

Abstract
Extracellular vesicles (EVs) derived from mesenchymal stem cells (MSCs) have emerged as important mediators of intercellular communication in response to cartilage damage. In this study, we sought to characterize the inhibitory role of microRNA (miR)-31 encapsulated in synovial MSC (SMSC)-derived EVs in knee osteoarthritis (OA). The expression of miR-31, lysine demethylase 2A (KDM2A), E2F transcription factor 1 (E2F1), and pituitary tumor transforming gene 1 (PTTG1) was validated in cartilage tissues of knee OA patients. Following SMSC-EV extraction and identification, chondrocytes with the miR-31 inhibitor were added with SMSC-EVs, whereupon the effects of miR-31 on proliferation and migration of chondrocytes were assessed. The interaction among miR-31, KDM2A, E2F1, and PTTG1 in chondrocyte activities was probed in vitro, along with an in vivo mouse knee OA model. We identified downregulated miR-31, E2F1, and PTTG1 and upregulated KDM2A in cartilage tissues of knee OA patients. SMSC-EV-packaged miR-31 potentiated chondrocyte proliferation and migration as well as cartilage formation by targeting KDM2A. Mechanistically, KDM2A bound to the transcription factor E2F1 and inhibited its transcriptional activity. Enrichment of E2F1 in the PTTG1 promoter region activated PTTG1 transcription, accelerating chondrocyte proliferation and migration. SMSC-EVs and EVs from miR-31-overexpressed SMSCs alleviated cartilage damage and inflammation in knee joints in vivo. SMSC-EV-encapsulated miR-31 ameliorates knee OA via the KDM2A/E2F1/PTTG1 axis.
Keywords: mesenchymal stem cell-derived extracellular vesicles, microRNA-31, KDM2A, E2F1, PTTG1, knee osteoarthritis
Graphical Abstract
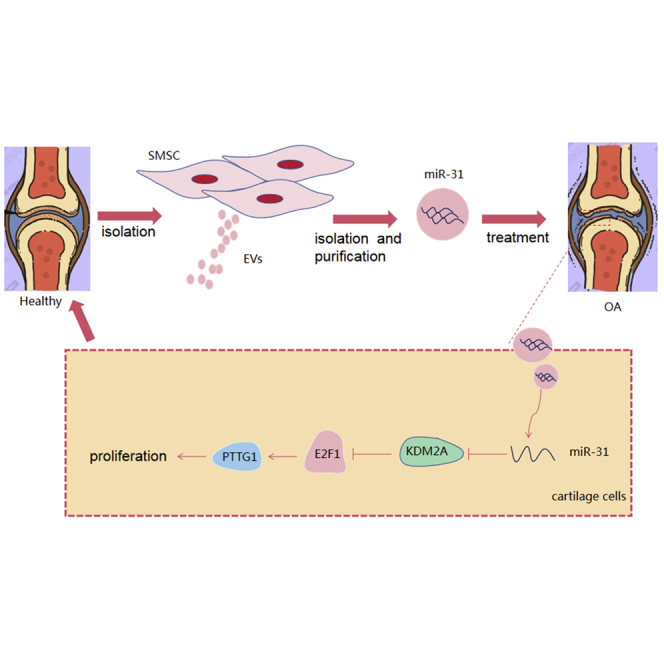
With the purpose of identifying the role of SMSC-secreted EVs containing miR-31 in knee OA, clinical samples, commercially purchased cell lines, and animals were used in a series of in vivo and in vitro experiments. The experimental data uncovered that SMSC-EV-encapsulated miR-31 ameliorates knee OA via the KDM2A/E2F1/PTTG1 axis.
Introduction
Osteoarthritis (OA) is a frequently occurring degenerative joint disease resulting in pain and dysfunction.1 OA represents a major cause of disability and social burden in older and obese populations,2 affecting nearly 30 million people all over the world and with no disease-modifying therapies.3 OA is characterized by progressive damage of articular cartilage, which is ascribed to the loss of chondrocytes in the articular cartilage.4 One factor in the present lack of disease-modifying managements may be attributed to inefficient delivery of drugs to target chondrocytes.3
Cell therapy is an attractive therapeutic platform for the treatment of a variety of diseases, including pulmonary, cardiovascular, and hepatic diseases.5 Among various types of cell therapies, stem cell therapy has been highlighted as a potential therapeutic approach, either using multi-potent or pluri-potent stem cells.6 The application of mesenchymal stem cells (MSCs) as therapeutic biological vehicles in cell therapy has gained substantial attention due to their advantages, which include immune silence, tumor tropism, easy and rapid isolation, ex vivo expansion, multilineage differentiation, and the ability to deliver various cargos of therapeutic agents.7,8 MSCs represent multi-potent precursors of connective tissue cells, which have emerged as a potential treatment option for joint disease and regeneration.9 Synovial MSCs (SMSCs) have remarkable proliferative and chondrogenic potential, thus imparting the possibility to repair cartilage injuries through accelerating proliferation of chondrocytes.10 Furthermore, prior evidence has indicated that extracellular vesicles (EVs) from bone marrow MSCs augment cartilage regeneration and alleviate OA.11 EVs isolated from human MSCs possess a powerful ability to improve joint repair and protect joints from degeneration after damage, thus constituting a therapeutic approach to joint damage and OA.12 EVs can participate in cellular communication by shuttling microRNAs (miRNAs or miRs), proteins, and messenger RNAs (mRNAs).13
Of note, miR-31 has been suggested to ameliorate OA due to its ability to strengthen chondrocyte viability and migration.14 In this study, the targeting relationship between miR-31 and lysine demethylase 2A (KDM2A) was first predicted by an initial bioinformatics analysis. Existing literature indicates that KDM2A can diminish the expression of trimethylate histone H3 at lysine 4 (H3K4me3) at the secreted frizzled-related protein 2 (SFRP2) promoter and thus, inhibits osteogenesis, thereby inducing the occurrence of OA.15 Moreover, KDM2A can bind to the transcription factor E2F transcription factor 1 (E2F1) and repress its transcriptional activity to regulate the occurrence and development of breast cancer cells.16 Interestingly, E2F1 has been implicated in the regulation mechanism of knee OA due to its potential in modulating articular chondrocytes.17 Moreover, E2F1 can elevate the transcription of its downstream target gene, pituitary tumor transforming gene 1 (PTTG1),18 which can promote OA progression.19 Therefore, we aimed to delineate a hypothesized mechanism by which miR-31-mediated effects on the KDM2A/E2F1/PTTG1 axis exert regenerative effects on cartilage during OA.
Results
SMSC-EV-Packaged miR-31 Stimulates Chondrocyte Proliferation In Vitro
We first isolated and characterized human SMSCs (Figures S1A and S1B) and identified the cellular immunophenotype of SMSCs, where CD44, CD90, and CD105 were positive, and CD14, CD34, CD45, and human leukocyte antigen (HLA)-DR were negative (Figure S1C). Then, we extracted EVs from SMSCs and further identified EVs using Transmission electron microscopy (TEM), nanoparticle tracking analysis (NTA), and western blot analysis. We found that SMSC-EVs had an average diameter of 95.01 ± 35.91 nm and expressed the EV maker proteins CD81, CD9, and CD63 but did not express calnexin protein (Figures S2A–S2C). Chondrocytes were stained with carboxyfluoresceinsuccinimidyl ester (CFSE), and SMSC-EVs were stained with CellMask Deep Red. Stained SMSC-EVs were added to CFSE-stained chondrocytes and cultured for 24 h. Observation with a fluorescence microscope revealed that SMSC-EVs were internalized or engulfed by chondrocytes (Figure 1A). miR-31 was highly expressed in SMSC-EVs (Figure 1B). To probe the effect of SMSC-EV-packaged miR-31 on chondrocyte proliferation and migration, we transfected chondrocytes with inhibitor-negative control (NC)/miR-31 inhibitor plasmids for 24 h and then treated chondrocytes with 10 μg of SMSC-EVs for 24 h, followed by determination of miR-31 expression in the cells and chondrocyte proliferation and migration. Relative to inhibitor-NC treatment, miR-31 inhibitor treatment reduced miR-31 expression in chondrocytes, while decreasing chondrocyte proliferation and migration ability. Following additional SMSC-EV treatment, miR-31 expression was elevated, and chondrocyte proliferation and migration ability also showed an upward trend (Figures 1C–1F). The aforementioned results suggest that SMSC-derived EVs can effectively promote chondrocyte proliferation.
Figure 1.

