

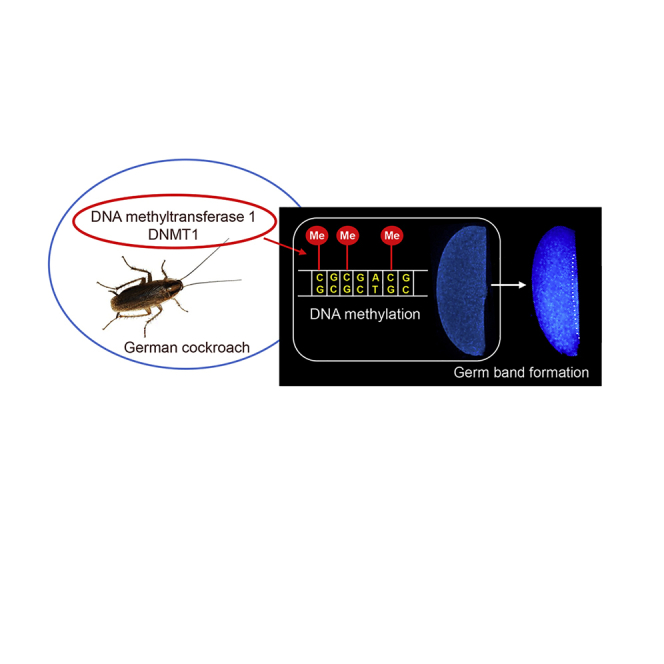
Summary
The influence of DNA methylation on gene behavior and its consequent phenotypic effects appear to be very important, but the details are not well understood. Insects offer a diversity of DNA methylation modes, making them an excellent lineage for comparative analyses. However, functional studies have tended to focus on quite specialized holometabolan species, such as wasps, bees, beetles, and flies. Here, we have studied DNA methylation in the hemimetabolan insect Blattella germanica. In this cockroach, a gene involved in DNA methylation, DNA methyltransferase 1 (DNMT1), is expressed in early embryogenesis. In our experiments, RNAi of DNMT1 reduces DNA methylation and impairs blastoderm formation. Using reduced representation bisulfite sequencing and transcriptome analyses, we observed that methylated genes are associated with metabolism and are highly expressed, whereas unmethylated genes are related to signaling and show low expression. Moreover, methylated genes show greater expression change and less expression variability than unmethylated genes.
Subject Areas: Developmental Genetics, Molecular Biology
Graphical Abstract
Highlights
-
•
Blattella germanica has DNMT1 and DNMT3 genes, which are expressed in early embryo
-
•
DNMT1 depletion reduces DNA methylation levels and impairs early embryo development
-
•
Methylated genes are highly expressed and are involved in metabolic processes
-
•
High DNA methylation is associated with low expression variability
Developmental Genetics; Molecular Biology
Introduction
DNA methylation is the covalent addition of a methyl group to a DNA nucleotide. In most of the animals studied, this only occurs in cytosines, particularly at CpG dinucleotide sites (He et al., 2011; Hunt et al., 2013; Bewick et al., 2017). It is a widespread epigenetic mechanism that contributes to gene expression regulation in eukaryotes (He et al., 2011; Jones, 2012; Sarda et al., 2012; Anastasiadi et al., 2018). In mammals, it has been associated with a number of biological processes, including embryo development, genomic imprinting, X-chromosome inactivation, and silencing of retrotransposons (He et al., 2011; Jones, 2012).
DNA methylation patterns differ between vertebrates and invertebrates. While in vertebrates, DNA methylation is typically localized in the 5′ regulatory regions and appears associated with gene inactivation (Anastasiadi et al., 2018), in invertebrates it is mainly localized in intragenic regions and seems associated with gene activation (Bonasio et al., 2012; Falckenhayn et al., 2013; Hunt et al., 2013; Wang et al., 2013; Glastad et al., 2014, 2016; Bewick et al., 2019). Moreover, DNA methylation levels in arthropods, particularly insects, are generally lower than in vertebrates (Bewick et al., 2017). Indeed, given the possibilities of comparing different orders that have distinct DNA methylation patterns (Bewick et al., 2017; Lewis et al., 2020), insects have become the model of choice for studying the functional significance of this DNA modification (Hunt et al., 2013).
Most insects develop through metamorphosis, which can be classified into two modes: hemimetabolan, or direct development through the embryo, nymph, and adult stages; and holometabolan, or diverging development through the embryo, larva, pupa, and adult stages (Belles, 2020). In this respect, although DNA methylation has been detected in the different insect groups, higher levels have been observed in hemimetabolan than holometabolan models (Falckenhayn et al., 2013; Bewick et al., 2017; Provataris et al., 2018). This, along with a comparative analysis between hemimetabolan and holometabolan insects, led us (Ylla et al., 2018) to hypothesize that DNA methylation could be instrumental in the type of embryo development, and the mode of metamorphosis. Many roles have been associated with DNA methylation in insects, including phenotypic plasticity and caste determination (Bonasio et al., 2012; Glastad et al., 2016; Robinson et al., 2016; Cardoso-Júnior et al., 2017; Li et al., 2018), alternative splicing (Bonasio et al., 2012; Glastad et al., 2014, 2016), and reproduction (Schulz et al., 2018; Bewick et al., 2019). However, there are few functional studies on the role of DNA methylation during early embryo development (Schulz et al., 2018; Bewick et al., 2019), despite this being the period when de novo DNA methylation is expected to occur (He et al., 2011).
DNA methylation is catalyzed by DNA-methyltransferases (DNMTs). In mammals, DNMTs are classified into DNMT3, which establishes new methylation (methylation de novo), and DNA methyltransferase 1 (DNMT1) which preferentially methylates hemimethylated DNA, maintaining methylation during successive cell generations (maintenance methylation) (He et al., 2011; Jones, 2012). Although a third DNMT was initially reported, further studies demonstrated that the so-called DNMT2 actually methylates tRNA, rather than DNA (Goll et al., 2006; Jurkowski et al., 2008; Lyko, 2018). Insects can possess either just DNMT1 (like the lepidopteran Bombyx mori and the coleopteran Tribolium castaneum), both DNMT1 and DNMT3 (like the hymenopteran Apis mellifera), or neither (like the dipteran, Drosophila melanogaster) due to secondary loss of both DNMT1 and DNMT3 (Bewick et al., 2017).
In a previous work, we reported the gene expression patterns in the German cockroach, Blattella germanica, on the basis of 11 transcriptomes representing key stages of embryonic and post-embryonic development (Ylla et al., 2018). One of the genes with the most characteristic profile was DNMT1, whose expression was concentrated in the first days of embryogenesis. At that time, we hypothesized that DNMT1 would catalyze DNA methylation and that it may play an important role in early embryo development. The present work was planned to study the DNMT1 expression pattern in detail, the relationships between DNMT1 and DNA methylation, and the possible role of DNMT1 in embryogenesis. Results obtained with quantitative PCR have confirmed that DNMT1expression concentrates in the first days of embryo development, and RNA interference (RNAi) and reduced representation bisulfite sequencing (RRBS) studies have revealed that DNMT1 promotes genome methylation and early embryo development in B. germanica.
