

Abstract
Drug discovery for disease‐modifying therapies for Alzheimer's disease and related dementias (ADRD) based on the traditional paradigm of experimental animal models has been disappointing. We describe the rationale and design of the Drug Repurposing for Effective Alzheimer's Medicines (DREAM) study, an innovative multidisciplinary alternative to traditional drug discovery. First, we use a systems biology perspective in the "hypothesis generation" phase to identify metabolic abnormalities that may either precede or interact with the accumulation of ADRD neuropathology, accelerating the expression of clinical symptoms of the disease. Second, in the "hypothesis refinement" phase we propose use of large patient cohorts to test whether drugs approved for other indications that also target metabolic drivers of ADRD pathogenesis might alter the trajectory of the disease. We emphasize key challenges in population‐based pharmacoepidemiologic studies aimed at quantifying the association between medication use and ADRD onset and outline robust causal inference principles to safeguard against common pitfalls. Candidate ADRD treatments emerging from this approach will hold promise as plausible disease‐modifying therapies for evaluation in randomized controlled trials.
Keywords: Alzheimer's disease, dementia, drug repurposing, pharmacoepidemiology
1. INTRODUCTION
Although rapid strides have been made in understanding the basic biology of Alzheimer's disease and related dementias (ADRD), this knowledge has not translated into effective treatments. The failures of phase III clinical trials 1 , 2 of disease‐modifying therapies for ADRD underscore the need to identify novel therapeutic targets for this neurodegenerative disease that currently affects > 5 million older Americans. 3 The current paradigm for drug discovery in ADRD relies predominantly on experimental animal models that recapitulate the features of ADRD pathology. 4 However, treatments targeting the amyloid plaque and/or neurofibrillary tangle pathologies identified in these model systems have not improved clinical outcomes for patients. 5 , 6 The high prevalence of plaque and tangles in cognitively normal older individuals suggests that targeting ADRD pathology rather than the early molecular triggers preceding accumulation of pathology and clinical symptoms may be ineffective. 7 , 8
An alternative approach to drug discovery in ADRD is hypothesis generation based on identifying existing treatments that target genetic or environmental regulators of early molecular determinants of ADRD pathology for therapeutic repurposing. 9 Pathways of interest may either trigger or enhance early ADRD neuropathology, accelerating the expression of clinical symptoms of disease. 10 The primary objectives of this review are to provide an overview of our "omics"‐based approach to identify dysregulated metabolic pathways that present plausible targets for drug repurposing in ADRD, review threats to validity in existing patient‐level pharmacoepidemiologic analyses of drugs for ADRD prevention or treatment, and propose solutions to overcome these threats. Collectively, the steps and principles described in this review form the rationale for the Drug Repurposing for Effective Alzheimer's Medicines (DREAM) study, which is an ongoing multidisciplinary collaborative study aimed at identifying drug repurposing candidates for ADRD (Figure 1).
FIGURE 1.
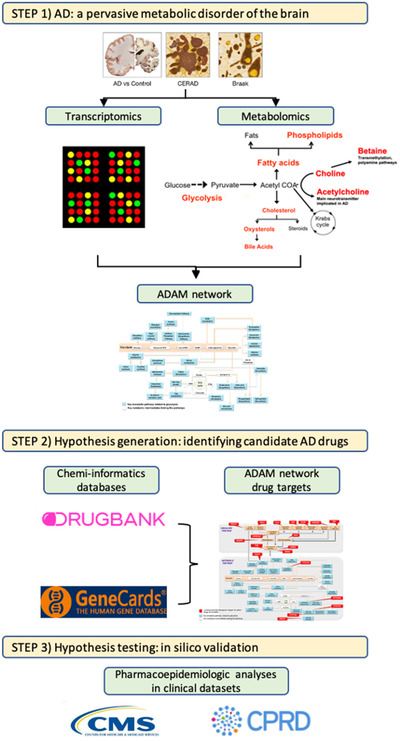
Drug Repurposing for Effective Alzheimer's Medicines (DREAM) study design. Schematic workflow of the DREAM study. Step 1: Alzheimer's disease (AD) is a pervasive metabolic disorder of the brain. Targeted metabolomics and transcriptomic analyses of brain tissue samples reveals dysregulation in multiple metabolic pathways related to abnormal glycolysis in AD. These pathways are proposed to be components of the Alzheimer's Disease Aberrant Metabolism (ADAM) network (see Figure 2a) and are associated with severity of AD pathology. Step 2: Hypotheses generation: identifying candidate drugs for Alzheimer's disease and related disorders (ADRD). Chemi‐informatics databases such as GeneCards and DRUGBANK are used to determine whether genetic regulators of biochemical reactions within the ADAM network (see Figure 2b) are targeted by approved drugs for non–ADRD‐related indications. Step 3: Hypotheses testing: in silico validation of candidate ADRD drugs. Pharmacoepidemiologic analyses in complementary population‐based clinical datasets (Centers for Medicare and Medicad Services, United States; Clinical Practice Research Datalink, United Kingdom) are used to test efficacy of candidate ADRD treatments
HIGHLIGHTS
The failures of phase III clinical trials of disease‐modifying therapies for Alzheimer's disease and related dementias (ADRD) underscore the need for exploring non‐traditional drug discovery approaches.
We propose an innovative approach for drug discovery focused on therapeutic repurposing in ADRD that integrates hypothesis generation based on metabolomics and transcriptomics analyses and hypothesis refinement based on pharmacoepidemiologic analyses.
We review threats to validity in existing patient‐level pharmacoepidemiologic analyses of drugs for ADRD prevention or treatment including immortal time bias, confounding, reverse causation, prevalent user bias, exposure, and outcome misclassification.
We further outline robust causal inference principles to safeguard against common pitfalls in non‐interventional studies of treatment effects on ADRD.
RESEARCH IN CONTEXT
Systematic review: We reviewed the literature using traditional (e.g., PubMed) sources to identify pharmacoepidemiologic studies and outlined common threats to validity including immortal time bias, confounding, reverse causation, prevalent user bias, exposure, and outcome misclassification in existing analyses focused on therapeutic repurposing of drugs for Alzheimer's disease and related dementias (ADRD) prevention or treatment.
Interpretation: We describe the rationale and design of the Drug Repurposing for Effective Alzheimer's Medicines (DREAM) study, an innovative multidisciplinary alternative to traditional drug discovery involving integration of hypothesis generation based on metabolomics and transcriptomics analyses and hypothesis refinement based on pharmacoepidemiologic analyses.
Future research: Repurposing candidates emerging from the DREAM study will merit confirmation in future mechanistic studies to elucidate potential mechanism of action and eventually in prospective randomized controlled trials of ADRD patients.