miR-31 Shuttled by SMSC-Derived EVs Stimulates Chondrocyte Proliferation In Vitro
(A) Representative immunofluorescence micrographs of CFSE (green)-labeled EVs internalized by chondrocytes, the nucleus of which was stained with 4′,6-diamidino-2-phenylindole (DAPI; blue) (×400, scale bar, 50 μm). (B) The expression of miR-31 determined by qRT-PCR in the supernatant and in SMSC-extracted EVs. ∗p < 0.05 compared with supernatant. (C) The expression of miR-31 determined by qRT-PCR in miR-31 inhibitor-treated chondrocytes with addition of SMSC-derived EVs. (D) Chondrocyte proliferation upon miR-31 inhibitor transfection and treatment with SMSC-derived EVs measured by the CCK-8 assay. (E) The number of migrated miR-31 inhibitor-transfected chondrocytes treated with SMSC-derived EVs measured by the Transwell assay. (F) Representative images of migrating chondrocytes (×200). ∗p < 0.05 compared with inhibitor-NC-transfected chondrocytes; #p < 0.05 compared with miR-31 inhibitor-transfected chondrocytes treated with PBS. Data are shown as mean ± standard deviation of three technical replicates. Unpaired t test was applied for comparison between two groups. Data comparison among multiple groups was performed using one-way ANOVA with Tukey’s post hoc test. Data comparison between groups at different time points was performed using repeated-measures ANOVA with Bonferroni correction.
KDM2A Is a Target of miR-31, and SMSC-EV-Packaged miR-31 Augments Cartilage Formation by Targeting KDM2A In Vitro
The downstream target genes of miR-31 were copredicted with RNA22, StarBase, and RNAInter of bioinformatics websites. An interaction network between miR-31 and mRNAs was obtained from the RNAInter website (Figure S3A). Following Venn diagram analysis of the aforementioned genes, 126 candidate genes were found at the intersection (Figure S3B). Analysis using the Coexpedia website found a coexpression relationship among the 126 genes (Figure S3C), and the correlation scores of the genes are shown in Table 1. KDM2A has been shown to diminish the expression of H3K4me3 at the SFRP2 promoter and thus inhibit osteogenesis, thereby promoting the occurrence of OA.15 At the same time, KDM2A emerged as one of the obtained candidate genes in the present study, leading us to focus on the relationship between KDM2A and miR-31. We first applied quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) and western blot analysis to determine the expression of miR-31 and KDM2A in clinical samples, which showed a linear relationship. Compared with articular cartilage tissues from non-OA subjects, samples from OA subjects showed lower miR-31 expression and higher KDM2A expression, and there was a negative regulatory relationship between the two markers (Figures S3D–S3F). Next, we further probed whether miR-31 could affect the occurrence of OA by modulating KDM2A. We first determined that KDM2A was a target gene of miR-31 through bioinformatic prediction (StarBase) (Figure S3G). A dual luciferase reporter gene assay further showed that cotransfection of miR-31 mimic with pGLO-KDM2A-wild-type (WT) diminished the luciferase activity, whereas there was no significant change in other treatment groups (Figure S3H). We employed mimic-NC/miR-31 mimic to transduce chondrocytes, finding that miR-31 mimic treatment downregulated KDM2A expression compared to mimic-NC treatment (Figures 2A and 2B). To further confirm that downregulation of KDM2A expression after EV treatment of chondrocytes was caused by miR-31 delivery to chondrocytes, we suppressed miR-31 in SMSCs and then extracted SMSC-EVs to treat the chondrocytes. This analysis showed that EV (inhibitor-NC) treatment led to elevated miR-31 expression and downregulated KDM2A expression, along with enhanced chondrocyte proliferation and migration. Whereas relative to treatment with EVs (inhibitor-NC), EV (miR-31 inhibitor) treatment resulted in diminished miR-31 expression, upregulated KDM2A expression, as well as repressed chondrocyte proliferation and migration (Figures 2C–2G). In addition, we also carried out the corresponding experiments in the isolated chondrocytes from OA patients. The results showed that chondrocytes overexpressing miR-31 showed downregulated KDM2A expression and meanwhile, that chondrocytes treated with EVs extracted from miR-31 inhibitor-transfected SMSCs exhibited upregulated KDM2A expression and reduced miR-31 expression, as well as attenuated cell proliferation and migration (Figure S4). The above results indicate that KDM2A is a target gene of miR-31, and miR-31-loaded EVs directly target KDM2A in chondrocytes to promote cartilage generation in vitro.
Table 1.
Coexpedia Website Analysis of Coexpression Relationship Scores among Genes
| Rank | Gene | Score |
|---|---|---|
| 1 | AK2 | 101.691 |
| 2 | ACADM | 68.124 |
| 3 | RBBP4 | 37.534 |
| 4 | PSMB2 | 35.276 |
| 5 | PPP1R12B | 30.895 |
| 6 | CDK1 | 26.551 |
| 7 | CSRP1 | 24.851 |
| 8 | ARPC5 | 23.382 |
| 9 | ATP2B4 | 23.214 |
| 10 | AHCYL1 | 23.153 |
| 11 | DDX21 | 20.968 |
| 12 | HDAC1 | 20.466 |
| 13 | PPT1 | 20.252 |
| 14 | STMN1 | 19.215 |
| 15 | GNAI3 | 15.976 |
| 16 | SAR1A | 14.501 |
| 17 | KDM2A | 14.331 |
| 18 | CELF1 | 14.118 |
| 19 | MAPKAPK2 | 12.851 |
| 20 | IDI1 | 12.309 |
Figure 2.

SMSC-EV-Packaged miR-31 Induces Cartilage Formation by Targeting KDM2A In Vitro
(A) The expression of miR-31 determined by qRT-PCR in chondrocytes transfected with miR-31 mimic. (B) The expression of KDM2A determined by qRT-PCR in chondrocytes transfected with miR-31 mimic. ∗p < 0.05 compared with chondrocytes transfected with mimic-NC. (C) The expression of miR-31 determined by qRT-PCR in chondrocytes treated with EVs from miR-31 inhibitor-transfected SMSCs. (D) The expression of KDM2A determined by qRT-PCR in chondrocytes treated with EVs from the miR-31 inhibitor transfected with SMSCs. (E) Chondrocyte proliferation measured by the CCK-8 assay in response to EVs from miR-31 inhibitor-transfected SMSCs. (F) The number of migrated chondrocytes measured by the Transwell assay in response to EVs from miR-31 inhibitor-transfected SMSCs. (G) Representative images of migrating chondrocytes in response to EVs from miR-31 inhibitor-transfected SMSCs (×200). ∗p < 0.05 compared with the control chondrocytes without any treatment; #p < 0.05 compared with chondrocytes treated with EVs from SMSCs transfected with inhibitor-NC. Data are shown as mean ± standard deviation of three technical replicates. Unpaired t test was applied for comparison between two groups. Data comparison among multiple groups was performed using one-way ANOVA with Tukey’s post hoc test. Data comparison between groups at different time points was performed using repeated-measures ANOVA with Bonferroni correction.
KDM2A Binds to Transcription Factor E2F1 and Inhibits Its Transcriptional Activity, Thereby Impeding Chondrocyte Proliferation In Vitro
We proceeded to predict downstream regulators of KDM2A using the websites hTFtarget and RNAInter to predict its downstream transcription factors. Analysis using the GeneCards database revealed 1,814 genes related to knee OA. Eight candidate genes (AR, MYC, ETS1, JUN, E2F1, HNF4A, SP1, and SMAD3) were obtained with the jvenn program through the intersection of prediction results and knee OA-related genes (Figure 3A). A previous study has shown that KDM2A can bind to transcription factor E2F1 and inhibit its transcriptional activity, thus regulating the occurrence and development of breast cancer cells.16 Meanwhile, E2F1 is involved in the regulation mechanism of OA.17 However, there are few reports on whether KDM2A regulates E2F1 and participates in knee OA. This led us to explore the regulatory mechanism of KDM2A and E2F1 in knee OA. A further search through the hTFtarget website confirmed the targeting relationship between KDM2A and E2F1 (Figure 3B).
Figure 3.