Beyond these findings, by analyzing the genome-wide methylation profiles in regions with a high CpG content, and looking at the relationships between methylation levels and gene expression, we discovered certain regularities that may be of more general interest. Comparing methylated and unmethylated genes, we found that the former are related to metabolism and are highly expressed throughout development, while the latter are more associated with signaling pathways and generally have low expression levels. Moreover, methylated genes present a relatively high degree of expression change after the DNMT1 peak, but with little expression variability, whereas unmethylated genes display the opposite properties.
Results
Blattella germanica Has DNMT1 and DNMT3 Genes that Express in the Early Embryo
By combining a BLAST search in B. germanica transcriptomes (Ylla et al., 2018), mapping of the resulting sequences in the B. germanica genome (Harrison et al., 2018), and PCR strategies, we obtained a cDNA of 4,662 nucleotides comprising the complete ORF (GenBank: MT881788), whose conceptual translation gave a 1,554 amino acid sequence that was highly similar to insect DNMT1 proteins. We also obtained a cDNA of 1,803 nucleotides, comprising the complete ORF (GenBank: MT881790), whose conceptual translation gave a 601 amino acid sequence that was highly similar to insect DNMT3 proteins. A phylogenetic analysis using DNMT1 and DNMT3 sequences from representative species showed that the DNMT1 and DNMT3 identified in B. germanica clustered at the DNMT1 and DNMT3 nodes, respectively (Figure S1), strongly suggesting that these were DNMT1 and DNMT3 orthologs.
With regard to protein organization, B. germanica DNMT1 contains all the characteristic DNMT1 domains described by Lyko (2018) (Figure 1A): a DNMT1-associated protein 1 (DMAP1) binding domain, which allows interaction with the transcriptional repressor DNMAP1 and the histone diacetylase HDAC2 (HD2); a replication foci targeting sequence (RFTS), which allows DNMT1 to target replication foci; a CXXC domain, which allows DNMT1 to bind unmethylated DNA; two bromo-adjacent homology domains, whose function is still unknown; and a catalytic domain at the C-terminal. B. germanica DNMT3 also contains all the characteristic DNMT3 domains described by Lyko (2018) (Figure 1A): a PWWP domain, which allows binding to histone H3 molecules that are trimethylated at lysine 36; an ATRX-DNMT3-DNMT3L (ADD) domain, which mediates targeting to histone H3 molecules with unmethylated lysine 4; and a catalytic domain, the C5-Cytosine-specific DNA methylase domain. In previous analyses, we also found a bona fide DNMT2 ortholog (Ylla et al., 2018). However, as DNMT2 methylates tRNA (Goll et al., 2006; Jurkowski et al., 2008; Lyko, 2018), the B. germanica ortholog has not been considered in this work, which focuses on DNA methylation.
Figure 1.

Blattella germanica DNMT1 and DNMT3, and Effects of Maternal RNAi
(A) Protein organization of DNMT1 and DNMT3; the domains follow the nomenclature established by Lyko (2018); numbers indicate the start and end amino acids of the different domains.
(B and C) (B) qRT-PCR mRNA levels of DNMT1 and DNMT3 during embryogenesis; NFE: non-fertilized egg; ED0 to ED16: embryo day 0 to embryo day 16; (C) Effects of DNMT1 (upper panels) and DNMT3 (lower panels) maternal RNAi on DNMT1 and DNMT3 transcript levels; dsDNMT1, dsDNMT3 or dsMock were injected into 5-day-old adult females, and measurements were taken on ED1.
(D–G) Phenotypes observed in unhatched oothecae from DNMT1-depleted embryos; D: Phenotype PA, embryos with development interrupted at the pre-blastoderm stage; E: Phenotype PB, embryos with malformed head and appendage-like structures; F: Phenotype PC, embryos around Tanaka stage 13, with no appendages and a narrower abdomen than normal; G: Phenotype PD, embryos at Tanaka stage 18, ready to hatch, but with darker coloration than normal.
(H) Number of individuals showing the phenotypes PA to PD; the total number of individuals studied was 289, and the number of individuals in each category is indicated at the top of each bar; the sample also includes the 100 nymphs hatched from 3 viable oothecae (N1).
(I–L) Phenotypes observed in ED4 from DNMT1-depleted embryos; I: Normal ED4 embryo (N); J: Phenotype PE, embryos with development interrupted at Tanaka stage 2; K: Phenotype PF, embryos with development interrupted at Tanaka stage 3; L: Phenotype PG, embryo with a general morphology similar to Tanaka stage 4, with the cephalic and thoracic segments delimited but incompletely developed, and the abdominal region amorphous and unsegmented.
(M) Number of embryos showing the phenotypes PN and PE to PG; the total number of embryos studied was 120, and the number of embryos in each category is indicated at the top of each bar. In D-G, and I-L, the upper part of each picture corresponds to the cephalic part of the embryos; the scale bars are equivalent to 500 μm in panels D-G, and 100 μm in panels I-L. In Figures B and C, each qRT-PCR value represents three biological replicates and is expressed as copies of mRNA per 1000 copies of Actin-5c mRNA (mean ± SEM); the triple asterisk indicates statistically significant differences with respect to controls (p < 0.001), calculated on the basis of a Pairwise Fixed Reallocation Randomization Test implemented in REST (Pfaffl, 2002).
By using real-time quantitative reverse transcription PCR (qRT-PCR), we studied the expression of DNMT1 and DNMT3 during B. germanica embryogenesis (embryo days 0, 1, 2, 4, 6, 7, 9, 11, 13, 14, and 16). The results show that both DNMT1 and DNMT3 are expressed between days 0 and 2 of embryogenesis (0–12% of embryo development), both showing an expression peak at day 1 (Figure 1B). However, the expression levels of DNMT3 are very low (maximum expression of 0.98 ± 0.46 copies per 1000 Actin-5c copies at day 1), not only when compared with that of DNMT1 (maximum expression of 86.06 ± 8.83 copies per 1000 Actin-5c copies at day 1) but also taking into account the very low quantity of absolute mRNA at this early embryo stage, including the absolute levels of Actin-5c mRNA.
