

Abstract
Local adaptation is particularly likely in invertebrate pests that typically have short generation times and large population sizes, but there are few studies on pest species investigating local adaptation and separating this process from contemporaneous and historical gene flow. Here, we use a population genomic approach to investigate evolutionary processes in the two most dominant spider mites in China, Tetranychus truncatus Ehara and Tetranychus pueraricola Ehara et Gotoh, which have wide distributions, short generation times, and large population sizes. We generated genome resequencing of 246 spider mites mostly from China, as well as Japan and Canada at a combined total depth of 3,133×. Based on demographic reconstruction, we found that both mite species likely originated from refugia in southwestern China and then spread to other regions, with the dominant T. truncatus spreading ~3,000 years later than T. pueraricola. Estimated changes in population sizes of the pests matched known periods of glaciation and reinforce the recent expansion of the dominant spider mites. T. truncatus showed a greater extent of local adaptation with more genes (76 vs. 17) associated with precipitation, including candidates involved in regulation of homeostasis of water and ions, signal transduction, and motor skills. In both species, many genes (135 in T. truncatus and 95 in T. pueraricola) also showed signatures of selection related to elevation, including G‐protein‐coupled receptors, cytochrome P450s, and ABC‐transporters. Our results point to historical expansion processes and climatic adaptation in these pests which could have contributed to their growing importance, particularly in the case of T. truncatus.
Keywords: invertebrate pests, local adaptation, range shifts, spider mites, whole‐genome resequencing
1. INTRODUCTION
Species range shifts are defined as any changes in the distribution of a species along latitude, longitude, and elevation or depth (marine ecosystems) over time (Lenoir & Svenning, 2015). The ranges of numerous species, as well as species composition and abundance in local communities, are shifting in response to climate change (Parmesan, 2006; Scheffers et al., 2016). A complex mosaic of local climate changes and associated biotic interactions involving temperature, precipitation, competitive release, and land‐use changes shape range shifts (Lenoir et al., 2010; VanDerWal et al., 2013). Changes in species abundance within a locality can represent intermediate states in an ongoing process of ranges shifting (Maggini et al., 2011). Species range shifts depend on inherent levels of species‐specific tolerance and the ability to which these can be modified by ongoing plastic changes and evolutionary adaptation (e.g., Atkinson, Siegel, Pakhomov, & Rothery, 2004; Jiguet et al., 2010; Moller, Rubolini, & Lehikoinen, 2008). Incorporating local adaptation in climate responses can greatly improve forecasts of species distribution and abundance under climate change (Bush et al., 2016; Peterson, Doak, & Morris, 2019).
Pest species particularly are expected to adapt to local climatic conditions given that short generation times and large population sizes in pests make rapid adaptation likely (Hoffmann, 2017). However, there is limited evidence for climatic adaptation in pests, with some notable exceptions being changes in diapause in planthoppers (Hou et al., 2016) and adaptive changes in thermal tolerance in earth mites (Hill, Chown, & Hoffmann, 2013). The importance of evolutionary factors in pest outbreaks is exemplified by adaptation to exogenous and endogenous toxins, such as pesticides (Kirk, Dorn, & Mazzi, 2013). On a longer evolutionary time scale, adaptation to host resources used by invertebrates has been linked to the expansion and contraction of gene families associated with detoxification (Rane et al., 2019).
While there is an increasing amount of information on the genomic basis of adaptation to pesticides and host plant toxins, there is much less information on adaptation in pests to other stresses. One reason why there is limited information on pest adaptation to climate is that the common garden and transplant experiments required to test for adaptation are rarely carried out, with only a few exceptions (Yadav, Stow, & Dudaniec, 2019). However, genomic data can also be used to obtain information on climatic adaptation (Rellstab, Gugerli, Eckert, Hancock, & Holderegger, 2015) particularly when a high density of markers is available across the genome, and samples are available across climatic gradients. Analyses of genomic variation are starting to be undertaken in pest species where invasion histories, range shifts, and demographic processes are understood and markers can then indicate signatures of selection. Knowledge of historical processes can help tracing the spread of invasive pests and the past conditions to which they have been exposed. This type of information can help in understanding and predicting potential pest invasion and outbreaks such as the likelihood of pest outbreaks occurring after invasion into new environments (Hoffmann, 2017).
Here, we undertake a detailed genomic study to investigate historical and current population processes affecting two agriculturally important and related spider mites, Tetranychus truncatus and Tetranychus pueraricola (Jin, Tian, Chen, & Hong, 2018). These spider mites have short generation times and large population sizes that should facilitate rapid adaptation across the range of climate conditions where they are found. The mites have genomes that are among the smallest in arthropods, making them suitable for whole‐genome resequencing (Grbic et al., 2011; Lasken, 2009). Both mite pests are common as well as being widespread: for instance, in a survey across the last ten years in mainland China, the occurrence frequency of T. truncatus on major crops reached 48.5%, followed by T. pueraricola at 21.4% (Jin et al., 2018), while in northern China, T. truncatus has become the main acarine pest of major crops including cotton, tomato beans and corn (Guo et al., 2013; Jin et al., 2018; Wang, Zhang, Wu, Xie, & Xu, 2013).
We report on whole‐genome sequencing of 173 T . truncatus, 67 T . pueraricola, and 6 individuals from sister groups. We investigated genomic diversity, population divergence, demographic history, and gene flow. We used these results to propose a scenario of how the spider mites originated and spread in China. We identified genes putatively associated with local adaptation to climate and elevation. We compared the number of genes putatively implicated in local adaptation for these two species with very different range sizes and abundances. The demographic and adaptation data in these two spider mites with different abundance were used to reveal important role that local adaptation may play in shaping range shifts.
2. MATERIALS AND METHODS
2.1. Sampling, amplification, and sequencing
We sampled large numbers of spider mites from fields in 2014–2017 across China. At each locality, we randomly collected up to three mites per plant, and plants were at least 1 m apart. We identified species through the nuclear ribosomal internal transcribed spacer (ITS) region (Ge, Ding, Zhang, & Hong, 2013). A total of 246 spider mites were selected for resequencing, including 173 T . truncatus, 67 T . pueraricola, and other Tetranychus species (Table S1).
Purified genomic DNA was extracted from female adult spider mites with DNeasy Blood & Tissue Kits following the manufacturer's protocol. In order to gain enough DNA for individual resequencing given the small size of mites (about 0.4 mm long; Fasulo & Denmark, 2000), whole‐genome amplification from purified genomic DNA was performed according to instructions of the QIAGEN REPLI‐g Ultrafast Mini WGA kit with some modifications. These included incubating the mixture of denaturation buffer and DNA at 95°C for 6 min and incubating the mixture of master mix and denatured DNA for multiple displacement amplification (MDA) at 30°C for 16 hr. Paired‐end 150 bp libraries with short inserts (~500 bp) were constructed according to the instructions of TruSeq Nano DNA Library Prep Kits. The whole‐genome sequencing was performed on an Illumina HiSeq 4,000 platform. Our target coverage was 10× per individual.
2.2. Alignment and SNP calling
Before mapping, raw reads were trimmed to obtain reliable clean reads using Trimmomatic 0.36 (Bolger, Lohse, & Usadel, 2014) under the following protocol: (a) cut adapter from the read; (b) perform a 4‐base wide sliding‐window trimming when the average quality per base drops below 15; (c) cut bases off both the start and the end of a read when below 5; and (d) drop reads below 50 bases long. The high‐quality reads were then aligned to the indexed T. urticae genome (Grbic et al., 2011) using bwa version 0.7.17 (Li & Durbin, 2009) with the mem command. The mapping results were transformed into BAM format and sorted using SAMtools v1.9 (Li et al., 2009). Instead of being removed, PCR duplicates were marked using the MarkDuplicates module of GATK version 4.0.12 (McKenna et al., 2010). We also used GATK4 to realign Indel regions of the indexed bam files and then identify variants across all individuals using the HaplotypeCaller module with default parameters. To reduce false variants, we applied hard filters to both SNPs and Indels in our raw variants based on the distribution of annotation values. For SNPs, we applied the following criteria: QD (variant confidence standardized by depth) <2.0; MQ (mapping quality of a SNP) <30.0; FS (strand bias in support for REF versus ALT allele calls) >60.0; and SOR (sequencing bias in which one DNA strand is favored over the other) >5.0. For Indels, we applied the following criteria: QD < 2.0; FS > 100.0; and SOR > 5.0. To get an analysis‐ready VCF file, we only kept SNP and Indel variants and biallelic sites.
2.3. Population structure
To infer evolutionary and genetic relationships of spider mites (e.g., T. truncatus, T. pueraricola, T. urticae, T. kanzawai, and T. piercei), we used a single VCF file with biallelic and high‐quality SNPs as an input into SNPhylo (Lee, Guo, Wang, Kim, & Paterson, 2014) to construct a maximum likelihood (ML) tree. Before determining a phylogenetic tree, we extracted SNP positions which met the criteria of minor allele frequency more than 0.01, missing rate less than 0.1 and R 2 < .6. Finally, we ended up with 16,251 SNPs for developing an ML tree with 1,000 bootstrap iterations and used T. piercei as the outgroup. To cluster spider mites by summarizing the major axis variation in allele frequencies, we conducted principal component analysis using the R package SNPRelate (Zheng et al., 2012) to obtain eigenvectors which could be plotted using the ggplot2 package (Wickham, 2016).
