

Abstract
Background
Although generally known as a human commensal, Staphylococcus epidermidis is also an opportunistic pathogen that can cause nosocomial infections related to foreign body materials and immunocompromized patients. Infections are often caused by multidrug-resistant (MDR) lineages that are difficult and costly to treat, and can have a major adverse impact on patients’ quality of life. Heterogeneity is a common phenomenon in both carriage and infection, but present methodology for detection of this is laborious or expensive.
In this study, we present a culture-independent method, labelled Epidome, based on an amplicon sequencing-approach to deliver information beyond species level on primary samples and to elucidate clonality, population structure and temporal stability or niche selection of S. epidermidis communities.
Results
Based on an assessment of > 800 genes from the S. epidermidis core genome, we identified genes with variable regions, which in combination facilitated the differentiation of phylogenetic clusters observed in silico, and allowed classification down to lineage level. A duplex PCR, combined with an amplicon sequencing protocol, and a downstream analysis pipeline were designed to provide subspecies information from primary samples. Additionally, a probe-based qPCR was designed to provide valuable absolute abundance quantification of S. epidermidis. The approach was validated on isolates representing skin commensals and on genomic mock communities with a sensitivity of < 10 copies/μL. The method was furthermore applied to a sample set of primary skin and nasal samples, revealing a high degree of heterogeneity in the S. epidermidis populations. Additionally, the qPCR showed a high degree of variation in absolute abundance of S. epidermidis.
Conclusions
The Epidome method is designed for use on primary samples to obtain important information on S. epidermidis abundance and diversity beyond species-level to answer questions regarding the emergence and dissemination of nosocomial lineages, investigating clonality of S. epidermidis communities, population dynamics, and niche selection. Our targeted-sequencing method allows rapid differentiation and identification of clinically important nosocomial lineages in low-biomass samples such as skin samples.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12866-020-02041-w.
Keywords: Targeted sequencing, Staphylococcus epidermidis, Sequencing, Population structure, Heterogeneity, Population dynamics, Clonal lineages, S. epidermidis, Amplicon sequence variant, Species-specific classification
Background
Staphylococci constitute an important group of human skin and mucosal commensals [1, 2], where coagulase-negative staphylococci (CoNS) generally are regarded to have a benign or symbiotic relationship with their hosts [3]. However, certain species are opportunistic human pathogens and play an important role in especially nosocomial infections [4, 5]. Among the CoNS, Staphylococcus epidermidis has recently gained much attention as a significant nosocomial pathogen associated with foreign body infections such as prosthetic joint infections (PJIs) [6, 7]. Furthermore, S. epidermidis is a common causative microorganism in infections in immunocompromized patients, including preterm infants and patients with hematological malignancies [4, 8, 9]. These infections result in large economic costs for society [10, 11], and can have major adverse impact on patients’ quality of life [12].
A large percentage of hospital S. epidermidis isolates are methicillin-resistant (MRSE), and especially multidrug-resistant S. epidermidis (MDRSE) lineages, such as sequence type (ST)2, ST5, ST23, and ST215, are spreading both locally and globally in hospital environments [6, 8, 13–17]. Identification of S. epidermidis as an invasive infectious pathogen is not trivial and distinguishing contamination from true infection can be challenging, and research suggests that high sub-species heterogeneity is common in healthy carriage [18, 19], but also in patients suffering from PJIs and atopic dermatitis [20, 21]. This observation is in contrast to the nature of Staphylococcus aureus communities that are known to be highly clonal [22–24].
Methods to elucidate clonality, population dynamics and temporal stability or niche selection of S. epidermidis communities due to antimicrobial prophylactic treatments currently include laborious culturing and characterization of multiple isolates or application of expensive metagenomics sequencing. In this paper we present the Epidome, a culture-independent method based on amplicon sequencing of two S. epidermidis-specific target genes. Similar amplicon-based methods exist to identify staphylococci at genus level [25], and to distinguish between various CoNS [26–28].
The Epidome method is designed for use on primary samples to obtain information on S. epidermidis abundance and diversity beyond species-level to answer important questions regarding the emergence and dissemination of nosocomial lineages. Here, we demonstrate our method on a sample set comprising nasal and skin swab samples from healthy community-dwelling adults.
Results
In order to establish the Epidome method, we designed and tested the specificity of primers for amplicon sequencing of two S. epidermidis specific targets. Furthermore, we set up a publicly available analysis pipeline for sequencing quality control, amplicon sequence variant (ASV) inference, and taxonomic classification (https://github.com/ssi-dk/epidome/). For the latter, we built two target-specific databases. A qPCR-based approach to complement the amplicon sequencing was also implemented. Below, we present our method and demonstrate the use of the analysis pipeline on an exemplary data set from human nasal and skin samples.
Population structure and Epidome gene target diversity
To infer clonal lineages of S. epidermidis, we investigated the population structure of the S. epidermidis population (n = 842) based on 86,139 SNPs identified in a 39% core (1,033,603 bp) using the high-quality requirements for retaining positions across the collection containing both assembled RefSeq genomes and data from 289 Swedish invasive and colonizing isolates (Fig. 1a). Definition of clonal groups by hierarchical clustering on pairwise SNP-dissimilarities defined 130 clonal groups.
Fig. 1.