Maternal RNAi of DNMT1 and DNMT3
To study the possible functions of DNMT1 and DNMT3 in the early embryo, we used maternal RNAi. Five-day-old adult females (AdD5) of B. germanica were injected with 3 μg of a dsRNA targeting DNMT1 (dsDNMT1) or DNMT3 (dsDNMT3). These females were then allowed to mate (fertilization was checked at the end of the experiment by examining the presence of spermatozoids in the spermatheca) and to produce the first ootheca. Control females were treated equivalently but with a non-specific dsRNA (dsMock). To estimate the efficiency of the RNAi, we measured the levels of the respective transcripts in 1-day-old embryos, the day of peak expression. In the dsDNMT1-treated females, the mRNA levels of DNMT1 were 87.5% lower than in the controls (Figure 1C), indicating that the maternal RNAi was remarkably efficient. In addition, the DNMT3 mRNA levels were similar in both groups, indicating that dsDNMT1 is specific and does not affect DNMT3 transcripts. In contrast, dsDNMT3 treatment did not significantly affect the mRNA levels of DNMT3, despite the high dose of dsRNA and the replication using three experimental batches containing 10 females each. Figure 1C illustrates representative results demonstrating that our dsDNMT3 treatments did not reduce DNMT3 mRNA levels. As we were unable to deplete DNMT3 transcript levels, we continued the functional studies with DNMT1.
Depletion of DNMT1 Impairs Embryo Development
A total of 10 control (dsMock-treated) females formed the first ootheca on day 8 of the adult stage, which hatched 19 days later, giving a total of 373 first instar nymphs (35–40 nymphs per ootheca, on average). The dsDNMT1-treated females (n = 10) also produced the first ootheca on day 8 but only 3 of 10 oothecae (30%) hatched 19 days later, giving a total of 100 first instar nymphs (30–35 nymphs per ootheca). No nymphs hatched from the remaining 7 oothecae (70%) produced by the dsDNMT1-treated females. The examination of the embryos in the 7 unviable oothecae, 20 days after the formation of the ootheca (n = 189 embryos), showed various phenotypes, which were classified into the following four categories. Phenotype PA (Figure 1D): embryos with development interrupted in a pre-blastoderm stage, thus, only white yolk was observed. Phenotype PB (Figure 1E): embryos that were completely transparent under the stereomicroscope, but for which 4′,6-diamidino-2-phenylindole (DAPI) staining revealed malformations of the head and appendages. Phenotype PC (Figure 1F): embryos at Tanaka stage 13 (Tanaka, 1976) (58% embryo development), but with no appendages, and narrower abdomens than the controls. Phenotype PD (Figure 1G): embryos at Tanaka stage 18, thus, just prior to hatching, but presenting a darker coloration than the controls. A total of 125 embryos of the 189 studied showed phenotype PA (66% of the abnormal embryos and 43% of all DNMT1-depleted embryos). Phenotype PB was represented by 50 embryos (26% of the abnormal embryos and 17% of all DNMT1-depleted embryos). Phenotypes PC and PD were the least frequent; 12 embryos presented phenotype PC (6% of the abnormal embryos and 4% of all DNMT1-depleted embryos), and only 2 embryos presented Phenotype PD (1% of the abnormal embryos and 0.7% of all DNMT1-depleted embryos) (Figure 1H).
Since most of the embryos from dsDNMT1-treated females died early in their development, coinciding with the temporal expression of DNMT1, we repeated the maternal RNAi experiment, but this time we examined the embryos 4 days after oviposition (ED4). We studied 120 embryos from 5 oothecae produced by control (dsMock-treated) females and 120 embryos from 5 oothecae produced by dsDNMT1-treated females. All the embryos from control females (100%) presented the normal aspect of an ED4 embryo, in other words, 20–25% embryo development and Tanaka stage 5–6 (Tanaka, 1976) (Figure 1I). A total of 62 of 120 embryos (51.7%) examined in oothecae from dsDNMT1-treated females, were normal embryos, similar to the controls. The remaining 58 embryos (48.3%) showed several different phenotypes that were classified into three categories, as follows. Phenotype PE (Figure 1J): embryos with development interrupted at Tanaka stage 2, when the germ band is delimited and slightly expanded on both sides (12% embryogenesis). Phenotype PF (Figure 1K): embryos with development interrupted at Tanaka stage 3 (16% development), when the germ band has started to expand on both sides. Phenotype PG (Figure 1L): embryos with a general morphology similar to Tanaka stage 4, at the start of abdominal segmentation and tail folding (17% embryogenesis) but presenting various malformations: cephalic and thoracic segments delimited but incompletely developed; and amorphous and unsegmented abdominal regions. Phenotype PE was represented by 50 embryos (86% of the abnormal embryos and 42% of all the embryos), while phenotypes PF and PG had 4 embryos each (7% of the abnormal embryos and 3% of all embryos, in both cases) (Figure 1M).
Depletion of DNMT1 Reduces DNA Methylation
To assess whether DNMT1 is required for DNA methylation in B. germanica, we studied DNA methylation levels in DNMT1-depleted and control embryos, following the RRBS method. For this purpose, we performed RRBS in two different conditions: 4-day-old control embryos (ED4C) and 4-day-old DNMT1-depleted embryos (ED4T), using four biological replicates per condition. We analyzed the levels of methylated cytosines within CG dinucleotides in these two conditions in different genomic features. Firstly, we considered all the regions available from the RRBS, then the sequences corresponding to intergenic regions, the genes (the region that is transcribed, including the UTRs), the promoter region (considering an arbitrary length of 2 Kb upstream of the transcription start site), the 5′ UTR, and the 3′ UTR. Moreover, we examined the levels of methylated cytosines in exonic and intronic regions. We considered all exons as a whole (including the 3′ UTR), the first exon, the last exon, all exons except the first and the last, and the exon of monoexonic genes. We performed an equivalent analysis for introns.
Considering the whole gene and different gene features, we observed that the 3′ UTR regions have the higher average levels of methylation (Table 1). Moreover, CG methylation levels are higher in genic regions than in intergenic regions; and within genes these are higher in exonic regions, particularly 3′ UTR regions. In intronic regions, there is a tendency to show higher levels of CG methylation toward the 3′ region (Table 1). Characteristically, the methylation density (Figure 2A) of the different genetic features shows two clear peaks, indicating that they are either very methylated (80–100% methylation), or have very low levels of methylation (0–4%), practically without any intermediate values. RRBS sequencing also revealed that the levels of CG methylation are lower in DNMT1-depleted embryos than in controls, irrespective of the genomic feature examined. In most cases, the reduction was between 50% and 60%. Greater reductions were observed in the first exon (63.8% reduction) and the single exon of monoexonic genes (73.7% reduction) (Table 1). Consequently, DNMT1 depletion modified the bimodal distribution of CG methylation since a significant proportion of intermediate values appeared, and the peak of high values (80–100% methylation) was reduced (Figure 2A).
Table 1.