KDM2A Binds to Transcription Factor E2F1 and Inhibits Its Transcriptional Activity, Thereby Impeding Chondrocyte Proliferation In Vitro
(A) Venn diagram of the predicted downstream transcription factors of KDM2A by the hTFtarget and RNAInter websites with the genes related to knee OA in the GeneCards database. (B) Targeting relationship between KDM2A and the transcription factor E2F1 verified by the hTFtarget website. (C) Immunohistochemistry of E2F1 protein in articular cartilage tissues of OA (N = 54) and non-OA (N = 36) subjects. ∗p < 0.05 compared with articular cartilage tissues of non-OA subjects. (D) KDM2A mRNA expression determined by qRT-PCR in chondrocytes overexpressing KDM2A. ∗p < 0.05 compared with overexpression of NC (oe-NC)-transfected chondrocytes. (E) Representative western blots of E2F1 protein and its quantitation in chondrocytes overexpressing KDM2A. ∗p < 0.05 compared with oe-NC-transfected chondrocytes. (F) Endogenous E2F1 in Myc-labeled, KDM2A-treated chondrocytes. Input is a positive control, and IgG is a negative control. (G) Representative western blots of total E2F1 protein and its quantitation in chondrocytes treated with CHX (100 ng/mL) at 0 h, 6 h, 12 h, and 24 h. ∗p < 0.05 compared with oe-KDM2A-treated chondrocytes. ∗p < 0.05 compared with control chondrocytes. (H) GST-KDM2A fusion protein and E2F1 protein translated in vitro were used to perform a GST pulldown assay to detect exogenous E2F1. Input is a positive control, and GST is a negative control. (I) In the luciferase assay, cells of each group were treated with different doses of KDM2A to inhibit E2F1-mediated E2.Luc transcription in a dose-dependent manner. ∗p < 0.05. (J) KDM2A mRNA expression determined by qRT-PCR in chondrocytes overexpressing KDM2A or combined with E2F1. (K) Representative western blots of E2F1 protein and its quantitation in chondrocytes overexpressing KDM2A or combined with E2F1. ∗p < 0.05 compared with oe-NC-transfected chondrocytes; #p < 0.05 compared with oe-KDM2A-transfected chondrocytes; &p < 0.05 compared with oe-E2F1-transfected chondrocytes. (L) Proliferation of chondrocytes overexpressing KDM2A or combined with E2F1 measured by the CCK-8 assay. (M) The number of migrated chondrocytes overexpressing KDM2A or combined with E2F1 measured by the Transwell assay. (N) Representative images of migrating chondrocytes (×200). Data are shown as mean ± standard deviation of three technical replicates. Unpaired t test was applied for comparison between two groups. Data comparison among multiple groups was performed using one-way ANOVA with Tukey’s post hoc test. Data comparison between groups at different time points was performed using repeated-measures ANOVA with Bonferroni correction.
The mean E2F1 level in articular cartilage tissues from clinical samples was lower than in the control samples, but the difference was not significant (Figure 3C). To further confirm the regulatory relationship between KDM2A and E2F1, we overexpressed (oe)-KDM2A in chondrocytes and found that E2F1 protein levels were reduced in response to oe-KDM2A treatment compared to oe-NC (Figures 3D and 3E). We performed an immunoprecipitation assay and glutathione S-transferase (GST) pulldown assay to detect whether KDM2A interacted directly with E2F1 at the protein level. Myc-labeled KDM2A was transduced in cells to detect endogenous E2F1 binding. Myc-KDM2A was immunoprecipitated with the anti-E2F1 antibody but was not immunoprecipitated by control immunoglobulin G (IgG) (Figure 3F), indicating that KDM2A interacts with endogenous E2F1 in chondrocytes. Chondrocytes overexpressing KDM2A were treated with cycloheximide (CHX), and total E2F1 protein was measured by western blot analysis at 0, 6, 12, and 24 h (Figure 3G). Relative to the control chondrocytes (without oe-KDM2A treatment), E2F1 protein degradation in chondrocytes overexpressing KDM2A increased with time. As shown in Figure 3H, a GST pulldown assay was performed using a GST-KDM2A fusion protein and an E2F1 protein translated in vitro, showing that GST protein alone could not bind E2F1 but that GST-KDM2A pulled down the E2F1 protein, thus indicating that KDM2A could bind to E2F1. We then performed transient E2F1 transfection in chondrocytes, which increased expression of the E2.Luc reporter gene (8 ± 0.5-fold increase). Cotransfection of KDM2A inhibited this E2F1-mediated transcription in a dose-dependent manner (Figure 3I), indicating that KDM2A can inhibit E2F1-mediated transcriptional activity.
We overexpressed KDM2A and E2F1 in chondrocytes, finding that oe-KDM2A treatment elevated KDM2A expression, downregulated E2F1 expression, and inhibited the proliferation and migration of chondrocytes, all relative to oe-NC. In the presence of oe-E2F1, KDM2A was essentially unchanged but was E2F1 elevated, as was the proliferation and migration of chondrocytes. Relative to the oe-E2F1 treatment, oe-KDM2A + oe-E2F1 resulted in increased KDM2A, reduced E2F1, and repressed proliferation and migration of chondrocytes (Figures 3J–3N).
E2F1 Elevates Transcriptional Activation of the PTTG1 Gene and Augments Chondrocyte Proliferation In Vitro
We confirmed the targeting relationship between E2F1 and PTTG1 through the website hTFtarget (Figure 4A). In articular cartilage tissues of clinical samples, PTTG1 expression was lower in OA subjects compared to non-OA subjects (Figure 4B). Next, two short hairpin RNA (shRNA)-PTTG1s were designed for transfection, of which sh-PTTG1-1 had higher transfection efficiency and was selected for subsequent experiments (Figure 4C). In the presence of sh-PTTG1 and oe-E2F1, the expression of E2F1 and PTTG1 and the proliferation and migration of chondrocytes were evaluated (Figures 4D–4H). Relative to oe-NC + sh-NC, oe-NC + sh-PTTG1 treatment had little effect on the expression of E2F1 but reduced the expression of PTTG1 and inhibited the proliferation and migration of chondrocytes. Relative to oe-NC + sh-PTTG1, oe-E2F1 + sh-PTTG1 treatment resulted in upregulation of E2F1 and PTTG1 expression and increased the proliferation and migration of chondrocytes. A chromatin immunoprecipitation (ChIP) assay was performed on E2F1 small interfering RNA (siRNA)-treated chondrocytes to confirm whether E2F1 regulates PTTG1 expression transcriptionally and to evaluate the function of endogenous E2F1 protein in chondrocytes. After transfection of chondrocytes with 3xFLAG-E2F1 vector, E2F1 protein was found to be highly enriched in the promoter region of the PTTG1 gene (Figure 4I). Therefore, endogenous E2F1 protein can bind to the promoter region of PTTG1 and elevate the expression of the PTTG1 gene.
Figure 4.

E2F1 Elevates Transcriptional Activation of the PTTG1 Gene and Augments Chondrocyte Proliferation In Vitro
(A) Targeting relationship between E2F1 and PTTG1 retrieved in the hTFtarget website. (B) Immunohistochemistry of PTTG1 protein in articular cartilage tissues of OA (N = 54) and non-OA (N = 36) subjects. ∗p < 0.05 articular cartilage tissues of non-OA subjects. (C) Transfection efficiency of the sh-PTTG1 determined by qRT-PCR in chondrocytes. ∗p < 0.05 compared with cells treated with sh-NC. (D) mRNA expression of E2F1 determined by qRT-PCR in chondrocytes in response to transfection with sh-PTTG1 or combined with oe-E2F1. (E) Representative western blots of PTTG1 protein and its quantitation in chondrocytes in response to transfection with sh-PTTG1 or combined with oe-E2F1. ∗p < 0.05 compared with chondrocytes transfected with oe-NC + sh-NC; #p < 0.05 compared with chondrocytes transfected with oe-E2F1 + sh-NC. (F) Chondrocyte proliferation measured by the CCK-8 assay in response to transfection with sh-PTTG1 or combined with oe-E2F1. ∗p < 0.05 compared with chondrocytes transfected with oe-NC + sh-NC; #p < 0.05 compared with chondrocytes transfected with oe-E2F1 + sh-NC. (G) The number of migrated chondrocytes measured by the Transwell assay in response to sh-PTTG1 or combined with oe-E2F1. ∗p < 0.05 compared with chondrocytes transfected with oe-NC + sh-NC; #p < 0.05 compared with chondrocytes transfected with oe-E2F1 + sh-NC. (H) Representative images of migrating chondrocytes (×200). (I) The promoter region of the PTTG1 gene analyzed by microarray. The top indicates a schematic of the PTTG1 promoter region. The location of amplified fragments by qRT-PCR was based on the number of nucleotides and related to the transcription start site (TSS) (amplicon). Primer pairs are used for qRT-PCR analysis (detailed information can be seen in Materials and Methods). The intermediate, crosslinked, and sheared chromatin was immunoprecipitated with anti-FLAG antibody (left) and anti-H3K9me2 antibody (right). When E2F1 was increased, H3K9me2 was decreased, with the signal displayed as the percentage of input chromatin (bottom). The average standard deviation of three independent experiments was 6. The p value was calculated by independent sample t test (∗p < 0.05). At the bottom, we employed the anti-H3K9me2 antibody to the ChIP experiment and treated the chondrocytes with si-NC and si-E2F1. The average standard deviation of three independent experiments was 6. The p value was calculated by independent sample t test (∗p < 0.05). Data are shown as mean ± standard deviation of three technical replicates. Data comparison among multiple groups was performed using one-way ANOVA with Tukey’s post hoc test. Data comparison between groups at different time points was performed using repeated-measures ANOVA with Bonferroni correction.
