

Abstract
Background
Extracts of red clover (Trifolium pratense L.) containing estrogenic and pro-estrogenic isoflavones are used in dietary supplements primarily for the management of menopausal symptoms in women.
Objective
A UHPLC-MS/MS assay was developed and validated for the quantitative analysis of the six major red clover isoflavones in dietary supplements and in human serum in support of clinical trials.
Methods
Enzymatic deconjugation of isoflavone glucuronides and sulfate conjugates in human serum specimens was carried out followed by protein precipitation. Isoflavones in red clover dietary supplements were acid hydrolyzed to release aglycons from glycosides. UHPLC separations (< 4 min) were combined with MS/MS using collision-induced dissociation, selective reaction monitoring and deuterated internal standards to measure biochanin A, formononetin, daidzein, genistein, irilone, and prunetin.
Results
The method was validated with respect to selectivity, specificity, accuracy, linearity, precision, LOD, and LOQ. The calibration curves for all analytes were linear (R2 > 0.998). The mean recovery for low-, medium- and high-quality control standards ranged between 80% and 108%. The precision of the method was assessed using coefficients of variation, which were <15%.
Conclusions
The UHPLC-MS/MS method is fast, precise, sensitive, selective, accurate, and applicable to the quantitative analysis of red clover isoflavones in different matrices.
Highlights
This validated UHPLC-MS/MS assay is applicable to the rapid quantitative analysis of red clover isoflavones in human serum and in dietary supplements.
Preparations of red clover (Trifolium pratense L.) are used primarily by women as dietary supplements to manage menopausal issues such as vasomotor symptoms and osteoporosis (1, 2). The demand for isoflavone-enriched dietary supplements is increasing as women seek natural alternatives to hormone therapy at menopause (3). The estrogenic isoflavones of red clover are concentrated in the aerial parts (4). Although the most estrogenic isoflavones in red clover are genistein and daidzein (Figure 1), the more abundant red clover isoflavones biochanin A and formononetin can be O-demethylated by cytochrome P450 enzymes to generate genistein and daidzein, respectively (5). Therefore, biochanin A and formononetin may be considered pro-estrogens. In addition to the estrogenic isoflavones in red clover, the isoflavones irilone and prunetin (Figure 1) have been reported to have progesterone-like activities (6, 7).
Figure 1.

Chemical structures of red clover isoflavones and deuterated internal standards.
Analytical methods for the quantitative analysis of isoflavones in red clover dietary supplements and in human serum have included HPLC-UV, HPLC-MS, and HPLC-MS/MS. For example, Clark et al. (8) reported an electrospray HPLC-MS method for red clover isoflavones that used a 28 min separation, did not include the analytes irilone and prunetin, and did not include human serum as a matrix. Similar HPLC methods in the literature also require long separations of up to 90 min (9), do not include all six major red clover isoflavones, and/or do not include human serum as a matrix (9–12). A recent method based on quantitative NMR included 6 red clover isoflavones, but lacked the sensitivity for human serum analysis and was not validated (13). In this same report, alternative UHPLC-UV analysis provided rapid 10min separation but was unable to measure irilone and prunetin (13). An unvalidated GC-MS/MS approach has also been reported for the measurement of all six red clover isoflavones in human serum, but required extensive sample preparation and silylation of the isoflavone hydroxyl groups (14).
Due to the limitations of existing analytical methods, there is a need for a new, fast and, validated assay for the measurement of red clover isoflavones. In this study, a UHPLC-MS/MS assay was developed and optimized for the measurement of six red clover isoflavones (Figure 1) in human serum and dietary supplements. A simple sample preparation protocol minimized many variables of previous protocols, and the sensitivity of the new method enabled quantification of pg/mL levels of isoflavones.
METHOD
Materials and Reagents
Genistein, daidzein, biochanin A, and formononetin (purity ≥ 98%) and internal standards d4-genistein and d4-daidzein (≥ 99% purity) were purchased from Cayman Chemicals (Ann Arbor, MI). Prunetin (≥ 98%) and β-glucuronidase/arylsulfatase (7.1 units/mL β-glucuronidase and 24 units/mL arylsulfatase) were purchased from Sigma-Aldrich (St. Louis, MO). Irilone (≥ 99%) was provided by the UIC Botanical Center (Chicago, IL). HPLC-MS-grade acetonitrile, methanol, and formic acid were purchased from Thermo Fisher Scientific (Waltham, MA). Ultrapure water was prepared using a Milli-Q water purification system (Millipore, MA). All other reagents and solvents were reagent grade or better and were purchased from VWR (Visalia, CA). The new method was applied to the analysis of five commercial red clover dietary supplements purchased from on-line suppliers as well as to the analysis of de-identified human serum samples obtained from our previous Phase I clinical trial of red clover safety and pharmacokinetics (1).