2. HYPOTHESIS GENERATION BASED ON THE ALZHEIMER'S DISEASE ABERRANT METABOLISM (ADAM) NETWORK
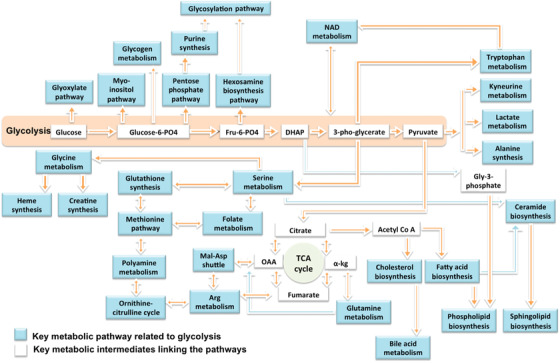
Studies of cohorts of older individuals, such as the Baltimore Longitudinal Study of Aging, have identified dysregulation in several metabolic pathways that are related to severity of ADRD neuropathology as well as symptomatic expression of the disease. 11 , 12 , 13 , 14 These studies suggest that ADRD is a pervasive metabolic disorder characterized by perturbations in multiple interacting biochemical pathways. Using quantitative metabolomic, proteomic, and transcriptomic analyses of brain tissue samples, we previously reported on dysregulation of glycolysis 11 and intermediate pathways linked to glycolytic substrates in ADRD. These biochemical pathways include phospholipid 14 and unsaturated fatty acid metabolism 13 as well as polyamine synthesis, transmethylation reactions, 12 and cholesterol catabolism through its conversion to oxysterols and bile acids. These findings have allowed us to define a broad spectrum of abnormal biochemical pathways originating from a primary defect in brain glycolysis during preclinical ADRD. In this paradigm, we hypothesize that abnormal glycolysis is a pivotal and "progenitor" metabolic perturbation that leads to secondary dysregulation within a diverse array of other biochemical pathways. This hypothesis has strong biologic plausibility given that intermediate metabolites of glycolysis enter many other biochemical pathways that have been well characterized and extensively studied in AD. 15 Thus, based on abnormal glycolysis as a primary progenitor pathway, we hypothesize that there may be > 20 related and distinct biochemical pathways that are dysregulated in the ADRD brain. We characterize these biochemical pathways as key components of the ADAM network (Figure 2a), defined by their relationship to abnormal brain glycolysis.
FIGURE 2.
A, The Alzheimer's Disease Aberrant Metabolism (ADAM) Network. A network of dysregulated metabolic pathways related to glycolysis and identified by targeted metabolomics and transcriptomics analyses in autopsy‐confirmed Alzheimer's disease and control brain samples. B, Drug targets in the ADAM Network. Genetic regulators of metabolic and signaling reactions in the ADAM network. Commonly prescribed approved drugs targeting these pathways will be tested by pharmacoepidemiologic analyses in the Drug Repurposing for Effective Alzheimer's Medicines study
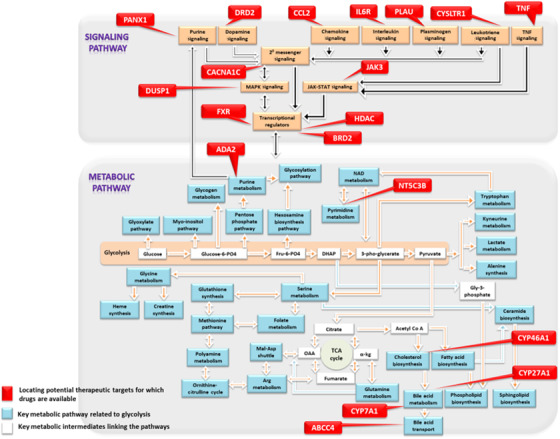
To further test the validity of our hypotheses about the role of perturbations in these metabolic pathways in ADRD, we generated a list of ≈ 250 genes that are known to regulate individual reactions within the component pathways of the ADAM network. Using two Gene Expression Omnibus (GEO) collections of autopsy‐confirmed AD (N = 29) and control brains (N = 56), we examined differential expression of these genes within the hippocampus and entorhinal cortex, two regions vulnerable to neurodegeneration where accumulation of pathology is believed to trigger onset of AD symptoms. We confirmed that 104 genes were differentially expressed in the AD hippocampus and/or entorhinal cortex (false discovery rate < 0.05) relative to controls, with several genes also validated with protein‐level data in brain tissue samples from the Baltimore Longitudinal Study of Aging. 16 , 17 This supports our conceptualization of ADRD as a pervasive metabolic disorder and allows us to undertake in silico drug repurposing studies in which the goal is to identify approved drugs that are known to target genetic regulators of biochemical reactions within abnormal metabolic pathways in the ADAM network.
3. IDENTIFYING FDA‐APPROVED DRUGS TARGETING ABNORMAL METABOLISM IN ADRD AS REPURPOSING CANDIDATES
We first categorized genetic regulators of biochemical reactions in the ADAM network as those involved in maintaining physiological levels of specific metabolites (ie, “metabolic pathway regulators”) and those participating in signaling cascades (ie, “signaling pathway regulators”; Figure 2b). Next, we queried two publicly available cheminformatics databases, DrugBank (https://www.drugbank.ca/) and GeneCards (https://www.genecards.org/), to identify U.S. Food and Drug Administration (FDA)‐approved drugs for non‐ADRD indications that target metabolic/signaling pathway regulators in the ADAM network. Using the ≈ 250 genetic regulators within the 20 dysregulated pathways in the ADAM network, we identified 35 FDA‐approved drugs as potential repurposing candidates.
4. PHARMACOEPIDEMIOLOGIC ANALYSES FOR HYPOTHESIS REFINEMENT USING ELECTRONIC PATIENT‐LEVEL HEALTH‐CARE DATA
Electronic patient‐level data collected during routine care are frequently used to generate actionable evidence regarding effectiveness, harm, use, and value of medications that supplements evidence generated in randomized controlled trials (RCTs), which are typically limited in scope owing to a relatively modest sample size, comparatively short follow‐up time, and underrepresentation of often the most relevant populations. 18 The unique strengths of routine health‐care data that make them ideal for evaluating hypotheses generated by molecular level predictions include their provision of large patient populations useful for detecting small differences, and the availability of a large number of patient factors recorded without recall bias, including demographics, comorbid conditions, and co‐medication use, that allow for high‐dimensional covariate adjustment to minimize confounding. 19 , 20 , 21 The DREAM study leverages two large population‐based data sources for pharmacoepidemiologic analyses: One is the insurance claims data from the Medicare program, a federal program in the United States that provides health insurance to people 65 years of age and older or disabled; and second is the Clinical Practice Research Datalink (CPRD) GOLD, which is a large UK primary care database containing de‐identified data on nearly 15 million people registered with >700 general practices from across the UK. 22 , 23 Separate implementation in complementary population‐based databases facilitates assessment of the robustness of the findings to differences in the underlying data structure (Medicare claims, CPRD electronic health records [EHRs]) and use patterns between different countries and health‐care systems (ie, United States vs UK). Of the 35 medications identified in the “hypothesis generation” phase, we selected 15 repurposing candidate medications (Table 1) for the pharmacoepidemiology‐based “hypothesis refinement” phase conditional on two key factors: (1) availability of sufficient patient data for meaningful evaluation (ie, exclusion of infrequently used medications), and (2) availability of an appropriate comparator treatment (as the reference group) that is used for the same underlying (non‐ADRD) indication as the repurposing candidate but not thought to be associated with ADRD.
TABLE 1.
Repurposing candidates in the Drug Repurposing for Effective Alzheimer's Medicines (DREAM) study
| Repurposing candidate | Target | Comparator drug | Original indications |
|---|---|---|---|
| Efavirenz a | CYP46A1 | Dolutegravir or nevirapine | Human immunodeficiency virus |
| Deferiprone/deferoxamine a | Iron | Deferasirox | Iron overload in thalassemia, myelodysplasia, sickle cell disease |
| Tofacitinib a | JAK3 | Abatacept | Rheumatoid arthritis |
| Tocilizumab a | IL‐6 | Abatacept | Rheumatoid arthritis |
| TNF‐inhibitors a | TNF | Abatacept | Rheumatoid arthritis |
| Dipyridamole b | ADA2 | Aspirin | Valve disorders for stroke prevention |
| Anastrozole | CYP27A1 | Exemestane and letrozole | Postmenopausal estrogen receptor‐positive breast cancer |
| PDE‐5 inhibitors | ABCC4 | Endothelin receptor antagonist | Pulmonary arterial hypertension |
| Valproic acid | HDAC | Lamotrigine | Epilepsy |
| Dihydropyridine calcium channel blockers | CACNA1C | Hydrochlorothiazide | Hypertension |
| Amiloride | PLAU | Triamterene | Hypertension |
| Salbutamol | DUSP1 | Long‐acting muscarinic antagonists | Chronic obstructive pulmonary disease |
| Probenecid a | PANX1 | Allopurinol | Gout |
| Montelukast | CYSLTR1 | Fluticasone | Asthma |
| Pentoxifylline | NT5C3B | Cilostazol | Peripheral arterial disease |
CPRD does not contain information on specialty medications and medications not approved by NHS; therefore, these analyses will only be conducted in Medicare claims.