SMSC-EV-Packaged miR-31 Targets KDM2A to Activate the E2F1/PTTG1 Axis, thereby Promoting the Proliferation and Migration of Chondrocytes In Vitro
We undertook further studies to show that miR-31 targets KDM2A and accelerates the activation of the E2F1/PTTG1 axis to modulate the proliferation and migration of chondrocytes. We extracted EVs from SMSCs (SMSC-EVs) transfected with inhibitor-NC/miR-31 inhibitor, which were added to sh-PTTG1-1-treated chondrocytes. The expressions of miR-31, KDM2A, E2F1, and PTTG1 (Figures 5A–5E) and the proliferation and migration of chondrocytes (Figures 5F and 5G) were examined. Versus sh-NC, sh-PTTG1 did not change the expression of miR-31, KDM2A, and E2F1 but reduced the expression of PTTG1 and inhibited the proliferation and migration of chondrocytes. Relative to sh-PTTG1, sh-PTTG1 + EVs (inhibitor-NC) elevated the expression of miR-31, downregulated KDM2A expression, elevated expression of E2F1 and PTTG1, and augmented proliferation and migration of chondrocytes. Relative to sh-PTTG1 + EVs (inhibitor-NC), sh-PTTG1 + EVs (miR-31 inhibitor) treatment led to diminished miR-31 expression, elevated KDM2A expression, diminished E2F1, PTTG1 expression, and impeded chondrocyte proliferation and migration (Figures 5A–5G).
Figure 5.

SMSC-EV-Packaged miR-31 Targets KDM2A to Activate the E2F1/PTTG1 Axis, Thereby Promoting the Proliferation and Migration of Chondrocytes In Vitro
sh-PTTG1-treated chondrocytes were treated with EVs from SMSCs transfected with the miR-31 inhibitor. (A) miR-31 expression determined by qRT-PCR in chondrocytes. (B) Representative western blots of KDM2A, E2F1, and PTTG1 proteins in chondrocytes. (C) KDM2A expression determined by qRT-PCR in chondrocytes. (D) E2F1 expression determined by qRT-PCR in chondrocytes. (E) PTTG1 expression determined by qRT-PCR in chondrocytes. ∗p < 0.05 compared with sh-NC-treated chondrocytes; #p < 0.05 compared with sh-PTTG1-treated chondrocytes; &p < 0.05 compared with sh-PTTG1 + EV (inhibitor-NC)-treated chondrocytes. (F) Chondrocyte proliferation measured by the CCK-8 assay. (G) The number of migrated chondrocytes measured by the Transwell assay. (H) Representative images of migrating chondrocytes (×200). Data are shown as mean ± standard deviation of three technical replicates. Data comparison among multiple groups was performed using one-way ANOVA with Tukey’s post hoc test. Data comparison between groups at different time points was performed using repeated-measures ANOVA with Bonferroni correction.
SMSC-EV-Packaged miR-31 Suppresses Knee OA through the KDM2A/E2F1/PTTG1 Axis In Vivo
We established a knee OA mouse model to confirm in vivo if SMSC-EV-packaged miR-31 targeted KDM2A to enhance activation of the E2F1/PTTG1 axis, thereby inhibiting the occurrence of knee OA. Cartilage tissue sections of the knee joint were stained with Safranin O-fast green and then morphologically observed. As depicted in Figures 6A and 6B, the cartilage structure of the sham-operated mice was normal. Relative to the sham-operated mice, the cartilage of OA mice was lightly stained, the surface layer was composed of damaged cartilage, and chondrocyte aggregation was reduced and irregular; these degenerative changes extended to deeper regions, indicating severe damage of the cartilage in OA mice. The degree of cartilage destruction in the OA mice treated with EVs recovered slightly but still showed moderate damage. The cartilage damage in the OA mice treated with EVs from miR-31 mimic-transfected SMSCs was significantly reduced, and the structure of the cartilage surface was restored, suggesting that miR-31 contained in SMSC-derived EVs prevented the OA damage. In addition, we determined the Osteoarthritis Research Society International (OARSI) scores of mice. The elevated knee cartilage OARSI score in OA mice decreased after 5 weeks of EV treatment (p < 0.05) and more significantly, in the OA mice treated with EVs from miR-31 mimic-transfected SMSCs (p < 0.01) (Figure 6C). These findings indicate that SMSC-EV-packaged miR-31 can prevent cartilage damage in knee OA.
Figure 6.

SMSC-EV-Packaged miR-31 Suppresses Knee OA through the KDM2A/E2F1/PTTG1 Axis In Vivo
(A) Safranin O and fast green staining (×200) of knee joint specimens of mice treated with EVs or EVs from miR-31 mimic-transfected SMSCs. (B) Statistical analysis of chondrocyte counts in a randomly selected high-power field of view. (C) Statistical analysis of OARSI score in mice treated with EVs or EVs from miR-31 mimic-transfected SMSCs. (D) miR-31 expression was determined by qRT-PCR in tissues of OA mice treated with EVs or EVs from miR-31 mimic-transfected SMSCs. (E) Representative western blots of KDM2A, E2F1, and PTTG1 proteins in tissues of OA mice treated with EVs or EVs from miR-31 mimic-transfected SMSCs. (F) Quantitation of KDM2A, E2F1, and PTTG1 proteins. (G) The concentrations of IL-1β, IL-6, and TNF-α were determined by ELISA in synovial fluid of OA mice treated with EVs or EVs from miR-31 mimic-transfected SMSCs. ∗p < 0.05 compared with the sham-operated mice; #p < 0.05 compared with OA mice treated with EVs. Data are shown as mean ± standard deviation of three technical replicates. Data comparison was performed using one-way ANOVA with Tukey’s post hoc test. n = 10 for mice following each treatment.
qRT-PCR and western blot analysis showed that compared with the sham-operated mice, miR-31, E2F1, and PTTG1 were underexpressed in OA mice, whereas KDM2A was overexpressed. In the OA mice treated with EVs, the expression of miR-31, E2F1, and PTTG1 was increased, whereas that of KDM2A was decreased. Similarly, in the OA mice treated with EVs from miR-31 mimic-transfected SMSCs, the expression of miR-31, E2F1, and PTTG1 was significantly increased, but that of KDM2A was suppressed (Figures 6D–6F).
Arthritis is usually accompanied by inflammation, and recent studies have shown that the concentrations of proinflammatory factors interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α increase during the development of OA.20,21 Therefore, we used enzyme-linked immunosorbent assay (ELISA) to measure the levels of inflammatory factors IL-1β, IL-6, and TNF-α in synovial fluid of OA mice, finding that compared with sham-operated mice, the levels of IL-1β, IL-6, and TNF-α were increased in OA mice. Relative to untreated OA mice, the levels of IL-1β, IL-6, and TNF-α were reduced in the OA mice treated with EVs, but the differences were not significant. In the OA mice treated with EVs from miR-31 mimic-transfected SMSCs, the levels of IL-1β, IL-6, and TNF-α were significantly reduced (Figure 6G). The above results indicate that SMSC-EV-packaged miR-31 can reduce cartilage damage and reduce inflammation through the KDM2A/E2F1/PTTG1 axis, suppressing the occurrence of knee OA.
Discussion
OA of the knee is the most common chronic musculoskeletal disorder and one of the leading causes of disability globally.22,23 The pathogenesis of OA is mainly attributed to cartilage damage, which is a vast obstacle for effective management, and delivery of beneficial gene vectors has been proposed as a targeted therapy.24 MSCs possess multilineage differentiation capacities, including osteogenesis and chondrogenesis.25 The alleviatory action of MSC-derived EVs for OA has been reported previously.26 Increasing miRNAs have a critical role to play in modulating chondrocyte behaviors and may potentially be used as therapeutic biomarkers.27 In this study, we expanded the understanding that miR-31 confers chondroprotective effects against knee OA by targeting the KDM2A/E2F1/PTTG1 axis.