Preparation of Internal Standard Solutions
d4-Genistein and d4-daidzein stock solutions (1 mg/mL) each were prepared by dissolving accurately weighed aliquots in dimethylsulfoxide. Working solutions were prepared by diluting each internal standard stock solution to a final concentration of 50 ng/mL in methanol–water (50:50; v/v).
Preparation of Standards and Quality Control Solutions
Stock solutions of red clover isoflavone standards in dimethylsulfoxide (1 mg/mL) were prepared and stored at −20°C until use. For the calibration curves, working solutions were prepared from the stock solutions of isoflavones in methanol–water (50:50; v/v) ranging from 0.001–13.12 µg/mL. The working solutions of the QC samples were also prepared in methanol–water (50:50; v/v).
Extraction of Red Clover Isoflavones
A 100 μL aliquot of blank human serum and 80 μL of internal standard mixture (50 ng/mL in aqueous methanol) were placed in a 1.5 mL micro-centrifuge tube, and standard solutions (20 µL) of varying concentrations of the red clover standards mixture were added (see final concentrations of the calibration curve standards in Table 1). Ice-cold methanol–ethanol (50:50; v/v, 800 µL) was added, and the mixture was vortexed for 2 min and allowed to stand in the freezer (−20°C) to precipitate the proteins. After centrifugation at 18 000×g and 4 °C for 30 min, the supernatant was transferred to a clean micro-centrifuge tube, dried under vacuum at 24 °C, and reconstituted in 200 µL of the initial mobile phase (water/acetonitrile containing 0.01% formic acid; 1:1, v/v). Aliquots of 3 µL were analyzed using UHPLC-MS/MS.
Table 1.
Retention times (RT), selected reaction monitoring (SRM) transitions, and collision energies (CE) used for UHPLC-MS/MS quantitative analysis and concentrations of calibration curve standards spiked into blank human seruma
| Isoflavone | RT, min | SRM transition, m/z (Polarity) | CE, V | Standard curve concentrations, ng/mL | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genistein | 2.06 | 271 → 153 (+) | 28 | 0.3 | 0.9 | 1.8 | 5.4 | 16.2 | 48.6 | 145.8 | 437.4 | 1312 |
| 269 → 133 (-) | 31 | |||||||||||
| Daidzein | 1.39 | 255 → 199 (+) | 24 | 0.07 | 0.2 | 0.6 | 1.8 | 5.4 | 16.2 | 48.6 | 145.8 | 437.4 |
| 255 → 137 (+) | 26 | |||||||||||
| Biochanin A | 3.76 | 285 → 213 (+) | 37 | 0.1 | 0.3 | 0.6 | 1.8 | 5.4 | 16.2 | 48.6 | 145.8 | 437.4 |
| 285 → 270 (+) | 25 | |||||||||||
| Irilone | 3.15 | 299 → 123 (+) | 36 | 0.3 | 0.9 | 1.8 | 5.4 | 16.2 | 48.6 | 145.8 | 437.4 | 1312 |
| 299 → 181 (+) | 26 | |||||||||||
| Prunetin | 3.74 | 285 → 242 (+) | 32 | 0.1 | 0.3 | 0.6 | 1.8 | 5.4 | 16.2 | 48.6 | 145.8 | 437.4 |
| 285 → 167 (+) | 28 | |||||||||||
| Formononetin | 2.69 | 269 → 197 (+) | 37 | 0.02 | 0.07 | 0.2 | 0.6 | 1.8 | 5.4 | 16.2 | 48.6 | 145.8 |
| 269 → 253 (+) | 28 | |||||||||||
| d4-Daidzein | 1.35 | 259 → 203 (+) | 25 | |||||||||
| 259 → 95 (+) | 38 | |||||||||||
| d4-Genistein | 2.01 | 275 → 219 (+) | 26 | |||||||||
| 273 → 137 (-) | 31 | |||||||||||
The first SRM transition was used as the quantifier while the second SRM transition was used as a qualifier.
Hydrolysis of Isoflavone Conjugates in Serum
A modification of the method of Doerge et al., was used (15) to deconjugate isoflavone glucuronic acid and sulfate conjugates in human serum. Briefly, human serum (0.1 mL) was added to a micro-centrifuge tube, and the pH was adjusted to 5.0 using 25 mM citrate buffer (pH 5.0). A mixture of β-glucuronidase/arylsulfatase (7.1 U/mL β-glucuronidase and 24 U/mL arylsulfatase) was added, and the solution was incubated at 37 °C for 1 h. Four volumes of ethanol–methanol (1:1, v/v) were added to precipitate proteins, and the unconjugated isoflavones were recovered as described above.