Medicare claims do not capture over‐the‐counter medications (aspirin); therefore, this analysis will only be conducted in CPRD.
Abbreviations: CPRD, Clinical Practice Research Datalink; NHS, National Health Service.
5. THREATS TO VALIDITY IN PHARMACOEPIDEMIOLOGIC STUDIES EVALUATING RISK OF ADRD
In pharmacoepidemiologic studies conducted with existing data recorded in electronic sources, selection of patients for comparison may seem straightforward (eg, whether or not individuals receive a given treatment); however, many commonly observed biases can be traced back to specific design decisions at the outset of the study. To conceptualize these potential biases, it is most helpful to consider a RCT one would conduct to answer whether prescription medications prevent the occurrence of ADRD. People free of ADRD would be randomized to a treatment or a placebo; they would be encouraged to adhere well to their treatment assignment; and would be followed for many years until the onset of ADRD, death, or exit from the study for other reasons. Envisioning this target trial 24 helps us to anticipate how deviations from this ideal design will lead to biases as described below.
5.1. Immortal time bias
Immortal time bias, also referred to as immune person‐time bias when studying outcomes other than mortality, 25 , 26 is introduced when membership in a treatment exposure group at study index (follow‐up beginning date) is defined based on exposure occurring at a future time point during follow‐up. This makes it impossible to observe the outcome between the follow‐up start date and the actual exposure date among the exposed. A recent study 27 compared the risk of ADRD between patients who were prescribed a tumor necrosis factor (TNF) inhibitor any time in the study period prior to ADRD diagnosis versus those who were never prescribed these drugs in the study period. These “ever user” versus “never user” comparisons may be affected by immortal time bias because patients who are diagnosed with ADRD early on in the follow‐up period have less opportunity to be classified into the treatment group and thus, more cases end up in the “never user” group (Figure 3). Misclassifying immortal person‐time as exposed time can lead to estimates suggesting an exaggerated beneficial effect. 25
FIGURE 3.
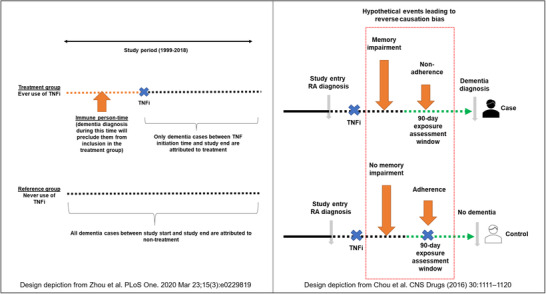
Immortal time bias (left) and reverse causation bias (right) in recent pharmacoepidemiologic studies evaluating associations between tumor necrosis factor (TNF)‐inhibitor use and the risk of Alzheimer's disease and related dementia (ADRD). Zhou et al. (left) compared the risk of ADRD between patients who were prescribed a TNF‐inhibitor any time in the study period prior to ADRD diagnosis versus those who were never prescribed these drugs in the study period. This “ever user” versus “never user” comparison is affected by immortal time bias because patients who are diagnosed with ADRD early on in the follow‐up period have less opportunity to be classified into the treatment group and thus, more cases end up in the “never user” group. Chou et al. (right) compared the risk of ADRD between individuals who received TNF‐inhibitor treatment versus those who did not in a 3‐month exposure assessment window before an AD diagnosis. This design is prone to substantial bias from reverse causation leading to spurious protective effect estimates because cognitive decline is plausibly accompanied by a decline in medication adherence
5.2. Confounding
To generate valid inferences about treatment effects in the absence of baseline randomization, one must account for the differences between treated patients and reference patients in the distributions of pre‐exposure confounding variables; that is, those variables that are risk factors for ADRD and affect the likelihood of treatment or treatment choice. 28 While confounding is a universal concern for all non‐interventional studies, comparisons of users of a particular treatment versus non‐users, which has been a common approach for estimating treatment effects on ADRD risk in previous studies, 27 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 are especially vulnerable because patient characteristics may substantially differ between treated and untreated patients. For instance, biologic disease‐modifying treatments for rheumatoid arthritis are highly effective but are also associated with serious adverse events such as infections; therefore, it is possible that in older age, these treatments are selectively used in “healthier” patients with longer life expectancy and withheld from patients with greater frailty or higher comorbidity burden. 38 Systematic differences resulting from such designs are likely to distort the association between treatment and ADRD risk.
5.3. Reverse causation
Reverse causation is an important consideration when studying ADRD because of a potentially prolonged preclinical phase of disease before diagnosis. 39 , 40 In research on treatments and ADRD risk, cognitive decline is plausibly accompanied by a decline in medication adherence. Therefore, comparisons of periods of medication use versus non‐use for the risk of ADRD could produce a misleading protective association. 41 In a case‐control study, Chou et al. 32 compared the risk of ADRD between individuals who received TNF‐inhibitor treatment versus those who did not in a 3‐month exposure assessment window before an AD diagnosis. This design is prone to substantial bias from reverse causation leading to spurious protective effect estimates due to reverse causation (Figure 3).
5.4. Prevalent user bias because of left‐truncated data
Prevalent users of a medication are often defined as those individuals who have been receiving treatment for some length of time before the start of follow‐up. 42 Including prevalent users in a study aimed at evaluating the association between a treatment and ADRD is similar to excluding the first few years of follow‐up after the start of randomization in an RCT of treatment initiators. It poses several threats to accurate effect estimation. First, if the treatment effect on the outcome of ADRD varies over time, any early effect—beneficial or harmful—shortly after starting the drug would be missed and the net overall treatment effect would be biased. 43 Second, confounding adjustment for risk factors measured at the beginning of the study may already be influenced by the patients’ treatment history and as such introduce bias in treatment effect estimation. 42 The direction and magnitude of bias due to inclusion of prevalent users vary depending on the context.
5.5. Exposure misclassification
Use of electronic health‐care data from routine clinical care to evaluate the risk of ADRD after exposure to a certain treatment has gained widespread use because of availability of medication dispensing or prescribing data, which eliminates the threat of recall bias. However, adherence to chronic medication is often suboptimal due to barriers such as costs, complexity of the regimen, and limited health literacy. 44 Therefore, studies in which follow‐up is continued indefinitely after treatment initiation has the potential to introduce substantial misclassification of non‐exposed person‐time as exposed time, which could dilute the treatment effect estimates. For example, in a study comparing the use of angiotensin receptor blockers (ARBs) versus non‐use, Chiu et al. 29 followed ARB initiators for an average of 9.4 years for the outcome of dementia; however, the average time on ARB treatment during follow‐up was only 2.9 years. This approach assumes that the purported benefit of ARB use on the risk of dementia is preserved long after treatment is discontinued.
