

Abstract
Compounds with novel modes of action are urgently needed to develop effective combination therapies for the treatment of tuberculosis. In this study, a series of compounds was evaluated for activity against replicating Mycobacterium tuberculosis and Vero cell line toxicity. Fourteen of the compounds with in vitro activities in the low micrometer range and a favorable selectivity index were classified using reporter strains of M. tuberculosis which showed that six interfered with cell wall metabolism and one disrupted DNA metabolism. Counter-screening against strains carrying mutations in promiscuous drug targets argued against DprE1 and MmpL3 as hits of any of the cell wall actives and eliminated the cytochrome bc 1 complex as a target of any of the compounds. Instead, whole-genome sequencing of spontaneous resistant mutants and/or counter-screening against common isoniazid-resistant mutants of M. tuberculosis revealed that four of the six cell wall-active compounds, all pyridine carboxamide analogues, were metabolized by KatG to form InhA inhibitors. Resistance to two of these compounds was associated with mutations in katG that did not confer cross-resistance to isoniazid. Of the remaining seven compounds, low-level resistance to one was associated with an inactivating mutation in Rv0678, the regulator of the MmpS5-MmpL5 system, which has been implicated in non-specific efflux of multiple chemotypes. Another mapped to the mycothiol-dependent reductase, Rv2466c, suggesting a prodrug mechanism of action in that case. The inability to isolate spontaneous resistant mutants to the seven remaining compounds suggests that they act via mechanisms which have yet to be elucidated.
Keywords: antimycobacterial, catalase, drug resistance, isoniazid, KaG, luciferase, tuberculosis
Introduction
Globally, it is estimated that the incidence of tuberculosis (TB) is declining by ~2% each year (WHO, 2019). However, the ongoing evolution and spread of strains of Mycobacterium tuberculosis (Mtb) resistant to first- and second-line anti-TB drugs and consequent increase in the incidence of drug-resistant TB threatens to diminish this decline and underscores the need for new drugs with novel mechanisms of action. In response to this need, a TB drug pipeline that is reasonably well-populated with new and repurposed anti-tubercular agents has been established (https://www.newtbdrugs.org/). Encouragingly, the discovery stage of the pipeline includes molecules that act on a more diverse range of essential cellular processes in Mtb which extends beyond the classical pathways of cell wall biosynthesis, transcription, energy metabolism, DNA replication, and protein synthesis targeted by existing first- and second-line TB drugs (Evans and Mizrahi, 2018; Wellington and Hung, 2018; Lienhardt and Raviglione, 2020). The challenges posed for compound penetration across the mycobacterial cell envelope, coupled with the ability of Mtb to metabolize and efflux small molecules have confounded target-led approaches to drug discovery. As a result, phenotypic approaches have predominated, with whole-cell assays having been developed and applied in high-throughput screens to identify molecules with inhibitory activity against Mtb under conditions which support bacillary replication or non-replicating persistence and are modeled on those encountered during human infection (Deb et al., 2009; Ioerger et al., 2013; Gold et al., 2015; Gold and Nathan, 2017; Huang et al., 2018; Parish, 2020). However, this approach is not without its own challenges, the most significant of which is mechanism-of-action (MoA) elucidation of compounds with whole-cell activity against Mtb. This process can be difficult and time-consuming (Singh and Mizrahi, 2017) and yet, is critically important for ensuring that the scarce resources available for hit expansion and/or hit-to-lead progression are deployed most judiciously. As a result, considerable effort has been placed on developing and applying biological assays that enable broad mechanistic classification—and hence, triage—of phenotypic screening hits that pass a certain threshold of cytotoxicity. Assays of this type are particularly useful for rapidly identifying chemotypes that act on promiscuous drug targets such as DprE1, MmpL3, Pks13, or QcrB for which multiple inhibitory scaffolds already exist (Cole, 2016; Lee and Pethe, 2018).
In this paper, we describe the application of this approach to the characterization of a set of 14 compounds selected on the basis of an anti-TB pharmacophore centered virtual screen conducted by the Division of AIDS at the NIAID.
Materials and Methods
Bacterial Strains, Culture Conditions, and Media
The strains employed in this study are described in Supplementary Table 1 . Mycobacterial strains were cultured in various media depending on the assay conducted. 7H9 OADC was prepared by supplementing Middlebrook 7H9 (Difco) with 100 ml oleic acid-albumin-dextrose-catalase (OADC) enrichment (Difco), 2g/L glycerol, and 2.5 ml 25% Tween 80. 7H9 ADC medium was prepared by supplementing Middlebrook 7H9 with 100 ml albumin-dextrose-catalase (ADC) enrichment (Difco), 2g/L glucose, and 2.5 ml 25% Tween 80. 7H9 CAS medium was prepared by supplementing Middlebrook 7H9 with 4g/L glucose, 0.3 g/L casitone, 0.81 g/L NaCl, and 2.5 ml 25% Tween 80. Glycerol-alanine-salts with iron (GAST/Fe) medium, pH 6.6, was supplemented with 0.3 g/L Bacto Casitone (Difco), 0.05g/L ferric ammonium citrate, 4 g/L dibasic potassium phosphate, 2 g/L citric acid, 1 g/L L-alanine, 1.2 g/L MgCl2, 0.6 g/L potassium sulfate, 2 g/L ammonium chloride, 1.8 ml of 10 g/L sodium hydroxide, 10 ml glycerol, and 2.5 ml 25% Tween 80. Cultures were incubated at 37°C in sealed culture flasks with no agitation. Cells were plated onto Middlebrook 7H10 agar plates with 19.47 g/L 7H10 agar base supplemented with 100 ml OADC and 5 ml glycerol. A GFP reporter strain, H37Rv-GFP (Chan et al., 2002; Abrahams et al., 2012), and Mtb bioluminescent reporter strains, PiniB-LUX and PrecA-LUX, were grown in 7H9 OADC with 20 µg/ml kanamycin (Naran et al., 2016). Spontaneous drug resistant mutants were grown in 7H9 OADC without antibiotic selection. Mtb strains with resistance-conferring mutations in mmpL3, dprE1, qcrB, katG, or upstream of inhA were grown in various media, dependent on the assay, without antibiotic selection. Similarly, the deletion mutants, Δtap and CydKO were grown in 7H9 CAS or 7H9 ADC. M. smegmatis was grown in 7H9 OADC media.
Source of Compounds
The compounds were selected from the National Cancer Institute (NCI) chemical database. A portion of the NCI database is available to the public at https://cactus.nci.nih.gov/ncidb2.2/ and several search functions are available in the NCI open database structures at this website. These compounds were selected from a large pool of compounds available in the database based on computerized substructure searching of TB active pharmacophores. The selection of the pharmacophores was facilitated by the existing NIAID chemical/biological database for HIV and TB at https://chemdb.niaid.nih.gov/. The corresponding NSC numbers for the tested compounds are listed: 127-09 (NSC37381), 127-17 (NSC351686), 127-21 (NSC624736), 127-23 (NSC641601), 127-11 (NSC123112), 127-13 (NSC270389), 127-14 (NSC275424), 127-15 (NSC293875), 127-18 (NSC353903), 127-19 (NSC369060), 127-20 (NSC603173), 127-22 (NSC635076), MN-6925 (NSC155263), and MN-9483 (NSC84486).