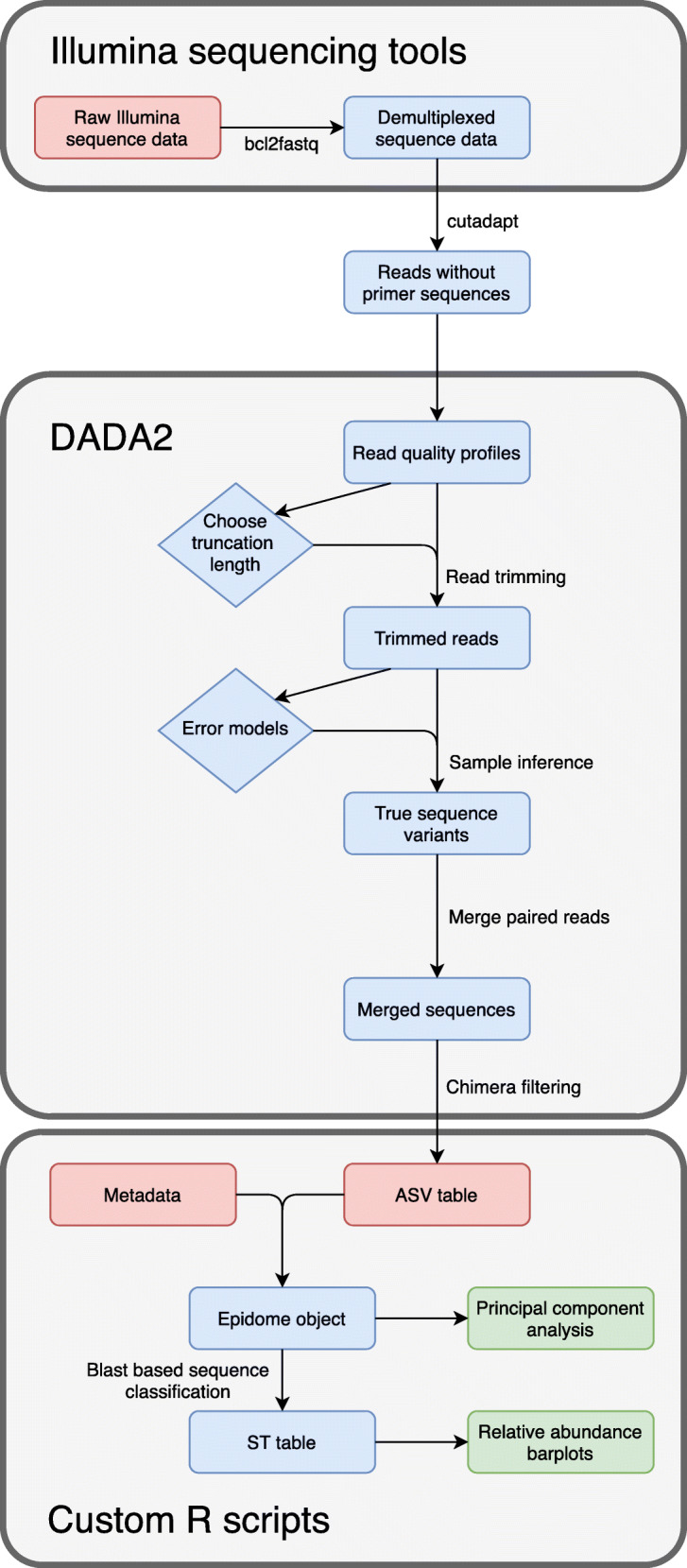
A schematic overview of the Epidome analysis pipeline from sequence reads to classification and figure outputs
Primer design for amplicon sequencing
Based on an assessment of the core genome among 50 S. epidermidis isolates representing the diversity observed in the core phylogeny, we identified a total of 1,900 candidate genes. When analyzing these for requirements as suitable targets for a species-specific amplicon-based approach (highly variable region flanked by conserved primer binding sites) we identified a total of 1088 primer pairs positioned in 89 different genes. Presence of the top candidate genes was then assessed across the entire population of 842 S. epidermidis genome sequences. Here, two targets (yycH and g216, locus tags SE_0020 and SE_0191) in S. epidermidis isolate ATCC 12228 (GenBank Accession ID AE015929) were chosen for a duplex PCR that in combination resembled the phylogenetic clustering observed on the basis of core genome SNPs (Fig. 1b). The g216 gene was the primary target as it offered the highest resolution of the population as a whole, whereas the combination with yycH allowed key endemic nosocomial lineages to be uniquely identified in silico.
Specificity testing and reproducibility of the Epidome method
Using DNA from S. epidermidis isolates (n = 26) representing 15 clonal groups including major hospital-associated lineages such as ST2, ST5, and ST215 as well as from other staphylococcal species (n = 27) and other common skin commensals (n = 21, see Supplementary Table 1), both primer sets were tested in separate PCR reactions. Here, the g216 PCR was found to be S. epidermidis specific, whereas the yycH PCR resulted in amplification of four non-S. epidermidis species out of the 48 tested (S. equorum, S. haemolyticus, S. pseudintermedius, and S. argenteus). Importantly, the sequences of these four species were distinct from S. epidermidis and were excluded from downstream analyses. The Epidome duplex PCR containing both the g126 and yycH primers was run under the same conditions as the singleplex reactions. To evaluate the capabilities of the multiplex PCR, sequencing and downstream analyses, the method was applied in triplicates of both even- and staggered genomic mock communities with six STs represented (Fig. 2). Even when present in low abundance down to 100 copies/μL, the STs were successfully detected with a high level of reproducibility for technical replicates (Fig. 2). However, for the staggered mocks a reliable detection sensitivity of ~ 1% was observed (Fig. 2), as illustrated by the lack of detection of the ST2 and ST218 in these with the present sequencing depth. In the mocks, ASVs classified as Others include ST307, ST252 and sequences matching isolates with novel sequence types. Due to the expected rate of read errors in Illumina sequencing, some ASVs matched with these sequence types despite not being present in the sample. ST307 was the most prominent of these misclassified types and represented up to 2% of reads from a sample, which indicates that classification of ASVs found at or below this level in a sample should be considered unreliable. Unclassified ASVs are sequences which had less than 98.5% similarity with any sequence in the database.
Fig. 2.

Sequencing of Staphylococcus epidermidis even and staggered mock communities. Data represent results of the amplicon sequencing of technical triplicates consisting of even mock communities comprised of 10,000 copies of S. epidermidis ST2, ST5, ST14, ST87, ST215, and ST218, and staggered mock communities with 10,000 copies of ST215 and ST87, 1000 copies of ST5 and ST14 and 100 copies of ST2 and ST218. The data is shown as relative abundance per sample. Category ‘Other’ includes clonal groups detected, but not expected in the mock samples
Amplicon sequence-preprocessing and taxonomic classification
For each of the two target genes, we set up a bioinformatic analysis pipeline comprising primer trimming, quality filtering and truncation of forward and reverse reads, Illumina error profile estimation, and inference of ASVs following amplicon sequencing. For each of the two amplicon targets, a custom blast database was set up containing all unique sequences found in the collection of 842 S. epidermidis genomes. Taxonomic classification was based on a combination of sequences from both target genes (see Methods section).