To infer ancestral proportions for each individual and suitable subgroups for populations of spider mites, ADMIXTURE (version 1.30; Alexander, Novembre, & Lange, 2009) analyses were performed with 10 replicate variant sets randomly down‐sampled to 10% of the high‐quality SNPs using different random seeds. Then, each replicate variant set was pruned to remove SNPs with minor allele frequency less than 1% and exclude SNPs above an r 2 threshold of 0.2 at sliding windows of 50 SNPs with a step size of 5 in PLINK v2.0 (Chang et al., 2015). Ten replicates of between 95,832 and 96,242 variants were used to carry out population structure analysis in ADMIXTURE 1.30 (Alexander et al., 2009). The number of assumed ancestral populations (K) ranged from 2 to 20 with fivefold cross‐validation and each K was repeated 10 times with different seeds based on the 10 replicate variant sets, resulting in a total of 100 runs for each value of K. Then, we used CLUMPAK (Kopelman, Mayzel, Jakobsson, Rosenberg, & Mayrose, 2015) with default settings to provide a better understanding of different solutions reported by ADMIXTURE. CLUMPAK generated average results of Q‐matrices for each value of K from all runs, so that we could plot ancestry proportions for all individuals.
To infer population‐level splits and mixtures for both T. truncatus and T. pueraricola populations, we filtered the high‐quality SNPs obtained by GATK with the following criteria: the max missing rate was 0.9, and r 2 should be less than 0.2. Then, we used Treemix 1.13 (Pickrell & Pritchard, 2012) to investigate the admixture between populations, with migration edges ranging from 1 to 20 and T. piercei as outgroup.
2.4. Analyses of genetic diversity, ROH, and LD
To reduce the influence of statistical bias caused by low coverage and unidentical sample sizes of different populations, we calculated population differentiation (F ST) and nucleotide diversity (θ) in ANGSD 0.929 (Korneliussen, Albrechtsen, & Nielsen, 2014). For the SFS computation, we implemented the following criteria: (a) only keep uniquely mapped reads; (b) use only sites with data from at least 80% individuals; (c) ensure the minimum total depth equals the sample sizes of the group; and (d) ensure both the minimum mapping quality and base quality equal 30. The ancestral state was assigned with the outgroup T. piercei to polarize allele state. Then, realSFS and thetaStat programs were used to estimate F ST and θ. The regions of homozygosity (ROH) for each population were calculated in the program BCFtools 1.9 (Narasimhan et al., 2016). Linkage disequilibrium (LD) decay analysis for each population/group was measured in PopLDdecay (Zhang, Dong, Xu, He, & Yang, 2018) with the following parameters: ‐MaxDist (maximum distance (kb) between two SNP) 200 and –MAF (minimum minor allele frequency filter) 0.01.
2.5. Demographic history
We used pairwise sequentially Markovian coalescent (PSMC) models to estimate variation in effective population size (N e) of different species of Tetranychus (T. urticae from Canada; T. kanzawai, T. truncatus, and T. pueraricola from China) based on density of heterozygotes (Li & Durbin, 2011). The parameters were set as follows: ‐N25 ‐t15 ‐r5 ‐p “4 + 25*2 + 4+6”. The description of each parameter and scripts is available on https://github.com/lh3/psmc. For Tetranychus, we assumed a mutation rate of 1 × 10−9 mutations per base pair per generation and a generation time of 1 month, modifying parameters from Drosophila (Keightley, Ness, Halligan, & Haddrill, 2014). The historical effect of recent effective population size was estimated with SMC++ which requires only unphased genomes and provides more accurate estimates for the recent past taking advantage of linkage disequilibrium information in coalescent hidden Markov models (Terhorst, Kamm, & Song, 2017). The default settings resulted in too much oscillation as well as overfitting of the curves for estimating recent effective population size of spider mites. To correct this issue, we experimented with various parameters suggested by SMC++ documentation to identify the settings best suited for our data. Finally, we specified polarization error of 0.5 as the identity of the ancestral allele is not known, and a regularization penalty of 0.5 to shrink too much oscillation of the estimated size history. To prevent overfitting, we specified a threshold of 1e‐2 for stopping the EM algorithm when the relative improvement in log‐likelihood became small. The estimation command was “‐‐cores 25 ‐‐timepoints 20 2e6 ‐knots 3 ‐‐thinning 600 ‐‐spline cubic ‐‐ftol 1e‐2 ‐‐polarization‐error 0.5 ‐rp 0.5 1e‐9”.
Inferences about the demographic history were made through a continuous‐time coalescent simulator fastsimocal2 (Excoffier, Dupanloup, Huerta‐Sánchez, Sousa, & Foll, 2013) which estimates parameters from a site frequency spectrum (SFS). To reduce the biases in SFS caused by sequencing error and coverage depth, we computed SFS based on the realigned bam files output from GATK4 using ANGSD 0.929 (Korneliussen et al., 2014). To avoid any kind of selection influence on demographic inference, we used only intergenic SNPs. After generating allele frequency likelihood files under GATK genotype likelihood model, we used realSFS program in ANGSD (Korneliussen et al., 2014) to estimate joint SFS. The joint SFS across different groups was used to estimate demographic parameters under 35 alternative models of phylogenetic relationships and historical events (Table S8). For each model, we ran fastsimcoal2 with 100,000 simulations to estimate the expected derived SFS, 40 conditional maximization (ECM) cycles and 50 times for estimating the parameters. SFS entries with less than 10 SNPs were pooled together. The best‐fitting model was assessed through the likelihood and Akaike information criterion (Excoffier et al., 2013). Parametric bootstrap estimates were obtained with 50 data sets simulated with estimates of parameters of the best model.
2.6. Local adaptation analysis
To detect genomic signatures of adaptation to the local climate and elevation, we performed a gene‐environment association study using the whole‐genome resequencing data. The climate and elevation data set (Table S10) were downloaded from CHELSA (Karger et al., 2017) and from USGS (Danielson & Gesch, 2011). We summarized the climatic variables by keeping half of the positive PCNMs (principal coordinates of neighbor matrices) for temperature variables and for precipitation variables. To get a genotype matrix, we used VCFtools v0.1.13 (Danecek et al., 2011) to preprocess SNPs by removing sites with MAF < 0.05, missing rate >0.1 and individuals with relatedness >0.0884. Then, the missing values of genotype were imputed with the program Beagle 5.0 (Browning, Zhou, & Browning, 2018) to improve the accuracy of the genotypes. After all these filtering steps, we had 120 individuals with 262,541 SNPs for T. truncatus and 51 individuals with 401,999 SNPs for T. pueraricola. To control the false discovery rate (FDR), we, respectively, chose appropriate value ranges of K = 12–16 and K = 4–7 for latent factors of T. truncatus and T. pueraricola based on the ADMIXTURE analysis (Figure S4). Then, we ran 5 repetitions of the latent factor mixed model to compute new calibrated P‐values in LFMM v1.5 (Frichot, Schoville, Bouchard, & François, 2013). The lists of candidate loci were obtained after controlling FDR at the level q = 0.5% using the Benjamini–Hochberg procedure. As low dispersal ability of spider mites might increase false‐positive rates with LFMM (Forester, Jones, Joost, Landguth, & Lasky, 2016), we also used redundancy analysis (RDA) to identify signatures of local adaptation following the methods and scripts referred in Capblancq, Luu, Blum, & Bazin (2018). Briefly, individual genotypes and PCNMs for climatic variables obtained from the above LFMM were used as the response matrix and explanatory matrix, respectively. The parameters used in the RDA analysis were the same as in Capblancq et al. (Capblancq et al., 2018) except for controlling FDR at the level q = 0.5% to identify outlier loci involved in local adaptation.
We reconstructed PCA for all candidate SNPs associated with local adaptation identified by LFMM to regroup populations of spider mites following the same PCA procedure described above for analyzing population structure. We used a sliding‐window approach (100‐kb windows sliding in 10‐kb steps) to calculate the genome‐wide distribution of genetic differentiation (F ST), pairwise nucleotide variation (θ π) ratios and selection statistics (Tajima's D) between different groups of the two species. These values were calculated with VCFtools v0.1.13 (Danecek et al., 2011) to detect selection signals for candidate loci.
3. RESULTS
3.1. Sequencing and variation
Our large‐scale sampling of spider mites from 39 locations in 2014–2017 mainly in mainland China (Figure S1) identified T. truncatus and T. pueraricola as the two most dominant species with different range shifts on crops, which agrees with a previous long‐term survey (Jin et al., 2018). After whole‐genome amplification and resequencing, we generated genomes of 173 T . truncatus from 17 populations, 67 T . pueraricola from 10 populations, and 6 individuals of outgroups (T. urticae, T. piercei, T. kanzawai, and T. parakanzawai; Figure S1 and Table S1). Most of our samples were collected from China, except for 6 T . truncatus from Japan and 2 T . urticae from Canada (Table S1). We generated a total of 285 Gb aligned high‐quality bases at a total of 3,133 × effective depth and an average of 13 × depth per individual (Table S2).
After filtering, we obtained 4.46 million SNPs for T. truncatus and 3.74 million SNPs for T. pueraricola with 0.95 million SNPs shared in these two species (Table S3). For T. truncatus, 3.58 million SNPs (80.3%) were located in non‐coding regions and 0.88 million SNPs (19.7%) were located in coding regions: 10.6% were synonymous and 7.5% were non‐synonymous, with a non‐synonymous/synonymous ratio of 0.707. For T. pueraricola, 3.02 million SNPs (80.7%) were located in non‐coding regions and 0.72 million SNPs (19.3%) were located in coding regions: 9.6% were synonymous and 7.8% were non‐synonymous, with a non‐synonymous/synonymous ratio of 0.815 (Table S4). To scale the distribution of SNPs across the T. urticae reference for all these 6 species, we calculated density of SNPs in bins of 100kb size and found that SNPs of all 6 spider mite species were mainly located in the first 50 scaffolds, while those of T. pueraricola were more commonly distributed in the rest of the scaffolds (Figure S2).