Drug Susceptibility Testing
Compound solutions were prepared as 10 mM stocks in DMSO and stored at −80°C. The MIC for each compound was determined by performing the microbroth dilution assay (Abrahams et al., 2012) and quantitatively analyzed with the colorimetric alamarBlue cell viability reagent (Merck). Briefly, each compound at a starting concentration of 50 µM was diluted two-fold in a clear-well, round-bottom 96-well microtiter plate. Mtb was grown to an OD600 of 0.5 (~ 108 cells/ml) and diluted 1,000-fold before adding an equal volume to each well, totaling 100 µl. The plate was sealed and incubated at 37°C for 13 days after which 10 µl of 0.01% alamarBlue was added and incubated for a further 24 h. Fluorescence was recorded using a SpectraMax i3x plate reader (Molecular Devices) at excitation and emission wavelengths of 540 and 590 nm, respectively, and the MIC90, the lowest drug concentration to inhibit growth by more than 90%, was determined from the dose-response curve.
Isolation and Characterization of Spontaneous Drug-Resistant Mutants and Analysis of Mutations
Mutants of Mtb resistant to the compounds under investigation were isolated as previously described (Singh et al., 2017). In brief, ~ 5×108–109 cells/ml from a late logarithmic-phase culture (OD600 = 0.8–1.0) was concentrated 100× and 100 µl spread onto 7H10 agar containing compound at 5×, 10×, 20×, or 50× the calculated MIC in liquid culture. Plates were incubated for 3–4 weeks. Individual colonies emerging on the plates were grown in drug-free 7H9 OADC to an OD600 of 0.5 and tested for drug susceptibility, as described above, to determine the level of resistance. A small number of individual colonies arose on the plates. For compounds 127-09, 127-17, 127-21, 127-23, 127-13, 127-14, 127-15, 127-18, 127-19, 127-20, 127-22, MN-6925, and MN-9483, the number of colonies picked for drug susceptibility testing were 5, 13, 8, 9, 17, 15, 11, 7, 6, 11, 11, 5, and 7 respectively. No spontaneous resistant mutants (SRMs) could be isolated in the case of compound 127-11. Confirmed mutant clones demonstrating heritable resistance, as determined by a ≥8-fold increase in MIC, were grown to an OD600 of 0.5 in 50 ml 7H9 OADC (in the absence of compound) and the cells harvested, and DNA extracted using the cetyltrimethylammonium bromide (CTAB) method in preparation for whole genome sequencing (WGS). Libraries were prepared using the TruSeq kit following the manufacturers protocol and sequenced on the Illumina NovaSeq at the Texas A&M University Genomics and Bioinformatics Service. Reads with an average read depth of 360-fold were mapped to the reference genome, Mtb H37RvMA (GenBank accession NZ_CM002884.1) using BWA (Li and Durbin, 2009) and polymorphisms (single nucleotide polymorphisms (SNPs) and insertions/deletions) were identified using variant-calling scripts developed in-house. PE_PGRS genes were excluded from the analysis. The sequencing data have been deposited in NCBI SRA under BioProject accession number PRJNA645974. The location of mutated residues in the respective proteins were analyzed using the protein modeling software Protein Homology/analogY Recognition Engine V2.0 (Phyre2) (Kelley et al., 2015).
Mammalian Cell Cytotoxicity Assay
Compounds were screened for cytotoxicity against the Vero cell line (kidney epithelial cells) using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT)-assay as previously described (Wilson et al., 2017). IC50 values were determined from the dose-response curves. The selectivity index (SI) of each compound was calculated by dividing the IC50 value by the MIC value.
GFP Release Assay
The assay was conducted as described previously (Kumar et al., 2012; Park et al., 2017). Briefly, H37Rv-GFP was grown to an OD600 of 0.2–0.3 in 7H9 OADC and exposed to compound at 1× or 10×MIC. Meropenem was used as the lytic drug control and linezolid as the non-lytic control. Every 24 h over a period of 6 days, 200 µl of culture was harvested and pelleted by centrifugation. Approximately 200 µl of the supernatant was transferred to a black, clear-bottom 96-well microtiter plate (Greiner CellStar®) and fluorescence (excitation 540 nm; emission 590 nm) measured using the SpectraMax i3x plate reader (Molecular Devices). Fluorescence intensity was normalized to the drug-free control.
Bioluminescence Assay
The assay was performed as previously described (Naran et al., 2016). Briefly, the PiniB-LUX or PrecA-LUX reporter strains of Mtb were grown to an OD600 ~ 0.4 and diluted to an OD600 ~ 0.04 in 7H9 OADC. The cells were inoculated into white, clear bottom 96-well microtiter plates (Greiner CellStar®) containing a two-fold serial dilution of each compound, prepared as described for the drug susceptibility testing assay. The plates were incubated at 37°C and the luminescence recorded every 24 h for 9 days using a SpectraMax i3x plate reader (Molecular Devices). The readings were plotted over time. Isoniazid (INH) and ciprofloxacin (CIP) were included as cell-wall-acting and genotoxic drug controls, respectively (Naran et al., 2016).
Results
Characterization of the Anti-TB Activity Profiles of Compounds Selected From a Pharmacophore-Based Virtual Screen
A set of 14 compounds, which were selected from the NCI database using a TB active pharmacophore substructure search, were obtained from the Division of AIDS at the NIAID. Three of the compounds share a pyridine-4-carboxamide motif (127-09, 127-21, and 127-23) and another contains a pyridine-3-carboxamide motif (127-17) with all four carrying a hydrazide, analogous to INH. One is structurally related to mitomycin C (127-11), and the other nine are structurally diverse ( Figure 1 ). The compounds were tested for activity against replicating Mtb H37Rv in different media ( Table 1 ) as specific carbon sources and/or media components are known to influence inhibitory activity of antimycobacterial agents (Franzblau et al., 2012). The 10 most active compounds had MIC values ranging from 0.4–3.1 µM in 7H9 OADC whereas the remaining four were less potent (MICs = 6.3–12.5 µM). Thirteen of the compounds showed ~5-fold increased potency in GAST/Fe (protein-free) media; the only outlier was 127-13, which showed an eight-fold lower activity in GAST/Fe. Otherwise, only minor (<4-fold) differences in MIC values were observed across the range of media tested. Compounds 127–11, 127–19, 127–20, and 127–22 also showed reasonably potent activity against Mycobacterium smegmatis, demonstrating MICs in the range of 0.4–6.3 µM ( Supplementary Table 2 ). All compounds, with the exception of 127-18 had a selectivity index (SI) above 10, indicative of effective antimycobacterial activity and (comparatively) low cytotoxicity against the mammalian Vero cell line ( Table 1 ) (Orme, 2001). Compounds 127-20, 127-17, 127-22 and MN-9483 exhibited the highest SI values (100 – >3000).
Figure 1.

Compounds exhibiting activity against H37Rv.
Table 1.