Effect of DNMT1 Depletion in Blattella germanica Embryos on CG Methylation Levels in Different Gene Regions
| Gene Region | Control | DNMT1-Depleted | Decrease (%) |
|---|---|---|---|
| All regions | 17.8 | 8.8 | 50.5 |
| Intergenic regions | 10.0 | 5.2 | 47.5 |
| Promoter region | 26.4 | 10.9 | 58.7 |
| Gene | 41.8 | 19.1 | 54.2 |
| 5′ UTR | 27.4 | 11.0 | 60.0 |
| All exons | 51.3 | 21.5 | 58.0 |
| First exon | 20.5 | 7.4 | 63.8 |
| Other exons | 62.7 | 27.2 | 56.7 |
| Last exon | 59.2 | 28.9 | 51.1 |
| Exon of monoexonic | 30.9 | 8.1 | 73.7 |
| All introns | 40.6 | 18.9 | 53.5 |
| First intron | 30.3 | 13.1 | 56.9 |
| Other introns | 45.8 | 21.4 | 53.2 |
| Last intron | 41.0 | 19.5 | 52.5 |
| Intron of monointronic | 23.3 | 11.3 | 51.3 |
| 3′UTR | 75.5 | 36.2 | 52.1 |
Measurements were taken on 4-day-old embryos, in controls and in DNMT1-depleted Insects. Results are expressed as a percentage of methylated CG.
Figure 2.

DNA Methylation in Blattella germanica and Effects of DNMT1 Depletion
(A) Kernel density plot of CG methylation in control and DNMT1-depleted 4-day-old embryos. The genomic features examined are the same as described in Table 1. In controls, the levels of CG methylation are generally either very high (80–100% methylation), or very low (0–4%), thus presenting a bimodal distribution; in DNMT1-depleted insects the bimodal distribution is modified as the peak of high values is reduced.
(B) Selection of GO terms of biological functions resulting from enrichment analyses carried out on methylated genes in 4-day-old control embryos. The 10 enriched biological functions with the lowest p values are shown for methylated and unmethylated genes. The p values were calculated according to Fisher's exact test.
Methylated Genes Are Associated with Metabolism and Are Highly Expressed, Whereas Unmethylated Genes Are Associated with Signaling and Show Low Expression Levels
To obtain information on the functions of the highly methylated genes (80–100% methylation, hereinafter referred to as “methylated” for simplicity) and practically unmethylated genes (0–4% methylation, hereinafter referred to as “unmethylated” for simplicity), we carried out a gene ontology (GO) enrichment analysis. The results (Figure 2B) indicate that methylated genes are enriched in biological functions related to metabolic processes, neurogenesis, and cytosolic transport. On the other hand, the potential biological functions of unmethylated genes appear to be related to signaling pathways, including neuropeptide signaling, cell surface receptor signaling, ion transport, signal transduction, detection of chemical stimulus, ecdysteroid metabolic processes, and leg patterning (Figure 2B).
Next, we examined the expression levels of the genes that had been designated as methylated or unmethylated in the 4-day-old embryonic stage in later stages of B. germanica. These stages were those associated to transcriptomes previously described (Ylla et al., 2018). The results show that the expression levels of the methylated genes are higher than those of the unmethylated genes in all the ontogenetic stages studied, the difference being more evident in early embryo stages, from ED0 to ED6 (Figure 3A).
Figure 3.

DNA Methylation and the Amount of Gene Expression in Blattella germanica
(A) Expression levels of methylated and unmethylated genes in the 11 embryo stages studied (NFE: non-fertilized egg, and ED0 to ED16: embryo day 0 to embryo day 16), four nymphal instars (N1, N3, N5, and N6), and the adult.
(B) Expression levels of methylated and unmethylated genes in 6-day-old embryos (ED6), considering gene expression levels in 6-day-old embryos (ED6), grouped by methylations status (unmethylated vs methylated) of various gene features. In all cases, expression is expressed as FPKM; the asterisks indicate statistically significant differences using the Mann-Whitney U test, adjusting p values by False Discovery Rate using the Benjamini-Hochberg method (∗ FDR <0.05; ∗∗ FDR <0.01; ∗∗∗ FDR <0.001; ∗∗∗∗ FDR <0.0001), non-significant differences (ns; FDR >0.05), are also indicated.
We then analyzed the differences in expression between methylated and unmethylated genes, considering the gene region where the methylation is located. For this analysis, we used the transcriptomic data corresponding to ED6 since this is the stage following the pulse of DNMT1 expression (Figure 1B). ED6 corresponds to Tanaka stage 8 (Tanaka, 1976), which precedes major developmental processes, like dorsal closure and organogenesis. The results show that the expression levels of methylated genes are significantly higher than those of unmethylated genes when methylation occurs in all the studied regions, except in the 5′ UTR or in the exon of monoexonic genes (Figure 3B). It is worth noting, however, that the number of annotated 5′ UTRs and monoexonic genes are relatively low, which could explain the lack of significant differences between methylated and unmethylated gene expression.
Methylated Genes Show Greater Expression Change Than Unmethylated Genes
Once again using the set of transcriptomes of Ylla et al. (2018), we examined the gene expression change between ED2 (when the peak expression of DNMT1 is already declining) and ED6 (4 days later) (Figure 1B). We determined the fold change (log2FC) of differentially expressed genes between these two stages, considering those having a | log2FC | ≥ 2 and false discovery rate of <0.05. In this way, we identified 1,599 genes, 553 of which were methylated and 1,046 of which were unmethylated. As shown in Figure 4A, both methylated and unmethylated genes increased or decreased their expression levels in similar proportions. Intriguingly, the change was less in methylated genes, regardless of whether this change was incremental or decremental, and this was more significant when the methylation was in the introns (Figure 4B). This notion can be condensed by expressing the coefficient of variation (CV) of gene expression between ED2 and ED6, which is significantly lower in methylated than in unmethylated genes (Figure 4C). The data suggest that the expression change of methylated genes has lower dispersion than that in unmethylated genes. This led us to analyze the CV of the expression levels of each gene between biological replicates at the same stage.
Figure 4.

DNA Methylation and Gene Expression Dynamics in Blattella germanica
(A) Expression increase or decrease between ED2 and ED6 in methylated and unmethylated genes; a minimum of Log2FC > 2 with FDR <0.05, was considered an increase or decrease.
(B and C) (B) Expression change (log2FC) between ED2 and ED6 of differentially upregulated or downregulated genes (Log2FC > 2 and FDR <0.05); in all cases, genetic features were classified as methylated or unmethylated; data outliers have been omitted for clarity; the gene features considered were those in Table 1 (C) Density plot and boxplot (inset) of the coefficient of variation (CV) of the gene expression between ED2 and ED6, considering methylated and unmethylated genes; the inset describes the mean CV between ED2 and ED6 in methylated and unmethylated genes; data outliers have been omitted for clarity. In all cases, the gray bars indicate methylated genes, and the white bars are unmethylated genes; in B and C, asterisks indicate statistically significant differences using the Mann-Whitney U test, adjusting p values by False Discovery Rate using the Benjamini-Hochberg method (∗ FDR <0.05; ∗∗ FDR <0.01; ∗∗∗ FDR <0.001; ∗∗∗∗ FDR <0.0001), non-significant differences (ns; FDR >0.05), are also indicated.