5.6. Outcome misclassification
Electronic health‐care data such as insurance claims or electronic medical records capture ADRD diagnosis with varying degrees of accuracy. Clinically, it is often difficult to pinpoint the precise time of ADRD onset, which manifests slowly with gradually increasing extent and may not get coded as ADRD by physicians until long into the disease course. Therefore, all pharmacoepidemiologic studies evaluating association between treatment use and ADRD risk are subject to bias to varying degrees due to outcome misclassification. For example, in studies comparing treatment users versus non‐users, this misclassification is likely differential between exposure groups because treated patients conceivably have greater contact with the health‐care system and, hence, greater probability of their symptoms being noted and recorded sooner by physicians than untreated patients. In studies comparing two active treatments, misclassification may still be present, but it is less likely differential between exposure groups. In the presence of differential misclassification, both absolute and relative measures of treatment effects are biased. Many previously published studies, 27 , 29 , 31 , 32 , 33 , 35 , 45 have taken ADRD recording in insurance claims or electronic medical records at face value without recognizing these limitations or varying outcome definitions in sensitivity analyses to investigate the impact of this misclassification on study results.
6. APPROACHES TO REDUCE BIAS IN NON‐INTERVENTIONAL STUDIES OF TREATMENT EFFECTS ON ADRD
The threats to validity described above have the potential to substantially distort study results. When multiple threats are present, it is impossible to gauge the magnitude of their impact on study results because they may bias the estimates in different directions (toward the null or away from the null). Therefore, adherence to robust causal inference principles is critically important for generating reliable and actionable evidence from pharmacoepidemiologic studies investigating the complex relationship between drug treatments and ADRD risk. In this section, we describe features of the DREAM study designed to mitigate bias threats. While most of these features are well described in general pharmacoepidemiology literature, we outline how each of these feature addresses challenges that are unique to ADRD drug repurposing research. Another key strength of the DREAM study is pre‐registration of study protocols on clinicaltrials.gov prior to data analysis for each comparison. 46 , 47 , 48 Pre‐registration of detailed study protocols, including hypotheses being tested and operational definitions of variable measurement, safeguards against publication bias and “data dredging” until a significant association is obtained, 49 , 50 both of which can undermine the credibility of non‐interventional studies of drug repurposing candidates conducted using existing health‐care data.
6.1. Basic study design
We will implement a new‐user, active‐comparator, cohort study design for all analyses. 51 This general study design framework can be conceptualized as emulating principles of a clinical trial comparing two treatments with observational data and is well suited for health‐care database analyses. 24 , 52 Focusing on new users allows evaluation of the effect of drug exposure on ADRD risk throughout the treatment course and precludes the threat from prevalent user bias. 42 By beginning follow‐up at treatment initiation and using an active comparison design, this design also safeguards against immortal‐time bias. 53 , 54 Further, appropriately selected active comparators minimize concerns related to confounding as well as differential outcome misclassification.
Figure 4 graphically summarizes the general study design. We will design a series of non‐interventional cohort studies using this design to assess the comparative risk of ADRD after exposure to medications of interest versus a pre‐specified reference treatment (unique to each analysis; Table 1). Patients will enter the cohort on a date of recorded use of either the treatment of interest or the reference treatment identified from Medicare claims or CPRD records and this date will be defined as the “cohort entry date.” Exposure to medications of interest will be defined based on dispensing records from Medicare claims and prescription orders recorded in CPRD. 23 Both data sources provide information regarding the date of prescription and quantity of medications, which will allow us to identify switching of treatments as well as discontinuation.
FIGURE 4.
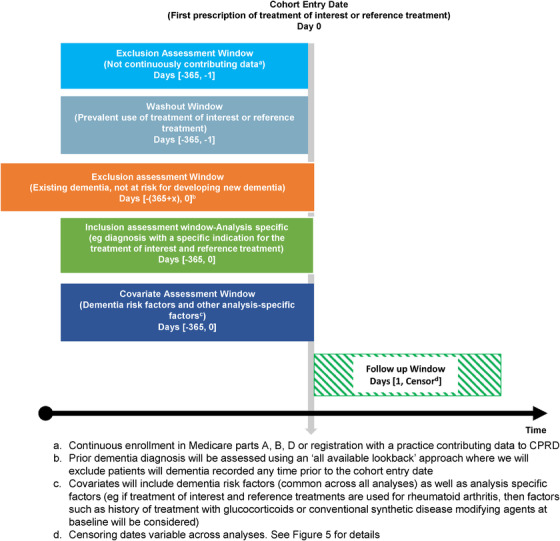
General study design for pharmacoepidemiologic analyses. Non‐interventional active comparator new‐user cohort study design is pictured. Patients enter the cohort on a date ("cohort entry date") of recorded use of either the treatment of interest or the reference treatment without previous use of these medications in a year prior. Patients with diagnosis codes indicating Alzheimer's disease and related dementia (ADRD) or medications for treatment of ADRD any time prior to the cohort entry date are excluded to solely enroll patients at risk of new‐onset ADRD. Finally, analysis‐specific inclusion criteria based on the primary indication for the treatment of interest and the reference treatment are implemented. For instance, if the treatment of interest and reference treatment are used for rheumatoid arthritis, we will only include patients with recorded rheumatoid arthritis diagnoses in the 365 days before the cohort entry date. Covariates of interest will also be measured in the 365 days before the cohort entry date. Outcome assessment is conducted after cohort entry date
Patients not continuously contributing data to Medicare or CPRD over the 365 days before cohort entry will be excluded. This exclusion will ensure availability of sufficient data regarding health‐care use to assess other inclusion and exclusion criteria as well as covariate information for all included patients. Next, we will focus on new users of the treatments of interest or the reference treatments by excluding patients with any record of these treatments in the 365 days before the cohort entry date. Further, we will also exclude patients with diagnosis codes indicating ADRD or medications for treatment of ADRD any time prior to the cohort entry date so as to solely enroll patients at risk of new‐onset ADRD. Finally, we will use analysis‐specific inclusion criteria based on the primary indication for the treatment of interest and the reference treatment. For instance, if the treatment of interest and reference treatment are used for rheumatoid arthritis, we will only include patients with recorded rheumatoid arthritis diagnoses in the 365 days before the cohort entry date. Covariates of interest will also be measured in the 365 days before the cohort entry date.
The outcome of ADRD will be defined in this study based on one inpatient or two outpatient claims using International Classification of Diseases (ICD) codes from Medicare claims and corresponding Read codes from CPRD. When validated against a structured in‐home dementia assessment, Medicare claims‐based dementia identification is reported to have a positive predictive value (PPV) in the range of 65% to 78%. 55 Corresponding Read code‐based definitions from CPRD that have been validated against information obtained from general practitioner questionnaires with PPVs ranging from 74% to 100%. 56 , 57 , 58 , 59 Of note, these measurement characteristics are for defining prevalent ADRD; their performance in identifying new‐onset ADRD, which is the outcome of interest in most pharmacoepidemiologic investigations, is unknown and presumably weaker.
6.2. Special design modifications to address specific bias threats
To accommodate various uncertainties involved in pharmacoepidemiologic investigations focused on ADRD risk, we will use four alternative analyses (Figure 5) with equal priority:
FIGURE 5.