Inhibitory activity against replicating Mtb and mammalian cell toxicity of compounds.
| Compound identification | Compound name | Other identifier | Log P | MIC (µM) in supplemented media | Inhibition and selectivity (Vero cell line) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 7H9 OADC |
7H9 ADC | 7H9 CAS | GAST/Fe | Cyto-toxicity IC50 (µM) | SI (IC50/MIC) | ||||
| 127-09 | N-[(E)-1-[(5-oxo-2-thioxo-imidazolidin-4-yl)methyl]hexylideneamino]pyridine-4-carboxamide | Pyridine carboxamide analogue | 2.18 | 1.6–3.1 | 1.6 | 0.4 | 0.4 | – | – |
| 127-17 | N-[(E)-(5-isopropenyl-2-methyl-cyclohex-2-en-1-ylidene)amino]pyridine-3-carboxamide | Pyridine carboxamide analogue |
2.94 | 1.6–3.1 | 6.3 | 1.6 | 1.0 | 420 | 280 |
| 127-21 | N-[1,1,4-trioxo-2-[4-(trifluoromethyl)phenyl]-1,3-thiazolidin-3-yl]pyridine-4-carboxamide | Pyridine carboxamide analogue | 1.72 | 1.6–3.1 | 6.3 | 1.6 | 0.5 | > 160 | > 25.4 |
| 127-23 | N-[(E)-[2-hydroxy-1-(6-nitro-3-oxo-4H-quinoxalin-2-yl)-2-(4-nitrophenyl)ethylidene]amino]pyridine-4-carboxamide | Pyridine carboxamide analogue | 3.74 | 3.1–6.3 | 12.5 | 3.1 | 3.1 | > 130 | > 10.4 |
| 127-11 | [amino-(3,4-dichlorobenzoyl)-methoxy-methyl-dioxo-yl]methyl carbamate | Mitomycin derivative T 56 | 1.03 | 0.4 | 0.1 | 0.8 | < 0.2 | 3.75 | 62.5 |
| 127-13 | benzyl N-[(E)-[(2E)-2-(carbamothioylhydrazono)propylidene]amino]carbamodithioate | 3.42 | 1.6–3.1 | 1.6 | 1.6 | 25 | 38.4 | 64 | |
| 127-14 | 2-[2-[(E)-[2-(5H-benzo[a]phenazin-7-yl)-4-oxo-1-naphthylidene]amino]anilino]naphthalene-1,4-dione | 5.45 | 12.5 | 6.3 | 6.3 | 6.3 | 98.5 | 15.6 | |
| 127-15 | 10H-3,10a-Epidithiopyrazino[1,2-a]indole-1,4-dione, 2,3-dihydro-2,3,10,10-tetramethyl- | 4.00 | 0.4–0.8 | 0.1 | 0.4 | < 0.2 | 19 | 31.6 | |
| 127-18 | 2,7-dimethyl-[1,4]benzodioxino[2,3-f][4,7]phenanthroline-4,5-diol | 2.81 | 12.5 | 25 | 25 | 1.4 | 163 | 6.52 | |
| 127-19 | 7-methyl-3-nitroso-2-phenyl-imidazo[1,2-a]pyridine | 3.11 | 0.8–1.6 | 0.7 | 0.1 | < 0.2 | 52.7 | 33 | |
| 127-20 | N1,N4-bis[4-(1-methylimidazo[1,2-a]pyridin-1-ium-2-yl)phenyl]terephthalamide | 5.80 | 1.6 | 1.4 | – | < 0.2 | > 4300 | > 3071 | |
| 127-22 | N,N’-bis[5-[[5-[(3-amino-3-imino-propyl)carbamoyl]-1-methyl-pyrrol-3-yl]carbamoyl]-1-methyl-pyrrol-3-yl]decanediamide | −2.06 | 3.1 | 1.6 | 12.5 | < 0.2 | 54.22 | 100 | |
| MN-6925 | [(E)-dibenzofuran-2-ylmethyleneamino]thiourea | 2.69 | 6.3–12.5 | 3.1 | 6.3 | 3.1 | 74 | > 23.9 | |
| MN-9483 | Naphtho[1’,2’:4,5]imidazo[1,2-a]pyridine-5,6-dione | 1.71 | 3.1 | 1.6 | 3.1 | 0.4 | > 252 | > 158 | |
Is not determined.
Drug-induced lytic activity was monitored by measuring the release of GFP from H37Rv-GFP treated with compound at 1×MIC or 10×MICs as described previously (Kumar et al., 2012; Park et al., 2017). Meropenem which induces rapid lysis of Mtb by inhibiting the cell wall remodeling enzymes, D,D-carboxypeptidase and L,D-transpeptidase was used as a lytic control whereas the bacteriostatic translation inhibitor, linezolid (Zhang et al., 2014), was used as a non-lytic control ( Figure 3 ). Meropenem, in both the 1×MIC and 10×MIC experiments, led to a steady increase in GFP fluorescence signal over time, whereas linezolid produced no fluorescence signal other than on day 6 in the 10×MIC experiment, when a weak signal was detected. Compound 127-18 was intrinsically fluorescent, yielding a drug-concentration-dependent, false positive signal in this assay. Furthermore, all compounds produced a late fluorescence signal at day 6 in the 10×MIC experiment, presumably attributable to cell death. Compound 127-11 led to GFP release from Mtb at both drug concentrations indicative of true lytic activity, albeit with markedly slower kinetics than the meropenem control. Of the putative cell wall targeting agents, none produced a significant fluorescence signal. Although INH and the pyridine carboxamides target the cell wall, none elicited a signal in the GFP release assay. One possible explanation is that the disruption in cellular metabolism due to inhibition of InhA activity may block growth earlier than the disruption in cell wall integrity, which is required for GFP release to occur.
Figure 3.
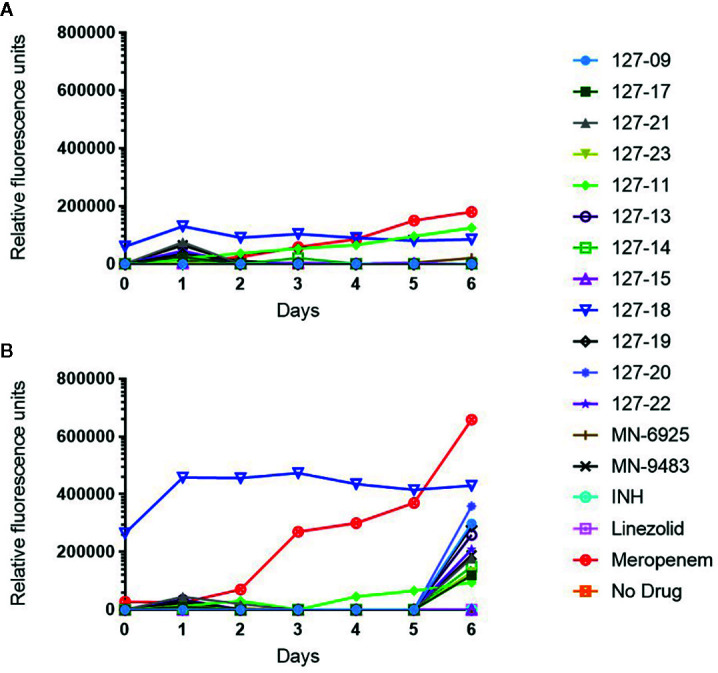
Measurement of released GFP in culture supernatant following exposure of H37Rv-GFP strain to 1× (A) or 10× (B) MIC of the respective compound. Note 127-18 is an intrinsically fluorescent compound.