Methylated Genes Have Less Expression Variance Than Unmethylated Genes
Using two biological replicates, each comprising a pool of specimens from the transcriptome set of Ylla et al. (2018), we first compared the gene expression levels between replicates at the different stages. The results showed that there are no differences between replicates (Figure S2). We then calculated the gene expression CV between the replicates in the same stage, comparing methylated with unmethylated genes at each stage. The results show that methylated genes present less expression variability, measured as covariance, between biological replicates than unmethylated genes do, a property that is more evident in earlier embryo stages (from ED0 to ED6) (Figure 5).
Figure 5.

DNA Methylation and Expression Variability in Blattella germanica
The coefficient of variation (CV) of gene expression for methylated and unmethylated genes between the two biological replicates generated from a pool of specimens for each of the developmental transcriptomes studied. Data outliers have been omitted for clarity; the developmental stages studied were: NFE: non-fertilized egg; ED0 to ED13: embryo day 0 to embryo day 13; N1-N6: first to sixth nymphal instar; and the adult (Ylla et al., 2018). The four asterisks indicate statistically significant differences using the Mann-Whitney U test, adjusting p values by False Discovery Rate using the Benjamini-Hochberg method (FDR <0.0001).
Discussion
The cockroach B. germanica has two DNMT genes, one coding for DNMT1 and one coding for DNMT3, which possess the functional motifs characteristic of these kinds of proteins, according to Lyko (2018). Quantitative determinations showed that DNMT1 and DNMT3 are expressed during the early embryo development (between 0% and 12% embryogenesis) of B. germanica. This suggests that both genes play roles in early embryogenesis, although DNMT3 expression levels are about 100 times lower than those of DNMT1. To study these roles, we used maternal RNAi, which efficiently knocked down the DNMT1, but not the DNMT3, whose mRNA levels were not reduced despite the relatively high doses of dsRNA used, and the three independent experimental batches employed. Although B. germanica is highly sensitive to RNAi (Belles, 2010), there are situations where this technique has been ineffective. For example, in the case of the lipophorin receptor, RNAi has proven highly efficient in the fatty body, where the gene is highly expressed, and much less efficient in the ovary, where it is expressed at low levels (Ciudad et al., 2007). In other cases, such as that of the yellow-g gene, the transience of its expression makes its depletion by RNAi impossible (Irles et al., 2009). We believe that the very low expression levels of DNMT3 (about 1 copy of mRNA per 1000 copies of Actin-5c mRNA at most) are very difficult to significantly lower any further using RNAi.
In the case of DNMT1, although the penetrance of the effects was not 100%, as is usual in maternal RNAi (Belles, 2010), the transcript decrease obtained and the phenotypes observed were clear. Indeed, the RNAi experiments and subsequent RRBS analyses showed that DNMT1 promotes DNA methylation in B. germanica, as observed in other insects, such as the milkweed bug Oncopeltus fasciatus (Bewick et al., 2019) and the beetle T. castaneum (Schulz et al., 2018), when implementing an equivalent approach. The RNAi experiments also revealed that DNMT1, and thus DNA methylation, promotes the formation of the germband in early embryogenesis, at 12% development. In the hymenopteran Nasonia vitripennis DNMT1-depleted embryos die at the onset of gastrulation (Zwier et al., 2012), at around 40% embryo development. In the beetle T. castaneum, although DNA methylation does not preferentially occur at CpG sites (Zemach et al., 2010; Feliciello et al., 2013; Song et al., 2017), DNMT1 is required in very early embryo development to progress beyond the first few cleavage cycles, in other words around 4% embryogenesis (Schulz et al., 2018). In the bug O. fasciatus, eggs laid by DNMT1-depleted females are inviable, although the stage at which development is interrupted has not been determined (Bewick et al., 2019). The phenotype of embryos that complete development but are incapable of hatching is reminiscent of what is observed in a percentage of embryos with depleted JH signaling (Fernandez-Nicolas and Belles, 2017). In the present case, this phenotype might correspond to embryos with alterations in the expression of hatching regulatory genes, alterations that could derive from a deficient state of methylation. The fact that DNMT1 is required for embryo development in vertebrates, including mice (Li et al., 1992; Jackson-Grusby et al., 2001), frogs (Stancheva et al., 2001), and zebrafish (Rai et al., 2006), may suggest that their functions in embryogenesis are conserved from insects to vertebrates (Zwier et al., 2012). However, DNMT1 depletion in the insects B. germanica, N. vitripennis, and T. castaneum affects different embryo stages, and DNMT1 depletion in mice, frogs, and zebrafish also elicits different phenotypes, resulting in the misexpression of genes that specify embryonic cell identity but with limited effects on early developmental mitosis (He et al., 2011). Thus, although DNA methylation is instrumental for embryogenesis in cockroaches up to mammals, the current evidence indicates that its specific action varies in different lineages, even within insects.
The matching expression patterns of DNMT1 and DNMT3 in B. germanica suggest that both act on the same early embryo stage, while the presence of a methyltransferase catalytic domain in DNMT1 and DNMT3 suggests that they both have the capacity to promote DNA methylation. However, when DNMT1 is depleted, a clear phenotype is observed in the embryo. This indicates that DNMT3 expression, which is not affected by DNMT1 RNAi, does not compensate for the DNMT1 deficiency. These lines of evidence point to the possibility that both proteins are functionally redundant, and if so, DNMT3, which has very low expression levels, could be dispensable.
Most mammals have two DNMT3, DNMT3A, and DNMT3B, which establish DNA methylation patterns. Even many rodent species have a third enzyme, DNMT3C that selectively methylate the promoters of young retrotransposon insertions in their germline (Molaro et al., 2020). In contrast, DNMT3 has been evolutionarily lost in a number of insect orders, including Odonata, Ephemeroptera, Orthoptera, Thysanoptera, Phthiraptera, Lepidoptera, Trichoptera, and Diptera (Bewick et al., 2017; Lewis et al., 2020). Moreover, Bewick et al. (2017) showed that the presence of DNMT1 correlates positively with DNA methylation, whereas that is not seen for DNMT3. These authors suggest that either DNMT3 is unnecessary for DNA methylation or that DNMT1 compensates for DNMT3. Finally, studies in vitro have demonstrated that DNMT1 can also act as a de novo methyltransferase (Fatemi et al., 2002). Taken together, the data suggest that B. germanica DNMT1 plays both the de novo and maintenance roles in DNA methylation, while DNMT3 has a minor role, and is possibly redundant with respect to DNMT1. It is worth noting that the DNMT3 sequence of B. germanica, especially the catalytic domain (Figure S3), is remarkably conserved with respect to other proven functional DNMT3, such as that of the honeybee A. mellifera (Wang et al., 2006), suggesting that B. germanica DNMT3 is functional, and thus natural selection maintains the conserved sequence.
