

Abstract
Purpose
To determine whether patients with amyotrophic lateral sclerosis (ALS) show retinal axon pathology.
Methods
Postmortem eyes from 10 patients with ALS were sectioned and compared with 10 age-matched controls. Retinal sections were evaluated with periodic acid Schiff and phosphorylated (P-NF) and nonphosphorylated (NP-NF) forms of neurofilament with SMI 31 and 32 antibodies. Spheroids identified in the retinal nerve fiber layer were counted and their overall density was calculated in central, peripheral, and peripapillary regions. P-NF intensity was quantified. Morphometric features of ALS cases were compared with age-matched controls using the exact Wilcoxon matched-pairs signed-rank test.
Results
Distinct periodic acid Schiff–positive round profiles were identified in the retinal nerve fiber layer of patients with ALS and were most commonly observed in the peripapillary and peripheral retina. The density of periodic acid Schiff–positive spheroids was significantly greater in patients with ALS compared with controls (P = 0.027), with increased density in the peripapillary region (P = 0.047). Spheroids positive for P-NF and NP-NF were detected. P-NF–positive spheroid density was significantly increased in patients with ALS (P = 0.004), while the density of NP-NF spheroids did not differ significantly between ALS and control groups (P > 0.05). P-NF immunoreactivity in the retinal nerve fiber layer was significantly greater in patients with ALS than in controls (P = 0.002).
Conclusions
Retinal spheroids and axon pathology discovered in patients with ALS, similar to hallmark findings in spinal cord motor neurons, point to disrupted axon transport as a shared pathogenesis. Retinal manifestations detected in ALS suggest a novel biomarker detectable by noninvasive retinal imaging to help to diagnose and monitor ALS disease.
Keywords: retinal nerve fiber layer, retinal ganglion cell, neurofilament, axonal transport, cytoskeleton, retina, immunofluorescence, confocal microscopy, retinal imaging
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive disorder of upper and lower motor neurons.1,2 It begins insidiously with focal weakness but spreads relentlessly to involve most muscles, including the diaphragm. Typically, death owing to respiratory paralysis occurs within 3 to 5 years of clinical onset. It is the most common form of adult human motor neuron disease, with an estimated global incidence rate of 2.08 per 100,000 population per year.3,4 With a mean age of onset of 55 to 65 years, it is the most frequent neurodegenerative disorder of midlife.1 In the United States alone, 800,000 persons who are now alive are expected to die from ALS.5 Currently, only two approved drugs, riluzole6 and edaravone, are available for the treatment of ALS, and both provide limited improvement in survival.7,8
ALS can be categorized according to its mode of onset, designated as lower spinal (lower somatic motoneurons) in 70% of cases, corticospinal (bulbar/upper motoneurons) in 25% of cases, and respiratory in 5% of cases. The bulbar and respiratory onset forms typically have the worst prognosis.9 Compelling clinical, imaging and neuropathologic data have shown heterogeneity in the central nervous system (CNS) features of ALS.2 Up to 50% of patients with ALS develop cognitive and behavioral impairment, and about 13% of patients have concomitant behavioral-variant frontotemporal dementia.10–12
The clinical heterogeneity of ALS complicates diagnosis and assessment of disease progression, which currently rely on indirect, invasive, and/or expensive techniques such as electromyography, nerve conduction study, magnetic resonance imaging, lumbar puncture, and muscle biopsy.13 The search for sensitive and noninvasive biomarkers for the disease is thus an important one, as clinicians require tools for rapid diagnosis, prognosis, and assessment of patient response to pharmacological interventions in clinical trials.14
Approximately 90% of ALS cases are sporadic and 10% are familial, with clear inheritance patterns.15 Functionally, the 30 genes associated with familial ALS can be grouped into three main pathophysiologic processes: RNA biology, protein turnover, and axonal transport, suggesting that deficits in these pathways are causal.1 In about 60% to 80% of patients with familial ALS, a mutation of large effect (presumably pathogenic) can be identified, of which C9orf72 (40%), SOD1 (20%), FUS (1%–5%), and TARBDP (1%–5%) are the most common.16
The causes of neurodegeneration in ALS are multifactorial and include oxidative stress, mitochondrial dysfunction, misfolded protein toxicity/autophagy defects, RNA toxicity, excitotoxicity, and defective axonal transport.17 Motor neurons have very long axons compared with other neurons and are particularly dependent on efficient axonal transport to maintain their structure and function. Axonal transport defects may be an important factor underlying their selective vulnerability in ALS.18