Broad Mechanistic Characterization Using Reporter Strains
Reporter strains of Mtb have been developed that enable rapid classification of antimycobacterial agents into three broad mechanistic classes: compounds that act on the cell wall, compounds that interfere with DNA metabolism, and inhibitors of mycobacterial energy metabolism. Another mutant strain has been developed to establish whether inhibitors are subject to efflux. These were used to screen the compounds.
A luminescence-based reporter assay (Naran et al., 2016) developed to detect cell wall stress or DNA damage induced by exposure of Mtb to antimycobacterial agents was used to establish which of the compounds fall into either of these broad mechanistic categories. This assay is reliant on the transcriptional responsiveness of the iniBAC promoter to cell wall stress (Alland et al., 2000) and the recA promoter to genotoxic stress (Brooks et al., 2001), and uses a luminescence-based readout. INH and CIP were included in all assays as positive controls for induction of iniBAC and recA induction, respectively. All compounds were tested in these assays at drug concentrations ranging from 0.125×MIC to 4×MIC. Consistent with previous observations, INH triggered a luminescence signal in PiniB-LUX peaking at 1×MIC (Naran et al., 2016), and also elicited a late, weak signal in PrecA-LUX at all concentrations Conversely, CIP generated a strong signal in PrecA-LUX, peaking at 0.125x−4xMIC, but elicited no response in PiniB-LUX at any of the concentrations tested. Six of the compounds elicited a positive iniBAC response, suggesting that they interfere with cell envelope biogenesis ( Figure 2 ; Supplementary Table 3 ). Compounds 127–09 and 127–23 elicited low-level signals, 3–5-fold lower than the INH control, peaking at day 3 at sub-inhibitory concentrations (0.25× and 0.5×MIC, respectively). 127-15 also induced a low-level signal in PiniB-LUX, peaking at day 3 at 1×MIC whereas 127-17 and 127-21 triggered responses 2-3-fold higher than INH at 2×MIC, also peaking at day 3. MN-6925 elicited the most distinctive response, reaching a level five-fold higher than the INH control and showing the strongest signal at sub-inhibitory concentrations of compound.
Figure 2.

Induction of iniBAC or recA bioluminescent reporter in response to antimycobacterial agents with unknown MoAs at concentrations ranging between 0.125-4× MIC, monitored over 9 days. For PiniB-LUX, INH was used as the positive control at a concentration of 0.39 µM (1× MIC); CIP was used as the negative control. The iniBAC response is shown for 127-09, 127-17, 127-21, 127-23, 127-13, 127-15, MN-6925, and the controls. For PrecA-LUX, CIP was used as the positive drug control at 3.13 µM (2× MIC), although concentrations in the range of 0.2–6.25 µM (0.125-4× MIC) all gave similar luminescence profiles; INH was the negative control. The recA response is shown for 127-11, 127-13 and the controls. RLU, relative luminescence unit.
To establish whether the compounds identified from the PiniB-LUX assay as putative “cell wall actives” act on two promiscuous cell envelope targets, DprE1 and MmpL3, they were tested in secondary screens against Mtb strains carrying known resistance-conferring mutations in dprE1 or mmpL3 ( Supplementary Table 4 ). The DprE1Y314C, DprE1Y314H, DprE1P116S, and MmpL3G253E mutants were tested in two media types. At least 15 inhibitors have been described that target DprE1 (Degiacomi et al., 2020). Resistance to one of these compounds, TCA1, maps to Y314C (Wang et al., 2013), a mutation which causes a structural change in the enzyme active site (Liu et al., 2017). Resistance to N-alkyl-5-hydroxypyrimidinone carboxamides was associated with the amino acid substitutions Y314C, Y314H, and P116S in DprE1 (Oh et al., 2018). Likewise, the G253E mutation in MmpL3 has been implicated in resistance to a wide range of MmpL3 inhibitors which include the tetrahydropyrazolopyrimidine THPP1, the 1,2-diamine SQ109, the adamantyl urea, AU1235, the indolecarboxamides NITD-304 and NITD-349 (Li et al., 2019), and other diverse inhibitors (Williams et al., 2019). None of the six compounds designated as cell wall active based on the PiniB-LUX assay showed cross-resistance to the DprE1 or MmpL3 mutants. Interestingly, however, MN-6925 showed increased potency against all four DprE1 and MmpL3 mutants, but only in 7H9 ADC media.
A strong sustained recA signal was triggered in response to compound 127-11 ( Figure 2 ; Supplementary Table 3 ). There was a clear concentration-dependent induction over the range of 0.125×–2×MIC, above which there was a drop in the luminescence signal, presumably due to cell death ( Figure 3 ). 127-11 is structurally related to mitomycin C, suggesting that this compound is a DNA crosslinking agent (Tomasz, 1995). The remaining compounds elicited a weak, late signal (around day 5) analogous to INH ( Figure 2 ; Supplementary Table 3 ), indicative of indirect DNA damage as a result of drug action, as suggested previously (Naran et al., 2016).
To ascertain whether any of the compounds acted on QcrB, a promiscuous target in mycobacterial energy metabolism, they were tested for hypersensitivity against the CydKO mutant of Mtb in which the cytochrome bd oxidase is functionally disrupted (Arora et al., 2014; Moosa et al., 2017). None of the compounds showed differential activity in this mutant compared to wildtype Mtb ( Supplementary Table 4 ). To determine whether any of the compounds was a substrate for efflux, they were tested against a knockout mutant in the efflux pump, Rv1258c (Tap) (Ramon-Garcia et al., 2012). Only MN-6925 showed an eight-fold reduction in MIC against the Δtap mutant; however, this potentiating effect was observed exclusively in 7H9 ADC media ( Supplementary Table 4 ).
Mechanistic Insights From Spontaneous Drug-Resistant Mutants
To elucidate mechanisms of action, Mtb was plated onto increasing concentrations of compound (5-50×MIC) to select for SRMs as described under Materials and Methods ( Table 2 ). SRMs displaying heritable resistance against 127-13, 127-15, 127-17, 127-21, and 127-23 were isolated at frequencies in the range of 10−6–10−7 ( Table 2 ). Two SRMs for each compound were chosen for WGS. WGS analysis of clones with high-level resistance against 127-17, 127-21, and 127-23 were all found to carry mutations in katG ( Table 3 ). Furthermore, cross resistance to these three compounds as well as 127-09 was observed in Mtb strains carrying the KatGS315T or inhA −15C→T mutations commonly observed in INH-resistant clinical isolates (Lempens et al., 2018). These results indicate, that all four are INH-like prodrugs, which are activated by KatG to target InhA ( Table 4 ). The S315T and S315R mutations commonly associated with INH resistance were found in mutants raised against compound 127-21; in contrast compounds 127-17 and 127-23 appeared to interact with KatG in a manner distinct from INH, as evidenced by the identification of the V544F, G285V, and *741R mutations in SRMs to these compounds, which have not been associated previously with INH resistance ( Table 3 ). The G285 residue is located on the surface at the mouth of the tunnel in relation to the active site heme, not far from S315, whereas V544 is located distal to this, buried within the C-terminal domain of KatG. The *741R mutation at the stop codon of katG is predicted to extend the protein product by creating a C-terminal fusion with the open reading frame of Rv1907c. The conclusion that 127-21, 127-17, and 127-23 interact with KatG differently from INH was corroborated by screening these mutants for cross-resistance to INH: while the KatGS315R mutation conferred cross-resistance to INH, the KatG*741R and KatGG285V mutations associated with resistance to 127-17 and 127-23, respectively, had no effect on the susceptibility of Mtb to INH. The KatG mutants that were sensitive to INH also retained sensitivity to 127-09 but were cross-resistant to the other pyridine carboxamides ( Table 4 ).