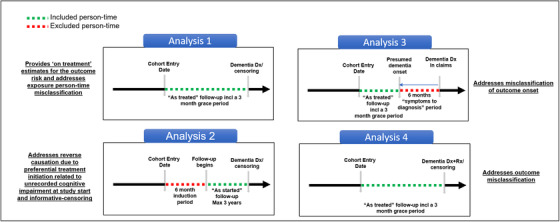
Specific design considerations to address unique challenges in using observational health‐care data sources to evaluate associations between medication use and the risk of Alzheimer's disease and related dementia (ADRD). Analysis 1 uses an “As‐treated” follow‐up approach, in which follow‐up will start on the day after the cohort entry date and censoring at follow‐up will occur at treatment discontinuation or switch (to comparator treatment) to provide estimates of “on treatment” risk of newly occurring ADRD for patients initiating the exposure of interest. Analysis 2 uses an “As‐started” follow‐up approach incorporating a 6‐month “induction” period, in which patients will be followed for a maximum of 3 years regardless of subsequent treatment changes or discontinuation, similar to an intent‐to‐treat approach in randomized trials. Analysis 3 incorporates a 6‐month “symptoms to diagnosis” period in which outcome date assigned is 6 months before the first recorded ADRD date to incorporate a “symptoms to diagnosis” period to address the potential bias due to misclassification of ADRD onset. Analysis 4 uses an alternative outcome definition including at least one diagnosis code combined with a prescription for a symptomatic treatment (donepezil, galantamine, rivastigmine, and memantine) to improve the specificity of outcome definition
6.2.1. Analysis 1: “As‐treated” follow‐up approach
In the first analysis, follow‐up will start on the day after the cohort entry date and censoring at follow‐up will occur at treatment discontinuation or switch (to comparator treatment) to provide estimates of “on treatment” risk of newly occurring ADRD for patients initiating the exposure of interest. 51 This approach minimizes the potential bias due to exposed person‐time misclassification. We will incorporate a 90‐day “grace period” after the end of the expected days‐supply of the most recently filled prescription to define the treatment discontinuation date to accommodate the possibility that patients may not immediately re‐fill their prescriptions due to availability of leftover medications from prior prescriptions as a result of suboptimal adherence.
6.2.2. Analysis 2: “As‐started” follow‐up approach incorporating a 6‐month “induction” period
While the “as treated” approach described in Analysis 1 addresses the issue of potential misclassification of exposed person‐time, it is vulnerable to bias due to reverse causation. Reverse causation can manifest in two ways in “as‐treated” analyses. First, if a treatment is preferentially started after problems with memory occurred, but before an ADRD diagnosis is recorded in the EHRs, then beginning follow‐up immediately after treatment initiation can lead to bias due to reverse causation. Second, if patients discontinue or if physicians de‐prescribe the treatments under consideration because of memory problems associated with ADRD, but the diagnosis is not recorded in the EHRs until after the end of follow‐up due to treatment‐related censoring, this informative censoring may also introduce bias due to reverse causation because events occurring in the period after censoring are not attributed to the treatment.
To address these challenges, we will conduct an “as‐started” analysis, in which patients will be followed for a maximum of 3 years regardless of subsequent treatment changes or discontinuation, similar to an intent‐to‐treat approach in RCTs. By not censoring on treatment changes, this design avoids reverse causation bias due to informative censoring. We will also incorporate a 6‐month induction period after the cohort entry date before beginning the follow‐up for ADRD. Incorporating a 6‐month induction period achieves two objectives. First, by excluding outcome events occurring in this period, this design avoids potential reverse causation bias due to preferential treatment initiation related to unrecorded cognitive impairment. Second, incorporating such an induction period in an “as‐started” analysis requires patients to be on treatment for at least 6 months before follow‐up begins, which limits misclassification of unexposed person‐time as exposed because patients who discontinue treatment after just one or two prescriptions do not contribute to the analysis.
6.2.3. Analysis 3: Incorporating a 6‐month “symptoms to diagnosis” period
In Analysis 3, we will assign an outcome date that is 6 months before the first recorded ADRD date to incorporate a “symptoms to diagnosis” period to address the potential bias due to misclassification of ADRD onset. For those who are censored without an event, we will move the censoring date 6 months prior to incorporate the possibility that these patients may have symptoms of ADRD but no diagnosis in this period.
6.2.4. Analysis 4: Alternative outcome definition
To reduce the possibility of bias due to outcome misclassification, in Analysis 4, we will use an alternative definition for incident ADRD including at least one diagnosis code combined with a prescription for a symptomatic treatment (donepezil, galantamine, rivastigmine, and memantine). Use of medication records to identify dementia has been reported to result in >95% PPV in a previous validation study. 60
6.3. Rigorous confounding adjustment
We will identify a large number of covariates for confounding adjustment as summarized in Table 2. This will include patient demographic factors such as age, sex, and race. Pre‐exposure risk factors for dementia identified in previous studies such as diabetes, stroke, and depression, 61 , 62 , 63 will be included in the risk adjustment to ensure a similar baseline risk for dementia between the treatment and reference groups. We will also adjust for lifestyle factors such as smoking as well as use of preventive services, including screening mammography and vaccinations, to account for healthy‐user effects. 64 We would expect certain lifestyle factors to be more completely recorded within CPRD. 65 Further, we will create measures for use of various health‐care services before cohort entry including number of distinct prescriptions filled, number of emergency department visits, hospitalizations, and number of physician office visits to account for patients’ general health and contact with the health‐care system to minimize the possibility of differential surveillance. 66 Frailty indicators based on composite scoring schemes 67 , 68 as well as factors such as falls and fractures will be considered for inclusion to address potential confounding by frailty.
TABLE 2.
General covariates for confounding adjustment in pharmacoepidemiologic investigations of Alzheimer's disease and related dementias
| Category | Potential confounders |
|---|---|
| Demographics | Age, sex, race, region, calendar year of index date |
| Dementia risk factors a , 61 , 62 , 63 | Diabetes, obesity, hyperlipidemia, hypertension, stroke, coronary artery disease, depression, anxiety, bipolar disorder, schizophrenia |
| Lifestyle factors and markers for healthy behavior | Smoking, drug abuse, alcohol abuse, mammography, colonoscopy, fecal occult blood test, influenza vaccination, pneumococcal vaccination, herpes zoster vaccination, BMD test |
| Health‐care use measures | Number of distinct generic agents used, number of emergency room visits, number of hospitalizations, number of physician office visits |
| Frailty markers | A composite frailty score, 67 , 68 cancer, osteoporosis, fractures, falls, use of supplemental oxygen, comorbidity score 81 , 82 |
| Socioeconomic status (SES) | Area‐level SES information covering occupation, income, wealth, education, and housing based on linking U.S. Census data with claims on zip‐code level in Medicare 70 ; Medicare‐Medicaid dual eligibility; English Index of Multiple Deprivation for CPRD |
Diagnosis codes and medications commonly used for these conditions will be identified (eg, benzodiazepine for anxiety, insulin and oral hypoglycemic agents for diabetes).
Abbreviations: BMD, bone mineral density; CPRD, Clinical Practice Research Datalink.
Confounding by socioeconomic status is important in studies focused on ADRD risk. Detailed information regarding patients’ socioeconomic factors is not recorded in administrative claims or EHR data. Therefore, neighborhood socioeconomic characteristics will be identified at the zip code level from the American Community Survey (ACS) data collected by the U.S. Census Bureau and linked to Medicare claims. 69 We will measure and adjust for the following characteristics: (1) percentage of unemployment, (2) percentage below the poverty line, (3) percentage of persons ages >25 years with < 12th‐grade education, (4) percentage of persons ages >25 years with at least 4 years of college, (5) median value of owner‐occupied homes, and (6) median household income. 70 Additionally, we will also use dual Medicaid‐Medicare eligibility as a proxy for lower socioeconomic status among the Medicare beneficiaries. For CPRD, neighborhood socioeconomic characteristics will be identified using the English Index of Multiple Deprivation (IMD). 71 IMD is a composite measure derived from indicators covering several domains of material deprivation, including income, employment, education and skills, health, housing, crime, access to services, and living environment. IMD is available for linkage to CPRD GOLD. 72
In addition to these factors that will be considered for inclusion in all analyses, to ensure similar disease severity at baseline between treatment groups, we will consider additional factors in the adjustment set for each analysis based on the primary indication of the repurposing candidate drug and the reference treatment. For instance, when the repurposing candidate is used for rheumatoid arthritis, rheumatoid arthritis disease activity‐related factors such as steroid use and steroid dose will be considered.