The experimental observations identified downregulated miR-31, E2F1, and PTTG1 and upregulated KDM2A in cartilage tissues of knee OA patients. This finding corroborates that of a previous study by Dai et al.14 which also identified miR-31 downregulation in OA and also demonstrated that artificially overexpressing miR-31 could enhance chondrocyte viability and migration through targeting CXCL12. However, the authors of that study only provided observations, without further probing the mechanism underlying the role of miR-31 in OA, without presenting results potentially translatable into the clinical context, even though articular cartilage damage represents a key event for the initiation and progression of OA.28 We first extracted SMSCs from human synovium and prepared SMSC-EVs, which served to deliver miR-31 to chondrocytes and intensify their proliferation and migration of chondrocytes. EVs are known to be powerful mediators of intercellular communication via their miRNA content. Their widespread distribution and propensity to be secreted and internalized by mammalian cells give them an important role in many complex physiological processes, as shown in many experiments.29 A prior bioinformatic analysis had unveiled that miR-31 was closely related to the osteogenic differentiation of MSCs, indicating it as a promising biomarker for bone-regeneration disorders.30 Moreover, MSCs have the capacity to repair damaged cells and diminish inflammation. Indeed, degeneration of joints is caused by dysfunction in chondrogenesis and inflammation, but coculture of MSCs and chondrocytes could alleviate these changes and promote cartilage regeneration in the joints.9
Further, the targeting relationship between miR-31 and KDM2A was determined by our initial bioinformatics prediction, followed by a confirmatory dual luciferase reporter assay. SMSC-EVs mediated the delivery of miR-31, which bound to KDM2A and augmented cartilage formation. KDM2A could enrich in the COX-2 promoter region and accelerate its transcriptional activation.31 Meanwhile, COX-2 expresses at high levels in OA-damaged cartilages.32 Therefore, it is reasonable to speculate that KDM2A may potentiate OA aggravation by favoring inflammatory pathways. Our mechanistic investigations uncovered that KDM2A bound to transcription factor E2F1 and inhibited its transcriptional activity, thereby impeding chondrocyte proliferation. Another study has suggested that the binding of KDM2A to E2F1 had an association with cell cycle distribution and that KDM2A diminished E2F1-dependent induction of cell proliferation.16 Consistent findings in other studies suggest that upregulated E2F1 in chondrocytes resulted in intensified cartilaginous callus formation, which promotes bone regeneration following fractures.33 Our study furthered the mechanistic exploration and unveiled that E2F1 elevated transcriptional activation of the PTTG1 gene, thus augmenting chondrocyte proliferation. Prior evidence has identified that PTTG1 was a direct E2F1 target and that E2F1 could repress the expression and biological function of PTTG1.18 A previously conducted bioinformatics analysis by Wang et al.19 and Wang et al.34 has identified the association of PTTG1 with OA pathogenesis and proposed it as a potential target for OA diagnosis and treatment. The present in vitro experiments confirmed that SMSC-EV-packaged miR-31 could target KDM2A to activate the E2F1/PTTG1 axis, thereby promoting the proliferation and migration of chondrocytes. After the induction of knee OA in mice, we found that SMSC-EVs and EVs from SMSCs overexpressing miR-31 downregulated KDM2A and activated the E2F1/PTTG1 axis, thereby alleviating cartilage damage and downregulating inflammatory factors IL-1β and TNF-α in the OA model. IL-1β and TNF-α are pivotal inflammatory factors in the initiation of OA and contribute to chondrocyte death.4 MSC-EVs have previously been found to curb TNF-α and IL release, thus facilitating cartilage regeneration.11
Overall, the findings obtained from this study suggest that miR-31 was downregulated in OA and that it could promote the proliferation and migration of chondrocytes. The miR-31-loaded SMSC-EVs bound to KDM2A and activated the E2F1/PTTG1 axis, thereby facilitating chondrocyte proliferation. Furthermore, miR-31, shuttled by SMSC-derived EVs, could retard cartilage damage by inhibiting KDM2A, upregulating the E2F1/PTTG1 axis, and downregulating the expression of inflammatory factors IL-1β, IL-6, and TNF-α to prevent the occurrence of knee OA. These findings highlighted that therapeutic strategies for knee OA should be directed toward the artificial overexpression of miR-31, which may potentially be clinically viable targets in the treatment of cartilage damage. A better understanding of molecular mechanisms behind the chondroprotective effects of miR-31 would not only help us to identify pathogenic causes of OA but possibly also enable new targeted therapies for cartilage defect-related diseases. Additionally, clinical translation of the aforementioned signaling axis will need confirmation in an animal model. Of course, clinical translation shall await toxicity studies in view of the pleiotropic effects of miRNAs and of downstream gene methylation proteins (KDM2A) or transcription factors (E2F1). However, the targeting of connective tissues with SMSC-derived EVs might reduce potential side effects of miRNA treatments, which can be used as a good drug target for in-depth discussion in the future.
Materials and Methods
Ethics Statement
The study was conducted with approval of the Ethics Committee of the Fourth Hospital of Harbin Medical University. All participating patients signed informed consent documentation prior to sample collection. The animal experimental processes were approved by the Ethics Committee of the Fourth Hospital of Harbin Medical University and conducted in strict accordance to the standard of the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Microarray-Based Gene-Expression Profiling
The downstream target genes of miR-31 were predicted using the RNA22 (https://cm.jefferson.edu/rna22/), StarBase (http://starbase.sysu.edu.cn/), and RNAInter (http://www.rna-society.org/rnainter/) websites. Next, the intersected genes were analyzed with the use of jvenn (http://jvenn.toulouse.inra.fr/app/example.html). The coexpression relationship between target genes was analyzed using the Coexpedia website (http://www.coexpedia.org/), and further prediction was conducted according to the correlation score. Subsequently, hTFtarget (http://bioinfo.life.hust.edu.cn/hTFtarget#!/) and RNAInter websites were applied to predict the regulatory factors of mRNAs. Additionally, knee OA-related genes were retrieved from the GeneCards database (https://www.genecards.org/). Finally, the predicted regulatory factors and knee OA-related genes were intersected by jvenn.
Clinical Sample Collection
Articular cartilage tissues were collected from 54 knee OA patients (30 males and 24 females; aged 50–66 years) who underwent total knee arthroplasty at the Fourth Hospital of Harbin Medical University from August 2015 to August 2017. During the same period, articular cartilage tissues were collected from 36 non-OA patients (22 males and 14 females; aged 55–68 years) who underwent surgical treatment of femoral or neck fractures at the Fourth Hospital of Harbin Medical University. Non-OA patients had no history of OA or rheumatoid arthritis.
Cell Culture and Transduction
Human chondrocyte cell line CHON-001 (CRL-2846; American Type Culture Collection [ATCC], Manassas, VA, USA) was cultured with Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium (DMEM/F12) containing 10% fetal bovine serum (FBS), 1% penicillin-streptomycin solution, and 25 μg/mL Amphotericin B in an incubator at 37°C and 5% CO2.
Lentiviral vectors expressing miR-31 mimic, sh-PTTG1-1, and sh-PTTG1-2 were obtained from GenePharma (Shanghai, P.R. China). Lentiviral transduction was carried out following the manufacturer’s instructions. The shRNA sequences used in this study are shown in Table S1. miR-31 inhibitor and NCs were purchased from Ribobio (Guangzhou, P.R. China). Oligonucleotide transduction was performed with riboFECT CP reagent (Ribobio, Guangzhou, P.R. China).
The supernatant obtained in the extraction procedure of EVs from SMSCs was harvested and used as a control for the extracted EVs. The chondrocytes were transduced with miR-31 mimic or inhibitor, oe-KDM2A alone or together with oe-E2F1, and sh-PTTG1 alone or together with oe-E2F1. Phosphate-buffered saline (PBS) served as a control for 10 μg SMSC-derived EVs (SMSC-EVs) in miR-31 inhibitor-treated chondrocytes. The EVs extracted from SMSCs, transduced with the miR-31 inhibitor or inhibitor-NC, were cocultured with untreated chondrocytes (EVs [miR-31 inhibitor] or EVs [inhibitor-NC]) or sh-PTTG1-transduced chondrocytes (sh-PTTG1 + EVs [miR-31 inhibitor] or sh-PTTG1 + EVs [inhibitor-NC]). A total of 100 nM mimic-NC and miR-31 mimic and 100 nM inhibitor-NC and miR-31 inhibitor were purchased from Shanghai GenePharma (Shanghai, P.R. China). These plasmids were transfected into chondrocytes or SMSCs using Lipofectamine 2000 reagents (Invitrogen, Carlsbad, CA, USA). After 6 h of transfection, the culture solution was renewed, followed by an additional 48 h of culture, after which, the cells were collected for subsequent experiments.