Twenty-two primary samples, collected from skin (n = 11) and anterior nares (n = 11) of healthy humans, were sequenced in technical duplicates. We obtained on average 14,927 initial reads of g216 gene sequences per sample. On average, 10,806 reads per sample (71% of the initial reads) passed quality filtering and had a read length exceeding the specified minimum. Of these reads, on average 10,482 reads per sample (99%) were non-chimeric reads. The read count of the two blank extraction controls after all filtering steps were 36 and 408, respectively.
YycH gene sequencing yielded on average 46,915 initial reads per sample. On average, 30,406 reads per sample (63% of the initial reads) passed quality and length filtering. Thereof, on average 28,999 reads per sample (98%) were non-chimeric reads. The two blank extraction controls had 707 and 1441 reads after all filtering steps, respectively. Read counts revealed an overall even distribution of numbers of Illumina reads for the yycH and g216 targets across all samples (Pearson’s correlation coefficient of 0.94, see Supplementary Fig. 1), illustrating the linear co-amplification across all samples with a higher efficiency of the yycH target.
To investigate whether any difference in read counts between samples were related to the detected richness of S. epidermidis, we generated rarefaction curves to display the number of observed ASVs over the library size per sample after quality filtering, see Supplementary Fig. 2A and 2B for g216 and yycH, respectively. A read count that was low (defined here as < 5000) in both technical replicates of a sample (e.g. P06_skin_1 (3885 reads) and P06_skin_2 (1771 reads)) indicated a low abundance of S. epidermidis in this sample (Supplementary Fig. 2A). This indication was reinforced in those samples where the rarefaction curve reached a plateau with regards to the number of observed ASVs despite a low read count. In the few samples where the read count was higher in one replicate than in the other, and where the replicate with the lower read count does not reach a plateau of observed ASVs (e.g. P03_skin_2 (11 ASVs in 3143 reads) vs P03_skin_1 (20 ASVs in 5827 reads)), the low read count is likely attributable to technical differences (Supplementary Fig. 2A). Overall, on the analyzed sample collection, 5000 filtered reads/sample seems sufficient to describe the richness of S. epidermidis on the skin and in the nares.
S. epidermidis heterogeneity in primary human skin and nasal samples
To validate the method on primary samples, it was applied to 22 swab samples from paired human skin (n = 11) and anterior nares (n = 11). We found that 20/22 (91%) of all primary samples were positive for S. epidermidis and that the samples generally showed a high degree of inter-sample heterogeneity regarding the presence of S. epidermidis lineages (Fig. 3). Samples were found to contain between two and 8 different clonal groups with an mean of five in each sample.
Fig. 3.

Carriage heterogeneity of Staphylococcus epidermidis on human skin and in nares. Results of the Epidome methodology with technical replicates on skin and anterior nasal samples from 11 individuals. a Relative abundance of detected clonal groups identified among the primary samples. The lineage legend highlight the 10 most prevalent clonal groups found across all samples. Reads not mapping to any sequences in the constructed reference database (Novel), mapping to sequences with no known ST (Unclassified), or to other characterized lineages (Other) are similarly presented. The median of the absolute abundance of S. epidermidis in the two primary samples using qPCR of the g216 gene target in triplicates are depicted below samples on a log10 scale. b Assessment of the reproducibility of the Epidome method on primary samples and between-sample diversity using a principal component analysis depicting the beta-diversity on the relative abundances of ASVs based on a two-sampling approach per site per individual
The majority of sequencing reads (76%) were distributed within 10 different clonal groups (ST2, ST5, ST14, ST59, ST73, ST87, ST130, ST215, ST218 and ST225). A subset of reads (5%) was assigned to other clonal groups while the rest of the reads either mapped to S. epidermidis sequences in the database with no known ST (Unclassified, 5%) or did not map to any sequences in the constructed reference database containing all presently available genome sequenced isolates of S. epidermidis (Novel, 13%).
We assessed the beta-diversity between the primary samples using a PCA which showed a high level of beta-diversity across all samples but also that the nose and skin had overlapping populations of S. epidermidis, see Supplementary Fig. 3. Importantly, pairwise clustering of technical duplicates of primary samples was observed, depicting the reproducibility of the Epidome methodology (Fig. 3b).
Absolute abundance of S. epidermidis assessed by qPCR
To extend the insights that compositional data generated by amplicon sequencing provides, we developed a complementing qPCR approach to measure absolute abundance. In the designed qPCR setup, we used the g216 target gene as a basis for which we successfully designed an MGB probe for qPCR detection and quantification of mixed S. epidermidis populations. Our analyses on serial 10-fold dilutions of g216 amplicons revealed an R2 of 0.99 across seven log10 dilutions with an efficiency of 70% and a limit of detection of < 10 copies/μL. Despite the low efficiency, the technical replicates showed excellent reproducibility (Supplementary Table 2). The qPCR was applied to our 22 primary samples in technical triplicates. S. epidermidis was detected in all but two samples with quantities per sample ranging from 2 to 962 copies/μL (Fig. 3a). Nasal samples generally exhibited a higher quantity of S. epidermidis than skin samples. Non-template controls were negative by qPCR. For samples with low absolute abundance, a higher degree of variation in the technical replicates was observed.
Discussion
We designed and validated a novel two-step method for identification and quantification of S. epidermidis from primary samples using targeted amplicon sequencing and qPCR.
While 16S rRNA gene sequencing is the most widely used tool for studying microbial communities, this method does not generally provide comprehensive information beyond species level. The approach described here, labelled the Epidome, is an alternative culture-independent method that rapidly provides high taxonomic resolution beyond species-level of heterogeneous populations of S. epidermidis without the cost associated with metagenomics and without being as time-consuming and laborious as cultivation-based methods.
Designing targets that unambiguously distinguish between all major lineages is however a major challenge as S. epidermidis is highly diverse and recombination is frequent in this species [15, 29]. Our approach was to include all available S. epidermidis genome sequences available in the public repository and to have a specific focus on resolving major S. epidermidis groups that are known to be the main causes of hospital-acquired infections; ST2, ST5, ST23, and ST215. Our method has proven successful in detecting and distinguishing between these selected groups as well as in providing insight into the overall S. epidermidis diversity. Thus we believe that the method will be a useful tool to study colonization rates, population dynamics and temporal stability or niche selection due to antimicrobial prophylactic treatments in relation to infections of this increasingly important nosocomial pathogen.
