [PubMed] [Google Scholar]
- Unissa A. N.; Subbian S.; Hanna L. E.; Selvakumar N. Overview on mechanisms of isoniazid action and resistance in Mycobacterium tuberculosis. Infect. Genet. Evol. 2016, 45, 474–492. 10.1016/j.meegid.2016.09.004. [DOI] [PubMed] [Google Scholar]
- Seaworth B. J.; Griffith D. E., Therapy of Multidrug-Resistant and Extensively Drug-Resistant Tuberculosis. Microbiol. Spectr. 2017,5 ( (2), ), 10.1128/microbiolspec.TNMI7-0042-2017. [DOI] [PubMed] [Google Scholar]
- Makarov V.; Salina E.; Reynolds R. C.; Kyaw Zin P. P.; Ekins S. Molecule Property Analyses of Active Compounds for Mycobacterium tuberculosis. J. Med. Chem. 2020, 63, 8917–8955. 10.1021/acs.jmedchem.9b02075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaradia L.; Lefebvre C.; Parra J.; Marcoux J.; Burlet-Schiltz O.; Etienne G.; Tropis M.; Daffe M. Dissecting the mycobacterial cell envelope and defining the composition of the native mycomembrane. Sci. Rep. 2017, 7, 12807. 10.1038/s41598-017-12718-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan P.; Tonge P. J. Targeting InhA, the FASII enoyl-ACP reductase: SAR studies on novel inhibitor scaffolds. Curr. Top. Med. Chem. 2012, 12, 672–693. 10.2174/156802612799984535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman M. S.; Cox J. S.; Jacobs W. R. Jr. A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol. Cell 2000, 5, 717–727. 10.1016/S1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- Dubnau E.; Chan J.; Raynaud C.; Mohan V. P.; Lanéelle M. A.; Yu K.; Quémard A.; Smith I.; Daffé M. Oxygenated mycolic acids are necessary for virulence of Mycobacterium tuberculosis in mice. Mol. Microbiol. 2000, 36, 630–637. 10.1046/j.1365-2958.2000.01882.x. [DOI] [PubMed] [Google Scholar]
- Bhatt A.; Molle V.; Besra G. S.; Jacobs W. R. Jr.; Kremer L. The Mycobacterium tuberculosis FAS-II condensing enzymes: their role in mycolic acid biosynthesis, acid-fastness, pathogenesis and in future drug development. Mol. Microbiol. 2007, 64, 1442–1454. 10.1111/j.1365-2958.2007.05761.x. [DOI] [PubMed] [Google Scholar]
- Kremer L.; Baulard A.R.; Besra G.S., Genetics of Mycolic Acid Biosynthesis. In Molecular Genetics of Mycobacteria, Hatfull G. F.; Jacobs W. R. Jr., Ed. ASM Press: 2000; 173–190. [Google Scholar]
- Vilchèze C.; Wang F.; Arai M.; Hazbón M. H.; Colangeli R.; Kremer L.; Weisbrod T. R.; Alland D.; Sacchettini J. C.; Jacobs W. R. Jr. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat. Med. 2006, 12, 1027–1029. 10.1038/nm1466. [DOI] [PubMed] [Google Scholar]
- Vilchéze C.; Morbidoni H. R.; Weisbrod T. R.; Iwamoto H.; Kuo M.; Sacchettini J. C.; Jacobs W. R. Jr. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J. Bacteriol. 2000, 182, 4059–4067. 10.1128/JB.182.14.4059-4067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath R. J.; Rock C. O. Fatty acid biosynthesis as a target for novel antibacterials. Curr. Opin. Investig. Drugs 2004, 5, 146–153. [PMC free article] [PubMed] [Google Scholar]
- Vilchéze C.; Hartman T.; Weinrick B.; Jain P.; Weisbrod T. R.; Leung L. W.; Freundlich J. S.; Jacobs W. R. Jr. Enhanced respiration prevents drug tolerance and drug resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 4495–4500. 10.1073/pnas.1704376114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Capodagli G. C.; Awasthi D.; Shrestha R.; Maharaja K.; Sukheja P.; Li S. G.; Inoyama D.; Zimmerman M.; Ho Liang H. P.; Sarathy J.; Mina M.; Rasic G.; Russo R.; Perryman A. L.; Richmann T.; Gupta A.; Singleton E.; Verma S.; Husain S.; Soteropoulos P.; Wang Z.; Morris R.; Porter G.; Agnihotri G.; Salgame P.; Ekins S.; Rhee K. Y.; Connell N.; Dartois V.; Neiditch M. B.; Freundlich J. S.; Alland D. Synergistic Lethality of a Binary Inhibitor of Mycobacterium tuberculosis KasA. MBio 2018, 9, e0210117 10.1128/mBio.02101-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.; Engels B. Clarification on the decarboxylation mechanism in KasA based on the protonation state of key residues in the acyl-enzyme state. J. Phys. Chem. B 2013, 117, 8095–8104. 10.1021/jp403067m. [DOI] [PubMed] [Google Scholar]