After defining these covariates, we will use a propensity score (PS) 73 –based approach to account for measured confounding in this study. The PS will be calculated as the predicted probability of initiating the exposure of interest (ie, the repurposing candidate) versus the reference drug conditional on baseline covariates using multivariable logistic regression constructed separately in each data source. On average, patients with similar PSs have similar distribution of potential confounders used to estimate the PS. Therefore, analyses conditioned on the PS provide effect estimates that are free from measured confounding. For all our analyses, initiators of each exposure of interest will be matched with initiators of the reference exposure based on their PS within each data source. 74 Pair matching will be conducted using a nearest‐neighbor algorithm, which seeks to minimize the distance between propensity scores in each pair of treated and reference patients, 75 and a caliper of 0.025 on the natural scale of the PS will be used to ensure similarity between the matched patients. 76 We will use multiple diagnostics for evaluating the adequacy of PS matching to control for confounding. First, we will evaluate the PS distributional overlap between two groups to ensure comparability of these groups. 77 In the case of substantial non‐overlap, we will seek to modify the design by either considering a different reference exposure group or by considering additional exclusion criteria to make the study populations more homogenous. 78 Next, we will assess balance in each individual covariate between two treatment groups using standardized differences and will assess the post‐matching c‐statistic as a global test of balance. 79 In the case of residual imbalances, we will attempt to reconstruct the PS model with higher‐order terms or interactions involving the imbalanced covariates and achieve balance.
6.4. Outcome analysis
Incidence rates for the outcome of ADRD will be estimated for the treatment and reference groups for each comparison separately in each database before and after PS matching. The competing risk of death could be of concern for the current set of analyses if mortality is frequent among patients included in the cohort and if differences in the risk of mortality between treatment and reference groups are substantial. In the PS‐matched sample, we will use cause‐specific hazard models 80 to provide hazard ratios averaged over the entire follow‐up period as well as interval specific hazard ratios (1, 2, and 3 years) for the association between the treatment of interest and risk of ADRD after considering all‐cause mortality as a competing event. Pre‐specified subgroup analyses will be conducted based on age, sex, and baseline cardiovascular disease. Each of these analyses will be conducted separately in Medicare and CPRD database and will be pooled using random effects meta‐analytic methods. Heterogeneity in estimates will be evaluated using I2 statistics and pooled estimates will be reported in the absence of substantial heterogeneity (no heterogeneity defined as I2 <40%).
7. COMMENT
The DREAM study aims to identify drug repurposing candidates for ADRD based on metabolic abnormalities that may precede or interact with the accumulation of neuropathology leading to the eventual expression of clinical symptoms. Integration of a hypothesis generation step based on metabolomics and transcriptomics analyses 11 , 12 , 13 , 14 and a subsequent hypothesis refinement step based on rigorous pharmacoepidemiologic analyses of two population‐based health‐care databases is a unique strength of this study.
Population‐based pharmacoepidemiologic analyses designed with robust causal inference principles will serve a critical role in prioritizing candidates for drug repurposing. The approach described here addresses key concerns observed in previous studies that may have led to misleading findings. Multiple design variations in the DREAM study as described above address a range of concerns and will together provide a robust assessment of the likely treatment effects. While we hope to identify a potential signal of treatment benefit, a convincing demonstration of no treatment benefit would be a useful addition to the literature to prevent wasteful resource use on future interventional studies testing unfounded hypotheses.
In summary, for diseases with a high need for disease modifying pharmacologic treatment such as ADRD, finding potential medication candidates among already marketed drugs for other indications remains a goal worthy of pursuing. The DREAM study represents a one‐of‐a‐kind initiative to integrate “omics”‐based drug discovery approaches with analyses of patient‐level data from large longitudinal health‐care databases using state‐of‐the‐art pharmacoepidemiologic methods to facilitate identification of drug repurposing candidates for ADRD.
FUNDING INFORMATION
The DREAM study is funded by the intramural program of the National Institute on Aging.
CONFLICTS OF INTEREST
Dr. Desai reports serving as principal investigator on research grants from Bayer, Vertex, and Novartis to the Brigham and Women's Hospital for unrelated projects. Dr. Gerhard reports grants from NIA during the conduct of the study; grants and personal fees from Bristol‐Myers Sqibb; personal fees from Merck, Pfizer, Lilly, IntraCellular Therapies, and Eisai. All are outside the submitted work. Dr. S. Varma does contract statistical analysis under the business name of “HiThru Analytics LLC”; currently he is working as a contractor, part time, with the National Institutes of Health (Bethesda, MD) and part time with Bristol Myers Squibbs (Lawrenceville, NJ). Dr. Schneeweiss is co‐principal investigator of investigator‐initiated grants to the Brigham and Women's Hospital from Boehringer Ingelheim unrelated to the topic of this study. He is a consultant to Aetion Inc., a software manufacturer of which he owns equity. His interests were declared, reviewed, and approved by the Brigham and Women's Hospital and Partners HealthCare System in accordance with their institutional compliance policies. Dr. Kim has received research grants to the Brigham and Women's Hospital from Pfizer, Roche, AbbVie, and Bristol‐Myers Squibb for unrelated topics. Other authors declare no conflicts.
Desai RJ, Varma VR, Gerhard T, et al. Targeting abnormal metabolism in Alzheimer's disease: The Drug Repurposing for Effective Alzheimer's Medicines (DREAM) study. Alzheimer's Dement. 2020;6:e12095 10.1002/trc2.12095
Contributor Information
Rishi J. Desai, Email: rdesai@bwh.harvard.edu.
Madhav Thambisetty, Email: thambisettym@mail.nih.gov.