Hydrolysis of Isoflavone Conjugates in Red Clover Dietary Supplements
Isoflavone conjugates in red clover dietary supplement were hydrolyzed using protocol based on Wu et al. (10). Red clover dietary supplement (50 mg) from capsules was placed in a 50 mL round-bottomed flask with ethanol (5 mL), water (2 mL), and concentrated hydrochloric acid (0.8 mL). The reaction was refluxed in an oil bath at 70 °C with stirring for 2 h. The mixture was transferred to a volumetric flask and diluted to 25 mL with methanol. After mixing, a 1 mL aliquot was transferred to a micro-centrifuge tube and centrifuged at 18 000×g and 4 °C for 10 min. For quantitative analysis, the supernatant was diluted to 10 µg/mL by adding internal standard (50 ng/mL) and initial UHPLC mobile phase.
UHPLC-MS/MS
Separations were carried out using a Shimadzu (Kyoto, Japan) Nexera UHPLC system fitted with an Advanced Chromatography Technologies (Aberdeen, Scotland) ACE Super C18 UHPLC column (1.7 μm, 90 Å, 2.1 mm × 100 mm). The autosampler temperature was 4 °C, and the column oven was 35 °C. The isoflavones were eluted using a 2 min linear gradient from 20% to 55% acetonitrile in water (both solvents containing 0.01% formic acid), followed by 2 min hold at 55% acetonitrile, and a 0.5 min gradient to 90% acetonitrile. The column was re-equilibrated to 20% acetonitrile for 1 min before the next injection. The flow rate was 0.4 mL/min, and the injection volume was 3 µL.
The UHPLC system was interfaced with a Shimadzu LCMS-8060 triple quadruple mass spectrometer equipped with electrospray. Nitrogen was used as the drying gas at a flow rate of 5 L/min and for nebulization at 3 L/min. The interface and desolvation line temperatures were 400 °C and 300 °C, respectively. The red clover isoflavones were measured using collision-induced dissociation with selected reaction monitoring (SRM). The collision gas pressure was 230 kPa. Electrospray ionization was used with polarity switching, and the SRM dwell time for each transition was 15 msec. Data acquisition, integration, and linear standard curves fitting were carried out using Shimadzu Lab Solutions software version 5.7. The coefficients of variation were calculated using Microsoft Excel software (Seattle, WA, USA).
High Resolution UHPLC-MS and MS/MS of Isoflavones
High-resolution mass spectra of each isoflavone and deuterated internal standard were obtained on a Shimadzu 9030 Q-ToF-MS/MS equipped with a Nexera UHPLC system. The electrospray ionization interface temperature was 300 °C, and the voltages were 4.5 kV and −3.5 kV for positive and negative ion mode, respectively. The heat block and desolvation line temperatures were 400 °C and 250 °C, respectively. Nitrogen was used as a drying gas at a flow rate of 10 L/min, for nebulization at 3 L/min and as a heating gas at 10 L/min. Mass spectra and product ion tandem mass spectra were acquired every 100 ms over the scan range of m/z 70–500. Product ion tandem mass spectra were obtained using a collision energy of 35 V with an energy spread of 17 V.
Validation of the UHPLC-MS/MS Analytical Method
The method was validated according to guidelines for both AOAC INTERNATIONAL single-laboratory validation of chemical methods for dietary supplements and botanicals (16) and U.S. FDA bioanalytical method validation (17). The method was validated for selectivity, specificity, sensitivity, linearity, LOD, LOQ, precision, and accuracy. Selectivity and specificity were evaluated by comparing retention times and ratios of SRM MS/MS responses (quantifier and qualifier signals) of standards in neat solution with standards spiked into blank human serum. Sensitivity and linearity were assessed by plotting the area ratio of the standard peak to that of internal standard versus the theoretical concentration of the standard.
Matrix effects were assessed using five different batches of human serum. Briefly, QC standards at medium and high concentrations were spiked into different batches of blank human serum that had been extracted with equimolar methanol–ethanol as described above. Internal standard solution was added, and the solvent was removed under vacuum. Separately, QC standards at medium and high concentrations in neat solution (methanol–water, 50:50; v/v) were mixed with four volumes of the methanol/ethanol mixture and processed similarly to the spiked samples. Isoflavones in all validation samples were measured using UHPLC-MS/MS.
The concentration ranges of the isoflavone standards are shown in Table 1. The LOD was determined using a signal-to-noise ratio of between three and five. The LOQ was determined similarly, but the signal-to-noise ratio was between five and 10. Method precision was determined from intraday and interday replicate assays (five replicates) at the LOQ, low, medium, and high concentrations. The accuracy was estimated through relative percentage recoveries of spiked standards in serum matrix at low, medium, and high analyte concentrations, measured in five replicates. The stability of analytes was evaluated at the auto-sampler temperature of 10 °C for 24 h and for two freeze-thaw cycles with a 14-day interval.