- Shetye G. S.; Franzblau S. G.; Cho S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl. Res. 2020, 220, 68–97. 10.1016/j.trsl.2020.03.007. [DOI] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Griffin J. E.; Gawronski J. D.; Dejesus M. A.; Ioerger T. R.; Akerley B. J.; Sassetti C. M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus M. A.; Gerrick E. R.; Xu W.; Park S. W.; Long J. E.; Boutte C. C.; Rubin E. J.; Schnappinger D.; Ehrt S.; Fortune S. M.; Sassetti C. M.; Ioerger T. R. Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. MBio 2017, 8, e0213316 10.1128/mBio.02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams K. A.; Chung C.-W.; Ghidelli-Disse S.; Rullas J.; Rebollo-López M. J.; Gurcha S. S.; Cox J. A. G.; Mendoza A.; Jiménez-Navarro E.; Martinez-Martinez M. S.; Neu M.; Shillings A.; Homes P.; Argyrou A.; Casanueva R.; Loman N. J.; Moynihan P. J.; Lelievre J.; Selenski C.; Axtman M.; Kremer L.; Bantscheff M.; Angulo-Barturen I.; Izquierdo M. C.; Cammack N. C.; Drewes G.; Ballell L.; Barros D.; Besra G. S.; Bates R. H. Identification of KasA as the cellular target of an anti-tubercular scaffold. Nat. Commun. 2016, 7, 12581. 10.1038/ncomms12581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager A. A.; Abood Z. S.; El-Amary W. M.; Bensaber S. M.; Al-Sadawe I. A.; Ermeli N. B.; Mohamed S. B.; Al-Forgany M.; Mrema I. A.; Erhuma M.; Hermann A.; Gbaj A. M. Design, Synthesis and Biological Evaluation of Some Triazole Schiff’s Base Derivatives as Potential Antitubercular Agents. Open Med. Chem. J. 2018, 12, 48–59. 10.2174/1874104501812010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. C.; Delorme V.; Bénarouche A.; Guy A.; Landry V.; Audebert S.; Pophillat M.; Camoin L.; Crauste C.; Galano J. M.; Durand T.; Brodin P.; Canaan S.; Cavalier J. F. Oxadiazolone derivatives, new promising multi-target inhibitors against M. tuberculosis. Bioorg. Chem. 2018, 81, 414–424. 10.1016/j.bioorg.2018.08.025. [DOI] [PubMed] [Google Scholar]
- Ballell L.; Bates R. H.; Young R. J.; Alvarez-Gomez D.; Alvarez-Ruiz E.; Barroso V.; Blanco D.; Crespo B.; Escribano J.; Gonzalez R.; Lozano S.; Huss S.; Santos-Villarejo A.; Martin-Plaza J. J.; Mendoza A.; Rebollo-Lopez M. J.; Remuinan-Blanco M.; Lavandera J. L.; Perez-Herran E.; Gamo-Benito F. J.; Garcia-Bustos J. F.; Barros D.; Castro J. P.; Cammack N. Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. ChemMedChem 2013, 8, 313–321. 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoyama D.; Awasthi D.; Capodagli G. C.; Tsotetsi K.; Sukheja P.; Zimmerman M.; Li S. G.; Jadhav R.; Russo R.; Wang X.; Grady C.; Richmann T.; Shrestha R.; Li L.; Ahn Y. M.; Ho Liang H. P.; Mina M.; Park S.; Perlin D. S.; Connell N.; Dartois V.; Alland D.; Neiditch M. B.; Kumar P.; Freundlich J. S. A Preclinical Candidate Targeting Mycobacterium tuberculosis KasA. Cell Chem. Biol. 2020, 27, 560–570. 10.1016/j.chembiol.2020.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto V. S.; Araujo J. S. C.; Silva R. C.; da Costa G. V.; Cruz J. N.; De A. N. M. F.; Campos J. M.; Santos C. B. R.; Leite F. H. A.; Junior M. C. S. In Silico Study to Identify New Antituberculosis Molecules from Natural Sources by Hierarchical Virtual Screening and Molecular Dynamics Simulations. Pharmaceuticals (Basel) 2019, 12, 36. 10.3390/ph12010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durairaj D. R.; Shanmughavel P. In Silico Drug Design of Thiolactomycin Derivatives Against Mtb-KasA Enzyme to Inhibit Multidrug Resistance of Mycobacterium tuberculosis. Interdiscip. Sci. 2019, 11, 215–225. 10.1007/s12539-017-0257-0. [DOI] [PubMed] [Google Scholar]
- Magnusson A. O.; Szekrenyi A.; Joosten H. J.; Finnigan J.; Charnock S.; Fessner W. D. nanoDSF as screening tool for enzyme libraries and biotechnology development. FEBS J. 2019, 286, 184–204. 10.1111/febs.14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel S. A. I.; Dijkman P. M.; Lea W. A.; van den Bogaart G.; Jerabek-Willemsen M.; Lazic A.; Joseph J. S.; Srinivasan P.; Baaske P.; Simeonov A.; Katritch I.; Melo F. A.; Ladbury J. E.; Schreiber G.; Watts A.; Braun D.; Duhr S. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 2013, 59, 301–315. 10.1016/j.ymeth.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerabek-Willemsen M.; André T.; Wanner R.; Roth H. M.; Duhr S.; Baaske P.; Breitsprecher D. MicroScale Thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 2014, 1077, 101–113. 10.1016/j.molstruc.2014.03.009. [DOI] [Google Scholar]