Axonal transport, an adenosine triphosphate–dependent and tightly regulated process, involves the trafficking of proteins and organelles along microtubules by motor proteins, such as kinesin and dynein, and adaptor proteins, such as dynactin. Several lines of evidence suggest that alterations in axonal transport contribute to motor neuron degeneration in ALS. First, genetic studies of patients with ALS found mutations of genes encoding key components of the transport machinery, including neurofilament heavy chain,19,20 kinesin family member 5A, kinesin-associated protein 3,21,22 dynactin-1,23,24 tubulin alpha-4A,25 and profilin-1.26,27 Second, the frequency of axonal transport perturbation in ALS mouse models—for example, mutant TDP43-linked ALS28,29 and SOD1-linked ALS30,31—suggests that trafficking alterations contribute to neuronal dysfunction in ALS. Third, early neuropathologic evidence of axonal transport defects in motor neurons in ALS came from postmortem studies of patients with ALS that showed abnormal spheroids composed of phosphorylated neurofilaments, mitochondria, and lysosomes in the axons of large motor neurons.32–35 Axonal spheroids and transport defects were also observed in several SOD1-mutant mouse models.30,36,37 Their early occurrence in disease progression, before symptom onset, suggests a role in the pathogenesis of ALS.38
Neurofilaments are a principal cargo of axonal transport machinery, comprised of three subunit proteins: neurofilament light, neurofilament middle, and neurofilament heavy (NF-H) chains. Neurofilament middle and NF-H are phosphorylated in axons, with NF-H being particularly heavily phosphorylated.39 Phosphorylated NF-H prevail in axons, whereas nonphosphorylated NF-H are preferentially localized to the cell body. Specific antibodies such as SMI 31 and SMI 32 detect phosphorylated and nonphosphorylated forms of NF-H, respectively.40,41 Axonal pathology in ALS is evidenced by the accumulation of phosphorylated and nonphosphorylated neurofilaments in axonal spheroids in motor neurons.42–47
ALS can no longer be considered simply a degenerative disorder of motor neurons, but rather a progressive neurodegenerative disorder in which motor neurons manifest by virtue of their susceptibility.2 The disease processes leading to axonal transport alterations in motor neurons in ALS may also affect neurons in other systems, such as the visual system. Retinal ganglion cells (RGCs) have long axons that convey visual information from the retinal nerve fiber layer (RNFL) to the lateral geniculate nucleus in the thalamus,48 making them particularly vulnerable to disruptions in axonal transport.48,49
We hypothesize that axonal transport alteration occurs in RGC axons in ALS and retinal manifestations include axonal spheroids and the abnormal neurofilament accumulation in the RNFL, as has been reported in motor neurons. In this study, we assess markers of axonal transport alteration in postmortem eye specimens from patients with ALS. We then seek to correlate our findings with aspects of the patients’ clinical profile. A retinal manifestation of ALS may form the basis of a novel biomarker for the disease that may be evaluated using noninvasive ocular imaging techniques, such as optical coherence tomography (OCT) and confocal scanning laser ophthalmoscopy.50
Methods
Following institutional research ethics board approval, postmortem human eye specimens were obtained from the Human Eye Biobank for Research at St. Michael's Hospital. Nineteen eyes from 10 patients with confirmed ALS (age 64.6 ± 9.0 years) and 17 eyes from 10 control patients (70.2 ± 10.7 years) were included in this study. The age difference between the two cohorts was not statistically significant (P = 0.22). All 10 patients with ALS had sporadic ALS and were tested for C9orf72, which was negative. Table 1 gives demographic and clinical information for each ALS patient: gender, age at death, disease duration, mode of disease onset (bulbar or nonbulbar), average bulbar score on the revised ALS functional rating scale,51 and rate of disease progression (slow, moderate, or fast); and for each control patient: gender, age at death, and cause of death. Exclusion criteria for controls were neurological diseases and blinding eye diseases including glaucoma, age-related macular degeneration, and advanced diabetic retinopathy. All eyes were removed within 24 hours of the patient's death and stored at 4˚C until they were immersion fixed in 10% formalin within 22 hours of enucleation.
Table 1.