Despite that a multitarget approach would have provided more discriminatory power, we used a two-gene target approach to distinguish between our defined S. epidermidis clonal groups to allow assignment to distinct branches of the S. epidermidis population while reducing the computation uncertainly of matching multiple, potential novel, targets in a heterogeneic sample material. In silico analysis against NCBI’s nucleotide collection (nr/nt) database showed no off-target amplification for any of the two primer sets. The g216 target was specific for S. epidermidis while the yycH target resulted in sporadic non-S. epidermidis-specific amplification. The amplicons of these other species were highly dissimilar compared to S. epidermidis making downstream exclusion easily feasible, and thus not a major limitation of the methodology. The g216 gene was selected as the qPCR target since it displayed high specificity for S. epidermidis. The qPCR showed sufficiently low detection limits and allowed highly accurate detection and quantification of mixed S. epidermidis populations.
The Epidome method allowed differentiation of S. epidermidis clonal groups in primary samples but also revealed a high number of novel sequences from the investigated primary samples that did not map to the reference databases. This suggests that either the diversity of naturally occurring S. epidermidis on the skin and in the nares of healthy individuals is higher than the sum of all published S. epidermidis genomes or that the high frequency of homologues recombination in S. epidermidis results in gene target combinations that are novel. In the nosocomial lineages, we observe that the targets are conserved, even though the genomic data originates from both temporally and spatially diverse collections.
Looking at the beta-diversity within the primary samples from skin and nares, our data indicates that the S. epidermidis community on the skin is more homogenous than in the nares, although a dedicated study is needed to validate this finding. In terms of reproducibility, our technical replicates of the mock communities and primary samples (Fig. 4a and Fig. 3b, respectively) show a high level of congruence. We believe that these results combined confirm that this method has potential as a tool to study in detail the population dynamics and diversity of S. epidermidis.
Fig. 4.

Phylogenetic clustering of the Staphylococcus epidermidis population and Epidome gene targets. a Unrooted phylogeny of 842 S. epidermidis genomes based on 86,139 core genome SNPs. Major lineages as well as clinical important multidrug-resistant lineages are highlighted. b Unrooted phylogeny of the concatenated sequence of the Epidome target genes across the S. epidermidis population depicting the lineage origin of all targets. Both phylogenies were annotated using iTol. Scale bars indicate substitutions per site
The accuracy of amplicon-sequencing based identification methods is naturally dependent on the underlying database used for classification. Importantly, the database used here, while extensive, is not expected to capture the entire diversity of the S. epidermidis population. Exactly how much of the diversity of S. epidermidis that still lack whole-genome sequenced data is hard to estimate. However, our observation that ~ 10% of S. epidermidis ASVs from nose and skin samples were labelled as Unclassified (not assignable to a single clonal group) or Novel (ASVs not matching any known variants in the database) provides an indication of the minimum level of unknown S. epidermidis variants we can expect to find in these environments. Our method thus also highlights the need to increase sequencing efforts to provide the taxonomic information necessary to classify the diverse S. epidermidis clonal groups which we now are able to identify. Other methods that rely on DNA fingerprinting such as PFGE or AFLP, would inherently be highly complex to analyze due to the heterogeneity observed in S. epidermidis populations and with limited ability to assign distinct lineages. Contrary, genome sequencing will naturally provide much more information on individual isolates in a clinical setting, but for research studies aimed at understanding heterogeneity and population dynamics, the Epidome approach will provide unparalleled insight.
In this approach we assigned all unique ASV sequences in our database to clonal groups. Given the incompleteness of the database, yet undescribed S. epidermidis ASV sequences may match equally well to multiple similar reference sequences, bearing a risk for misassignment. However, it is most likely that those sequences would remain Novel, or be misassigned to a closely related clonal group.
It should also be noted that the whole-genome sequencing data currently available on S. epidermidis is largely dominated by isolates from human nasal and skin samples as well as infections – particularly infections from indwelling medical devices. Other niches may be populated by a very different population of S. epidermidis, in which case the accuracy of sequence type classification will inevitably be lowered. Regardless of the completeness of the database, unbiased analysis based on ASVs identified with DADA2 can always be performed and can provide valuable information on the stability of S. epidermidis communities, temporal changes in composition, and similarities between sampling sites.
Conclusions
We successfully designed a PCR- and amplicon sequencing-based approach - the Epidome – to provide insight into S. epidermidis quantity and diversity beyond species-level. Our targeted sequencing method allows reproducible identification of important epidemic nosocomial lineages in primary samples even when present in low abundance. To strengthen interpretation of the sequencing data we designed and validated a qPCR that provides an important quantification of S. epidermidis population in primary samples. Our analyses on primary samples also highlight the heterogeneity of S. epidermidis found on the skin and in the nose of healthy individuals. Importantly, our analysis pipeline, underlying database, and supporting documentation for investigating the population of S. epidermidis are publicly available.
Using the Epidome, a number of questions can be answered without the need for laborious and biased cultivation and without using costly methods such as metagenomics. We believe that the approach shows great potential especially for studying the important issues of heterogeneity of S. epidermidis communities, population dynamics and temporal stability or niche selection due to antimicrobial prophylaxis or treatments.
Methods
To investigate the diversity of S. epidermidis at various sampling sites such as the skin and nose, we aimed at designing a simple quantitative approach using parallel sequencing as offered by next generation sequencing technology on species-specific targets that represent the population diversity of the species, see Fig. 1. The method was designed and validated on primary samples representing body sites of colonization.
Population structure of S. epidermidis
The dataset used for identification of suitable amplicon targets consisted of all 553 S. epidermidis genomic assemblies obtained from the NCBI’s Reference Sequence Database (RefSeq, https://www.ncbi.nlm.nih.gov/refseq/, accessed September 2019) and 289 SPAdes [30] assembled genomes from a recent Swedish study of S. epidermidis and PJIs [31]. Importantly, the Swedish collection included ST215/ST434 MDRSE isolates but also a diverse population of antibiotic susceptible isolates from healthy carriers.