3.2. Population structure of the most two dominant spider mites
The phylogenetic and ADMIXTURE analyses clearly separated the two species (T. truncatus and T. pueraricola; Figure 1a). A few individuals exhibited ancestral proportions from both species; one JXtp individual of T. pueraricola had the largest ancestral proportion of T. truncatus when K = 2 (7%; Figure S3). The results of cross‐validation error suggested that the best K was 15, which makes it complicated to classify mite populations (Figure S3). The ancestral proportions of separate ADMIXTURE analyses (Figure S4) were consistent with the combined analyses (Figure S3), with the best K of 12 and 5 for T. truncatus and T. pueraricola, respectively. As 4 paraphyletic groups of T. truncatus and the 2 paraphyletic groups of T. pueraricola are suggested by phylogenetic tree (Figure 1a), we used the K = 6 ADMIXTURE result to cluster all populations of the spider mites in China into six main groups that were geographically associated. We used these six clusters in demographic analyses as well. Group I – Group IV comprised T. truncatus populations from southern China (provinces Jiangxi, Hunan, Fujian, and Guangdong; JX, HN, FJ, and GD), eastern China (provinces Shanxi, Hebei, Shandong, Jiangsu; SXT, HBB, SD, and JS) and Japan (JA), midland and northwestern China(provinces Gansu, Shaanxi, Xinjiang, and Inner Mongolia; GS, SXX, XJ, and NMG), northeastern China (provinces Hebei, Liaoning, Sichuan, and Heilongjiang; HBH, LN, SC, and HLJ); Group V and Group VI comprised T. pueraricola populations from southwestern China (provinces Yunnan, Sichuan, and Guangxi; YNL, SCC, YNY, GXtp, and SCB) and other areas of China (provinces Gansu, Jiangxi, Jiangsu, Liaoning, and Heilongjiang; GStp, JXtp, JStp, LNtp, and HLJtp).
FIGURE 1.
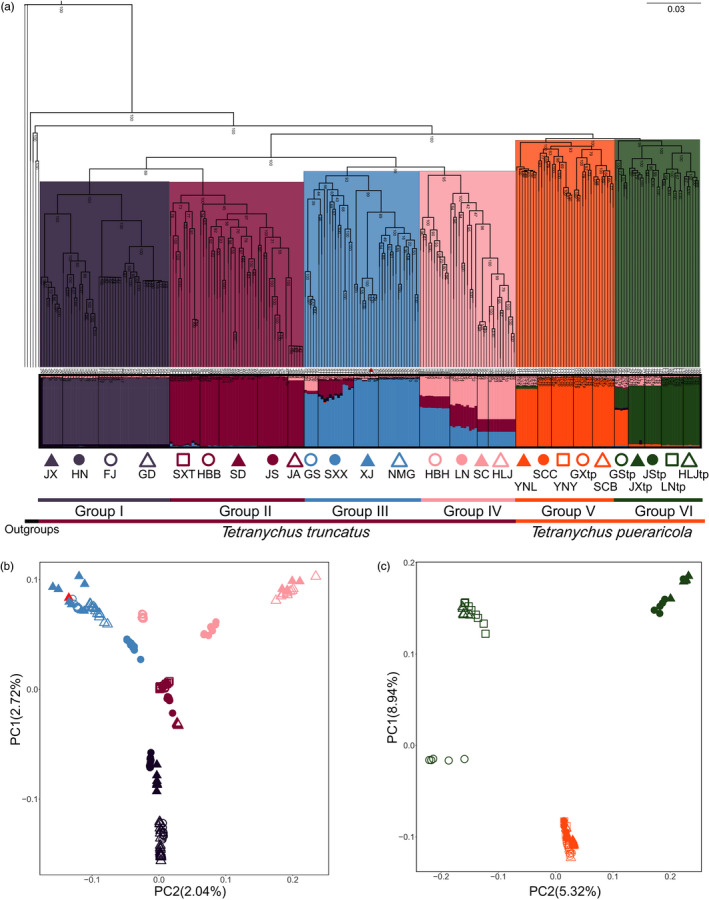
Population genetics analyses of T. truncatus and T. pueraricola. (a) Phylogenetic tree (maximum likelihood) and the ADMIXTURE analysis (K = 6) inferred from whole‐genome SNPs of spider mites, with T. piercei, T. kanzawai, T. parakanzawai, and T. urticae as outgroups. In ADMIXTURE analysis, each horizontal bar represents an individual. All populations are represented by open and filled symbols which are used in the PCA in the next panel. (b) Principal components analysis (PCA) of T. truncatus and (c) T. pueraricola. An outlying individual of the SC population denoted as a solid triangle may reflect a sampling error
The principal component analysis (PCA) supported the grouping results (Figure 1b,c). The first axis of the PCA explained a high degree of variance for the overall data set including T. truncatus and T. pueraricola (41.32%, Figure S5a) indicating the clear differences between the species. Both the PCA of T. truncatus and T. pueraricola mirrored the geography of the sampled populations (Figure 2a,b), which highlighted historical migrations or reflected “isolation by distance” (Reich, Price, & Patterson, 2008). PC1 of T. truncatus aligned along the south/north direction of China and accounted for 2.72% of the variation. PC1 of T. pueraricola aligned in a southwest/northeast direction and accounted for approximately three times as much of variation as the first axis in T. truncatus (8.94% vs. 2.72%).
FIGURE 2.
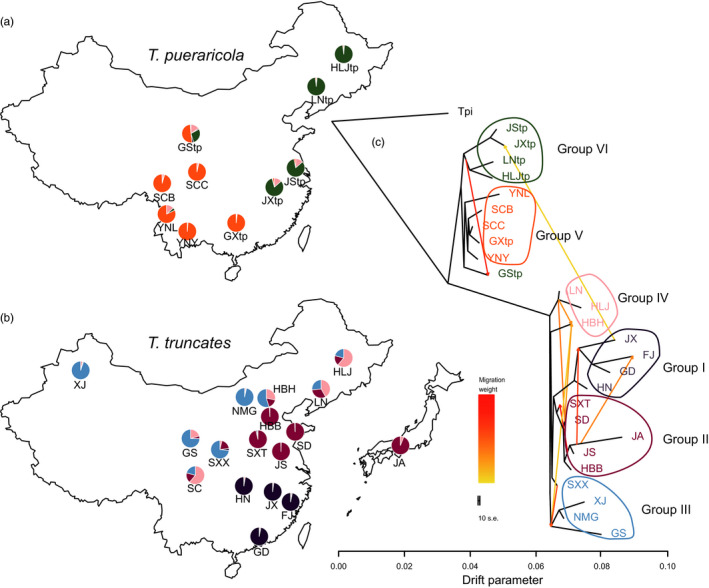
Maps and admixture events of the spider mites. Geographic locations of 27 populations for T. pueraricola (a) and T. truncatus (b) with ancestral proportions (K = 6) inferred from ADMIXTURE. (c) The relationships among populations of these two dominated spider mites. Each population is colored according to its corresponding group, and each arrow indicates a migration event which is colored with migration weights. The scale bar shows 10× the average standard error of the entries in the sample covariance matrix
The ML tree inferred from Treemix explained up to 99.99% of the variance between populations when being added with 10 migration edges (Figure 2c). Among 10 migration events, half were related to Group II, including within Group II, from Group II to Group I, and from Group II to Group IV. The lower genetic differentiation (F ST) between Group II and other groups (Table S7) also supported higher gene flow. There was introgression between T. truncatus and T. pueraricola in Jiangxi (JX and JXtp).
3.3. Nucleotide diversity, population differentiation, and linkage disequilibrium
The average nucleotide diversity (θπ) for the 4 spider mites (T. truncatus, T. pueraricola, T. piercei, and T. urticae) was 5.54 × 10−4. The genome‐wide θπ value ranged from 2.74 × 10−4 to 6.66 × 10−4 and 3.59 × 10−4 to 1.06 × 10−3 for T. truncatus and T. pueraricola, respectively, (Table S5). Despite a wider geographic distribution and a higher abundance (Jin et al., 2018), T. truncatus had lower genetic diversity than T. pueraricola as a whole (Figure S6a,b). The pattern of genetic diversity remained the same in species comparisons from the same locations where both species were sampled (GS, JX, JS, LN, and HLJ).
The F ST values among different populations of the two spider mites varied markedly (T. truncatus, 0.041‐0.809; T. pueraricola, 0.115‐0.748; Table S7). The FST values between remote populations were higher than between adjacent populations, suggesting the absence of long‐distance migration in most instances. The regions of homozygosity (ROH) were fewer and shorter for both T. truncatus and T. pueraricola in average number and size in southern (Group I) and southwestern (Group V) China (Table S6 and Figure S6c–f), indicative of larger populations or admixture (Ceballos, Joshi, Clark, Ramsay, & Wilson, 2018). The range of LD decay distances of T. truncatus populations was wider than that of T. pueraricola populations due to one outlier population, but otherwise, the two species showed similar levels of decay (Figure S7). These genetic parameters suggest distinct evolutionary and demographic histories for some T. truncatus and T. pueraricola populations.