Results and Discussion
Selection and Extraction of Red Clover Isoflavones
The selection of the six isoflavones for measurement (Figure 1) was based on their pro-estrogenic activity and high abundance in the aerial parts of red clover (formononetin and biochanin A), estrogenic activity (daizein and genistein), and progesterone-like activity (irilone and prunetin). The internal standards d4-genistein and d4-daidzein were used to control for variation in sample preparation and analysis and thereby enhance accuracy and precision of the quantitative method. Extraction was optimized to minimize sample preparation steps while maximizing analyte recovery. Although acetonitrile alone is often preferred for serum protein precipitation (18), an equimolar mixture of methanol and ethanol was found to provide the best recovery of all six isoflavones (Table 2).
Table 2.
Percent recovery of red clover isoflavones spiked into blank human serum
| Isoflavone | Concentration, ng/mL |
|||||
|---|---|---|---|---|---|---|
| Low | Recovery, % | Medium | Recovery, % | High | Recovery, % | |
| Genistein | 0.9 | 108.4 | 16.2 | 100.6 | 437.4 | 100.1 |
| Daidzein | 0.6 | 99.6 | 5.4 | 96.6 | 145.8 | 94.5 |
| Biochanin A | 0.3 | 105.6 | 5.4 | 98.4 | 145.8 | 94.5 |
| Irilone | 0.9 | 106.5 | 16.2 | 82.2 | 437.4 | 82.0 |
| Prunetin | 0.1 | 95.6 | 5.4 | 98.9 | 145.8 | 94.0 |
| Formononetin | 0.3 | 98.2 | 1.8 | 99.4 | 48.6 | 95.4 |
UHPLC-MS/MS Optimization
Three reversed phase UHPLC columns were compared for the separation of isoflavones (Advanced Chromatography Technologies ACE column and Waters’ Cortecs and Acquity columns), and the ACE column provided the best combination of separation and speed. Using this column, the separation of red clover isoflavones was further optimized by testing different mobile phase compositions as follows: water containing 0.1% formic acid and methanol (without formic acid); 0.1% formic acid in water and acetonitrile (without formic acid); and 3.5 mM ammonium formate (pH 3.5) in water and acetonitrile (without ammonium formate). The mobile phases containing acetonitrile provided similar isoflavone separation and were both superior to the mobile phase containing methanol. Further optimization showed that 0.01% formic acid in both water and acetonitrile provided optimal peak resolution and MS/MS intensity (Figure 2). Note that no analyte carry over was observed between injections using this optimized solvent system and column.
Figure 2.
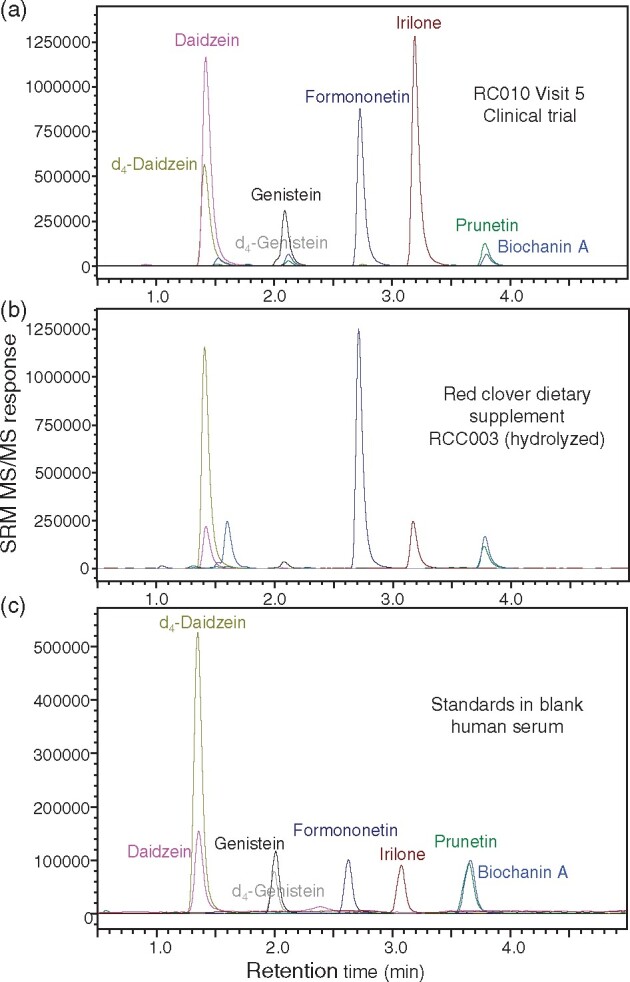
UHPLC-MS/MS chromatograms of red clover isoflavones extracted from (A) hydrolyzed human serum obtained from a woman volunteer 5 h after consuming an ethanolic extract of the aerial parts of red clover that had been standardized to 120 mg isoflavones; (B) a commercial dietary supplement (RCC003); and (C) isoflavone standards spiked into blank human serum at the medium QC level.