Table 2.
Drug resistant mutants isolated against selected compounds.
| Compound | MIC against H37Rv (µM) | MIC against SRM (µM) | Fold shift in SRM MIC (over H37Rv) | Mutation frequency |
|---|---|---|---|---|
| 127-17R SRM-1 | 1.6–3.1 | 50 | 31× | 2.0×10−7 |
| 127-21R SRM-1 | 1.6–3.1 | 50 | 31× | 2.0×10−7 |
| 127-21R SRM-2 | 1.6–3.1 | 50 | 31× | 2.0×10−7 |
| 127-23R SRM-1 | 3.1–6.3 | 50 | 16× | 1.5×10−7 |
| 127-23R SRM-2 | 3.1–6.3 | 25 | 8× | 1.5×10−7 |
| 127-13R SRM-1 127-13R SRM-2 |
1.6–3.1 | 12.5 | 8× | 9.0×10−6 |
| 127-15R SRM-1 127-15R SRM-2 |
0.4–0.8 | 6.3 | 8× | 2.5×10−6 |
MICs in 7H9 OADC media.
Table 3.
Mutations identified by WGS of Mtb mutants spontaneously resistant to selected compounds.
| Compound | Gene | Mutation/s | Product |
|---|---|---|---|
| 127-17R SRM-1 |
katG
fadD26 |
*741R G74* |
Catalase-peroxidase-peroxynitritase Fatty-acid-AMP ligase |
| 127-21R SRM-1 |
katG
ppsC Rv3755c |
Y597N; S315Na
Q1085* D100G |
Catalase-peroxidase-peroxynitritase Phenolpthiocerol synthesis type-I polyketide synthase Unknown (conserved protein) |
| 127-21R SRM-2 | katG | S315R | Catalase-peroxidase-peroxynitritase |
| 127-23R SRM-1 |
katG
ppsB |
V544F Q945P |
Catalase-peroxidase-peroxynitritase Phenolpthiocerol synthesis type-I polyketide synthase |
| 127-23R SRM-2 |
katG
ppsC phoP |
G285V +c insertion at nt 680 IS6110 insertion at nt 210 |
Catalase-peroxidase-peroxynitritase Phenolphthiocerol synthesis type-I polyketide synthase Possible two component system response transcriptional positive regulator PhoP |
| 127-13R SRM-1 127-13R SRM-2 |
Rv0678 fadD26 Rv3479 |
-g deletion at nt 198 S553* N831K |
Regulator of MmpS5-MmpL5 efflux pump Fatty-acid-AMP ligase Transmembrane protein |
| 127-15R SRM-1 127-15R SRM-2 |
Rv2466c fadD26 |
-t deletion at nt 151 G74* |
Mycothiol dependent reductase, D Fatty-acid-AMP ligase |
*stop codon; nt, nucleotide.
aSince these sites were heterogeneous, 127-21R SRM-1 was probably a mixture of 2 mutants, each with a different KatG mutation.
Table 4.
Evaluation of cross resistance of pyridine carboxamide analogues against KatGS315T, inhA −15C→T and spontaneous drug resistant mutants.
| Mtb strain | MIC (µM) | |||||
|---|---|---|---|---|---|---|
| H37Rv | KatGS315T | inhA −15C→T | 127-17R SRM-1 | 127-21R SRM-2 | 127-23R SRM-2 | |
| Compound | ||||||
| 127-09 | 1.6-3.1 | 25 | 6.3 | 6.3 | 12.5 | 3.1 |
| 127-17 | 1.6-3.1 | 50 | 25 | 50 | 50 | 50 |
| 127-21 | 1.6-3.1 | 50 | 12.5 | 25 | 50 | 50 |
| 127-23 | 3.1-6.3 | 50 | 25 | 50 | 50 | 50 |
| INH | 0.1 | 1.6 | 0.4 | 0.1 | 6.3 | 0.1 |
Tested against 127-17R SRM-1 *741R, 127-21R SRM-2 S315R and 127-23R SRM G285V.
SRMs displaying intermediate-level resistance (~8-fold higher MIC than wildtype) were also isolated against compounds 127-13 and 127-15 ( Table 2 ). WGS analysis of two SRMs to 127-13 identified a frameshift mutation in Rv0678, the regulator of the MmpS5-MmpL5 efflux pump, whereas two SRMs to 127-15 carried a frameshift mutation in dsbA, which encodes a mycothiol dependent reductase ( Table 3 ). In addition to these probable resistance-conferring mutations, all SRMs sequenced in this study also carried mutations in genes involved in PDIM biosynthesis (fadD26, ppsB, ppsC). Mutations in the PDIM locus which affect the ability to produce this abundant virulence lipid are commonly observed when Mtb is passaged in vitro and are unlikely to be related to the MoAs of these compounds (Domenech and Reed, 2009). Finally, unlike the six compounds described above, SRMs could not be isolated on solid media for the remaining eight compounds under any of the conditions tested.
Discussion
In this study, we describe the application of a set of phenotypic assays to elucidate the MoAs of 14 compounds with activity against replicating Mtb in the 0.1–25 µM range and selectivity indices ranging from 6 to >3,000. A SciFinder (Scifinder, 2020) search found an exact match to 127-09, which had been reported more than six decades ago as a compound that displayed good activity against Mtb and was less toxic than INH in a mouse model (Archer et al., 1956). To the best of our knowledge, the 13 other compounds have not been previously reported or characterized in terms of antitubercular activity. Among the most potent compounds, 127-11, demonstrated genotoxic and lytic activity against Mtb, consistent with its structural similarity to mitomycin C, a drug commonly used for the treatment of certain cancers, with known activity against growing and hypoxic cultures of Mycobacterium bovis (Peh et al., 2001) and efficacy against other bacterial persisters (Kwan et al., 2015). However, supporting a non-specific MoA involving DNA cross-linking, 127-11 displayed significant mammalian cell toxicity and hence, a modest SI.
Six of the compounds were classified as cell wall active based on responsiveness of the iniBAC promoter upon exposure of Mtb to these compounds. Of the six, three (127-09, 127-21, and 127-23) contained a pyridine-4-carboxamide motif shared with INH, and another (127-17) contained a related pyridine-3-carboxamide motif. All four compounds share a common MoA with INH, as inferred from WGS of spontaneous resistant mutants and/or cross-screening against INH-resistant mutants carrying the common resistance-conferring mutations, KatGS315T and inhA −15C→T. Therefore, these compounds are most likely prodrugs that are biotransformed by KatG to metabolites that inhibit InhA. However, the KatG*741R mutation selected by resistance to 127-17, and KatGG285V mutation selected by resistance to 127-23 did not confer resistance to INH. This lack of cross-resistance is most likely attributable to the involvement of additional residues in binding these bulky INH analogues to the KatG active site. In a recent study, a 2-pyridone bicyclic scaffold compound was shown to re-sensitize an INH resistant mutant to INH by countering oxidative stress tolerance (Flentie et al., 2019). It would be interesting to establish whether this compound could also restore sensitivity of katG mutants to these pyridine carboxamide analogues. It would likewise be useful to assess whether the compounds differ from INH in terms of activity in models of slow- or non-replicating persistence induced by low oxygen, mild hypoxia (Cho et al., 2015; Zheng et al., 2017), carbon starvation (Grant et al., 2013), or a combination of multiple stresses (Deb et al., 2009; Gold et al., 2015). Extensive effort has been made to improve the antitubercular potency while lowering the cytotoxicity of INH, albeit with limited success (Martins et al., 2014; Oliveira et al., 2017; Dragostin et al., 2019). Consistent with this, the pyridine carboxamides investigated here are significantly less potent against Mtb and have lower SI values than INH (SI>2941 based on Vero cell line toxicity) (Phillips et al., 2012).