As in other species, the CG methylation levels in B. germanica present a bimodal distribution, being either very high or very low. In insects, this has also been reported in the locust Schistocerca gregaria (Falckenhayn et al., 2013) and the wasp N. vitripennis (Wang et al., 2013). Moreover, CG methylation in B. germanica tends to concentrate toward the 3′ region of the gene, in line with general DNA methylation trends in insects (Bewick et al., 2017; Lewis et al., 2020). In holometabolan species, DNA methylation appears to be biased toward the exons close to the 5′ region of the gene (Bonasio et al., 2012; Hunt et al., 2013; Wang et al., 2013; Glastad et al., 2016), while in hemimetabolans it presents higher levels toward the 3′ region of the gene coding part (Glastad et al., 2016; Bewick et al., 2019). In this sense, the DNA methylation pattern of hemimetabolans is similar to that of vertebrates, where the first intron and first exon are less methylated than the remaining regions in different tissues, species, and developmental stages (Anastasiadi et al., 2018). Furthermore, DNA methylation is biased toward exons rather than introns in some hemimetabolan insects, like the locust S. gregaria (Falckenhayn et al., 2013), the termite Zootermosis nevadensis (Glastad et al., 2016), and the bug O. fasciatus (Bewick et al., 2019), as is also the case in B. germanica. In general, our findings in embryos are similar to those observed by Bewick et al. (2019) in B. germanica adults, using whole-genome bisulfite sequencing data. These authors found similar general levels of CG methylation, with the highest being observed in intragenic regions rather than intergenic regions, and which tended to concentrate toward the 3′ UTR (Bewick et al., 2019; Lewis et al., 2020).
With respect to DNA methylation and gene functions, GO enrichment analyses revealed that methylated genes are mainly involved in metabolic processes, and are more highly expressed than unmethylated genes, which are instead related to signaling pathways. In other insects, like the ant Camponotus floridanus (Bonasio et al., 2012) and the wasp N. vitripennis (Wang et al., 2013), both holometabolan insects, GO terms analyses performed on methylated genes revealed that they were enriched for housekeeping functions. Furthermore, it has recently been found that putatively methylated genes are under stronger purifying selection in both hemimetabolan and holometabolan insects (Ylla et al., 2020), highlighting the evolutionary importance of those genes undergoing DNA methylation.
A controversial aspect of DNA methylation is whether it can stimulate or repress gene expression. In vertebrates, a negative correlation between DNA methylation and gene expression has been reported, especially when methylation is located in the promoters, first intron, and first exon (Anastasiadi et al., 2018). In insects, a number of studies report a positive correlation between DNA methylation in intragenic regions and gene expression, such as in the termite Z. nevadensis (hemimetabolan) (Glastad et al., 2016), the ants C. floridanus and Harpegnathos saltator (Bonasio et al., 2012), and the wasp N. vitripennis (Wang et al., 2013) (holometabolans). However, in other insects like the desert locust S. gregaria (Falckenhayn et al., 2013), and the bug O. fasciatus (Bewick et al., 2019) (both hemimetabolans), no relationships have been found between DNA methylation and gene expression, although in the migratory locust, Locusta migratoria, alternative solitary or gregarious phases are associated with the methylation status of genes that are differentially expressed in these two phases, and in the expression of genes involved in DNA methylation (Robinson et al., 2016). Our observations indicate that methylated genes are significantly more expressed than unmethylated genes, especially in early embryogenesis (from ED0 to ED6), regardless of the methylation location in the gene.
The transcriptomic analysis of the methylated and unmethylated genes between ED2 and ED6 (i.e., after the DNMT1 expression pulse) showed that the percentage of methylated genes with increased expression was higher than the percentage of those with decreased expression (61% vs. 39%). Nevertheless, the unmethylated genes behaved similarly (78% vs. 22%). Comparing the magnitude of change, in other words, how much the gene expression increased or decreased between ED2 and ED6, and the amount of expression variability (in terms of CV), revealed more that high DNA methylation levels are associated with high expression levels. At the same time, the amount of change between ED2 and ED6 is lower in methylated genes, regardless of whether the change involved increasing or decreasing expression. This fits with the results of our GO enrichment analyses, as signaling factors are rarely expressed at high levels, but suffer higher expression variations, whereas high expression levels and low expression variation of housekeeping genes, and increased metabolism, is typical in early embryo development (Miyazawa and Aulehla, 2018). Our results are reminiscent of those obtained for N. vitripennis, where methylated genes were shown to have higher median expression levels and lower expression variation across developmental stages than unmethylated genes (Wang et al., 2013).
Finally, comparing the gene expression between biological replicates at the different stages of B. germanica development revealed that methylated genes show lower expression variability than unmethylated genes. This indicates that the expression of the methylated genes is tightly regulated, a feature that fits with the essential roles identified for these genes. The amount of intrinsic expression variability between individuals has been considered an inherent property of genes (Alemu et al., 2014; De Jong et al., 2019), representing a layer of gene regulation information that is just as important as changes in the mean expression levels (Wang and Zhang, 2011). Expression variability has been shown to be low in genes involved in growth, general metabolism, and universal functions, whereas it is high in genes involved in environmental responses and non-housekeeping functions, in general, thus affecting gene network functioning by lowering noise (Alemu et al., 2014; De Jong et al., 2019).
Several features pertaining to the genomic, epigenomic, regulatory, polymorphic, functional, structural, and network characteristics of the gene, have been correlated with expression variability (Alemu et al., 2014). In the epigenomic context, a long-standing hypothesis posits that DNA methylation in gene regions reduces transcriptional noise, although the mechanisms involved are unclear (Bird, 1995; Suzuki et al., 2007). However, the information supporting this hypothesis is scarce and it focuses on human tissues. Using nucleotide-resolution data on genomic DNA methylation and microarray data for human brain and blood tissues, Huh et al. (2013) showed that gene body methylation appears to lower expression variability. Further studies of human brain tissues, using Illumina sequencing, have indicated that genes with low and high expression variability are likely to have low and medium gene methylation, respectively, whereas non-variable genes are likely to be highly methylated (Bashkeel et al., 2019). Also in plants, recent studies of Arabidopsis thaliana indicate that genes with high expression variability are depleted in DNA methylation (Cortijo et al., 2019). Our data on B. germanica, based on RRBS sequencing and transcriptomic data during embryo development, when DNA methylases are expressed and DNA methylation occurs, afford the first association between high DNA methylation and low expression variability in an insect.
Limitations of the Study
Despite our attempts, we have not been able to reduce the levels of DNMT3 transcripts. This leaves open the question of the possible functions of DNMT3 in relation to DNMT1. Furthermore, the association between high DNA methylation and low expression variability seems very relevant, but our evidence is based on the two replicates of gene expression values from our 11 stage-specific transcriptomes. This association would deserve further extensive research, not only collecting more data from a single model but also covering other animal and plant models, which may lead to the conclusion that reducing transcriptional noise is a universal property of DNA methylation.