3.4. Demographic history
With single representatives of each species sequenced at higher coverage, we estimated historical changes in the effective population size (N e) by pairwise sequentially Markovian coalescent analysis (Li & Durbin, 2011). The historical N e of T. urticae sampled in Canada exhibited different demographic patterns to other Asian spider mites (Figure 3a). The ancestors of all spider mites reached their peak N e at about 0.21 Mya (million years ago; Figure 3a) during interglacial periods before the Penultimate glaciation. Then, in the period of Penultimate glaciation, both North American and Asian populations experienced bottleneck events which were more dramatic in Asian populations. At about 80 Kya (thousand years ago), the N e of North American and Asian populations reached their lowest point and then began to increase. Since the last glaciation 40 Kya, the North American population has been declining, whereas even across the LGM (Last Glacial Maximum, 26.5 Kya BP), Asian populations have been gradually expanding (Figure 3a).
FIGURE 3.
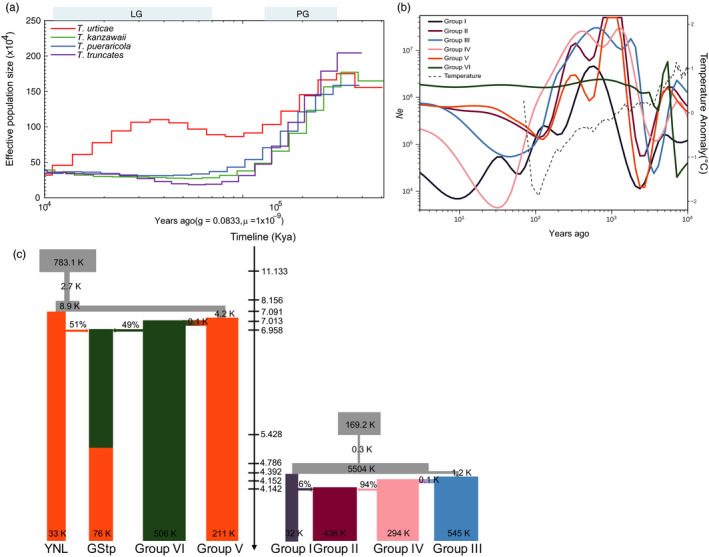
Demographic history of the spider mites. (a) PSMC analysis performed on the representatives of each species sequenced at high coverage to indicate variation in N e over the last 105 years. LG and PG represent period of last glaciation and penultimate glaciation separately. (b) Demographic changes of N e for six groups of T. truncatus and T. pueraricola in the recent 104 years using SMC++. Note that the SC population has been excluded from Group IV because of sampling uncertainty. Northern hemispheric (90‐30N) temperature anomaly from the mean temperature of AD 1962–1990. (c) The best demographic scenarios for T. truncatus and T. pueraricola inferred by fastsimcoal2. The gray rectangles represent ancestral populations, and the arrows indicate admixture events. To better understand demographic history of T. pueraricola, populations of YNL and GStp are assigned as single groups
As PSMC analysis has limited resolution in the recent past, we implemented SMC++ (Terhorst et al., 2017) to infer recent N e history of the 6 groups of T. truncatus and T. pueraricola. The default settings in SMC++ did not suit our species well, producing oscillation in N e. By experimenting with various parameters, we obtained a clearer recent historical pattern of N e which partly mirrored fluctuations in northern hemispheric temperature (Figure 3b; Marcott, Shakun, Clark, & Mix, 2013). This pattern suggests that each group experienced bottleneck events during 4–2 Kya and then increased to at least 106 , followed by a sharp decrease over hundreds of years.
Based on the population structure analysis (Figure 1), we assigned YNL and GStp populations as single groups to infer demographic history. Among a total of 35 alternative models of demographic diffusion detected by fastsimcoal2 (Excoffier et al., 2013), we found that model 15 for T. truncatus and model 14 for T. pueraricola achieved the maximum log‐likelihood values and these were selected as the optimal models (Table S8). According to these models, the migratory directions of T. truncatus and T. pueraricola, from south to north and southwest to northeast, were consistent with the first PCs (Figure 1c,d). The T. pueraricola populations from eastern and northern China were predicted to have diverged from the southwestern lineage, estimated as ~7,031 years ago, soon after the initial divergence among the original southwestern populations, estimated as ~7,091 years ago. There was a comparably higher effective population size (N e = 506K) in colonized regions. Meanwhile, a hybrid population, GStp, in midland China was derived from admixture of the ancestors of YNL (43%) and Group VI (57%) in a short time (Figure 3c). The T. truncatus populations from midland and northern (Group III) China diverged ~4,392 years ago from the lineage ancestral to contemporary southern populations, with an effective population size of 545K. Then, the southern lineage admixed with northern populations which diverged an estimated ~4,152 years ago from Group III eastern populations (Group II) and emerged as the eastern lineage Group II.
3.5. Genomic signatures of local adaptation
To investigate genetic basis of local adaptation, we identified genomic variants associated with temperature, precipitation, and elevation using latent factor mixed models which accounted for background structure (Frichot et al., 2013). After several runs, we chose K = 16 and K = 7, respectively, for T. truncatus and T. pueraricola for which the estimates of genomic inflation factors (λ) were close to 1.0 (Figure S8). However, the unsmooth histograms of p‐values (Figure S8) indicated that confounding effects in T. pueraricola could not be well controlled. The p‐value histograms of all variables for T. truncatus appear flat except for a peak close to 1 which may reflect genotypes at these sites differing in only one or two populations. In T. truncatus, we found 329, 705, and 1,493 SNPs associated with temperature, precipitation, and elevation, respectively (expected FDR = 0.5%; Figure 4, Figure S9). In T. pueraricola, the equivalent figures were 2, 208 and 1,241 SNPs, respectively. Most of the SNPs associated with local adaptation located in the upstream and downstream regions (5kb) followed by the exon region (Table S11). The RDA analysis also identified more outlier loci involved in local adaptation for T. truncatus, including 72% of the candidate loci detected by LFMM, than for T. pueraricola (4,090 vs. 0 SNPs; Figure S10). As LFMM is more powerful for detecting outliers under weak selection (Forester et al., 2016), we paid more attention to the candidate loci detected by LFMM. The functional variants caused amino acid substitutions in 51, 76, and 135 genes in T. truncatus associated with temperature, precipitation, and elevation, respectively, and 0, 17, and 95 genes in T. pueraricola associated with these factors, respectively (Tables S12–S14). Amino acid substitutions affect the activity, folding, or assembly of the protein and may have measurable effects on fitness. These type of changes may contribute to local adaptation in this study.
FIGURE 4.
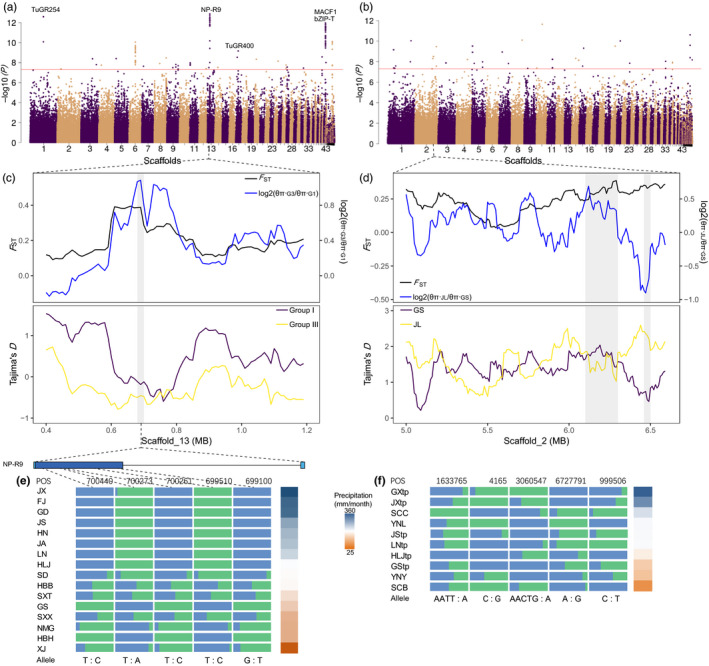
Signatures of local adaptation in the genome of spider mites. Manhattan plots of adjusted p‐values using the LFMM for association between SNPs and precipitation in T. truncatus (a) and T. pueraricola (b). The genome‐wide significance threshold (‐log10(5e‐8)) is indicated by the red horizontal line. Those genes with non‐synonymous substitution caused by significantly associated SNPs (adjusted p‐value > 5e‐8) were marked in the Manhattan plots. (c) FST, log2(θ π ratio), and Tajima's D values around the strongest associated gene NP‐R9 of T. truncatus. G1 and G3 are the abbreviations for Group I and Group III. (d) FST, log2(θ π ratio), and Tajima's D values around significantly associated genomic regions in scaffold_2 of T. pueraricola. JL represents JStp and LNtp populations. GS represents GXtp and SCB populations. (e) Allele frequencies of five non‐synonymous mutations within the NP‐R9 gene across the populations of T. truncatus. Based on the precipitation of the wettest month, the relatively moist regions include FJ, GD, JX, and HN and the relative arid regions include XJ, NMG, GS, and HBH. (f) Allele frequencies of the five most associated SNPs with the genomic positions of scaffold_10:1633765, scaffold_230:4165, scaffold_8:3060547, scaffold_1:6727791, and scaffold_21:999506 across the populations of T. pueraricola
The LFMM analysis indicated a number of SNPs that were significantly associated with precipitation (adjusted p‐value ≤ 5e‐8) in T. truncatus, higher than for T. pueraricola (Figure 4a,b). There were also several contiguous regions with adaptive loci detected in T. truncatus, but not in T. pueraricola. The PCA based on candidate SNPs associated with precipitation showed that T. truncatus was clearly clustered to moist (Group I, precipitation of wettest month > 220 mm/month) and arid (Group III, precipitation of wettest month < 130 mm/month) regions, while clusters for T. pueraricola were not related to precipitation (Figure S11 and Table S10). The first PC of T. truncatus accounted for almost fivefold as much of the variance as the second PC (33.77% versus. 7.02%) and that of T. pueraricola was 1.7 × higher (25.13% vs. 15.03%), which suggested that precipitation may play a more important role in local adaptation by T. truncatus.