To evaluate the population structure of S. epidermidis, single nucleotide polymorphisms (SNPs) from the core genome across all 842 genomes were obtained with the Northern Arizona SNP Pipeline (NASP) [32]. Here, paired-end Illumina reads from the 289 isolates from the Swedish collection and genomic assemblies from the 553 RefSeq isolates were mapped to the reference chromosome of S. epidermidis isolate ATCC 12228 (GenBank Accession ID AE015929) [33]. Positions with less than 10x coverage or less than 90% consensus were excluded from the core genome. An unrooted phylogeny of the population was obtained using the maximum likelihood approximation implemented in FastTree v2.1.5 using the GTR substitution model and both nearest neighbor Interchange (NNI) and subtree pruning and re-grafting (SPR) for hierarchical search methods [34]. iTol v5.5 (https://itol.embl.de) was used for visualization of the phylogeny. Based on pairwise SNP-dissimilarity between isolates, hierarchical clustering was performed using complete-linkage, where clonal groups were defined using a cutoff of 1000 SNPs and categorized according to their most prevalent sequence type.
Identification of amplicon targets and PCR design
For identification of suitable S. epidermidis gene targets for PCR analyses, 50 isolates were selected, representing diverse clusters including commensal lineages and key hospital-associated lineages. The 50 genomes were annotated using Prokka v1.12 [35] and core genes identified using Roary v3.11.2 [36] with default parameters. Core genes were parsed for sequence variability to identify genes in which highly variable regions were flanked by conserved regions (> 95% sequence similarity) making them suitable PCR targets. Using Primer3 v2.3.7 (http://bioinfo.ut.ee/primer3/), all conserved regions flanking a variable region < 1500 bp were analyzed to find potential primers. These candidate primer targets were extracted from all 842 genomes using BLASTN and evaluated based on their potential for discriminating between clonal clusters. No single target sufficiently distinguished all major clusters, therefore two targets were selected; a primary target, g216 (521 bp), with enough resolution to distinguish between most key clusters – and a secondary target, yycH (493 bp), adding resolution to identify additional key clusters. The yycH, gene along with yycL, functions as a regulator for the expression of the WalK/WalR two-component system which in turn regulates cell wall metabolism through expression of autolysins. Regarding the conserved g216 gene, it is so far uncharacterized in Staphylococcus species and no information could be found on potential homologues in other species. The primer sequences for amplification of the two targets were: 5′-CGATGCKAAAGTGCCGAATA-3′/5′-CTTCATTTAAGAAGCCACCWTGACT-3′ and 5′-TGGGTATGRCAATCACTTTACA-3′/5′-GCATCAAAAGCACTCTCATTACC-3′ for yccH and g216, respectively. An unrooted phylogeny of the concatenated gene sequences of the two targets across the population was derived using IQ-TREE v1.6.12 [37] with model selection and 100 bootstrap replicates. Illumina adapter sequences were added to the 5′-end of all the primers to allow for downstream indexing and sequencing.
For determination of the absolute abundance of S. epidermidis, a minor groove binder (MGB) probe (TaqMan, Thermo Fisher Scientific) was designed to hybridize to the g216 S. epidermidis-specific target (5′-FAM-CATGCCAGATATGAAT-MGB-3′). The MGB probe target sequence was selected based on an alignment of the g216 gene from all included S. epidermidis genome sequences using Qiagen’s CLC Genomics Workbench v11 with subsequent validation in Primer Express v3.0 (Thermo Fisher).
Collection of primary samples and DNA extraction
For testing of primer specificity, we used DNA from S. epidermidis isolates (n = 26) representing 15 clusters including major hospital-associated lineages such as ST2, ST5 and ST215, as well as from other staphylococcal species (n = 27) and other common skin commensals (n = 21), see Supplementary Table 1. Additionally, purified DNA from six S. epidermidis isolates with known STs was used to create genomic mock communities in both even and staggered design for method validation. The even mock contained equal copy numbers (10,000) of ST2, ST5, ST14, ST87, ST215, and ST218, whereas the staggered mock contained 10,000 copies of ST87 and ST215, 1000 copies of ST5 and ST14 and 100 copies of ST2 and ST218.
From the isolates, DNA was extracted using an enzymatic pre-lysis step followed by extraction using the Qiagen Blood and Tissue kit (Qiagen). The pre-lysis step was carried out by dissolving bacterial cultures in a lysis buffer containing 20 nM Tris, 2 nM EDTA, 1.2% Triton X and PCR Grade water. Enzymes were added to reach a final concentration of 20 μg/μL lysozyme and 5.56 μg/mL lysostaphin. The enzymatic reaction was incubated for 30 min at 37 °C before processing with the Qiagen Blood and Tissue protocol. DNA concentrations were measured using either Qubit dsDNA BR Assay Kit or Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific).
Primary samples were obtained from 11 healthy community dwelling individuals in Copenhagen, Denmark, from both nose and skin using ESwabs (Copan). Samples from the nares were collected by rotating the swab in one of the anterior nares three times, while skin samples were collected by rubbing the swab against the skin of the participant’s arm (either the volar forearm or antecubital fossa) for 30s. All samples were stored immediately at − 80 °C before processing. DNA was purified from the 22 primary samples, as well as from two blank controls using an enzymatic pre-lysis step before automated purification on a MagNA Pure 96 (Roche Diagnostics A/S) using the MagNA Pure 96 DNA and Viral NA Small Volume Kit and the DNA Blood ds SV 2.0 protocol (Roche Diagnostics A/S). The pre-lysis step was carried out using 200 μL ESwab sample. Enzymes were added to reach a final concentration of 8.4 mol/L of lysozyme, 0.001 U/μL lysostaphin and 0.1 U/μL mytanolysin in 50 μl TE buffer. The enzymatic reaction was incubated for 30 min at 37 °C before addition of 20 μL Proteinase K followed by a 30 min incubation step at 56 °C. All primary samples were purified in technical duplicates to allow subsequent assessment of reproducibility.
PCR and sequencing
The duplex amplification PCR was carried out using 2x KAPA HiFi (Kapa Biosystems) with 0.2 μM of each primer and 10 μL template in a final volume of 25 μL on a 2720 Thermal Cycler (Applied Biosystems) using the following PCR conditions: 3 min at 95 °C, 25 cycles of 20s at 98 °C, 15 s at 60 °C and 45 s at 72 °C with a final extension step of 5 min at 72 °C. The indexing PCR was carried out using 2x KAPA HiFi, Nextera XT DNA Library Prep Kit v2 for indexing (Illumina) and with 2 μL amplicon product from the amplification PCR in final volume of 25 μL on a 2720 Thermal Cycler (Applied Biosystems) using the following conditions: 3 min at 95 °C, 20 cycles of 20s at 98 °C, 15 s at 55 °C and 45 s at 72 °C, and final 5 min elongation step at 72 °C. After indexing, post-PCR cleanup was performed using a 1:1 ratio of AMPure XP beads (Beckman Coulter) following manufacturer’s instructions before quantification using AccuClear Ultra High Sensitivity dsDNA Quantification Kit (Biotium) following manufacturer’s instructions. Subsequently, samples were pooled in equimolar concentrations and quantified using the Qubit dsDNA HS assay kit (Thermo Fisher Scientific) prior to sequencing on a MiSeq with a 600-cycle MiSeq Reagent Kit v3 (Illumina) and a pool of libraries loaded at 10 pM final concentrations.