Resource Availability
Lead Contact
Any questions or requests should be addressed to the Lead Contact (xavier.belles@ibe.upf-csic.es).
Materials Availability
We used the transcriptomes data as mentioned below. This study did not generate new reagents.
Data and Code Availability
The accession numbers for the two sequences, DNMT1 and DNMT3, reported in this paper are GenBank: MT881788, and GenBank: MT881790, respectively. The transcriptomic analyses were carried out on the RNA-seq libraries produced in our laboratory (Ylla et al., 2018) and available at Gene Expression Omnibus with accession number GSE99785.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by the Spanish Ministry of Economy and Competitiveness (grants CGL2012-36251, CGL2015-64727-P and PID2019-104483GB-I00 to XB, including FEDER funds), the CSIC (grant 2019AEP029), and the Catalan Government (grants 2014 SGR 619 and 2017 SGR 1030).
Author Contributions
X.B. designed the research; A.V.-A. X.B., J.C.M and G.Y. performed the research; GY and J.C.M performed the bioinformatics analyses; X.B., A.V.-A., G.Y. and J.C.M discussed and interpreted the results; A.V.-A. and X.B. wrote the paper.
Declarations of Interests
The authors declare no competing interests.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101778.
Supplemental Information
References
- Alemu E.Y., Carl J.W., Jr., Corrada Bravo H., Hannenhalli S. Determinants of expression variability. Nucleic Acids Res. 2014;42:3503–3514. doi: 10.1093/nar/gkt1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasiadi D., Esteve-Codina A., Piferrer F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin. 2018;11:37. doi: 10.1186/s13072-018-0205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashkeel N., Perkins T.J., Kærn M., Lee J.M. Human gene expression variability and its dependence on methylation and aging. BMC Genomics. 2019;20:941. doi: 10.1186/s12864-019-6308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belles X. Beyond Drosophila: RNAi in vivo and functional genomics in insects. Annu. Rev. Entomol. 2010;55:111–128. doi: 10.1146/annurev-ento-112408-085301. [DOI] [PubMed] [Google Scholar]
- Belles X. Academic Press; 2020. Insect Metamorphosis. From Natural History to Regulation of Development and Evolution. [Google Scholar]
- Bewick A.J., Sanchez Z., McKinney E.C., Moore A.J., Moore P.J., Schmitz R.J. Dnmt1 is essential for egg production and embryo viability in the large milkweed bug, Oncopeltus fasciatus. Epigenetics Chromatin. 2019;12:6. doi: 10.1186/s13072-018-0246-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewick A.J., Vogel K.J., Moore A.J., Schmitz R.J. Evolution of DNA methylation across insects. Mol. Biol. Evol. 2017;34:654–665. doi: 10.1093/molbev/msw264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A.P. Gene number, noise reduction and biological complexity. Trends Genet. 1995;11:94–100. doi: 10.1016/S0168-9525(00)89009-5. [DOI] [PubMed] [Google Scholar]
- Bonasio R., Li Q., Lian J., Mutti N.S., Jin L., Zhao H., Zhang P., Wen P., Xiang H., Ding Y. Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr. Biol. 2012;22:1755–1764. doi: 10.1016/j.cub.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso-Júnior C.A., Fujimura P.T., Santos-Júnior C.D., Borges N.A., Ueira-Vieira C., Hartfelder K., Goulart L.R., Bonetti A.M. Epigenetic modifications and their relation to caste and sex determination and adult division of labor in the stingless bee Melipona scutellaris. Genet. Mol. Biol. 2017;40:61–68. doi: 10.1590/1678-4685-GMB-2016-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciudad L., Bellés X., Piulachs M.D. Structural and RNAi characterization of the German cockroach lipophorin receptor, and the evolutionary relationships of lipoprotein receptors. BMC Mol. Biol. 2007;8:53. doi: 10.1186/1471-2199-8-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortijo S., Aydin Z., Ahnert S., Locke J.C. Widespread inter-individual gene expression variability in Arabidopsis thaliana. Mol. Syst. Biol. 2019;15:e8591. doi: 10.15252/msb.20188591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falckenhayn C., Boerjan B., Raddatz G., Frohme M., Schoofs L., Lyko F. Characterization of genome methylation patterns in the desert locust Schistocerca gregaria. J. Exp. Biol. 2013;216:1423–1429. doi: 10.1242/jeb.080754. [DOI] [PubMed] [Google Scholar]
- Fatemi M., Hermann A., Gowher H., Jeltsch A. Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. Eur. J. Biochem. 2002;269:4981–4984. doi: 10.1046/j.1432-1033.2002.03198.x. [DOI] [PubMed] [Google Scholar]
- Feliciello I., Parazajder J., Akrap I., Ugarković D. First evidence of DNA methylation in insect Tribolium castaneum: environmental regulation of DNA methylation within heterochromatin. Epigenetics. 2013;8:534–541. doi: 10.4161/epi.24507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Nicolas A., Belles X. Juvenile hormone signaling in short germ-band hemimetabolan embryos. Development. 2017;144:4637–4644. doi: 10.1242/dev.152827. [DOI] [PubMed] [Google Scholar]
- Glastad K.M., Gokhale K., Liebig J., Goodisman M.A.D. The caste- and sex-specific DNA methylome of the termite Zootermopsis nevadensis. Sci. Rep. 2016;6:37110. doi: 10.1038/srep37110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glastad K.M., Hunt B.G., Goodisman M.A.D. Evolutionary insights into DNA methylation in insects. Curr. Opin. Insect Sci. 2014;1:25–30. doi: 10.1016/j.cois.2014.04.001. [DOI] [PubMed] [Google Scholar]
- Goll M.G., Kirpeka r. F., Maggert K.A., Yoder J.A., Hsieh C.L., Zhang X., Golic K.G., Jacobsen S.E., Bestor T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311:395–398. doi: 10.1126/science.1120976. [DOI] [PubMed] [Google Scholar]
- Harrison M.C., Jongepier E., Robertson H.M., Arning N., Bitard-Feildel T., Chao H., Childers C.P., Dinh H., Doddapaneni H., Dugan S. Hemimetabolous genomes reveal molecular basis of termite eusociality. Nat. Ecol. Evol. 2018;2:557–566. doi: 10.1038/s41559-017-0459-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.J., Chen T., Zhu J.K. Regulation and function of DNA methylation in plants and animals. Cell Res. 2011;21:442–465. doi: 10.1038/cr.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh I., Zeng J., Park T., Yi S.V. DNA methylation and transcriptional noise. Epigenetics Chromatin. 2013;6:9. doi: 10.1186/1756-8935-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt B.G., Glastad K.M., Yi S.V., Goodisman M.A.D. The function of intragenic DNA methylation: insights from insect epigenomes. Integr. Comp. Biol. 2013;53:319–328. doi: 10.1093/icb/ict003. [DOI] [PubMed] [Google Scholar]