Using high resolution product ion MS/MS, SRM transitions and optimum collision energies were selected for each isoflavone based on the abundances of product ions formed during positive ion and negative ion electrospray MS/MS. The most abundant product ion for each isoflavone was used as the quantifier SRM transition while the second most abundant product ion was used as the qualifier SRM transition. Positive ion electrospray was used for the quantifier SRM transitions of all analytes. Negative ion electrospray was used only for the qualifier SRM transitions of genistein and d4-genistein, due to the higher abundance and consistency compared with alternative positive ion SRM transitions. The SRM transitions and their corresponding collision energies are shown in Table 1.
Constitutional isomers, prunetin and biochanin A, differ by the position of the methoxy group (Figure 1). Although these isomers were only partially separated (prunetin eluted at 3.7 min while biochanin A eluted at 3.8 min, Figure 2), optimization of the collision energies for each during positive electrospray facilitated the selective detection of biochanin A using the abundant SRM transition of m/z 285 to m/z 213 and the selective detection of prunetin using the SRM transition of m/z 285 to m/z 242 (See Supplemental Figure S1).
Validation of UHPLC-MS/MS Method
During electrospray UHPLC-MS/MS, analyte ion suppression or enhancement can occur due to matrix effects caused by co-eluting compounds. Sample preparation, chromatographic separation, and optimization of ion source parameters can mitigate such interference. In this study, the post-extraction spike method was utilized for evaluation of matrix effects. The matrix effect did not exceed the U.S. FDA recommended value (< 20%) in five different serum batches tested for all six isoflavones.
The recoveries at low, medium, and high concentrations of QC standards spiked in blank human serum were determined using UHPLC-MS/MS (Table 2). The recoveries ranged from 96–108% at low isoflavone concentrations and 82–100% at high concentrations. The recoveries of irilone were the lowest at 82% at medium and high concentrations, and the recoveries of genistein were the highest at 108% at low concentration and 100% for both medium and high concentrations. The accuracy of the method was determined for each isoflavone at LOQ, low, medium, and high concentrations. The accuracies, reported as percent coefficient of variation (CV%) ranged between 1.3% and 11.8%, except genistein which was 19% at LOQ (Table 3).
Table 3.
LOD, calibration curve linear range, the coefficient of determination (R2), accuracy, and precision for the UHPLC-MS/MS assay of 6 red clover isoflavonesa
| Precision CV% |
|||||||
|---|---|---|---|---|---|---|---|
| Analyte | LLOD, ng/mL | Linear range, ng/mL | R2 | QC, ng/mL | Accuracy CV% (n = 5) | Intraday (n = 5) | Interday n ≥ 8 |
| Genistein | 0.2 | 0.9–1312 | 0.9997 | 0.3 | 11.8 | 0.9 | 0.8 |
| 0.9 | 9.7 | 1.0 | 0.8 | ||||
| 16.2 | 2.2 | 3.1 | 3.4 | ||||
| 437.4 | 3.4 | 4.9 | 5.9 | ||||
| Daidzein | 0.07 | 0.2–437.4 | 0.9992 | 0.2 | 8.9 | 6.4 | 6.3 |
| 0.6 | 9.5 | 14.9 | 10.3 | ||||
| 5.4 | 2.8 | 4.5 | 4.8 | ||||
| 145.8 | 1.3 | 4.6 | 2.6 | ||||
| Biochanin A | 0.07 | 0.3–437.4 | 0.9989 | 0.1 | 19 | 0.8 | 0.5 |
| 0.3 | 4.9 | 3.0 | 3.8 | ||||
| 5.4 | 2.7 | 5.9 | 5.2 | ||||
| 145.8 | 2.1 | 6.8 | 4.2 | ||||
| Irilone | 0.1 | 0.3–1312 | 0.9994 | 0.3 | 7.2 | 1.1 | 1.5 |
| 0.9 | 2.5 | 1.9 | 0.5 | ||||
| 16.2 | 10.1 | 12.8 | 4.3 | ||||
| 437.4 | 4.7 | 11.5 | 3.9 | ||||
| Prunetin | 0.07 | 0.1–437.4 | 0.9988 | 0.1 | 10.5 | 0.4 | 0.4 |
| 0.3 | 6.1 | 1.2 | 0.8 | ||||
| 5.4 | 3.3 | 4.9 | 4.3 | ||||
| 145.8 | 2.2 | 7.0 | 4.1 | ||||
| Formononetin | 0.07 | 0.1– 145.8 | 0.9996 | 0.1 | 2.7 | 0.9 | 1.0 |
| 0.3 | 3.6 | 3.5 | 2.8 | ||||
| 1.8 | 5.4 | 8.5 | 9.8 | ||||
| 48.6 | 3.2 | 5.4 | 2.3 | ||||
Accuracy and precision were determined at LOQ, low, medium, and high concentrations of QC samples, and are expressed as coefficient of variation (CV).