Displaying a sub-micromolar MIC, 127-15 was one of the most potent compounds investigated in this study. It was designated as cell wall active based on early induction of the iniBAC promoter; however, the signal was low and not sustained, perhaps as a result of rapid cell death. Moreover, this compound showed no cross-resistance to the four DprE1 or MmpL3 mutants tested arguing against the involvement of these promiscuous targets in the MoA of 127-15. In contrast, MN-6925 induced a very strong iniBAC signal unambiguously identifying it as an inhibitor of cell envelope biogenesis. Interestingly, MN-6925 was distinguished by the fact that it was the only compound that showed increased potency against the MmpL3G235E, DprE1Y314H, DprE1Y314C, DprE1P116S, and Δtap mutants in 7H9 ADC media. While the mechanistic basis of this observation is unclear, it is worth noting that mutations in MmpL3 can result in alterations in the hydrophobicity of the mycobacterial cell wall (McNeil et al., 2017) and thereby potentially increase compound permeability, and that disruption of Tap results in generalized repression of genes involved in cell wall biosynthesis during stationary phase (Ramon-Garcia et al., 2012).
A deletion in Rv0678, a negative regulator of the MmpL5-MmpS5 efflux system led to intermediate-level resistance to compound 127-13. Mutations in this MarR-like regulator were initially identified in azole-resistant mutants of Mtb (Milano et al., 2009) and then in clofazimine- as well as bedaquiline-resistant mutants (Andries et al., 2014; Hartkoorn et al., 2014). Mutations in this regulator have been found to confer resistance to other chemotypes (Ioerger et al., 2013), consistent with non-specific compound efflux via induction of the MmpL5-MmpS5 system as a generalized mechanism of resistance in Mtb.
An eight-fold increase in MIC was also observed for drug resistant mutants isolated against compound 127-15. This was associated with a loss-of-function mutation in Rv2466c, encoding a mycothiol-dependent reductase, DsbA, which is essential for Mtb survival under oxidative stress (Albesa-Jové et al., 2014; Rosado et al., 2017). This enzyme has been implicated in the activation of the prodrug TP053, a thienopyrimidine derivative, by a dithiol-disulfide mechanism (Albesa-Jové et al., 2014). The compound 127-15 is therefore likely to be metabolized by Rv2466c by reduction of the disulfide bond to generate an active metabolite that could inhibit multiple cellular targets in Mtb.
In summary, this study has demonstrated the utility of reporter strains of Mtb for rapidly classifying phenotypic screening hits into broad mechanistic categories. This approach provides a means of judiciously applying scarce resource to hit expansion or hit-to-lead activities while gaining further mechanistic insight into the most promising hits. Of the 14 compounds investigated in this study, five were found to be prodrugs that require activation via mechanisms described previously for other prodrugs, including INH. These were readily identified by sequencing of resistant mutants that arose at comparatively high frequencies. This finding is consistent with the over-representation of prodrugs among new and existing TB drugs, and further underscores Mtb-mediated biotransformation as a prominent feature of anti-tubercular drug MoAs (Awasthi and Freundlich, 2017). Half of the compounds proved refractory to MoA elucidation using the assays applied here. Therefore, while phenotypic screening provides a convenient means of identifying new inhibitory scaffolds, MoA elucidation can remain a challenging undertaking (Singh and Mizrahi, 2017). It will be interesting to determine whether high-throughput chemical-genetic approaches, such as the PROSPECT system for screening hypomorph libraries (Johnson et al., 2019), could be informative in such cases.
Data Availability Statement
The sequencing data have been deposited in NCBI SRA under BioProject accession number PRJNA645974.
Author Contributions
VM, DW, and MC conceived and designed the experiments. MN provided the compounds, SF performed MIC and cytotoxicity assays, AJ and RS performed MIC assays, TI performed the WGS analyses, and MC performed all other experiments. MC and VM wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by grants from the South African Medical Research Council (to VM), the Strategic Health Innovations Partnerships (SHIP) Unit of the SAMRC (to DW and VM), the National Research Foundation of South Africa (to VM), the Oppenheimer Memorial Trust (to VM), the Bill & Melinda Gates Foundation (OPP1158806, via subaward from the FNIH) and an Early-Career Researcher Award from the University of Cape Town (to MC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Peter Kim, Division of AIDS-NIAID for his help in the initiation of the collaboration between DAIDS and University of Cape Town. We thank Vinayak Singh, Robin Warren, and Jose A. Aínsa for providing strains, Helena Boshoff for helpful discussions, and members of the MMRU for advice and assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.582416/full#supplementary-material
References
- Abrahams G. L., Kumar A., Savvi S., Hung A. W., Wen S., Abell C., et al. (2012). Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem. Biol. 19, 844–854. 10.1016/j.chembiol.2012.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albesa-Jové D., Chiarelli L. R., Makarov V., Pasca M. R., Urresti S., Mori G., et al. (2014). Rv2466c Mediates the Activation of TP053 To Kill Replicating and Non-replicating Mycobacterium tuberculosis . ACS Chem. Biol. 9, 1567–1575. 10.1021/cb500149m [DOI] [PubMed] [Google Scholar]
- Alland D., Steyn A. J., Weisbrod T., Aldrich K., Jacobs W. R., Jr. (2000). Characterization of the Mycobacterium tuberculosis iniBAC promoter, a promoter that responds to cell wall biosynthesis inhibition. J. Bacteriol. 182, 1802–1811. 10.1128/JB.182.7.1802-1811.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andries K., Villellas C., Coeck N., Thys K., Gevers T., Vranckx L., et al. (2014). Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS One 9, e102135. 10.1371/journal.pone.0102135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer S., Unser M. J., Froelich E. (1956). Some 5-(Oxoalkyl)-2-thiohydantoins and Their Derivatives. J. Am. Chem. Soc. 78, 6182–6185. 10.1021/ja01604a064 [DOI] [Google Scholar]