Absolute abundance using qPCR
The S. epidermidis-specific g216 gene was used as basis for a qPCR setup. A standard curve ranging from 1000,000 to 1 copies/μL was generated using AMPure XP bead-purified (× 0,8 ratio, Beckman Coulter) g216 amplicon generated as described above on DNA purified from an S. epidermidis ST2 isolate. The qPCR was performed on a QuantStudio 5 Real-Time PCR system (Thermo Fisher) using PerfeCTA qPCR ToughMix Low ROX (Quantabio) with a 0.5 μM concentration of g216 primers, 0.15 μM of the g216 MGB probe, an additional 2 mM MgCl2, and 5 μL template in a total volume of 30 μL. qPCR conditions were as follows: 3 min at 95 °C, followed by 40 cycles of 30s at 95 °C, 60s at 60 °C and 60s at 72 °C, with a final 5 min extension step at 72 °C.
Epidome target gene sequence pre-processing
We used the bcl2fastq Conversion Software (Illumina) to demultiplex raw sequence reads, generating one forward and one reverse FASTQ file for each sample with primer trimming using the Cutadapt software v2.3 [38]. The sequences were processed twice, i.e. the respective primer pairs for each of the two targets were trimmed separately (https://github.com/ssi-dk/epidome/tree/master/scripts). Primer sequences in each read were matched at a tolerated maximum error rate of 6% which corresponds to one mismatch using Cutadapt. To keep a read pair, primers also had to be found in both reads. In order to infer amplicon sequence variants (ASVs) from the trimmed reads, the R package DADA2 v1.12.1 was used [39]. The truncation lengths were adjusted to the expected amplicon lengths of the two target genes, i.e. to obtain ASVs based on the g216 gene, forward reads were truncated at 276 bp, and reverse reads at 237 bp, aiming at a total length of 513 bp corresponding to an amplicon length of 493 bp with 20 bp overlap for merging. Similarly, for ASV inference based on the yycH gene, forward reads were truncated at 278 bp, and reverse reads at 262 bp, aiming at a total length of 540 bp (amplicon length of 520 bp with 20 bp overlap to allow merging). Accordingly, the data was processed with DADA2 separately for each target with default settings except truncation lengths (https://github.com/ssi-dk/epidome/tree/master/scripts). We used the removeBimeraDenovo function (method “consensus”) to identify chimeras on a per-sample basis and subsequently removed them from all samples globally. An overview of the per-sample read counts after each filtering step can be found at https://github.com/ssi-dk/epidome/tree/master/example_data.
Taxonomic database setup and classification
Custom databases of all unique g216 and yycH target sequences can be found at https://github.com/ssi-dk/epidome/tree/master/DB. We formatted our g216 and yycH gene databases to be compatible with DADA2’s assignTaxonomy function and used it to classify the S. epidermidis ASVs with the RDP naive Bayesian classifier method (https://github.com/ssi-dk/epidome/tree/master/scripts).
ST classification of samples was performed using the g216 target sequence as the primary identifier. All g216 sequences unique to a single clonal cluster in the database were immediately classified as the matching clone, and in cases were the g216 sequence matched multiple clones, the secondary yycH target sequences were parsed to determine which clone was present. When this classification failed to resolve due to multiple potential combinations of sequences, ASVs were categorized as “Unclassified”. Similarly, g216 sequences not found in the database were labelled as “Novel”. The resulting taxonomic count tables are provided at https://github.com/ssi-dk/epidome/tree/master/example_data.
Statistical analyses
Statistical analyses and generation of graphs were performed in R v3.6.0 (R Core Team 2019). All R scripts documenting the analyses are provided at https://github.com/ssi-dk/epidome/tree/master/scripts. Rarefaction curves were generated by utilizing the R packages phyloseq and ranacapa v0.1.0 [40, 41], and plots were generated with ggplot2 v3.3.1 [42]. The qPCR data was analyzed using QuantStudio Design and Analysis Software (Thermo Fisher). The scatterplot examining correlation between read counts from the two primer pairs was created with ggplot2 v3.3.1 and correlation was tested using Pearson’s correlation. Presentations of the relative abundances of clinical and mock samples were presented in barplots created with ggplot2 v3.3.1 and principal component analysis (PCA) was performed on Euclidean distances between samples based on relative abundances of ASVs. Comparisons between groups were made using ANOSIM tests with 1000 permutations based on the Euclidean distances.
Supplementary information
Supplementary Figure 1. Read counts after trimming and chimera filtering of the g216 and yych gene targets across all analyzed primary samples. The analyses revealed a higher level of yycH read counts compared to g216, however with an overall even distribution of across all samples.
Supplementary Figure 2. Rarefaction curves to investigate the dependency of Staphylococcus epidermidis abundance/lineages richness on sample library size in mock communities and primary samples. The generated rarefaction curves display the number of observed ASVs over the library size per sample after quality filtering, for g216 and yycH in panel A and B, respectively.
Supplementary Figure 3. Beta-diversity of the Staphylococcus epidermidis population across sample sites. Principal component analysis depicting the beta-diversity across all primary samples from skin and nares indicate overlapping populations highlighting samples sites compared to Fig. 4b.
Supplementary Table 1. Bacterial isolates used for laboratory validation of the method. Included are sequence types for all Staphylococcus epidermidis isolates, as well as a total of 48 different bacterial species commonly identified as skin commensals that were used for specificity testing; 27 staphylococcal species and 21 non-staphylococcal species.
Supplementary Table 2. qPCR results on standards, as well as primary skin and nasal samples. Presented are the threshold cycle (Ct) values with standard deviation as well as equivalent counts.