Demographic and Clinical Information for Patients With and Without ALS
| Gender | Age (Years) | Disease Duration (Years) | Bulbar vs. Nonbulbar | Average Bulbar ALSFRS-R Score* | Disease Progression | |
|---|---|---|---|---|---|---|
| ALS case | ||||||
| 1 | Female | 46 | 15.5 | Nonbulbar | Unknown | Unknown |
| 2 | Female | 57 | 3 | Bulbar | 0 | Moderate |
| 3 | Female | 58 | 2.8 | Nonbulbar | 1 | Fast |
| 4 | Male | 62 | 6 | Nonbulbar | 2.7 | Slow |
| 5 | Female | 66 | 5 | Bulbar | 1 | Moderate |
| 6 | Female | 67 | 2.8 | Bulbar | 0.3 | Moderate |
| 7 | Male | 70 | 18 | Nonbulbar | 2 | Slow |
| 8 | Female | 71 | 23 | Nonbulbar | 3.7 | Slow |
| 9 | Male | 74 | 1.1 | Bulbar | 0.7 | Fast |
| 10 | Male | 75 | 4 | Bulbar | 0 | Moderate |
| Control case | Gender | Age (Years) | Cause of Death | |||
| 1 | Female | 50 | Asthma | |||
| 2 | Female | 65 | Pneumonia | |||
| 3 | Male | 65 | Myocardial infarction | |||
| 4 | Female | 67 | Liver failure | |||
| 5 | Male | 69 | Respiratory failure | |||
| 6 | Male | 69 | Subdural hemorrhage | |||
| 7 | Female | 73 | Chronic obstructive pulmonary disease | |||
| 8 | Male | 75 | Cancer | |||
| 9 | Female | 77 | Myocardial infarction | |||
| 10 | Male | 92 | Myelodysplastic syndrome | |||
Each patient received a score from 0 (complete disability) to 4 (normal function) for speech, salivation, and swallowing. The average of these three scores is reported here.
Histopathology and Immunofluorescence
Eyes were processed and embedded in paraffin and cut into 8 µm sections. Sections from each case were deparaffinized and stained with periodic acid Schiff (PAS) and imaged with an Aperio AT Turbo scanner (Leica Biosystems, Nussloch, Germany).
For immunofluorescence, sections were deparaffinized and rehydrated using a Leica Autostainer XL (Leica Biosystems). Sections were heated at 65˚C for 10 minutes, then underwent one 3-minute wash in xylene, two 2-minute washes in xylene, two 1-minute washes in 100% alcohol, and one 1-minute wash in 95% alcohol. Formalin-fixed paraffin-embedded sections underwent heat-induced antigen retrieval to improve the immunostaining.52,53 Sections were placed in a staining dish filled with 10 mM citric acid, pH 6, and the dish was immersed in 500 mL distilled water in a Biocare Medical Decloaking Chamber (Biocare Medical, LLC, Concord, CA). Sections were heated at 95˚C for 20 minutes, followed by 90˚C for 10 seconds. Sections were then allowed to cool on the benchtop for 20 minutes immersed in PBS. Sections were rinsed twice in PBS, for 5 minutes each, and then incubated in blocking solution (2% goat serum, 0.3% TritonX-100 in PBS) for 40 minutes at room temperature. Incubations in primary antibodies (diluted in blocking solution) were carried out overnight at 4˚C. Sections were stained with either SMI 31 (monoclonal mouse IgG1, 1:500; BioLegend, San Diego, CA) or SMI 32 (monoclonal mouse IgG1, 1:2500; BioLegend). SMI 31 reacts with phosphorylated high molecular weight NF (P-NF);54 SMI 32 reacts with nonphosphorylated high molecular weight NF (NP-NF).55 Negative controls without primary antibody were processed in parallel. SMI 31 antibody can detect axonal spheroids in motor neurons of patients with ALS45,56 and mouse models.33,34 Sections were then rinsed three times in PBS for 5 minutes each. Sections were incubated in Alexa-Fluor 555 goat anti-mouse IgG (H+L) (1:1000; Thermo Fisher Scientific, Waltham, MA) and 4’,6’-diamino-2-phenylindole (DAPI, 1:1000; Thermo Fisher Scientific) for 1 hour at room temperature, protected from light. Sections were then rinsed three times in PBS for 5 minutes each and coverslips were mounted using Dako Fluorescent Mounting Medium (Dako, North America, Inc., Carpinteria, CA). Slides were allowed to dry for two hours in the fume hood, protected from light, and were then stored at 4˚C overnight. Slides were imaged using a Zeiss LSM 700 confocal scanning laser microscope (Carl Zeiss MicroImaging, Göttingen, Germany). Images were captured in the central, peripheral, and peripapillary retina. The AlexaFluor 555 channel was used to visualize P-NF (laser power: 2.0%; gain: 600) and NP-NF (laser power: 5.0%; gain: 750). The DAPI channel was used to visualize cell nuclei (laser power: 2.0%; gain: 600). Images were analyzed in a masked manner.