- Arora K., Ochoa-Montaño B., Tsang P. S., Blundell T. L., Dawes S. S., Mizrahi V., et al. (2014). Respiratory flexibility in response to inhibition of cytochrome C oxidase in Mycobacterium tuberculosis. Antimicrobial Agents Chemother. 58, 6962–6965. 10.1128/AAC.03486-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi D., Freundlich J. S. (2017). Antimycobacterial Metabolism: Illuminating Mycobacterium tuberculosis Biology and Drug Discovery. Trends Microbiol. 25, 756–767. 10.1016/j.tim.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks P. C., Movahedzadeh F., Davis E. O. (2001). Identification of some DNA damage-inducible genes of Mycobacterium tuberculosis: apparent lack of correlation with LexA binding. J. Bacteriol. 183, 4459–4467. 10.1128/JB.183.15.4459-4467.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K., Knaak T., Satkamp L., Humbert O., Falkow S., Ramakrishnan L. (2002). Complex pattern of Mycobacterium marinum gene expression during long-term granulomatous infection. Proc. Natl. Acad. Sci. U. S. A. 99, 3920–3925. 10.1073/pnas.002024599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S., Lee H. S., Franzblau S. (2015). Microplate Alamar Blue Assay (MABA) and Low Oxygen Recovery Assay (LORA) for Mycobacterium tuberculosis. Methods Mol. Biol. 1285, 281–292. 10.1007/978-1-4939-2450-9_17 [DOI] [PubMed] [Google Scholar]
- Cole S. T. (2016). Inhibiting Mycobacterium tuberculosis within and without. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 371, 20150506. 10.1098/rstb.2015.0506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb C., Lee C.-M., Dubey V. S., Daniel J., Abomoelak B., Sirakova T. D., et al. (2009). A Novel In Vitro Multiple-Stress Dormancy Model for Mycobacterium tuberculosis Generates a Lipid-Loaded, Drug-Tolerant, Dormant Pathogen. PLoS One 4, e6077. 10.1371/journal.pone.0006077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degiacomi G., Belardinelli J. M., Pasca M. R., De Rossi E., Riccardi G., Chiarelli L. R. (2020). Promiscuous Targets for Antitubercular Drug Discovery: The Paradigm of DprE1 and MmpL3. Appl. Sci. 10, 623. 10.3390/app10020623 [DOI] [Google Scholar]
- Domenech P., Reed M. B. (2009). Rapid and spontaneous loss of phthiocerol dimycocerosate (PDIM) from Mycobacterium tuberculosis grown in vitro: implications for virulence studies. Microbiology 155, 3532–3543. 10.1099/mic.0.029199-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragostin I., Dragostin O. M., Samal S. K., Dash S., Tatia R., Dragan M., et al. (2019). New isoniazid derivatives with improved pharmaco-toxicological profile: Obtaining, characterization and biological evaluation. Eur. J. Pharm. Sci. 137, 104974. 10.1016/j.ejps.2019.104974 [DOI] [PubMed] [Google Scholar]
- Evans J. C., Mizrahi V. (2018). Priming the tuberculosis drug pipeline: new antimycobacterial targets and agents. Curr. Opin. Microbiol. 45, 39–46. 10.1016/j.mib.2018.02.006 [DOI] [PubMed] [Google Scholar]
- Flentie K., Harrison G. A., Tükenmez H., Livny J., Good J. A. D., Sarkar S., et al. (2019). Chemical disarming of isoniazid resistance in Mycobacterium tuberculosis . Proc. Natl. Acad. Sci. 116, 10510. 10.1073/pnas.1818009116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzblau S. G., Degroote M. A., Cho S. H., Andries K., Nuermberger E., Orme I. M., et al. (2012). Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis . Tuberculosis 92, 453–488. 10.1016/j.tube.2012.07.003 [DOI] [PubMed] [Google Scholar]
- Gold B., Nathan C. (2017). Targeting Phenotypically Tolerant Mycobacterium tuberculosis . Microbiol. Spectr. 5, 1–69. 10.1128/microbiolspec.TBTB1122-0031-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold B., Warrier T., Nathan C. (2015). A multi-stress model for high throughput screening against non-replicating Mycobacterium tuberculosis. Methods Mol. Biol. 1285, 293–315. 10.1007/978-1-4939-2450-9_18 [DOI] [PubMed] [Google Scholar]
- Grant S. S., Kawate T., Nag P. P., Silvis M. R., Gordon K., Stanley S. A., et al. (2013). Identification of Novel Inhibitors of Nonreplicating Mycobacterium tuberculosis Using a Carbon Starvation Model. ACS Chem. Biol. 8, 2224–2234. 10.1021/cb4004817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartkoorn R. C., Uplekar S., Cole S. T. (2014). Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 2979–2981. 10.1128/AAC.00037-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Kushner N. L., Theriault M. E., Pisu D., Tan S., Mcnamara C. W., et al. (2018). The Deconstructed Granuloma: A Complex High-Throughput Drug Screening Platform for the Discovery of Host-Directed Therapeutics Against Tuberculosis. Front. Cell Infect. Microbiol. 8, 275. 10.3389/fcimb.2018.00275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioerger T. R., O’malley T., Liao R., Guinn K. M., Hickey M. J., Mohaideen N., et al. (2013). Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS One 8, e75245. 10.1371/journal.pone.0075245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson E. O., Laverriere E., Office E., Stanley M., Meyer E., Kawate T., et al. (2019). Large-scale chemical-genetics yields new M. tuberculosis inhibitor classes. Nature 571, 72–78. 10.1038/s41586-019-1315-z [DOI] [PubMed] [Google Scholar]
- Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P., Arora K., Lloyd J. R., Lee I. Y., Nair V., Fischer E., et al. (2012). Meropenem inhibits D,D-carboxypeptidase activity in Mycobacterium tuberculosis. Mol. Microbiol. 86, 367–381. 10.1111/j.1365-2958.2012.08199.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan B. W., Chowdhury N., Wood T. K. (2015). Combatting bacterial infections by killing persister cells with mitomycin C. Environ. Microbiol. 17, 4406–4414. 10.1111/1462-2920.12873 [DOI] [PubMed] [Google Scholar]
- Lee B. S., Pethe K. (2018). Therapeutic potential of promiscuous targets in Mycobacterium tuberculosis. Curr. Opin. Pharmacol. 42, 22–26. 10.1016/j.coph.2018.06.006 [DOI] [PubMed] [Google Scholar]
- Lempens P., Meehan C. J., Vandelannoote K., Fissette K., De Rijk P., Van Deun A., et al. (2018). Isoniazid resistance levels of Mycobacterium tuberculosis can largely be predicted by high-confidence resistance-conferring mutations. Sci. Rep. 8, 3246. 10.1038/s41598-018-21378-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Stevens C. M., Pandya A. N., Darzynkiewicz Z., Bhattarai P., Tong W., et al. (2019). Direct Inhibition of MmpL3 by Novel Antitubercular Compounds. ACS Infect. Dis. 5, 1001–1012. 10.1021/acsinfecdis.9b00048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienhardt C., Raviglione M. C. (2020). TB Elimination Requires Discovery and Development of Transformational Agents. Appl. Sci. 10, 2605. 10.3390/app10072605 [DOI] [Google Scholar]
- Liu R., Lyu X., Batt S. M., Hsu M. H., Harbut M. B., Vilchèze C., et al. (2017). Determinants of the Inhibition of DprE1 and CYP2C9 by Antitubercular Thiophenes. Angew Chem. Int. Ed. Engl. 56, 13011–13015. 10.1002/anie.201707324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins F., Santos S., Ventura C., Elvas-Leitão R., Santos L., Vitorino S., et al. (2014). Design, synthesis and biological evaluation of novel isoniazid derivatives with potent antitubercular activity. Eur. J. Med. Chem. 81, 119–138. 10.1016/j.ejmech.2014.04.077 [DOI] [PubMed] [Google Scholar]