Acknowledgements
We would like to thank Ditte Marie Brix, Elvira Chapka, Christian Høiby Aumayr and Emine Yüksel Coskun at the Department of Bacteria, Parasites and Fungi at Statens Serum Institut for providing excellent laboratory assistance.
Authors’ contributions
AKR, TBJ, EM, BS, PSA and MS designed the study; SE collected clinical samples; AKR and SI performed the laboratory experiments, TBJ, ACI and SB contributed analytic tools; AKR, TBJ, ACI, SB, JSJ, and MS analyzed the data; AKR, TBJ, ACI and MS wrote the manuscript; All authors edited the manuscript. All authors have read and approved the manuscript as submitted.
Funding
This work was supported by Statens Serum Institut, Copenhagen, Denmark, the University Hospital of Örebro, Örebro, Sweden, and the National Institute of Allergy and Infectious Diseases, National Institutes of Health (1R01AI125562–01).
Availability of data and materials
The amplicon sequences are available through the European Nucleotide Archive (ENA) at the European Bioinformatics Institute (EBI) (PRJEB39895). The R scripts for the sequence pre-processing and taxonomy assignment, including the resulting datasets, as well as our custom data bases and R scripts for statistical analyses are available from https://github.com/ssi-dk/epidome/.
Ethics approval and consent to participate
Collection of nasal and skin samples from healthy adults was approved by the local ethical committee (Videnskabetisk Komite Region-Hovedstaden, project id: H-16023435) and the Danish Data Protection Agency. All participants provided written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that there is no conflict of interest regarding the publication of this paper.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. 2018;16(3):143–155. doi: 10.1038/nrmicro.2017.157. [DOI] [PubMed] [Google Scholar]
- 2.Stacy A, Belkaid Y. Microbial guardians of skin health. Science. 2019;363(6424):227–228. doi: 10.1126/science.aat4326. [DOI] [PubMed] [Google Scholar]
- 3.Heikens E, Fleer A, Paauw A, Florijn A, Fluit AC. Comparison of genotypic and phenotypic methods for species-level identification of clinical isolates of coagulase-negative staphylococci. J Clin Microbiol. 2005;43(5):2286–2290. doi: 10.1128/JCM.43.5.2286-2290.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker K, Heilmann C, Peters G. Coagulase-negative staphylococci. Clin Microbiol Rev. 2014;27(4):870–926. doi: 10.1128/CMR.00109-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohanty SS, Kay PR. Infection in total joint replacements. Why we screen MRSA when MRSE is the problem? J Bone Joint Surg Br. 2004;86(2):266–268. doi: 10.1302/0301-620X.86B2.14129. [DOI] [PubMed] [Google Scholar]
- 6.Hellmark B, Soderquist B, Unemo M, Nilsdotter-Augustinsson A. Comparison of Staphylococcus epidermidis isolated from prosthetic joint infections and commensal isolates in regard to antibiotic susceptibility, agr type, biofilm production, and epidemiology. Int J Med Microbiol. 2013;303(1):32–39. doi: 10.1016/j.ijmm.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Widerstrom M. Significance of Staphylococcus epidermidis in health care-associated infections, from contaminant to clinically relevant pathogen: this is a wake-up call! J Clin Microbiol. 2016;54(7):1679–1681. doi: 10.1128/JCM.00743-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahlstrand E, Hellmark B, Svensson K, Soderquist B. Long-term molecular epidemiology of Staphylococcus epidermidis blood culture isolates from patients with hematological malignancies. PLoS One. 2014;9(6):e99045. doi: 10.1371/journal.pone.0099045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimmerli W, Trampuz A, Ochsner PE. Prosthetic-joint infections. N Engl J Med. 2004;351(16):1645–1654. doi: 10.1056/NEJMra040181. [DOI] [PubMed] [Google Scholar]
- 10.Kurtz SM, Lau E, Watson H, Schmier JK, Parvizi J. Economic burden of periprosthetic joint infection in the United States. J Arthroplast. 2012;27(8 Suppl):61–65. doi: 10.1016/j.arth.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 11.Puhto T, Puhto AP, Vielma M, Syrjala H. Infection triples the cost of a primary joint arthroplasty. Infect Dis (Lond) 2019;51(5):348–355. doi: 10.1080/23744235.2019.1572219. [DOI] [PubMed] [Google Scholar]
- 12.Moore AJ, Blom AW, Whitehouse MR, Gooberman-Hill R. Deep prosthetic joint infection: a qualitative study of the impact on patients and their experiences of revision surgery. BMJ Open. 2015;5(12):e009495. doi: 10.1136/bmjopen-2015-009495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JYH, Monk IR. Goncalves da Silva a, Seemann T, Chua KYL, Kearns a, Hill R, Woodford N, Bartels MD, Strommenger B et al: global spread of three multidrug-resistant lineages of Staphylococcus epidermidis. Nat Microbiol. 2018;3(10):1175–1185. doi: 10.1038/s41564-018-0230-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li M, Wang X, Gao Q, Lu Y. Molecular characterization of Staphylococcus epidermidis strains isolated from a teaching hospital in Shanghai, China. J Med Microbiol. 2009;58(Pt 4):456–461. doi: 10.1099/jmm.0.007567-0. [DOI] [PubMed] [Google Scholar]
- 15.Miragaia M, Thomas JC, Couto I, Enright MC, de Lencastre H. Inferring a population structure for Staphylococcus epidermidis from multilocus sequence typing data. J Bacteriol. 2007;189(6):2540–2552. doi: 10.1128/JB.01484-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordon RJ, Miragaia M, Weinberg AD, Lee CJ, Rolo J, Giacalone JC, Slaughter MS, Pappas P, Naka Y, Tector AJ, et al. Staphylococcus epidermidis colonization is highly clonal across US cardiac centers. J Infect Dis. 2012;205(9):1391–1398. doi: 10.1093/infdis/jis218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Arias CA, Aitken SL, Galloway Pena J, Panesso D, Chang M, Diaz L, Rios R, Numan Y, Ghaoui S, et al. Clonal emergence of invasive multidrug-resistant Staphylococcus epidermidis Deconvoluted via a combination of whole-genome sequencing and microbiome analyses. Clin Infect Dis. 2018;67(3):398–406. doi: 10.1093/cid/ciy089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh J, Byrd AL, Deming C, Conlan S, Program NCS, Kong HH, Segre JA. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014;514(7520):59–64. doi: 10.1038/nature13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu L, Umeda A, Amako K. Typing of Staphylococcus epidermidis colonizing in human nares by pulsed-field gel electrophoresis. Microbiol Immunol. 1995;39(5):315–319. doi: 10.1111/j.1348-0421.1995.tb02207.x. [DOI] [PubMed] [Google Scholar]