The specificity and selectivity were determined by comparing the chromatograms of standards in neat solution with those spiked into blank human serum. Six batches of blank human serum were used in this study. All isoflavones eluted within 5 min without co-eluting peaks from the matrix that exceeded 20% of the area of the analytes at the LOQ level. Additionally, there were no co-eluting peaks exceeding 5% of the area of any internal standard (See Supplemental Figure S2).
The linearity of the method was established by constructing standard curves for each isoflavone. The standard curves for most of the isoflavones had a wide linear range. For example, genistein had a linear range from 0.9–1312 ng/mL with a correlation coefficient (R2) of 0.9997. In contrast, the signal for the protonated molecule of formononetin saturated the ion detector by 500 ng/mL, so that the linear range of the calibration curve ranged from 0.1 to just 145.8 ng/mL. All of the calibration curves were linear with R2 values > 0.99 (See Table 3 and Supplemental Figures S3–S8). The LOD and LOQ were also determined and ranged from 0.07 ng/mL for biochanin A and daidzein to 0.2 ng/mL for genistein (Table 3).
The precision of the method was determined for intraday and interday assays using QC samples at four different concentrations (LOQ, low, medium, and high). The percent coefficient of variation (CV%) was calculated for each analyte (Table 3). The intraday and interday precision for genistein had the lowest CV% at all QC concentrations, which ranged from 0.9% to 4.9% and from 0.8% to 5.9%, respectively. Daidzein showed the highest CV% ranging from 4.5% to 14.9% for intraday precision and 2.6% to 10.3% for interday precision. All the isoflavones at all QC concentrations satisfied the <15% CV% requirement by the U.S. FDA guidelines for analytical method development (17)
All isoflavones were stable in the autosampler at 10 °C for at least 24 h, which was sufficient for UHPLC-MS/MS analysis of the processed samples. Freeze-thaw studies indicated that analyte concentrations were stable through one freeze-thaw cycle but decreased significantly after the second free-thaw cycle (Table 4). In the current study, samples were processed without the need for multiple freeze-thaw cycles.
Table 4.
Stability of red clover isoflavones during storage and handling, measured using UHPLC-MS/MS
| Stability, % |
|||
|---|---|---|---|
| Analyte (n ≥ 3) | Autosampler 10 °C (24 h) | Freeze-thaw cycle |
|
| 1 | 2 | ||
| Genistein | 104 ± 4 | 97.9 ± 4.3 | 84.2 ± 17.2 |
| Daidzein | 105 ± 8 | 97.5 ± 3.6 | 94.4 ± 6.4 |
| Biochanin A | 104 ± 6 | 98.0 ± 5.4 | 79.3 ± 28.1 |
| Irilone | 93.9 ± 5.7 | 91.9 ± 13.5 | 70.1 ± 3.1 |
| Prunetin | 105 ± 4 | 90.6 ± 9.4 | 77.8 ± 28.4 |
| Formononetin | 103 ± 4 | 101 ± 1 | 78.5 ± 19.3 |
Analysis of Red Clover Isoflavones in Serum from a Clinical Trial
The new UHPLC-MS/MS assay was applied to the determination of six red clover isoflavones in human serum obtained from a Phase I pharmacokinetics clinical trial (1). Serum samples represented three different human subjects, two different dosages of red clover extract (80 mg total isoflavones and 120 mg isoflavones), and three different time points post dosing (1 h, 5 h, and 11 h) (Table 5). Serum isoflavones were deconjugated prior to UHPLC-MS/MS measurement. Note that numerous methods describing enzymatic deconjugation of phase II metabolites have been reported (15, 19–21), and the method by Doerge et al. was chosen due to its short reaction time and extent of hydrolysis.
Table 5.