- Lane T.; Russo D. P.; Zorn K. M.; Clark A. M.; Korotcov A.; Tkachenko V.; Reynolds R. C.; Perryman A. L.; Freundlich J. S.; Ekins S. Comparing and Validating Machine Learning Models for Mycobacterium tuberculosis Drug Discovery. Mol. Pharmaceutics 2018, 15, 4346–4360. 10.1021/acs.molpharmaceut.8b00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiga M.; Agarwal N.; Ammerman N. C.; Gupta R.; Guo H.; Maiga M. C.; Lun S.; Bishai W. R. Successful shortening of tuberculosis treatment using adjuvant host-directed therapy with FDA-approved phosphodiesterase inhibitors in the mouse model. PLoS One 2012, 7, e30749 10.1371/journal.pone.0030749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiga M.; Ammerman N. C.; Maiga M. C.; Tounkara A.; Siddiqui S.; Polis M.; Murphy R.; Bishai W. R. Adjuvant host-directed therapy with types 3 and 5 but not type 4 phosphodiesterase inhibitors shortens the duration of tuberculosis treatment. J. Infect. Dis. 2013, 208, 512–519. 10.1093/infdis/jit187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec J.; Vilchéze C.; Lun S.; Perryman A. L.; Wang X.; Freundlich J. S.; Bishai W.; Jacobs W. R. Jr.; Kozikowski A. P. Biological evaluation of potent triclosan-derived inhibitors of the enoyl-acyl carrier protein reductase InhA in drug-sensitive and drug-resistant strains of Mycobacterium tuberculosis. ChemMedChem 2014, 9, 2528–2537. 10.1002/cmdc.201402255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh S. L.; Xiao G.; Tonge P. J. Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochemistry 2000, 39, 7645–7650. 10.1021/bi0008940. [DOI] [PubMed] [Google Scholar]
- Kremer L.; Dover L. G.; Morbidoni H. R.; Vilchéze C.; Maughan W. N.; Baulard A.; Tu S. C.; Honore N.; Deretic V.; Sacchettini J. C.; Locht C.; Jacobs W. R. Jr.; Besra G. S. Inhibition of InhA activity, but not KasA activity, induces formation of a KasA-containing complex in mycobacteria. J. Biol. Chem. 2003, 278, 20547–20554. 10.1074/jbc.M302435200. [DOI] [PubMed] [Google Scholar]
- Freundlich J. S.; Wang F.; Vilchéze C.; Gulten G.; Langley R.; Schiehser G. A.; Jacobus D. P.; Jacobs W. R. Jr.; Sacchettini J. C. Triclosan derivatives: towards potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. ChemMedChem 2009, 4, 241–248. 10.1002/cmdc.200800261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozzo R.; Kuo M.; Sidhu A. B. S.; Valiyaveettil J. T.; Bittman R.; Jacobs W. R. Jr.; Fidock D. A.; Sacchettini J. C. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J. Biol. Chem. 2002, 277, 13106–13114. 10.1074/jbc.M112000200. [DOI] [PubMed] [Google Scholar]
- Pantoliano M. W.; Petrella E. C.; Kwasnoski J. D.; Lobanov V. S.; Myslik J.; Graf E.; Carver T.; Asel E.; Springer B. A.; Lane P.; Salemme F. R. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen 2001, 6, 429–440. 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Olechno J.; Williams A. J. Dispensing processes impact apparent biological activity as determined by computational and statistical analyses. PLoS One 2013, 8, e62325 10.1371/journal.pone.0062325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meslamani J.; Li J.; Sutter J.; Stevens A.; Bertrand H. O.; Rognan D. Protein-ligand-based pharmacophores: generation and utility assessment in computational ligand profiling. J. Chem. Inf. Model. 2012, 52, 943–955. 10.1021/ci300083r. [DOI] [PubMed] [Google Scholar]
- Lamichhane G.; Freundlich J. S.; Ekins S.; Wickramaratne N.; Nolan S. T.; Bishai W. R. Essential metabolites of Mycobacterium tuberculosis and their mimics. MBio 2011, 2, 301–310. 10.1128/mBio.00301-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K.; Oerlemans R.; Groves M. R. Theory and applications of differential scanning fluorimetry in early-stage drug discovery. Biophys. Rev. 2020, 12 (1), 85–104. 10.1007/s12551-020-00619-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.