Isolation and Identification of SMSCs
SMSCs were isolated from the synovium of human knee joints. To identify multiple differentiation potentials, differentiation of corresponding cell types was induced by switching to osteogenic (alizarin red staining), adipogenic (oil red O staining), or chondrogenic (Alcian blue staining) differentiation media.
The SMSCs at passage 3 were resuspended and adjusted to a density of 1 × 109 cells/L, followed by incubation with fluorescent dye-labeled antibodies against CD44, CD90, CD105, CD14, CD34, CD45, and HLA-DR (BD Biosciences, San Diego, CA, USA) in the dark. Flow cytometry (FACSVantage, Becton Dickinson, USA) was applied for cell fluorescence detection and data analysis.
Isolation and Identification of EVs from SMSCs
EVs were isolated from the conditioned medium of SMSCs. Cell debris was removed by centrifugation at 300 × g for 30 min. The particle size distribution of EVs was measured by dynamic light scattering (DLS). The microparticles (500–1,000 nm) were centrifuged at 10,000 × g for 20 min. Next, the EVs (50–500 nm) were sedimented by ultracentrifugation at 100,000 × g for 20 min. The EVs were then washed with 25 mL PBS and recentrifuged for 1 h at 100,000 × g, after which, the supernatant was removed, and the EVs were resuspended in 400 μL PBS for immediate use or storage at −80°C. TEM was used to identify EVs.35 EV pellets resuspended in PBS were subjected to NTA using a NanoSight NS-300 instrument (Malvern, Worcestershire, UK) at 405 nm. Total RNA and protein isolation kits (Invitrogen, Carlsbad, CA, USA) were applied to extract RNA and proteins from EVs for further analysis.
Immunofluorescence Assay for Uptake of EVs by Chondrocytes
EVs were stained with CellMask Deep Red (Thermo Fisher Scientific) with an excitation/emission wavelength of 649/666 nm. The EVs were then centrifuged at 100,000 × g for 1 h, and then the pellet was suspended in PBS, and the protein concentration was determined using a bicinchoninic acid (BCA) protein detection kit. Cells were stained with CellTrace CFSE (Life Technologies, Carlsbad, CA) with an excitation/emission of 492/517 nm. CFSE-stained cells were incubated with EVs at different time points and observed with a fluorescence microscope.
RNA Isolation and Quantitation
Total RNA was isolated from tissues, cells, and EVs using TRIzol reagents (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s protocol. Complementary DNA (cDNA) reverse transcription and qRT-PCR were performed as previously described.36 The relative expression of mRNA was normalized to endogenous β-actin, and that of miRNA was normalized to U6 (Table S1).
Cell Counting Kit-8 (CCK-8) Assay and Transwell Assays for Chondrocyte Proliferation and Migration
Transfected chondrocytes or chondrocytes treated with EVs for 24 h were seeded into a 96-well plate at a density of 2.5 × 104 cells/well overnight. Then, 10 μL of CCK-8 solution was added to each Petri dish and incubated at 37°C. At 0, 24, 48, and 72 h, cell proliferation was measured using a CCK-8 kit (Dojindo Molecular Technologies, Kyushu, Japan) and quantified with a plate reader at 450 nm. Three replicates were set for each sample, and three biological replicates were made.
The Transwell assay was applied to evaluate the migration of chondrocytes treated with EVs or infected with lentivirus. After digestion and counting, approximately 5 × 104 cells were seeded into the upper chamber of a 24-well plate. Next, 600 μL of chondrocyte medium containing EVs was incubated in the lower chamber of a Transwell plate at 37°C for 12 h. The upper chamber was then fixed with 4% paraformaldehyde and stained with 0.5% crystal violet. Cells on the upper surface of the upper chamber that had not migrated to the lower surface were carefully wiped off with a cotton swab. Five randomly selected fields per well were photographed using a Leica microscope and evaluated by two pathologists, who were blinded to the identity of the assays.
Western Blot Analysis
Cells were digested in radioimmunoprecipitation assay (RIPA) lysis buffer (Shanghai Beyotime Biotechnology, Shanghai, P.R. China). The total protein concentration was quantified using the BCA assay kit (Shanghai Beyotime Biotechnology, Shanghai, P.R. China). The protein was then separated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA). The membrane was blocked with 5% skimmed milk powder containing 0.1% Tween 20. Next, blots were probed with primary antibodies rabbit anti-KDM2A (ab191387), E2F1 (ab218527), PTTG1 (ab128040), rabbit anti-β-actin (ab8226, 1:5,000), rabbit anti-CD9 (ab92726, 1:2,000), rabbit anti-CD63 (ab134045, 1:1,000), rabbit anti-CD81 (ab109201, 1:5,000), and rabbit anti-calnexin (ab92573, 1:20,000) overnight at 4°C. After washing, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibody IgG (ab6721, 1:5,000) for 2 h. All antibodies were from Abcam (Cambridge, UK). An enhanced chemiluminescence reagent (catalog number [cat. no.] BB-3501; Amersham, Little Chalfont, UK) was employed to visualize the protein band and an imaging system (Bio-Rad, Hercules, CA, USA) for image acquisition and analysis.
Dual Luciferase Reporter Gene Assay
The binding site of KDM2A and miR-31 was predicted using the StarBase website, and the dual luciferase reporter gene assay was applied to verify whether KDM2A was a direct target of miR-31. Potential binding fragments of miR-31 in the KDM2A 3′ untranslated region (3′ UTR) and mutant (MUT) KDM2A 3′ UTR fragment with binding-site mutation were cloned into separate pGLO vectors, namely pGLO-KDM2A-WT and pGLO-KDM2A-MUT. These two reporter plasmids were cotransfected with miR-31 mimic or mimic-NC plasmids into HEK293T cells (Invitrogen, Carlsbad, CA, USA), respectively. After 24 h, the supernatant of HEK293T cells was collected, and the luciferase activity was measured using a dual luciferase reporter gene detection system (E1910; Promega, Madison, WI, USA). The relative luciferase (RLU) activity was calculated as the ratio of the RLU activity of firefly luciferase to that of Renilla luciferase.
Transient transfection was performed following a previous study.11 Chondrocytes were cotransfected with 1 μg E2F1 and KDM2A at different concentrations (0.5, 1, and 2 μg). A 0.5 μg sample of the pRL construct containing the glomerulonecoccal luciferase gene was used as a standardized control. By adding empty vector pGL3, the total DNA in each well was adjusted to the same level. The luciferase assay was performed using a dual luciferase assay system (Promega). RLU activity was defined as the ratio of activity of firefly luciferase to that of Renilla luciferase obtained from three independent experiments.37
Immunoprecipitation Assay
Coimmunoprecipitation, using anti-myc antibodies, was performed according to the kit manufacturer’s instructions. Anti-FLAG and anti-myc monoclonal antibodies were purchased from Sigma (St. Louis, MO, USA). Chondrocytes were lysed with ice-cold cell lysis buffer. Cell lysate was sonicated on ice and centrifuged at 4°C for 10 min. Supernatant (200 μL) and primary anti-Myc antibody (2 μL) were incubated overnight at 4°C, then mixed with 30 μL of 50% protein A-agarose beads (Upstate, Waltham, MA, USA), and shaken at 4°C for 1–3 h. Beads were washed five times with 500 μL of 1 × cell lysate. The protein was suspended in 20–50 μL of SDS sample buffer and analyzed by western blot analysis.
CHX Assay to Assess Protein Degradation
In chondrocytes overexpressing KDM2A, protein degradation was determined by CHX experiments, according to kit instructions. CHX was purchased from Sigma (St. Louis, MO, USA) and used at a concentration of 100 ng/mL. Western blot analysis was applied to measure total E2F1 protein at 0, 6, 12, and 24 h.
GST Pulldown Assay
GST and GST-KDM2A were purified from bacterial culture and bound with glutathione-Sepharose beads. The beads were washed 3 times with PBS, and protein synthesis was assessed by PAGE and Coomassie blue staining. 35S-methionine-labeled E2F1 was prepared using a rabbit reticulocyte lysate translation system, according to the manufacturer’s instructions (Promega, USA). Labeled lysate (10 μL) was incubated with an equal amount of GST or GST-KDM2A beads in protein-binding buffer. The samples were incubated at 4°C for 2 h and then washed six times in binding buffer. The bound protein was eluted in a gel-loading buffer and separated by SDS-PAGE, after which, the band was visualized by autoradiography. The amount of protein in the control input lane was approximately one-fifth of the total protein used in the binding assay.