We estimated population genetics parameters of the extreme populations (Group I and Group III of T. truncatus; JStp, LNtp, and GXtp, SCB populations of T. pueraricola) based on the PCA of these two spider mites (Figure S11 and S12). The strongest association (adjusted p‐value = 1.3e‐13; Figure 4a) between genotype and precipitation in T. truncatus was adjacent to the NP‐R9 gene (tetur13g01650). The target gene NP‐R9 exhibited markedly higher F STand log2(θ π ratio) but lower Tajima's D values (Figure 4c) than adjacent genomic regions, suggesting a selective sweep. Within NP‐R9, allele frequencies of several non‐synonymous substitutions varied with precipitation (Figure 4e), suggesting an important role for this gene in local adaptation. However, the associated loci of T. pueraricola detected by LFMM exhibited little evidence for population genetic signatures of natural selection. For example, the F ST, log2(θ π ratio), and Tajima's D values of associated loci were not always significantly different from adjacent regions (Figure 4d). The allele frequencies of the top 5 hit SNPs showed unclear patterns of distribution among the populations of T. pueraricola (Figure 4f).
For elevation, large numbers of SNPs showed associations in both spider mites (Figure S9). The reconstructed PCA of elevation‐associated SNPs clustered with the elevation gradient alongside PC1 for both species (Figure S12, Table S10), with PC1 for T. pueraricola accounting for a similar amount of variation as for T. truncatus (34.83% vs. 27.73%). The genome areas associated mostly with significant loci were upstream of the neuropeptide receptor NP‐R9 (Figure S9c) which could play a central role in evolutionary adaptation of T. truncatus. For T. pueraricola, the most significantly associated SNPs resulting in amino acid replacements were inconsistent with T. truncatus and had potential functional effects on gustatory receptors, X‐prolyl aminopeptidase, immunoglobulin, and laminin.
4. DISCUSSION
Our genomic study provides information on the evolutionary histories of the two dominant mites in China including the possible origin of the pests, their range shifts, demographic histories, and potential genes involved in local adaptation. Although having lower genetic diversity and spreading to colonize new areas ~3,000 years later than T. pueraricola, T. truncatus has become more prevalent and more abundant after expanding its range to arid northern China. T. pueraricola remains common in areas close to its origin rather than in expanded ranges.
A species difference in the degree of local adaptation may have shaped the extent of range shifts in these spider mites and changes in species abundance. Our comparative genome‐environment association analyses uncovered significantly different F ST, log2(θ π ratio), and Tajima's D values of candidate loci in T. truncatus along with more genes strongly associated with precipitation, while adaptation to precipitation in T. pueraricola appeared weaker. These results suggest that adaptation to climate might also influence the relative ranges of these pests in the future. Understanding how pests respond to climate stresses can aid in predicting future invasion and outbreaks. For instance, an invasive distribution range may be under‐predicted when temperature adaptation occurs, as in the case of Halotydeus and T. evansi mites (Hill et al., 2013; Migeon et al., 2009). Local adaptation may result in spider mites being pests in areas where they have previously not been observed, resulting in the need for additional control measures in those areas.
4.1. Linking demographic and evolutionary histories to range shifts of spider mites
The favored models of population expansion (Figure 3c) suggested that both T. truncatus and T. pueraricola in China originated from southern and southwestern China. Southwestern China and northern Vietnam have provided long‐term stable refugia for many relict plants (Tang et al., 2018), and have been proposed as possible origin regions for some species such as pears and tigers (Liu et al., 2018; Wu et al., 2018). Despite its absence in southwest China, T. truncatus may also originate from refugia near southwestern China because of identical fluctuation of ancestral effective population size during LGM (Figure 3a). The long‐term climatically stable refugia probably provided host plants and suitable environments for protecting ancient populations of Asian spider mites. The species richness of spider mites in southern China is also higher than that in northern China (Jin et al., 2018), which likely reflects the fact that the southwestern area provided refugia for many invertebrates. Although there are parallel patterns of expansion between the mite species, the early expansion of T. urticae is intriguing and may indicate a higher tolerance of conditions existing during glaciation periods or movement of this species from other areas that aided population expansion.
We had expected the value of best K in the ADMIXTURE analysis to be 2 (Figure S3), reflecting two distinct species of Tetranychus. The resulting best K as 15 for 27 populations of T. truncatus and T. pueraricola reflects the substantial genetic differentiation between populations within species, while with 2 subgroups assumed there are departures from Hardy–Weinberg equilibrium. The favored models (Figure 3c) suggested migration directions of south to north and southwest to northeast in China for T. truncatus and T. pueraricola, respectively. The importance of these directions in the demographic history of these spider mites was supported by the PC1 axis of PCA (Figure 1b,c). Some population expansion patterns supported by the favored models were also supported by Treemix, for example, migration event from ancestors of Group VI to GStp (Figure 2c). However, the paraphyletic topology of the mite species suggested by the ML tree (Figure 1a) demonstrated unclear evolutionary relationships between the groups, probably arising from long‐term isolation. Intriguingly, we note that T. truncatus in Japan probably originated from locations near Jiangsu in China, from which small brown planthoppers in Japan are also thought to have originated (Otuka et al., 2010). The association between Jiangsu and Japan could be further tested by characterizing additional Japanese populations. In addition, we note a close genetic relationship between geographically distant populations of T. truncatus (SC and HLJ; Figure 1a and Table S7). These close affinities likely represent the result of human‐mediated movement such as through trade.
After experiencing a dramatic decrease, N e of T. truncatus in northern and midland China (Group II, Group III, and Group IV) began to increase almost 30 years ago (Figure 3b), which is consistent with historical reports. Since the mid‐1980s, with the expansion of wheat, corn, and soybean plantings, combined with warm and drought conditions, T. truncatus has increased from being a minor pest to a major pest (Hong, 2012). Populations of T. truncatus from Group I had the lowest level of genetic diversity (Figure S6a) and the greatest estimated fluctuations in N e based on a coalescence analysis of LD patterns (Figure 3b). Note that this Group also showed the highest positive Tajima's D relative to other groups (Figure S13) which indicates population contraction.
The average genome‐wide θπ (5.54 × 10−4) of spider mites was lower than estimates for most animals and plants that have been characterized, such as the giant panda (1.24 × 10−3; Zhao et al., 2013), Anopheles mosquito (1.5 × 10−2; Consortium, 2017), and cultivated rice (5.4 × 10−3; Xu et al., 2011). The relatively lower genetic diversity of T. truncatus may represent recent differences in population processes in this species compared to T. pueraricola. Range‐limit theory proposes that abiotic factors form high‐latitude/altitude limits, whereas biotic interactions create lower limits (Siren & Morelli, 2020). Perhaps the high standing genetic diversity of T. pueraricola relates to biotic factors such as competition, predation, and pathogens producing heterogeneous environments which has been hypothesized for a long time as facilitating the maintenance of genetic variation (McDonald & Ayala, 1974), and contribute to the widespread distribution of this species in low latitude areas. A relatively low genetic diversity does not appear to have impeded the spread of T. truncatus to high‐latitude areas where conditions are more homogeneous and directional selection associated with abiotic factors (e.g., cold climate) is expected to be intense. Given that high genomic diversity can facilitate adaptive shifts, T. pueraricola in Gansu with high diversity (Figure S6a) may adapt to future conditions and lead to a range expansion of this acarid pest, particularly if rare alleles are involved in the adaptive shifts.
According to field surveys (Guo et al., 2013; Jin et al., 2018; Wang et al., 2013), T. truncatus was the most abundant and widespread acarid pest in areas of Group II, Group III, and Group IV; T. pueraricola was the most prevalent in areas of Group V. The different patterns of range shifts of these two spider mites may be linked to several factors. First, monoculture farming in northern China increased ecological homogeneity, leading to the possibility of T. truncatus to spread. Low levels of biodiversity caused by agricultural intensification generally benefit agricultural pests by reducing natural enemies and supplying abundant resources (Benton, Vickery, & Wilson, 2003; Hong, 2012; Sotherton & Self, 2000). A simplified landscape may allow for the rapid growth of pest populations, which can facilitate rapid adaptation (Reznick & Ghalambor, 2001). Second, T. truncatus in its prevalent regions (Group II, Group III, and Group IV) had higher gene flow (Figure 2c), which could have contributed to a higher level of genetic variability (Table S5) and more rapid range shifts of those regions. Third, results here suggested that T. truncatus were locally adapted to climatic conditions which could benefit range shifts. Although many factors interact with climate change to alter range shifts, the potential role of local adaptation has been highlighted theoretically in driving nonintuitive patterns of range shifts (Louthan, Doak, & Angert, 2015; Suttle, Thomsen, & Power, 2007). Strong local adaptation may lead an expanded range of climate tolerances for a species as a whole. In southern China which likely represents the region where T. truncatus originated, a relatively low N e (Figure 3c) may be the result of weak adaptation to the hot‐humid environment and limited gene flow or others factors such as competition with other spider mites. In contrast, local adaptation together with high gene flow of T. truncatus in northern China may contribute to relatively high performance under arid conditions. The genetic parameters of genome regions around candidate loci also suggest adaptation to dry conditions in T. truncatus but less clear cut signals of adaptation for T. pueraricola. However, these remain conjectures and need to be tested. In particular, quantitative genetic experiments are required to assess the heritability of traits linked to tolerating different climate conditions. And common garden experiments could indicate the extent of local adaptation across the species to see whether these match expectations based on the genomic data.