REFERENCES
- 1. Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug‐development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yiannopoulou KG, Anastasiou AI, Zachariou V, Pelidou SH. Reasons for failed trials of disease‐modifying treatments for Alzheimer disease and their contribution in recent research. Biomedicines. 2019;7(4):97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010‐2050) estimated using the 2010 census. Neurology. 2013;80(19):1778‐1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Godyn J, Jonczyk J, Panek D, Malawska B. Therapeutic strategies for Alzheimer's disease in clinical trials. Pharmacol Rep. 2016;68(1):127‐138. [DOI] [PubMed] [Google Scholar]
- 5. Iqbal K, Liu F, Gong CX. Alzheimer disease therapeutics: focus on the disease and not just plaques and tangles. Biochem Pharmacol. 2014;88(4):631‐639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Panza F, Logroscino G, Imbimbo BP, Solfrizzi V. Is there still any hope for amyloid‐based immunotherapy for Alzheimer's disease. Curr Opin Psychiatry. 2014;27(2):128‐137. [DOI] [PubMed] [Google Scholar]
- 7. Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community‐based studies. Neurology. 2006;66(12):1837‐1844. [DOI] [PubMed] [Google Scholar]
- 8. Castellani RJ, Perry G. The complexities of the pathology‐pathogenesis relationship in Alzheimer disease. Biochem Pharmacol. 2014;88(4):671‐676. [DOI] [PubMed] [Google Scholar]
- 9. Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18(1):41‐58. [DOI] [PubMed] [Google Scholar]
- 10. Thambisetty M. Understanding mechanisms and seeking cures for Alzheimer's disease: why we must be “extraordinarily diverse”. Am J Physiol Cell Physiol. 2017;313(4):C353‐C361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. An Y, Varma VR, Varma S, et al. Evidence for brain glucose dysregulation in Alzheimer's disease. Alzheimers Dement. 2018;14(3):318‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mahajan UV, Varma VR, Griswold ME, et al. Dysregulation of multiple metabolic networks related to brain transmethylation and polyamine pathways in Alzheimer disease: a targeted metabolomic and transcriptomic study. PLoS Med. 2020;17(1):e1003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Snowden SG, Ebshiana AA, Hye A, et al. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: a nontargeted metabolomic study. PLoS Med. 2017;14(3):e1002266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Varma VR, Oommen AM, Varma S, et al. Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: a targeted metabolomics study. PLoS Med. 2018;15(1):e1002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berg JM, Tymoczko JL, Stryer L. Biochemistry. New York: Freeman; 2003. [Google Scholar]
- 16. Johnson ECB, Dammer EB, Duong DM, et al. Large‐scale proteomic analysis of Alzheimer's disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. 2020;26(5):769‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson ECB, Dammer EB, Duong DM, et al. Deep proteomic network analysis of Alzheimer's disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol Neurodegener. 2018;13(1):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schneeweiss S, Eichler HG, Garcia‐Altes A, et al. Real world data in adaptive biomedical innovation: a framework for generating evidence fit for decision‐making. Clin Pharmacol Ther. 2016;100(6):633‐646. [DOI] [PubMed] [Google Scholar]
- 19. Schneeweiss S, Avorn J. A review of uses of health care utilization databases for epidemiologic research on therapeutics. J Clin Epidemiol. 2005;58(4):323‐337. [DOI] [PubMed] [Google Scholar]
- 20. Schneeweiss S, Rassen JA, Glynn RJ, Avorn J, Mogun H, Brookhart MA. High‐dimensional propensity score adjustment in studies of treatment effects using health care claims data. Epidemiology. 2009;20(4):512‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brilliant MH, Vaziri K. Mining retrospective data for virtual prospective drug repurposing: l‐DOPA and age‐related macular degeneration. Am J Med. 2016;129(3):292‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wishart DS, Wu A. Using drugbank for in silico drug exploration and discovery. Curr Protoc Bioinformatics. 2016;54:14.. [DOI] [PubMed] [Google Scholar]
- 23. Herrett E, Gallagher AM, Bhaskaran K, et al. Data resource profile: clinical Practice Research Datalink (CPRD). Int J Epidemiol. 2015;44(3):827‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hernan MA, Sauer BC, Hernandez‐Diaz S, Platt R, Shrier I. Specifying a target trial prevents immortal time bias and other self‐inflicted injuries in observational analyses. J Clin Epidemiol. 2016;79:70‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suissa S. Immortal time bias in pharmacoepidemiology. Am J Epidemiol. 2007;167(4):492‐499. [DOI] [PubMed] [Google Scholar]
- 26. Lash TL, Cole SR. Immortal person‐time in studies of cancer outcomes. J Clin Oncol. 2009;27(23):e55‐e56. [DOI] [PubMed] [Google Scholar]
- 27. Zhou M, Xu R, Kaelber DC, Gurney ME. Tumor Necrosis Factor (TNF) blocking agents are associated with lower risk for Alzheimer's disease in patients with rheumatoid arthritis and psoriasis. PLoS One. 2020;15(3):e0229819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Walker AM. Confounding by indication. Epidemiology. 1996;7(4):335‐336. [PubMed] [Google Scholar]
- 29. Chiu W‐C, Ho W‐C, Lin M‐H, et al. Angiotension receptor blockers reduce the risk of dementia. J Hypertens. 2014;32(4):938‐947. [DOI] [PubMed] [Google Scholar]
- 30. Yasar S, Xia J, Yao W, et al. Antihypertensive drugs decrease risk of Alzheimer disease: ginkgo evaluation of memory Study. Neurology. 2013;81(10):896‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsu C‐C, Wahlqvist ML, Lee M‐S, Tsai H‐N. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimers Dis. 2011;24(3):485‐493. [DOI] [PubMed] [Google Scholar]
- 32. Chou RC, Kane M, Ghimire S, Gautam S, Gui J. Treatment for rheumatoid arthritis and risk of Alzheimer's disease: a nested case‐control analysis. CNS Drugs. 2016;30(11):1111‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Judge A, Garriga C, Arden NK, et al. Protective effect of antirheumatic drugs on dementia in rheumatoid arthritis patients. Alzheimers Dement (N Y). 2017;3(4):612‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Szwast S, Hendrie H, Lane K, et al. Association of statin use with cognitive decline in elderly African Americans. Neurology. 2007;69(19):1873‐1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tai S‐Y, Chien C‐Y, Chang Y‐H, Yang Y‐H. Cilostazol use is associated with reduced risk of dementia: a nationwide cohort study. Neurotherapeutics. 2017;14(3):784‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee C, Lin C‐L, Sung F‐C, Liang J‐A, Kao C‐H. Antidepressant treatment and risk of dementia: a population‐based, retrospective case‐control study. J Clin Psychiatry. 2016;77(1):117‐122.. [DOI] [PubMed] [Google Scholar]
- 37. Gomm W, von Holt K, Thomé F, et al. Association of proton pump inhibitors with risk of dementia: a pharmacoepidemiological claims data analysis. JAMA Neurol. 2016;73(4):410‐416. [DOI] [PubMed] [Google Scholar]
- 38. Desai RJ, Rao JK, Hansen RA, Fang G, Maciejewski ML, Farley JF. Predictors of treatment initiation with tumor necrosis factor‐α inhibitors in patients with rheumatoid arthritis. J Manag Care Pharm. 2014;20(11):1110‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357‐367. [DOI] [PubMed] [Google Scholar]
- 40. Rajan KB, Wilson RS, Weuve J, Barnes LL, Evans DA. Cognitive impairment 18 years before clinical diagnosis of Alzheimer disease dementia. Neurology. 2015;85(10):898‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Power MC, Weuve J, Sharrett AR, Blacker D, Gottesman RF. Statins, cognition, and dementia—systematic review and methodological commentary. Nat Rev Neurol. 2015;11(4):220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ray WA. Evaluating medication effects outside of clinical trials: new‐user designs. Am J Epidemiol. 2003;158(9):915‐920. [DOI] [PubMed] [Google Scholar]
- 43. Hernán MA. Counterpoint: epidemiology to guide decision‐making: moving away from practice‐free research. Am J Epidemiol. 2015;182(10):834‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lauffenburger JC, Isaac T, Bhattacharya R, Sequist TD, Gopalakrishnan C, Choudhry NK. Prevalence and impact of having multiple barriers to medication adherence in nonadherent patients with poorly controlled cardiometabolic disease. Am J Cardiol. 2020;125(3):376‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li N‐C, Lee A, Whitmer RA, et al. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ. 2010;340:b5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Data Analysis for Drug Repurposing for Effective Alzheimer's Medicines (DREAM)—Tocilizumab vs Abatacept. In: https://ClinicalTrials.gov/show/NCT04529863.
- 47. Data Analysis for Drug Repurposing for Effective Alzheimer's Medicines (DREAM)—Tofacitinib vs Abatacept. In: https://ClinicalTrials.gov/show/NCT04529876.
- 48. Data Analysis for Drug Repurposing for Effective Alzheimer's Medicines—(DREAM) TNFi vs Abatacept. In: https://ClinicalTrials.gov/show/NCT04529902.