ChIP Assay
ChIP analysis was performed using the ChIP kit (Millipore, Billerica, MA, USA), according to the protocols of the kit manufacturer. In brief, after cells were transfected with pCAGGS-n3fc (mock)/pCAGGS-n3FC-KDM3A (FLAG-E2F1) and siRNAs for 48 h, antibodies against FLAG and histone H3 Lys9 dimethylation (H3K9me2) (ab1220; Abcam, Cambridge, UK) were used to immunoprecipitate fragments of E2F1 and chromatin complex. Next, the bound DNA fragments were eluted and subjected to qRT-PCR analysis.
Establishment of Knee OA Models in Mice
Twenty male C57 mice (25–30 g, 8 weeks old) were subcutaneously anesthetized with sodium pentobarbital (8 mg/mL, 1 mL/100 g). Under a stereoscopic microscope, an OA model was established by completely transecting the medial collateral ligament and medial meniscus, cutting the meniscus at the narrowest point without damaging the tibial surface, and transecting the anterior cruciate ligament.36 The mice were randomly divided into 4 groups: (1) the sham group, which was unoperated and with saline injection in the articular cavity; (2) the OA group, which was treated on the first day of each week from the 5th to the 8th week after operation with saline injection in the joint cavity; (3) the OA + SMSC-EV group, which received articular cavity injection of 5 μL SMSC-EV particles per mL; and (4) the OA + EV (miR-31 mimic) group (mice received 5 μL miR-31 mimic-transduced SMSC-EV particles per milliliter). In each of the four groups, 10 knee joints of 5 mice were injected, thus n = 10 joints. Mice were injected every 3 days for 4 weeks. 12 weeks after the operation, the mice were euthanized by high concentrations of CO2, and knee samples were collected to assess disease progression. The knee with the more-pronounced OA was tested.
ELISA
Synovial fluid in the joint cavity of mice was collected at the time when the mice were euthanized. With the use of a cytokine ELISA kit (R&D Systems, MN, USA), the expression of IL-1β (ab100768; Abcam, Cambridge, UK), IL-6 (ab119548; Abcam, Cambridge, UK), and TNF-α (ab100785; Abcam, Cambridge, UK) was measured according to the manufacturer’s instructions.
Histological Staining
Mouse knee samples were collected at the 12th week after surgery in mice, and cartilage tissues were isolated and sectioned for immunohistochemistry. The knee joint bone was then fixed with neutral formalin (containing 4% formaldehyde) for 24 h and decalcified in ethylenediaminetetraacetic acid for 21 days (changed once a day), followed by dehydration with gradient ethanol, clearing with xylene, embedding in paraffin, and tissue sectioning (5 μm). The overall joint morphology was assessed with Safranin O and fast green staining. Based on Safranin O-fast green staining, an OARSI score21 was assigned.
Immunohistochemistry
Clinical samples were collected and processed to isolate cartilage tissues, and the tissue sections were subjected to immunohistochemical detection of KDM2A-, E2F1-, and PTTG1-positive expression rates. Antigens were retrieved from tissue sections by boiling in sodium citrate buffer. Immunohistochemistry was carried out by incubating sections with primary antibodies rabbit anti-KDM2A (ab191387; Abcam, Cambridge, UK), E2F1 (ab218527; Abcam, Cambridge, UK), and PTTG1 (ab128040; Abcam, Cambridge, UK). Normal rabbit serum was employed as NC instead of primary antibody. Next, the sections were incubated with HRP-secondary antibody and then exposed to 3,3′-diaminobenzidine (DAB) reagent prior to microscopic observation. KDM2A, E2F1, and PTTG1 expression was positive with brownish-yellow particles in chondrocytes. Specimens were scored according to the intensity of the dye color and the number of positive cells.38
Statistical Analysis
Statistical analysis was performed using SPSS 21.0 software (IBM, Armonk, NY, USA). The measurement data were expressed as mean ± standard deviation. Unpaired t test was applied for comparison between two groups. Data comparison among multiple groups was performed using one-way analysis of variance (ANOVA) with Tukey’s post hoc test. Data comparison between groups at different time points was performed using repeated-measures ANOVA with Bonferroni correction. Pearson correlation analysis was adopted to evaluate the correlation between miR-31 expression and KDM2A expression in clinical samples. p <0.05 indicated statistical significance.
Author Contributions
K.W., Feng Li, Y.Y., and L.S. designed the study. K.W., Y.C., and J.Q. analyzed and produced the initial draft of the manuscript. Feng Li and L.S. contributed to drafting the manuscript. All authors have read and approved the final submitted manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We acknowledge and appreciate our colleagues for their valuable efforts and comments on this paper. The primary data for this study are available from the authors on direct request. This study was supported by Scientific Research Project of Heilongjiang Health Commission (No.2020-088)
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.09.014.
Supplemental Information
References
- 1.Li H., Guo H., Lei C., Liu L., Xu L., Feng Y., Ke J., Fang W., Song H., Xu C. Nanotherapy in Joints: Increasing Endogenous Hyaluronan Production by Delivering Hyaluronan Synthase 2. Adv. Mater. 2019;31:e1904535. doi: 10.1002/adma.201904535. [DOI] [PubMed] [Google Scholar]
- 2.Hunter D.J., Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393:1745–1759. doi: 10.1016/S0140-6736(19)30417-9. [DOI] [PubMed] [Google Scholar]
- 3.Geiger B.C., Wang S., Padera R.F., Jr., Grodzinsky A.J., Hammond P.T. Cartilage-penetrating nanocarriers improve delivery and efficacy of growth factor treatment of osteoarthritis. Sci. Transl. Med. 2018;10:eaat8800. doi: 10.1126/scitranslmed.aat8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hosseinzadeh A., Kamrava S.K., Joghataei M.T., Darabi R., Shakeri-Zadeh A., Shahriari M., Reiter R.J., Ghaznavi H., Mehrzadi S. Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. J. Pineal Res. 2016;61:411–425. doi: 10.1111/jpi.12362. [DOI] [PubMed] [Google Scholar]
- 5.Mirzaei H., Sahebkar A., Sichani L.S., Moridikia A., Nazari S., Sadri Nahand J., Salehi H., Stenvang J., Masoudifar A., Mirzaei H.R., Jaafari M.R. Therapeutic application of multipotent stem cells. J. Cell. Physiol. 2018;233:2815–2823. doi: 10.1002/jcp.25990. [DOI] [PubMed] [Google Scholar]
- 6.Goradel N.H., Hour F.G., Negahdari B., Malekshahi Z.V., Hashemzehi M., Masoudifar A., Mirzaei H. Stem Cell Therapy: A New Therapeutic Option for Cardiovascular Diseases. J. Cell. Biochem. 2018;119:95–104. doi: 10.1002/jcb.26169. [DOI] [PubMed] [Google Scholar]
- 7.Mohammadi M., Jaafari M.R., Mirzaei H.R., Mirzaei H. Mesenchymal stem cell: a new horizon in cancer gene therapy. Cancer Gene Ther. 2016;23:285–286. doi: 10.1038/cgt.2016.35. [DOI] [PubMed] [Google Scholar]
- 8.Mirzaei H., Sahebkar A., Avan A., Jaafari M.R., Salehi R., Salehi H., Baharvand H., Rezaei A., Hadjati J., Pawelek J.M., Mirzaei H.R. Application of Mesenchymal Stem Cells in Melanoma: A Potential Therapeutic Strategy for Delivery of Targeted Agents. Curr. Med. Chem. 2016;23:455–463. doi: 10.2174/0929867323666151217122033. [DOI] [PubMed] [Google Scholar]
- 9.Barry F., Murphy M. Mesenchymal stem cells in joint disease and repair. Nat. Rev. Rheumatol. 2013;9:584–594. doi: 10.1038/nrrheum.2013.109. [DOI] [PubMed] [Google Scholar]