- Irles P., Bellés X., Piulachs M.D. Identifying genes related to choriogenesis in insect panoistic ovaries by Suppression Subtractive Hybridization. BMC Genomics. 2009;10:206. doi: 10.1186/1471-2164-10-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Grusby L., Beard C., Possemato R., Tudor M., Fambrough D., Csankovszki G., Dausman J., Lee P., Wilson C., Lander E., Jaenisch R. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat. Genet. 2001;27:31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- De Jong T.V., Moshkin Y.M., Guryev V. Gene expression variability: the other dimension in transcriptome analysis. Physiol. Genomics. 2019;51:145–158. doi: 10.1152/physiolgenomics.00128.2018. [DOI] [PubMed] [Google Scholar]
- Jurkowski T.P., Meusburger M., Phalke S., Helm M., Nellen W., Reuter G., Jeltsch A. Human DNMT2 methylates tRNAAsp molecules using a DNA methyltransferase-like catalytic mechanism. RNA. 2008;14:1663–1670. doi: 10.1261/rna.970408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis S.H., Ross L., Bain S.A., Pahita E., Smith S.A., Cordaux R., Miska E.A., Lenhard B., Jiggins F.M., Sarkies P. Widespread conservation and lineage-specific diversification of genome-wide DNA methylation patterns across arthropods. PLoS Genet. 2020;16:e1008864. doi: 10.1371/journal.pgen.1008864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Hou L., Zhu D., Xu X., An S., Wang X. Identification and caste-dependent expression patterns of DNA methylation associated genes in Bombus terrestris. Sci. Rep. 2018;8:2332. doi: 10.1038/s41598-018-20831-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E., Bestor T.H., Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018;19:81–92. doi: 10.1038/nrg.2017.80. [DOI] [PubMed] [Google Scholar]
- Miyazawa H., Aulehla A. Revisiting the role of metabolism during development. Development. 2018;145:dev131110. doi: 10.1242/dev.131110. [DOI] [PubMed] [Google Scholar]
- Molaro A., Malik H.S., Bourc’his D. Dynamic evolution of de novo DNA methyltransferases in rodent and primate genomes. Mol. Biol. Evol. 2020;37:1882–1892. doi: 10.1093/molbev/msaa044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provataris P., Meusemann K., Niehuis O., Grath S., Misof B. Signatures of DNA methylation across insects suggest reduced DNA methylation levels in Holometabola. Genome Biol. Evol. 2018;10:1185–1197. doi: 10.1093/gbe/evy066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl M.W. Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai K., Nadauld L.D., Chideste r. S., Manos E.J., James S.R., Karpf A.R., Cairns B.R., Jones D.A. Zebra fish Dnmt1 and Suv39h1 regulate organ-specific terminal differentiation during development. Mol. Cell. Biol. 2006;26:7077–7085. doi: 10.1128/MCB.00312-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson K.L., Tohidi-Esfahani D., Ponton F., Simpson S.J., Sword G.A., Lo N. Alternative migratory locust phenotypes are associated with differences in the expression of genes encoding the methylation machinery. Insect Mol. Biol. 2016;25:105–115. doi: 10.1111/imb.12203. [DOI] [PubMed] [Google Scholar]
- Sarda S., Zeng J., Hunt B.G., Yi S.V. The evolution of invertebrate gene body methylation. Mol. Biol. Evol. 2012;29:1907–1916. doi: 10.1093/molbev/mss062. [DOI] [PubMed] [Google Scholar]
- Schulz N.K.E., Wagner C.I., Ebeling J., Raddatz G., Diddens-de Buhr M.F., Lyko F., Kurtz J. Dnmt1 has an essential function despite the absence of CpG DNA methylation in the red flour beetle Tribolium castaneum. Sci. Rep. 2018;8:16462. doi: 10.1038/s41598-018-34701-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X., Huang F., Liu J., Li C., Gao S., Wu W., Zhai M., Yu X., Xiong W., Xie J., Li B. Genome-wide DNA methylomes from discrete developmental stages reveal the predominance of non-CpG methylation in Tribolium castaneum. DNA Res. 2017;24:445–458. doi: 10.1093/dnares/dsx016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancheva I., Hensey C., Meehan R.R. Loss of the maintenance methyltransferase, xDnmt1, induces apoptosis in Xenopus embryos. EMBO J. 2001;20:1963–1973. doi: 10.1093/emboj/20.8.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M.M., Kerr A.R.W., De Sousa D., Bird A. CpG methylation is targeted to transcription units in an invertebrate genome. Genome Res. 2007;17:625–631. doi: 10.1101/gr.6163007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A. Stages in the Embryonic Development of the German Cockroach, Blattella germanica Linné (Blattaria, Blattellidae) Kontyû, Tokyo. 1976;44:1703–1714. [Google Scholar]
- Wang X., Wheeler D., Avery A., Rago A., Choi J.H., Colbourne J.K., Clark A.G., Werren J.H. Function and evolution of DNA methylation in Nasonia vitripennis. PLoS Genet. 2013;9:1003872. doi: 10.1371/journal.pgen.1003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Jorda M., Jones P.L., Maleszka R., Ling X., Robertson H.M., Mizzen C.A., Peinado M.A., Robinson G.E. Functional CpG methylation system in a social insect. Science. 2006;314:645–647. doi: 10.1126/science.1135213. [DOI] [PubMed] [Google Scholar]
- Wang Z., Zhang J. Impact of gene expression noise on organismal fitness and the efficacy of natural selection. Proc. Natl. Acad. Sci. U S A. 2011;108:E67–E76. doi: 10.1073/pnas.1100059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylla G., Piulachs M.D., Belles X. Comparative transcriptomics in two extreme neopterans reveals general trends in the evolution of modern insects. iScience. 2018;4:164–179. doi: 10.1016/j.isci.2018.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylla G., Nakamura T., Itoh T., Kajitani R., Toyoda A., Tomonari S., Bando T., Ishimaru Y., Watanabe T., Fuketa M. Cricket genomes: the genomes of future food. bioRxiv. 2020 doi: 10.1101/2020.07.07.191841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A., McDaniel I.E., Silva P., Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- Zwier M.V., Verhulst E.C., Zwahlen R.D., Beukeboom L.W., Van De Zande L. DNA methylation plays a crucial role during early Nasonia development. Insect Mol. Biol. 2012;21:129–138. doi: 10.1111/j.1365-2583.2011.01121.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the two sequences, DNMT1 and DNMT3, reported in this paper are GenBank: MT881788, and GenBank: MT881790, respectively. The transcriptomic analyses were carried out on the RNA-seq libraries produced in our laboratory (Ylla et al., 2018) and available at Gene Expression Omnibus with accession number GSE99785.