- 20.De-la-Fuente M, Martinez-Perez M, Gonzalez-Pallares I, Esteban J. Detection of Polyclonality among clinical isolates from prosthetic joint infections. J Clin Microbiol. 2015;53(12):3766–3772. doi: 10.1128/JCM.01018-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrd AL, Deming C, Cassidy SKB, Harrison OJ, Ng WI, Conlan S, Program NCS, Belkaid Y, Segre JA, Kong HH. Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci Transl Med. 2017;9(397):1–12. [DOI] [PMC free article] [PubMed]
- 22.Cespedes C, Said-Salim B, Miller M, Lo SH, Kreiswirth BN, Gordon RJ, Vavagiakis P, Klein RS, Lowy FD. The clonality of Staphylococcus aureus nasal carriage. J Infect Dis. 2005;191(3):444–452. doi: 10.1086/427240. [DOI] [PubMed] [Google Scholar]
- 23.Votintseva AA, Miller RR, Fung R, Knox K, Godwin H, Peto TE, Crook DW, Bowden R, Walker AS. Multiple-strain colonization in nasal carriers of Staphylococcus aureus. J Clin Microbiol. 2014;52(4):1192–1200. doi: 10.1128/JCM.03254-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu L, Umeda A, Kondo S, Amako K. Typing of Staphylococcus aureus colonising human nasal carriers by pulsed-field gel electrophoresis. J Med Microbiol. 1995;42(2):127–132. doi: 10.1099/00222615-42-2-127. [DOI] [PubMed] [Google Scholar]
- 25.Martineau F, Picard FJ, Ke D, Paradis S, Roy PH, Ouellette M, Bergeron MG. Development of a PCR assay for identification of staphylococci at genus and species levels. J Clin Microbiol. 2001;39(7):2541–2547. doi: 10.1128/JCM.39.7.2541-2547.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poyart C, Quesne G, Boumaila C, Trieu-Cuot P. Rapid and accurate species-level identification of coagulase-negative staphylococci by using the sodA gene as a target. J Clin Microbiol. 2001;39(12):4296–4301. doi: 10.1128/JCM.39.12.4296-4301.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwase T, Seki K, Shinji H, Mizunoe Y, Masuda S. Development of a real-time PCR assay for the detection and identification of Staphylococcus capitis, Staphylococcus haemolyticus and Staphylococcus warneri. J Med Microbiol. 2007;56(Pt 10):1346–1349. doi: 10.1099/jmm.0.47235-0. [DOI] [PubMed] [Google Scholar]
- 28.Kim J, Hong J, Lim JA, Heu S, Roh E. Improved multiplex PCR primers for rapid identification of coagulase-negative staphylococci. Arch Microbiol. 2018;200(1):73–83. doi: 10.1007/s00203-017-1415-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meric G, Miragaia M, de Been M, Yahara K, Pascoe B, Mageiros L, Mikhail J, Harris LG, Wilkinson TS, Rolo J, et al. Ecological overlap and horizontal gene transfer in Staphylococcus aureus and Staphylococcus epidermidis. Genome Biol Evol. 2015;7(5):1313–1328. doi: 10.1093/gbe/evv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Månsson E, Johannesen TB, Nilsdotter Å, Söderquist B, Stegger M: Comparative genomics of Staphylococcus epidermidis from prosthetic joint infections and nares highlights genetic traits associated to antimicrobial resistance, not virulence. Submitted 2020. [DOI] [PMC free article] [PubMed]
- 32.Sahl JW, Lemmer D, Travis J, Schupp JM, Gillece JD, Aziz M, Driebe EM, Drees KP, Hicks ND, Williamson CHD, et al. NASP: an accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microb Genom. 2016;2(8):e000074. doi: 10.1099/mgen.0.000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacLea KS, Trachtenberg AM. Complete Genome Sequence of Staphylococcus epidermidis ATCC 12228 Chromosome and Plasmids, Generated by Long-Read Sequencing. Genome Announc. 2017;5(36):1–2. [DOI] [PMC free article] [PubMed]
- 34.Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 36.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, Parkhill J. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31(22):3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;1:10–12. [Google Scholar]
- 39.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kandlikar GS, Gold ZJ, Cowen MC, Meyer RS, Freise AC, Kraft NJB, Moberg-Parker J, Sprague J, Kushner DJ, Curd EE. ranacapa: An R package and Shiny web app to explore environmental DNA data with exploratory statistics and interactive visualizations. F1000Res. 2018;7:1734. doi: 10.12688/f1000research.16680.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag; 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Read counts after trimming and chimera filtering of the g216 and yych gene targets across all analyzed primary samples. The analyses revealed a higher level of yycH read counts compared to g216, however with an overall even distribution of across all samples.
Supplementary Figure 2. Rarefaction curves to investigate the dependency of Staphylococcus epidermidis abundance/lineages richness on sample library size in mock communities and primary samples. The generated rarefaction curves display the number of observed ASVs over the library size per sample after quality filtering, for g216 and yycH in panel A and B, respectively.
Supplementary Figure 3. Beta-diversity of the Staphylococcus epidermidis population across sample sites. Principal component analysis depicting the beta-diversity across all primary samples from skin and nares indicate overlapping populations highlighting samples sites compared to Fig. 4b.
Supplementary Table 1. Bacterial isolates used for laboratory validation of the method. Included are sequence types for all Staphylococcus epidermidis isolates, as well as a total of 48 different bacterial species commonly identified as skin commensals that were used for specificity testing; 27 staphylococcal species and 21 non-staphylococcal species.
Supplementary Table 2. qPCR results on standards, as well as primary skin and nasal samples. Presented are the threshold cycle (Ct) values with standard deviation as well as equivalent counts.
Data Availability Statement
The amplicon sequences are available through the European Nucleotide Archive (ENA) at the European Bioinformatics Institute (EBI) (PRJEB39895). The R scripts for the sequence pre-processing and taxonomy assignment, including the resulting datasets, as well as our custom data bases and R scripts for statistical analyses are available from https://github.com/ssi-dk/epidome/.