- 10.Wadowsky R.M., Butler L.J., Cook M.K., Verma S.M., Paul M.A., Fields B.S., Keleti G., Sykora J.L., Yee R.B. Growth-supporting activity for Legionella pneumophila in tap water cultures and implication of hartmannellid amoebae as growth factors. Appl. Environ. Microbiol. 1988;54:2677–2682. doi: 10.1128/aem.54.11.2677-2682.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vonk L.A., van Dooremalen S.F.J., Liv N., Klumperman J., Coffer P.J., Saris D.B.F., Lorenowicz M.J. Mesenchymal Stromal/stem Cell-derived Extracellular Vesicles Promote Human Cartilage Regeneration In Vitro. Theranostics. 2018;8:906–920. doi: 10.7150/thno.20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mianehsaz E., Mirzaei H.R., Mahjoubin-Tehran M., Rezaee A., Sahebnasagh R., Pourhanifeh M.H., Mirzaei H., Hamblin M.R. Mesenchymal stem cell-derived exosomes: a new therapeutic approach to osteoarthritis? Stem Cell Res. Ther. 2019;10:340. doi: 10.1186/s13287-019-1445-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Withrow J., Murphy C., Liu Y., Hunter M., Fulzele S., Hamrick M.W. Extracellular vesicles in the pathogenesis of rheumatoid arthritis and osteoarthritis. Arthritis Res. Ther. 2016;18:286. doi: 10.1186/s13075-016-1178-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai Y., Liu S., Xie X., Ding M., Zhou Q., Zhou X. MicroRNA-31 promotes chondrocyte proliferation by targeting C-X-C motif chemokine ligand 12. Mol. Med. Rep. 2019;19:2231–2237. doi: 10.3892/mmr.2019.9859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu G., Wang J., Lin X., Diao S., Cao Y., Dong R., Wang L., Wang S., Fan Z. Demethylation of SFRP2 by histone demethylase KDM2A regulated osteo-/dentinogenic differentiation of stem cells of the apical papilla. Cell Prolif. 2016;49:330–340. doi: 10.1111/cpr.12256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rizwani W., Schaal C., Kunigal S., Coppola D., Chellappan S. Mammalian lysine histone demethylase KDM2A regulates E2F1-mediated gene transcription in breast cancer cells. PLoS ONE. 2014;9:e100888. doi: 10.1371/journal.pone.0100888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pellicelli M., Picard C., Wang D., Lavigne P., Moreau A. E2F1 and TFDP1 Regulate PITX1 Expression in Normal and Osteoarthritic Articular Chondrocytes. PLoS ONE. 2016;11:e0165951. doi: 10.1371/journal.pone.0165951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou C., Wawrowsky K., Bannykh S., Gutman S., Melmed S. E2F1 induces pituitary tumor transforming gene (PTTG1) expression in human pituitary tumors. Mol. Endocrinol. 2009;23:2000–2012. doi: 10.1210/me.2009-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q., Li Y., Zhang Z., Fang Y., Li X., Sun Y., Xiong C., Yan L., Zhao J. Bioinformatics analysis of gene expression profiles of osteoarthritis. Acta Histochem. 2015;117:40–46. doi: 10.1016/j.acthis.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 20.Peng K.T., Liu J.F., Chiang Y.C., Chen P.C., Chiang M.H., Shih H.N., Chang P.J., Lee C.W. Particulate matter exposure aggravates osteoarthritis severity. Clin. Sci. (Lond.) 2019;133:2171–2187. doi: 10.1042/CS20190458. [DOI] [PubMed] [Google Scholar]
- 21.Gao G., Ding H., Zhuang C., Fan W. Effects of Hesperidin on H2O2-Treated Chondrocytes and Cartilage in a Rat Osteoarthritis Model. Med. Sci. Monit. 2018;24:9177–9186. doi: 10.12659/MSM.913726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tachmazidou I., Hatzikotoulas K., Southam L., Esparza-Gordillo J., Haberland V., Zheng J., Johnson T., Koprulu M., Zengini E., Steinberg J., arcOGEN Consortium Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat. Genet. 2019;51:230–236. doi: 10.1038/s41588-018-0327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Englund M. Osteoarthritis: Replacing the meniscus to prevent knee OA--fact or fiction? Nat. Rev. Rheumatol. 2015;11:448–449. doi: 10.1038/nrrheum.2015.87. [DOI] [PubMed] [Google Scholar]
- 24.Cucchiarini M., Madry H. Biomaterial-guided delivery of gene vectors for targeted articular cartilage repair. Nat. Rev. Rheumatol. 2019;15:18–29. doi: 10.1038/s41584-018-0125-2. [DOI] [PubMed] [Google Scholar]
- 25.Wang S.H., Gou G.H., Wu C.C., Shen H.C., Lin L.C., Pan R.Y. Increased COUP-TFII Expression Mediates the Differentiation Imbalance of Bone Marrow-Derived Mesenchymal Stem Cells in Femoral Head Osteonecrosis. BioMed Res. Int. 2019;2019:9262430. doi: 10.1155/2019/9262430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y., Yu D., Liu Z., Zhou F., Dai J., Wu B., Zhou J., Heng B.C., Zou X.H., Ouyang H., Liu H. Exosomes from embryonic mesenchymal stem cells alleviate osteoarthritis through balancing synthesis and degradation of cartilage extracellular matrix. Stem Cell Res. Ther. 2017;8:189. doi: 10.1186/s13287-017-0632-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheleschi S., De Palma A., Pecorelli A., Pascarelli N.A., Valacchi G., Belmonte G., Carta S., Galeazzi M., Fioravanti A. Hydrostatic Pressure Regulates MicroRNA Expression Levels in Osteoarthritic Chondrocyte Cultures via the Wnt/β-Catenin Pathway. Int. J. Mol. Sci. 2017;18:133. doi: 10.3390/ijms18010133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oláh T., Reinhard J., Gao L., Haberkamp S., Goebel L.K.H., Cucchiarini M., Madry H. Topographic modeling of early human osteoarthritis in sheep. Sci. Transl. Med. 2019;11:eaax6775. doi: 10.1126/scitranslmed.aax6775. [DOI] [PubMed] [Google Scholar]
- 29.Lovett J.A.C., Durcan P.J., Myburgh K.H. Investigation of Circulating Extracellular Vesicle MicroRNA Following Two Consecutive Bouts of Muscle-Damaging Exercise. Front. Physiol. 2018;9:1149. doi: 10.3389/fphys.2018.01149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia B., Zhang Z., Qiu X., Chu H., Sun X., Zheng X., Zhao J., Li Q. Analysis of the miRNA and mRNA involved in osteogenesis of adipose-derived mesenchymal stem cells. Exp. Ther. Med. 2018;16:1111–1120. doi: 10.3892/etm.2018.6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu S., Yang Y., Du Y., Cao L.L., Li M., Shen C., Hou T., Zhao Y., Wang H., Deng D. The transcription factor c-Fos coordinates with histone lysine-specific demethylase 2A to activate the expression of cyclooxygenase-2. Oncotarget. 2015;6:34704–34717. doi: 10.18632/oncotarget.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amin A.R., Attur M., Patel R.N., Thakker G.D., Marshall P.J., Rediske J., Stuchin S.A., Patel I.R., Abramson S.B. Superinduction of cyclooxygenase-2 activity in human osteoarthritis-affected cartilage. Influence of nitric oxide. J. Clin. Invest. 1997;99:1231–1237. doi: 10.1172/JCI119280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Premnath P., Zhu Y., Besler B.A., Boyd S., Krawetz R. Overexpression of E2F1 in chondrocytes increases cartilaginous callus formation and consequent bone regeneration after fracture. Osteoarthritis and Cartilage. 2018;26 (Supplement 1), S91–S92. [Google Scholar]
- 34.Wang Y.X., Wang R.L., Zhao L.L., Xiong G.Y., Xu J., Dian W. Expression of PTTG-1 and MMP-2 and their biological significance in synovium of rheumatoid arthritis. Chinese Journal of Clinical and Experimental Pathology. 2012;28:166–169. [Google Scholar]
- 35.Shentu T.P., Huang T.S., Cernelc-Kohan M., Chan J., Wong S.S., Espinoza C.R., Tan C., Gramaglia I., van der Heyde H., Chien S., Hagood J.S. Thy-1 dependent uptake of mesenchymal stem cell-derived extracellular vesicles blocks myofibroblastic differentiation. Sci. Rep. 2017;7:18052. doi: 10.1038/s41598-017-18288-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tao S.C., Yuan T., Zhang Y.L., Yin W.J., Guo S.C., Zhang C.Q. Exosomes derived from miR-140-5p-overexpressing human synovial mesenchymal stem cells enhance cartilage tissue regeneration and prevent osteoarthritis of the knee in a rat model. Theranostics. 2017;7:180–195. doi: 10.7150/thno.17133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rastogi S., Rizwani W., Joshi B., Kunigal S., Chellappan S.P. TNF-α response of vascular endothelial and vascular smooth muscle cells involve differential utilization of ASK1 kinase and p73. Cell Death Differ. 2012;19:274–283. doi: 10.1038/cdd.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riegger J., Brenner R.E. Evidence of necroptosis in osteoarthritic disease: investigation of blunt mechanical impact as possible trigger in regulated necrosis. Cell Death Dis. 2019;10:683. doi: 10.1038/s41419-019-1930-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.