4.2. Genes involved in local adaptation
Many SNPs were significantly associated with climate and elevation in T. truncatus and T. pueraricola, and we focused on the genes with amino acid substitutions. There were less SNPs genome‐wide significantly associated (adjusted p‐value ≤ 5e‐8) with temperature than with precipitation and elevation in both mites (Figure S9). Along with the recent historical N e which partly reflected past temperature, these may indicate spider mites are susceptible to temperature selection. There has been much research on temperature effects on development, fecundity, longevity, and pesticide impacts in spider mites (Auger, Guichou, & Kreiter, 2003; Margolies & Wrensch, 1996; Riahi, Shishehbor, Nemati, & Saeidi, 2013). Under intensifying global warming, spider mites will increasingly threaten agricultural production (Migeon et al., 2009). Our results provide a framework for genome‐wide association studies to locate key genes that may be responsible for further adaptation to temperature changes in spider mites.
Many genes were found to be significantly associated with precipitation in T. truncatus. Three genes with amino acid replacements, NP‐R9, TuGR254, and TuGR400 (Figure 4a), belong to members of the G‐protein‐coupled receptor (GPCR) family. When interacting with insect neurohormones, these genes could play a role in the control of development, behavior, feeding, reproduction, and many other physiological processes (Hauser et al., 2008; Martin, Boto, Gomez‐Diaz, & Alcorta, 2013). A vasopressin‐like GPCR found in Tribolium might help this xerophilous insect to effectively control water reabsorption (Hauser et al., 2008). Variation in these GPCRs might contribute to the regulation of homeostasis of water and ions (Gäde, 2004), and thereby be involved in adaptation to the dry environment in northern China. Other significantly associated genes including tetur43g00160 and tetur43g00520 were annotated to bZIP‐T and MACF1 (Figure 4a). The bZIP‐T gene encodes a basic leucine zipper‐type protein which participate in an abscisic acid‐dependent signal transduction pathway when Arabidopsis face drought and high‐salinity environments (Uno et al., 2000). The reduction of MACF1 caused by heterozygous duplication resulted in periodic hypotonia, lax muscles and diminished motor skills in human (Jørgensen et al., 2014). The NP‐R9 and its upstream are involved in adapting to precipitation and elevation, and may serve as target regions for further functional experiment. However, these genes need to be further characterized through functional studies.
Although both T. truncatus and T. pueraricola showed many genes associated with elevation, many of them are not well annotated (Table S14). A dense region of significantly associated SNPs on scaffold_8 (Figure S9c) included amino acid replacement in 4 genes which encoded a hypothetical protein, Agrin, and Myc‐type transcription factors. A cytochrome P450 gene, CYP389C5, had a non‐synonymous substitution, C100840T, which only appeared in high‐elevation populations (GS, SXX, NMG, and HBH). Cytochrome P450 enzymes have been related to high‐elevation adaptation in Tibetan human populations (Simonson et al., 2010). Other strongly associated genes that had amino acid replacements, such as TuABCH‐21 (ABC‐transporter) and HR96‐like h (HR96‐like nuclear receptor h), have not been previously linked to high‐elevation conditions and need further study. The results here suggest that the related mite species have different strategies when adapting to high elevations.
The higher number of SNPs associated with local adaptation and the lower θπ (Table S5) in T. truncatus vs. T. pueraricola populations from the same location suggest that T. truncatus may have undergone stronger purifying selection despite having lower levels of genomic variation. Experimental studies suggest that rates of adaptation are linked to genome‐wide genetic diversity (Ørsted, Hoffmann, Sverrisdóttir, Nielsen, & Kristensen, 2019) but T. truncatus appears to have adapted despite lower diversity. It will be interesting to carry out quantitative assessments of genetically based fitness differences across populations to assess the extent to which populations from the two species are locally adapted and whether the molecular data might link to trait differentiation within and across populations (e.g., Drosophila melanogaster and Carabus japonicus; Weeks, McKechnie, & Hoffmann, 2002; Komurai, Fujisawa, Okuzaki, & Sota, 2017). The candidate genes we mentioned here are likely to provide insights into the mechanisms involved in local adaptation in spider mites, but many other significantly associated genes (Tables S12–S14) were not annotated clearly and need future exploration.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
X.Y.H., L.C., and J.T.S. designed the research. L.C., P.Y.J., and D.S.Z. collected materials. L.C. performed all experiments and analyses. J.T.S., P.Y.J., X.L.B., X.F.X., and A.H. took part in designing a few analyses and explaining the results. L.C., X.Y.H, and A.H. wrote the manuscript.
Supporting information
Fig S1‐S13
Table S1‐S14
ACKNOWLEDGEMENTS
We sincerely thank Yu‐Xi Zhu, Xu Zhang, Kun Yang, and Xue Xia for their aid in sampling of spider mites. We are also grateful to Professor Tetsuo Gotoh of Ibaraki University, Japan, and Nicolas Bensoussan of Grbic's lab, Western University, Canada for samples. The analyses in this study were also supported by the high‐performance computing platform of Bioinformatics Center, Nanjing Agricultural University. This study was supported in part by a grant‐in‐aid for Scientific Research (31672035, 32020103011, 31871976) from the National Natural Science Foundation of China.
[Correction added on 30 September 2020, after first online publication: an additional grant number has been added to the Acknowledgements section.]
Chen L, Sun J‐T, Jin P‐Y, et al. Population genomic data in spider mites point to a role for local adaptation in shaping range shifts. Evol Appl. 2020;13:2821–2835. 10.1111/eva.13086
DATA AVAILABILITY STATEMENT
All sequencing data in this study have been deposited in the NCBI database under BioProject accession PRJNA578957 (http://www.ncbi.nlm.nih.gov/bioproject/578957). Scripts used in this article are available on Github (https://github.com/Chenleinice/Codes‐for‐population‐genomic‐of‐spider‐mites).
REFERENCES
- Alexander, D. H. , Novembre, J. , & Lange, K. (2009). Fast model‐based estimation of ancestry in unrelated individuals. Genome Research, 19(9), 1655–1664. 10.1101/gr.094052.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson, A. , Siegel, V. , Pakhomov, E. , & Rothery, P. (2004). Long‐term decline in krill stock and increase in salps within the Southern Ocean. Nature, 432(7013), 100–103. 10.1038/nature02996 [DOI] [PubMed] [Google Scholar]
- Auger, P. , Guichou, S. , & Kreiter, S. (2003). Variations in acaricidal effect of wettable sulfur on Tetranychus urticae (Acari: Tetranychidae): Effect of temperature, humidity and life stage. Pest Management Science, 59(5), 559–565. 10.1002/ps.665 [DOI] [PubMed] [Google Scholar]
- Benton, T. G. , Vickery, J. A. , & Wilson, J. D. (2003). Farmland biodiversity: Is habitat heterogeneity the key? Trends in Ecology & Evolution, 18(4), 182–188. 10.1016/S0169-5347(03)00011-9 [DOI] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning, B. L. , Zhou, Y. , & Browning, S. R. (2018). A one‐penny imputed genome from next‐generation reference panels. The American Journal of Human Genetics, 103(3), 338–348. 10.1016/j.ajhg.2018.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush, A. , Mokany, K. , Catullo, R. , Hoffmann, A. , Kellermann, V. , Sgrò, C. , … Ferrier, S. (2016). Incorporating evolutionary adaptation in species distribution modelling reduces projected vulnerability to climate change. Ecology Letters, 19(12), 1468–1478. 10.1111/ele.12696 [DOI] [PubMed] [Google Scholar]
- Capblancq, T. , Luu, K. , Blum, M. G. B. , & Bazin, E. (2018). Evaluation of redundancy analysis to identify signatures of local adaptation. Molecular Ecology Resources, 18(6), 1223–1233. 10.1111/1755-0998.12906 [DOI] [PubMed] [Google Scholar]
- Ceballos, F. C. , Joshi, P. K. , Clark, D. W. , Ramsay, M. , & Wilson, J. F. (2018). Runs of homozygosity: Windows into population history and trait architecture. Nature Reviews Genetics, 19(4), 220 10.1038/nrg.2017.109 [DOI] [PubMed] [Google Scholar]
- Chang, C. C. , Chow, C. C. , Tellier, L. C. , Vattikuti, S. , Purcell, S. M. , & Lee, J. J. (2015). Second‐generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience, 4, 7 10.1186/s13742-015-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, A. gambiae 1000 G . (2017). Genetic diversity of the African malaria vector Anopheles gambiae . Nature, 552(7683), 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , … Durbin, R. (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson, J. J. , & Gesch, D. B. (2011). Global multi‐resolution terrain elevation data 2010 (GMTED2010). U.S. Geological Survey Open‐File Report 2011–1073. Earth Resources Observation and Science (EROS) Center, U.S. Geological Survey, Sioux Falls, South Dakota, USA. 10.3133/ofr20111073 [DOI]