- 49. Berger ML, Sox H, Willke RJ, et al. Good practices for real‐world data studies of treatment and/or comparative effectiveness: recommendations from the joint ISPOR‐ISPE special task force on real‐world evidence in health care decision making. Value Health. 2017;20(8):1003‐1008. [DOI] [PubMed] [Google Scholar]
- 50. Smith GD, Ebrahim S. Data dredging, bias, or confounding: They can all get you into the BMJ and the Friday papers. In: British Medical Journal Publishing Group; 2002. [DOI] [PMC free article] [PubMed]
- 51. Schneeweiss S. A basic study design for expedited safety signal evaluation based on electronic healthcare data. Pharmacoepidemiol Drug Saf. 2010;19(8):858‐868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lund JL, Richardson DB, Stürmer T. The active comparator, new user study design in pharmacoepidemiology: historical foundations and contemporary application. Curr Epidemiol Rep. 2015;2(4):221‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Suissa S. Immortal time bias in pharmaco‐epidemiology. Am J Epidemiol. 2008;167(4):492‐499. [DOI] [PubMed] [Google Scholar]
- 54. Yoshida K, Solomon DH, Kim SC. Active‐comparator design and new‐user design in observational studies. Nat Rev Rheumatol. 2015;11(7):437‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Taylor DH, Jr, Østbye T, Langa KM, Weir D, Plassman BL. The accuracy of medicare claims as an epidemiological tool: the case of dementia revisited. J Alzheimers Dis. 2009;17(4):807‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dunn N, Holmes C, Mullee M. Does lithium therapy protect against the onset of dementia?. Alzheimer Dis Assoc Disord. 2005;19(1):20‐22. [DOI] [PubMed] [Google Scholar]
- 57. Dunn N, Mullee M, Perry VH, Holmes C. Association between dementia and infectious disease: evidence from a case‐control study. Alzheimer Dis Assoc Disord. 2005;19(2):91‐94. [DOI] [PubMed] [Google Scholar]
- 58. Imfeld P, Bodmer M, Jick SS, Meier CR. Benzodiazepine use and risk of developing Alzheimer's disease or vascular dementia: a case‐control analysis. Drug Saf. 2015;38(10):909‐919. [DOI] [PubMed] [Google Scholar]
- 59. Imfeld P, Brauchli Pernus YB, Jick SS, Meier CR. Epidemiology, co‐morbidities, and medication use of patients with Alzheimer's disease or vascular dementia in the UK. J Alzheimers Dis. 2013;35(3):565‐573. [DOI] [PubMed] [Google Scholar]
- 60. Solomon A, Ngandu T, Soininen H, Hallikainen MM, Kivipelto M, Laatikainen T. Validity of dementia and Alzheimer's disease diagnoses in finnish national registers. Alzheimer's Dement. 2014;10(3):303‐309. [DOI] [PubMed] [Google Scholar]
- 61. Kivipelto M, Ngandu T, Laatikainen T, Winblad B, Soininen H, Tuomilehto J. Risk score for the prediction of dementia risk in 20 years among middle aged people: a longitudinal, population‐based study. Lancet Neurol. 2006;5(9):735‐741. [DOI] [PubMed] [Google Scholar]
- 62. Barnes DE, Beiser AS, Lee A, et al. Development and validation of a brief dementia screening indicator for primary care. Alzheimers Dement. 2014;10(6):656‐665. e651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Albrecht JS, Hanna M, Kim D, Perfetto EM. Predicting diagnosis of Alzheimer's disease and related dementias using administrative claims. J Manag Care Spec Pharm. 2018;24(11):1138‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Brookhart MA, Patrick AR, Dormuth C, et al. Adherence to lipid‐lowering therapy and the use of preventive health services: an investigation of the healthy user effect. Am J Epidemiol. 2007;166(3):348‐354. [DOI] [PubMed] [Google Scholar]
- 65. Horton DB, Bhullar H, Carty L, et al. Electronic health record databases. Pharmacoepidemiology. 2019:241‐289. [Google Scholar]
- 66. Schneeweiss S, Seeger JD, Maclure M, Wang PS, Avorn J, Glynn RJ. Performance of comorbidity scores to control for confounding in epidemiologic studies using claims data. Am J Epidemiol. 2001;154(9):854‐864. [DOI] [PubMed] [Google Scholar]
- 67. Kim DH, Schneeweiss S, Glynn RJ, Lipsitz LA, Rockwood K, Avorn J. Measuring frailty in medicare data: development and validation of a claims‐based frailty index. J Gerontol A Biol Sci Med Sci. 2017;73(7):980‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Segal JB, Chang H‐Y, Du Y, Walston J, Carlson M, Varadhan R. Development of a claims‐based frailty indicator anchored to a well‐established frailty phenotype. Med Care. 2017;55(7):716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gopalakrishnan C, Gagne JJ, Sarpatwari A, et al. Evaluation of socioeconomic status indicators for confounding adjustment in observational studies of medication use. Clin Pharmacol Ther. 2019;105(6):1513‐1521. [DOI] [PubMed] [Google Scholar]
- 70. Bonito A, Bann C, Eicheldinger C, Carpenter L. Creation of New Race‐Ethnicity Codes and Socioeconomic Status (SES) Indicators for Medicare Beneficiaries. Final Report, Sub‐Task 2. (Prepared by RTI International for the Centers for Medicare and Medicaid Services through an interagency agreement with the Agency for Healthcare Research and Policy, under Contract No. 500‐00‐0024, Task No. 21) AHRQ Publication No. 08‐0029‐EF. Rockville, MD: Agency for Healthcare Research and Quality; 2008. [Google Scholar]
- 71. Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci USA. 2007;104(21):8685‐8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ray WA, Meredith S, Thapa PB, Meador KG, Hall K, Murray KT. Antipsychotics and the risk of sudden cardiac death. Arch Gen Psychiatry. 2001;58(12):1161‐1167. [DOI] [PubMed] [Google Scholar]
- 73. Rosenbaum PR, Rubin DB. The central role of the propensity score in observational studies for causal effects. Biometrika. 1983;70(1):41‐55. [Google Scholar]
- 74. Rassen JA, Avorn J, Schneeweiss S. Multivariate‐adjusted pharmacoepidemiologic analyses of confidential information pooled from multiple health care utilization databases. Pharmacoepidemiol Drug Saf. 2010;19(8):848‐857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rassen JA, Shelat AA, Myers J, Glynn RJ, Rothman KJ, Schneeweiss S. One‐to‐many propensity score matching in cohort studies. Pharmacoepidemiol Drug Saf. 2012;21(Suppl 2):69‐80. [DOI] [PubMed] [Google Scholar]
- 76. Austin PC. Some methods of propensity‐score matching had superior performance to others: results of an empirical investigation and monte carlo simulations. Biom J. 2009;51(1):171‐184. [DOI] [PubMed] [Google Scholar]
- 77. AM Walker AM, Patrick A, Lauer M, et al. Tool for assessing the feasibility of comparative effectiveness research. Comp Effect Res. 2013;3:11‐20. [Google Scholar]
- 78. Schneeweiss S, Patrick AR, Stürmer T, et al. Increasing levels of restriction in pharmacoepidemiologic database studies of elderly and comparison with randomized trial results. Med Care. 2007;45(10 SUPL):S131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Franklin JM, Rassen JA, Ackermann D, Bartels DB, Schneeweiss S. Metrics for covariate balance in cohort studies of causal effects. Stat Med. 2014;33(10):1685‐1699. [DOI] [PubMed] [Google Scholar]
- 80. Austin PC, Lee DS, Fine JP. Introduction to the analysis of survival data in the presence of competing risks. Circulation. 2016;133(6):601‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gagne JJ, Glynn RJ, Avorn J, Levin R, Schneeweiss S. A combined comorbidity score predicted mortality in elderly patients better than existing scores. J Clin Epidemiol. 2011;64(7):749‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Khan NF, Perera R, Harper S, Rose PW. Adaptation and validation of the charlson index for Read/OXMIS coded databases. BMC Fam Pract. 2010;11(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]