- McNeil M. B., Dennison D., Parish T. (2017). Mutations in MmpL3 alter membrane potential, hydrophobicity and antibiotic susceptibility in Mycobacterium smegmatis . Microbiol. (Reading Engl.) 163, 1065–1070. 10.1099/mic.0.000498 [DOI] [PubMed] [Google Scholar]
- Milano A., Pasca M. R., Provvedi R., Lucarelli A. P., Manina G., Ribeiro A. L., et al. (2009). Azole resistance in Mycobacterium tuberculosis is mediated by the MmpS5-MmpL5 efflux system. Tuberc. (Edinb) 89, 84–90. 10.1016/j.tube.2008.08.003 [DOI] [PubMed] [Google Scholar]
- Moosa A., Lamprecht D. A., Arora K., Barry C. E., Boshoff H. I. M., Ioerger T. R., et al. (2017). Susceptibility of Mycobacterium tuberculosis Cytochrome bd Oxidase Mutants to Compounds Targeting the Terminal Respiratory Oxidase, Cytochrome c. Antimicrobial Agents Chemother. 61, e01338–e01317. 10.1128/AAC.01338-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naran K., Moosa A., Barry C. E.,. 3., Boshoff H. I. M., Mizrahi V., Warner D. F. (2016). Bioluminescent Reporters for Rapid Mechanism of Action Assessment in Tuberculosis Drug Discovery. Antimicrobial Agents Chemother. 60, 6748–6757. 10.1128/AAC.01178-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S., Park Y., Engelhart C. A., Wallach J. B., Schnappinger D., Arora K., et al. (2018). Discovery and Structure-Activity-Relationship Study of N-Alkyl-5-hydroxypyrimidinone Carboxamides as Novel Antitubercular Agents Targeting Decaprenylphosphoryl-β-d-ribose 2’-Oxidase. J. Med. Chem. 61, 9952–9965. 10.1021/acs.jmedchem.8b00883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira P. F. M., Guidetti B., Chamayou A., Andre-Barres C., Madacki J., Kordulakova J., et al. (2017). Mechanochemical Synthesis and Biological Evaluation of Novel Isoniazid Derivatives with Potent Antitubercular Activity. Molecules 22, art.1457 – 1427p. 10.3390/molecules22091457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orme I. (2001). Search for new drugs for treatment of tuberculosis. Antimicrobial Agents Chemother. 45, 1943–1946. 10.1128/AAC.45.7.1943-1946.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish T. (2020). In vitro drug discovery models for Mycobacterium tuberculosis relevant for host infection. Expert Opin. Drug Discov. 15, 349–358. 10.1080/17460441.2020.1707801 [DOI] [PubMed] [Google Scholar]
- Park Y., Pacitto A., Bayliss T., Cleghorn L. A. T., Wang Z., Hartman T., et al. (2017). Essential but Not Vulnerable: Indazole Sulfonamides Targeting Inosine Monophosphate Dehydrogenase as Potential Leads against Mycobacterium tuberculosis . ACS Infect. Dis. 3, 18–33. 10.1021/acsinfecdis.6b00103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peh H. L., Toh A., Murugasu-Oei B., Dick T. (2001). In vitro activities of mitomycin C against growing and hypoxic dormant tubercle bacilli. Antimicrobial Agents Chemother. 45, 2403–2404. 10.1128/AAC.45.8.2403-2404.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips O. A., Udo E. E., Varghese R. (2012). Antimycobacterial Activities of Novel 5-(1H-1,2,3-Triazolyl)Methyl Oxazolidinones. Tuberc. Res. Treat 2012, 289136–289136. 10.1155/2012/289136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon-Garcia S., Mick V., Dainese E., Martin C., Thompson C. J., De Rossi E., et al. (2012). Functional and genetic characterization of the tap efflux pump in Mycobacterium bovis BCG. Antimicrob. Agents Chemother. 56, 2074–2083. 10.1128/AAC.05946-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado L. A., Wahni K., Degiacomi G., Pedre B., Young D., De La Rubia A. G., et al. (2017). The antibacterial prodrug activator Rv2466c is a mycothiol-dependent reductase in the oxidative stress response of Mycobacterium tuberculosis . J. Biol. Chem. 292, 13097–13110. 10.1074/jbc.M117.797837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scifinder (2020).
- Singh V., Mizrahi V. (2017). Identification and validation of novel drug targets in Mycobacterium tuberculosis. Drug Discov. Today 22, 503–509. 10.1016/j.drudis.2016.09.01 [DOI] [PubMed] [Google Scholar]
- Singh V., Donini S., Pacitto A., Sala C., Hartkoorn R. C., Dhar N., et al. (2017). The Inosine Monophosphate Dehydrogenase, GuaB2, Is a Vulnerable New Bactericidal Drug Target for Tuberculosis. ACS Infect. Dis. 3, 5–17. 10.1021/acsinfecdis.6b00102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasz M. (1995). Mitomycin C: small, fast and deadly (but very selective). Chem. Biol. 2, 575–579. 10.1016/1074-5521(95)90120-5 [DOI] [PubMed] [Google Scholar]
- Wang F., Sambandan D., Halder R., Wang J., Batt S. M., Weinrick B., et al. (2013). Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 110, E2510–E2517. 10.1073/pnas.1309171110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellington S., Hung D. T. (2018). The Expanding Diversity of Mycobacterium tuberculosis Drug Targets. ACS Infect. Dis. 4, 696–714. 10.1021/acsinfecdis.7b00255 [DOI] [PubMed] [Google Scholar]
- WHO (2019). Global Tuberculosis Report 2019 (Geneva, Switzerland: World Health Organization; ). Available at: https://apps.who.int/iris/bitstream/handle/10665/329368/9789241565714-eng.pdf?ua=1 (Accessed 26 May 2020). [Google Scholar]
- Williams J. T., Haiderer E. R., Coulson G. B., Conner K. N., Ellsworth E., Chen C., et al. (2019). Identification of New MmpL3 Inhibitors by Untargeted and Targeted Mutant Screens Defines MmpL3 Domains with Differential Resistance. Antimicrobial Agents Chemother. 63, e00547–e00519. 10.1128/AAC.00547-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. R., Gessner R. K., Moosa A., Seldon R., Warner D. F., Mizrahi V., et al. (2017). Novel Antitubercular 6-Dialkylaminopyrimidine Carboxamides from Phenotypic Whole-Cell High Throughput Screening of a SoftFocus Library: Structure-Activity Relationship and Target Identification Studies. J. Med. Chem. 60, 10118–10134. 10.1021/acs.jmedchem.7b01347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Sala C., Dhar N., Vocat A., Sambandamurthy V. K., Sharma S., et al. (2014). In vitro and in vivo activities of three oxazolidinones against nonreplicating Mycobacterium tuberculosis . Antimicrobial Agents Chemother. 58, 3217–3223. 10.1128/AAC.02410-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H., Colvin C. J., Johnson B. K., Kirchhoff P. D., Wilson M., Jorgensen-Muga K., et al. (2017). Inhibitors of Mycobacterium tuberculosis DosRST signaling and persistence. Nat. Chem. Biol. 13, 218–225. 10.1038/nchembio.2259 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data have been deposited in NCBI SRA under BioProject accession number PRJNA645974.