- Excoffier, L. , Dupanloup, I. , Huerta‐Sánchez, E. , Sousa, V. C. , & Foll, M. (2013). Robust demographic inference from genomic and SNP data. PLoS Genetics, 9(10), e1003905 10.1371/journal.pgen.1003905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasulo, T. R. , & Denmark, H. A. (2000). Twospotted Spider Mite, Tetranychus urticae Koch (Arachnida: Acari: Tetranychidae). Retrieved from http://edis.ifas.ufl.edu/in307 [Google Scholar]
- Forester, B. R. , Jones, M. R. , Joost, S. , Landguth, E. L. , & Lasky, J. R. (2016). Detecting spatial genetic signatures of local adaptation in heterogeneous landscapes. Molecular Biology and Evolution, 25(1), 104–120. 10.1111/mec.13476 [DOI] [PubMed] [Google Scholar]
- Frichot, E. , Schoville, S. D. , Bouchard, G. , & François, O. (2013). Testing for associations between loci and environmental gradients using latent factor mixed models. Molecular Biology and Evolution, 30(7), 1687–1699. 10.1093/molbev/mst063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gäde, G. (2004). Regulation of intermediary metabolism and water balance of insects by neuropeptides. Annual Review of Entomology, 49(1), 93–113. [DOI] [PubMed] [Google Scholar]
- Ge, C. , Ding, X.‐L. , Zhang, J.‐P. , & Hong, X.‐Y. (2013). Tetranychus urticae (green form) on Gossypium hirsutum in China: Two records confirmed by aedeagus morphology and RFLP analysis. Systematic and Applied Acarology, 18(3), 239–245. [Google Scholar]
- Grbić, M. , Van Leeuwen, T. , Clark, R. M. , Rombauts, S. , Rouzé, P. , Grbić, V. , … Van de Peer, Y. (2011). The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature, 479(7374), 487–492. 10.1038/nature10640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y. L. , Jiao, X. D. , Yang, S. , Li, G. Y. , Xia, W. , & Zhang, J. P. (2013). Effect of host plants on the population dynamics and host selection of Tetranychus turkestani and Tetranychus truncatus . Journal of Environmental Entomology, 35, 140–147. [Google Scholar]
- Hauser, F. , Cazzamali, G. , Williamson, M. , Park, Y. , Li, B. , Tanaka, Y. , … Grimmelikhuijzen, C. J. P. (2008). A genome‐wide inventory of neurohormone GPCRs in the red flour beetle Tribolium castaneum . Frontiers in Neuroendocrinology, 29(1), 142–165. 10.1016/j.yfrne.2007.10.003 [DOI] [PubMed] [Google Scholar]
- Hill, M. P. , Chown, S. L. , & Hoffmann, A. A. (2013). A predicted niche shift corresponds with increased thermal resistance in an invasive mite, Halotydeus destructor . Global Ecology and Biogeography, 22(8), 942–951. [Google Scholar]
- Hoffmann, A. A. (2017). Rapid adaptation of invertebrate pests to climatic stress? Current Opinion in Insect Science, 21, 7–13. 10.1016/j.cois.2017.04.009 [DOI] [PubMed] [Google Scholar]
- Hong, X.‐Y. (2012). Acarid pests of grain crops In Agricultural acarology (pp. 186–189). Beijing: China Agriculture Press. [Google Scholar]
- Hou, Y.‐Y. , Xu, L.‐Z. , Wu, Y. , Wang, P. , Shi, J.‐J. , & Zhai, B.‐P. (2016). Geographic variation of diapause and sensitive stages of photoperiodic response in Laodelphax striatellus Fallén (Hemiptera: Delphacidae). Journal of Insect Science, 16(1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiguet, F. , Devictor, V. , Ottvall, R. , Van Turnhout, C. , der Jeugd, H. , & Lindstrom, A. (2010). Bird population trends are linearly affected by climate change along species thermal ranges. Proceedings of the Royal Society B‐Biological Sciences, 277(1700), 3601–3608. 10.1098/rspb.2010.0796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, P.‐Y. , Tian, L. , Chen, L. , & Hong, X.‐Y. (2018). Spider mites of agricultural importance in China, with focus on species composition during the last decade (2008–2017). Systematic and Applied Acarology, 23(11), 2087–2099. 10.11158/saa.23.11.1 [DOI] [Google Scholar]
- Jørgensen, L. H. , Mosbech, M.‐B. , Færgeman, N. J. , Graakjaer, J. , Jacobsen, S. V. , & Schrøder, H. D. (2014). Duplication in the microtubule‐actin cross‐linking factor 1 gene causes a novel neuromuscular condition. Scientific Reports, 4, 5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karger, D. N. , Conrad, O. , Böhner, J. , Kawohl, T. , Kreft, H. , Soria‐Auza, R. W. , … Kessler, M. (2017). Climatologies at high resolution for the earth’s land surface areas. Scientific Data, 4, 170122 10.1038/sdata.2017.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D. , Ness, R. W. , Halligan, D. L. , & Haddrill, P. R. (2014). Estimation of the spontaneous mutation rate per nucleotide site in a Drosophila melanogaster full‐sib family. Genetics, 196(1), 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk, H. , Dorn, S. , & Mazzi, D. (2013). Molecular genetics and genomics generate new insights into invertebrate pest invasions. Evolutionary Applications, 6(5), 842–856. 10.1111/eva.12071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komurai, R. , Fujisawa, T. , Okuzaki, Y. , & Sota, T. (2017). Genomic regions and genes related to inter‐population differences in body size in the ground beetle Carabus japonicus . Scientific Reports, 7(1), 7773 10.1038/s41598-017-08362-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopelman, N. M. , Mayzel, J. , Jakobsson, M. , Rosenberg, N. A. , & Mayrose, I. (2015). Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Molecular Ecology Resources, 15(5), 1179–1191. 10.1111/1755-0998.12387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneliussen, T. S. , Albrechtsen, A. , & Nielsen, R. (2014). ANGSD: Analysis of next generation sequencing data. BMC Bioinformatics, 15(1), 356 10.1186/s12859-014-0356-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasken, R. S. (2009). Genomic DNA amplification by the multiple displacement amplification (MDA) method. Biochemical Society Transactions, 37(2), 450–453. 10.1042/BST0370450 [DOI] [PubMed] [Google Scholar]
- Lee, T.‐H. , Guo, H. , Wang, X. , Kim, C. , & Paterson, A. H. (2014). SNPhylo: A pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics, 15(1), 162 10.1186/1471-2164-15-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenoir, J. , Gégout, J.‐C. , Guisan, A. , Vittoz, P. , Wohlgemuth, T. , Zimmermann, N. E. , … Svenning, J.‐C. (2010). Going against the flow: Potential mechanisms for unexpected downslope range shifts in a warming climate. Ecography, 33(2), 295–303. 10.1111/j.1600-0587.2010.06279.x [DOI] [Google Scholar]
- Lenoir, J. , & Svenning, J.‐C. (2015). Climate‐related range shifts ‐ a global multidimensional synthesis and new research directions. Ecography, 38(1), 15–28. 10.1111/ecog.00967 [DOI] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2011). Inference of human population history from individual whole‐genome sequences. Nature, 475(7357), 493 10.1038/nature10231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , … Durbin, R. (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y.‐C. , Sun, X. , Driscoll, C. , Miquelle, D. G. , Xu, X. , Martelli, P. , … Luo, S.‐J. (2018). Genome‐Wide Evolutionary Analysis of Natural History and Adaptation in the World’s Tigers Report Genome‐Wide Evolutionary Analysis of Natural History and Adaptation in the World’s Tigers. Current Biology, 28(23), 1–10. 10.1016/j.cub.2018.09.019 [DOI] [PubMed] [Google Scholar]
- Louthan, A. M. , Doak, D. F. , & Angert, A. L. (2015). Where and when do species interactions set range limits? Trends in Ecology & Evolution, 30(12), 780–792. 10.1016/j.tree.2015.09.011 [DOI] [PubMed] [Google Scholar]
- Maggini, R. , Lehmann, A. , Kery, M. , Schmid, H. , Beniston, M. , Jenni, L. , & Zbinden, N. (2011). Are Swiss birds tracking climate change? Detecting elevational shifts using response curve shapes. Ecological Modelling, 222(1), 21–32. 10.1016/j.ecolmodel.2010.09.010 [DOI] [Google Scholar]
- Marcott, S. A. , Shakun, J. D. , Clark, P. U. , & Mix, A. C. (2013). A reconstruction of regional and global temperature for the past 11,300 years. Science, 339(6124), 1198–1201. [DOI] [PubMed] [Google Scholar]
- Margolies, D. C. , & Wrensch, D. L. (1996). Temperature‐induced changes in spider mite fitness: Offsetting effects of development time, fecundity, and sex ratio. Entomologia Experimentalis Et Applicata, 78(1), 111–118. 10.1111/j.1570-7458.1996.tb00770.x [DOI] [Google Scholar]
- Martin, F. , Boto, T. , Gomez‐Diaz, C. , & Alcorta, E. (2013). Elements of olfactory reception in adult Drosophila melanogaster . The Anatomical Record, 296(9), 1477–1488. [DOI] [PubMed] [Google Scholar]