Quantitative analysis of red clover isoflavones in serum obtained at different time points from women participating in a Phase I pharmacokinetics study (1)a
| Isoflavone | Concentration, ng/mL |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Subject RC010 (120 mg) |
Subject RC011 (80 mg) |
Subject RC012 (80 mg) |
|||||||||
| 1 h | 5 h | 11 h | 1 h | 5 h | 11 h | 1 h | 5 h | 11 h | |||
| Genistein | 40.6 ± 3.2 | 75.2 ± 5.1 | 45.2 ± 4.2 | 14.5 ± 0.5 | 14.7 ± 1.2 | 14.9 ± 0.7 | 15.2 ± 0.5 | 27.0 ± 2.6 | 13.9 ± 0.5 | ||
| Daidzein | 13.5 ± 0.2 | 31.9 ± 0.6 | 19.6 ± 0.8 | 0.36 ± 0.03 | 0.58 ± 0.08 | 0.49 ± 0.04 | 0.41 ± 0.02 | 0.19 ± 0.03 | 0.53 ± 0.12 | ||
| Biochanin A | 2.58 ± 0.29 | 8.92 ± 0.79 | 2.99 ± 0.30 | 0.60 ± 0.03 | 0.60 ± 0.05 | 0.62 ± 0.07 | 0.60 ± 0.02 | 0.59 ± 0.04 | 0.70 ± 0.15 | ||
| Irilone | 41.6 ± 4.9 | 138 ± 14 | 56.6 ± 9.6 | 4.19 ± 0.10 | 4.06 ± 0.12 | 3.94 ± 0.22 | 4.20 ± 0.01 | 4.10 ± 0.18 | 3.75 ± 0.50 | ||
| Prunetin | 4.74 ± 0.55 | 15.5 ± 1.7 | 3.95 ± 0.68 | 0.29 ± 0.01 | 0.25 ± 0.06 | 0.29 ± 0.06 | 0.32 ± 0.01 | 0.34 ± 0.04 | 0.47 ± 0.29 | ||
| Formononetin | 10.5 ± 2.9 | 33.3 ± 7.7 | 10.5 ± 2.1 | 0.96 ± 0.16 | 0.95 ± 0.06 | 1.09 ± 0.37 | 0.96 ± 0.04 | 0.97 ± 0.09 | 1.04 ± 0.16 | ||
Each subject consumed a single dose of a red clover dietary supplement containing either 80 mg or 120 mg total isoflavones.
Although red clover contains more formononetin and biochanin A than genistein and daidzein [(22) and Table 6] there was comparatively more genistein and daidzein in the serum samples. This is consistent with the known metabolic conversion of biochanin A and formononetin to genistein and daidzein, respectively (5). For example, the relative amounts of formononetin and biochanin A in human serum (Table 5, subject RC011) were 9.2% and 2.3% at the 1 h time point, whereas the red clover extract administered to this subject contained 32% and 39% formononetin and biochanin A, respectively (1).
Table 6.
Quantitative analysis of isoflavones in red clover dietary supplements
| Botanical dietary supplement, mg/g extract |
|||||
|---|---|---|---|---|---|
| Isoflavones | RCC001 | RCC002 | RCC003 | RCC004 | RCC005 |
| Genistein | 1.77 | 2.36 | 1.43 | <LOQ | 1.57 |
| Daidzein | 0.07 | 0.01 | 0.23 | 0.07 | 0.79 |
| Biochanin A | 13.5 | 3.96 | 2.24 | 0.05 | 2.55 |
| Irilone | 2.26 | 0.36 | 2.53 | 0.42 | 7.24 |
| Prunetin | 7.07 | 3.18 | 1.55 | 0.10 | 2.08 |
| Formononetin | 10.0 | 2.43 | 5.53 | 0.10 | 15.4 |
Analysis of Isoflavones in Red Clover Dietary Supplements
Five commercial red clover supplements were analyzed using the validated UHPLC-MS/MS assay. To facilitate the quantification of total isoflavones in each supplement, glycosides were converted to aglycons under acidic conditions. Acidic hydrolysis targets the bond between the isoflavone and the glycosidic group (9), unlike some alkaline hydrolysis methods that hydrolyze primarily ester bonds.
The supplement labels indicated that the products contained dried aerial parts of red clover. Specifically, product RCC001 contained red clover blossoms and leaf, RCC002 contained stem, leaf, and flower, RCC003 contained blossoms, RCC004 had aerial parts, and RCC005 contained stem and leaf. None of the dietary supplement products indicated they were extracts and were thus considered ground raw material. Major variations in the isoflavone content among the five supplements were observed (Table 6). Notably, RCC004 had the lowest isoflavone content. As expected (22), the biochanin A and formononetin levels were higher than other isoflavones, including genistein and daidzein.
Conclusions
A method based on UHPLC-MS/MS analysis was developed and validated for the quantitative analysis of six major red clover isoflavones in human serum and commercial dietary supplements. Sample preparation consisted of simple protein precipitation, and fast UHPLC separation (<5 min). This new assay is suitable for the support of clinical trials of the safety and efficacy of red clover dietary supplements as well as for the chemical standardization of red clover botanical dietary supplements.
Supplementary Material
Acknowledgments
The authors would like to thank Shimadzu for providing the UHPLC-MS/MS and high resolution Q-ToF-MS/MS instrumentation used in this study. This work was supported by an administrative supplement to grant P50AT000155 from the NIH National Center for Complementary and Integrative Health.
Supplemental Information
Supplemental information is available on the J. AOAC Int. website.