- McDonald, J. F. , & Ayala, F. J. (1974). Genetic response to environmental heterogeneity. Nature, 250(467), 572–574. 10.1038/250572a0 [DOI] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , … DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migeon, A. , Ferragut, F. , Escudero‐Colomar, L. A. , Fiaboe, K. , Knapp, M. , de Moraes, G. J. , … Navajas, M. (2009). Modelling the potential distribution of the invasive tomato red spider mite, Tetranychus evansi (Acari: Tetranychidae). Experimental and Applied Acarology, 48(3), 199–212. 10.1007/s10493-008-9229-8 [DOI] [PubMed] [Google Scholar]
- Moller, A. P. , Rubolini, D. , & Lehikoinen, E. (2008). Populations of migratory bird species that did not show a phenological response to climate change are declining. Proceedings of the National Academy of Sciences of the United States of America, 105(42), 16195–16200. 10.1073/pnas.0803825105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan, V. , Danecek, P. , Scally, A. , Xue, Y. , Tyler‐Smith, C. , & Durbin, R. (2016). BCFtools/RoH: A hidden Markov model approach for detecting autozygosity from next‐generation sequencing data. Bioinformatics, 32(11), 1749–1751. 10.1093/bioinformatics/btw044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ørsted, M. , Hoffmann, A. A. , Sverrisdóttir, E. , Nielsen, K. L. , & Kristensen, T. N. (2019). Genomic variation predicts adaptive evolutionary responses better than population bottleneck history. PLoS Genetics, 15(6), e1008205 10.1371/journal.pgen.1008205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otuka, A. , Matsumura, M. , Sanada‐Morimura, S. , Takeuchi, H. , Watanabe, T. , Ohtsu, R. , & Inoue, H. (2010). The 2008 overseas mass migration of the small brown planthopper, Laodelphax striatellus, and subsequent outbreak of rice stripe disease in western Japan. Applied Entomology and Zoology, 45(2), 259–266. 10.1303/aez.2010.259 [DOI] [Google Scholar]
- Parmesan, C. (2006). Ecological and evolutionary responses to recent climate change. Annual Review of Ecology Evolution and Systematics, 37, 637–669. 10.1146/annurev.ecolsys.37.091305.110100 [DOI] [Google Scholar]
- Peterson, M. L. , Doak, D. F. , & Morris, W. F. (2019). Incorporating local adaptation into forecasts of species’ distribution and abundance under climate change. Global Change Biology, 25(3), 775–793. 10.1111/gcb.14562 [DOI] [PubMed] [Google Scholar]
- Pickrell, J. K. , & Pritchard, J. K. (2012). Inference of population splits and mixtures from genome‐wide allele frequency data. PLoS Genetics, 8(11), e1002967 10.1371/journal.pgen.1002967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane, R. V. , Ghodke, A. B. , Hoffmann, A. A. , Edwards, O. R. , Walsh, T. K. , & Oakeshott, J. G. (2019). Detoxifying enzyme complements and host use phenotypes in 160 insect species. Current Opinion in Insect Science, 31, 1–8. 10.1016/j.cois.2018.12.008 [DOI] [PubMed] [Google Scholar]
- Reich, D. , Price, A. L. , & Patterson, N. (2008). Principal component analysis of genetic data. Nature Genetics, 40(5), 491 10.1038/ng0508-491 [DOI] [PubMed] [Google Scholar]
- Rellstab, C. , Gugerli, F. , Eckert, A. J. , Hancock, A. M. , & Holderegger, R. (2015). A practical guide to environmental association analysis in landscape genomics. Molecular Ecology, 24(17), 4348–4370. 10.1111/mec.13322 [DOI] [PubMed] [Google Scholar]
- Reznick, D. N. , & Ghalambor, C. K. (2001). The population ecology of contemporary adaptations: what empirical studies reveal about the conditions that promote adaptive evolution In Hendry A. P. & Kinnison M. T. (Eds.), Microevolution rate, pattern, process (pp. 183–198). Dordrecht, Netherlands: Springer. [PubMed] [Google Scholar]
- Riahi, E. , Shishehbor, P. , Nemati, A. R. , & Saeidi, Z. (2013). Temperature effects on development and life table parameters of Tetranychus urticae (Acari: Tetranychidae). Journal of Agricultural Science and Technology, 15, 661–672. [Google Scholar]
- Scheffers, B. R. , De Meester, L. , Bridge, T. C. L. , Hoffmann, A. A. , Pandolfi, J. M. , Corlett, R. T. , … Watson, J. E. M. (2016). The broad footprint of climate change from genes to biomes to people. Science, 354(6313), aaf7671 10.1126/science.aaf7671 [DOI] [PubMed] [Google Scholar]
- Simonson, T. S. , Yang, Y. , Huff, C. D. , Yun, H. , Qin, G. , Witherspoon, D. J. , … Jorde, L. B. (2010). Genetic evidence for high‐altitude adaptation in Tibet. Science, 329(5987), 72–75. [DOI] [PubMed] [Google Scholar]
- Siren, A. P. K. , & Morelli, T. L. (2020). Interactive range‐limit theory (iRLT): An extension for predicting range shifts. Journal of Animal Ecology, 89(4), 940–954. 10.1111/1365-2656.13150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotherton, N. W. , & Self, M. J. (2000). Changes in plant and arthropod diversity on lowland farmland: An overview In Aebischer N. J., Evans A. D., & Grice P. A. (Eds.), Ecology and conservation of lowland farmland birds (pp. 26–35). University of Southampton, UK: British Ornithologists Union. [Google Scholar]
- Suttle, K. B. , Thomsen, M. A. , & Power, M. E. (2007). Species interactions reverse grassland responses to changing climate. Science, 315(5812), 640–642. 10.1126/science.1136401 [DOI] [PubMed] [Google Scholar]
- Tang, C. Q. , Matsui, T. , Ohashi, H. , Dong, Y.‐F. , Momohara, A. , Herrando‐Moraira, S. , … López‐Pujol, J. (2018). Identifying long‐term stable refugia for relict plant species in East Asia. Nature Communications, 9(1), 4488 10.1038/s41467-018-06837-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhorst, J. , Kamm, J. A. , & Song, Y. S. (2017). Robust and scalable inference of population history from hundreds of unphased whole genomes. Nature Genetics, 49(2), 303–309. 10.1038/ng.3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno, Y. , Furihata, T. , Abe, H. , Yoshida, R. , Shinozaki, K. , & Yamaguchi‐Shinozaki, K. (2000). Arabidopsis basic leucine zipper transcription factors involved in an abscisic acid‐dependent signal transduction pathway under drought and high‐salinity conditions. Proceedings of the National Academy of Sciences of the United States of America, 97(21), 11632–11637. 10.1073/pnas.190309197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDerWal, J. , Murphy, H. T. , Kutt, A. S. , Perkins, G. C. , Bateman, B. L. , Perry, J. J. , & Reside, A. E. (2013). Focus on poleward shifts in species’ distribution underestimates the fingerprint of climate change. Nature Climate Change, 3(3), 239–243. 10.1038/NCLIMATE1688 [DOI] [Google Scholar]
- Wang, S.‐L. , Zhang, Y.‐J. , Wu, Q.‐J. , Xie, W. , & Xu, B.‐Y. (2013). Dominant species identification of spider mites on vegetables in some areas in Beijing and Hebei. Journal of Environmental Entomology, 36, 481–486. [Google Scholar]
- Weeks, A. R. , McKechnie, S. W. , & Hoffmann, A. A. (2002). Dissecting adaptive clinal variation: Markers, inversions and size/stress associations in Drosophila melanogaster from a central field population. Ecology Letters, 5(6), 756–763. 10.1046/j.1461-0248.2002.00380.x [DOI] [Google Scholar]
- Wickham, H. (2016). ggplot2: Elegant Graphics for Data Analysis. Cham, Switzerland: Springer; Retrieved from http://ggplot2.org [Google Scholar]
- Wu, J. , Wang, Y. , Xu, J. , Korban, S. S. , Fei, Z. , Tao, S. , … Zhang, S. (2018). Diversification and independent domestication of Asian and European pears. Genome Biology, 19(1), 77 10.1186/s13059-018-1452-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. , Liu, X. , Ge, S. , Jensen, J. D. , Hu, F. , Li, X. , … Wang, W. (2011). Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nature Biotechnology, 30(1), 105 10.1038/nbt.2050 [DOI] [PubMed] [Google Scholar]
- Yadav, S. , Stow, A. J. , & Dudaniec, R. Y. (2019). Detection of environmental and morphological adaptation despite high landscape genetic connectivity in a pest grasshopper (Phaulacridium vittatum). Molecular Ecology. 10.1111/mec.15146 [DOI] [PubMed] [Google Scholar]
- Zhang, C. , Dong, S.‐S. , Xu, J.‐Y. , He, W.‐M. , & Yang, T.‐L. (2018). PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics, 35(10), 1786–1788. 10.1093/bioinformatics/bty875 [DOI] [PubMed] [Google Scholar]
- Zhao, S. , Zheng, P. , Dong, S. , Zhan, X. , Wu, Q. I. , Guo, X. , … Wei, F. (2013). Whole‐genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nature Genetics, 45(1), 67 10.1038/ng.2494 [DOI] [PubMed] [Google Scholar]
- Zheng, X. , Levine, D. , Shen, J. , Gogarten, S. M. , Laurie, C. , & Weir, B. S. (2012). A high‐performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics, 28(24), 3326–3328. 10.1093/bioinformatics/bts606 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S13
Table S1‐S14
Data Availability Statement
All sequencing data in this study have been deposited in the NCBI database under BioProject accession PRJNA578957 (http://www.ncbi.nlm.nih.gov/bioproject/578957). Scripts used in this article are available on Github (https://github.com/Chenleinice/Codes‐for‐population‐genomic‐of‐spider‐mites).