References
- 1. Piersen C., Booth N., Sun Y., Liang W., Burdette J., Breemen R.B., Geller S., Gu C., Banuvar S., Shulman L., Bolton J., Farnsworth N. (2004) Curr Med Chem. 11, 1361–1374. doi:10.2174/0929867043365134 [DOI] [PubMed] [Google Scholar]
- 2. Kolodziejczyk-Czepas J. (2012) J. Ethnopharmacol. 143, 14–23. doi:10.1016/j.jep.2012.06.048 [DOI] [PubMed] [Google Scholar]
- 3. Kolada G. (2002) Race to Fill Void in Menopause-Drug Market, The New York Times, New York, NY. https://www.nytimes.com/2002/09/01/us/rush-to-fill-void-in-menopause-drug-market.html? searchResultPosition=1 (accessed 2020) [Google Scholar]
- 4. Booth N.L., Piersen C.E., Banuvar S., Geller S.E., Shulman L.P., Farnsworth N.R. (2006) Menopause 13, 251–264. doi:10.1097/01.gme.0000198297.40269.f7 [DOI] [PubMed] [Google Scholar]
- 5. Tolleson W.H., Doerge D.R., Churchwell M.I., Marques M.M., Roberts D.W. (2002) J. Agric. Food Chem. 50, 4783–4790. doi:10.1021/jf025549r [DOI] [PubMed] [Google Scholar]
- 6. Lee J.-H., Dean M., Austin J.R., Burdette J.E., Murphy B.T. (2018) J. Nat. Prod. 81, 1962–1967. doi:10.1021/acs.jnatprod.8b00131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Toh M.F., Sohn J., Chen S.N., Yao P., Bolton J.L., Burdette J.E. (2012) Steroids 77, 765–773. doi:10.1016/j.steroids.2012.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clarke D.B., Bailey V., Lloyd A.S. (2008) Food Addit. Contam. Part A 25, 534–547. doi:10.1080/02652030701658340 [DOI] [PubMed] [Google Scholar]
- 9. Delmonte P., Perry J., Rader J.I. (2006) J. Chromatogr. 1107, 59–69. doi:10.1016/j.chroma.2005.11.060 [DOI] [PubMed] [Google Scholar]
- 10. Wu Q., Wang M., Simon J.E. (2003) J. Chromatogr. 1016, 195–209. doi:10.1016/j.chroma.2003.08.001 [DOI] [PubMed] [Google Scholar]
- 11. Ramos G.P., Dias P.M.B., Morais C.B., Fröehlich P.E., Dall’Agnol M., Zuanazzi J.A.S. (2008) Chromatographia. 67, 125–129. doi:10.1365/s10337-007-0450-0 [Google Scholar]
- 12. Burdette C.Q., Marcus R.K. (2013) J. AOAC Int. 96, 925–932 [DOI] [PubMed] [Google Scholar]
- 13. Phansalkar R.S., Simmler C., Bisson J., Chen S.-N., Lankin D.C., McAlpine J.B., Niemitz M., Pauli G.F. (2017) J. Nat. Prod. 80, 634–647. doi:10.1021/acs.jnatprod.6b00923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maul R., Kulling S.E. (2010) Br. J. Nutr. 103, 1569–1572. doi:10.1017/S0007114509993564 [DOI] [PubMed] [Google Scholar]
- 15. Doerge D.R., Churchwell M.I., Delclos K.B. (2000) Rapid Commun. Mass Spectrom. 14, 673–678. doi:10.1002/(SICI)1097-0231(20000430)14:8<673::AID-RCM935>3.0.CO; 2-F [DOI] [PubMed] [Google Scholar]
- 16.Appendix K: Guidelines for Dietary Supplements and Botanicals (2013) AOAC Int. Gaithersburg, MD. www.eoma.aoac.org (accessed 2020)
- 17.US FDA Guidance for Industry Bioanalytical Method Validation (2018) Silver Spring, MD. www.fda.gov (accessed 2020)
- 18. Polson C., Sarkar P., Incledon B., Raguvaran V., Grant R. (2003) J. Chromatogr. B 785, 263–275. doi:10.1016/j.jpba.2018.10.012 [DOI] [PubMed] [Google Scholar]
- 19. Cohen L.A., Crespin J.S., Wolper C., Zang E.A., Pittman B., Zhao Z., Holt P.R. (2007) In Vivo 21, 507–512 [PubMed] [Google Scholar]
- 20. Kuklenyik Z., Ye X., Reich J.A., Needham L.L., Calafat A.M. (2004) J. Chromatogr. Sci. 42, 495–500. doi:10.1093/chromsci/42.9.495 [DOI] [PubMed] [Google Scholar]
- 21. Holder C.L., Churchwell M.I., Doerge D.R. (1999) J. Agric. Food Chem. 47, 3764–3770. doi:10.1021/jf9902651 [DOI] [PubMed] [Google Scholar]
- 22. Hu M., Krausz K., Chen J., Ge X., Li J., Gelboin H.L., Gonzalez F.J. (2003) Drug Metab. Dispos. 31, 924–931. doi:10.1124/dmd.31.7.924 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.