Quantification and Statistical Analysis
One entire PAS-stained retina section from each eye was visualized using the Aperio scanner under 20× magnification. All round PAS-positive profiles in the RNFL with a solid pink–purple color were counted in the central, peripheral, and peripapillary regions of the retina, and their diameter was measured using the Aperio scanner software's measurement tool. The dot sampling method of the scanner software was used to estimate the square area of the RNFL in the peripapillary, central, and peripheral regions of the retina, as well as the total RNFL area in each section. A grid with squares measuring 100 × 100 µm was superimposed on the image; any square corner falling on the RNFL was counted and the total count was multiplied by one square area to estimate the total area. The central retina was defined as the region temporal to the optic nerve head extending from the border of the peripapillary retina to a distance of 10 mm, including the macula. The peripheral retina was defined as the region extending anteriorly from the anterior border of the central retina on the temporal side, and from the border of the peripapillary retina on the nasal side. The peripapillary retina was defined as the region extending from the peripapillary border of the choroid to a radius of 1.5 mm from the center of the optic nerve head. The diameter of each PAS-positive profile was measured using the measurement tool of the scanner software (Aperio). Quantile regression was used to examine the difference in profile size between patients with ALS and controls at different locations (10th, 25th, 50th, 75th, 90th, 95th, and 99th percentiles) in the size distribution of profiles. At the 95th percentile, after adjusting for age and gender, profiles detected in patients with ALS had significantly larger diameters than profiles detected in control patients (P < 0.01). The adjusted median and the 95% confidence interval for controls were derived at the 95th percentile: 10.63 µm (9.07 µm, 12.19 µm). The lower confidence limit (9.07 µm) was used as the cut-point to define PAS-positive pathologic spheroids thereafter. All available eyes were used to determine the cut-off point. The number of PAS-positive spheroids larger than 9.07 µm in diameter was divided by the square area of the RNFL to determine the density of PAS-positive spheroids in the total retina, and the central, peripheral, and peripapillary retina individually.
Immunofluorescence-stained sections from patients with ALS and controls were visualized using confocal microscopy under 20× magnification. One section per eye was analyzed for each primary antibody. The total number of P-NF–positive spheroids and NP-NF–positive spheroids in each section was counted and divided by the square area of the RNFL to determine the density. P-NF immunoreactivity in the RNFL was quantified using ImageJ (version 2.00-rc-69/1.52p; NIH, Bethesda, MD). In each image, a region of interest was constructed around the RNFL, and the mean pixel intensity of P-NF immunoreactivity within this region was calculated. The maximum pixel intensity in a corresponding negative control image was used as the lower threshold for mean pixel intensity measurements. For each eye, the mean pixel intensity was measured in the central, peripapillary, and peripheral RNFL, and an average value in arbitrary units (AU) was calculated (weighted according to the surface area of RNFL in each image). There was no difference in MPI between left eyes and right eyes. Age-adjusted analysis of the median difference between left eyes and right eyes assessed using quantile regression at the median showed no significant differences (P = 0.957).
For all statistical analyses, one eye was randomly selected from each patient. Owing to the small sample sizes and the non-normality of the data to be analyzed (as assessed by the Shapiro–Wilk test), nonparametric statistical tests were performed.
The exact Wilcoxon matched-pairs signed-rank test was used to assess whether there was a statistically significant difference in the density of PAS-positive spheroids, P-NF–positive spheroids, and NP-NF–positive spheroids in patients with ALS compared with age-matched controls, and to assess differences in the mean pixel intensity of P-NF immunoreactivity between groups. The Spearman rank-order correlation coefficient was used to measure the extent of the association between the clinical characteristics of patients with ALS and the density of PAS-positive spheroids, P-NF–positive spheroids, and NP-NF–positive spheroids, and the mean pixel intensity of P-NF immunoreactivity. P values were considered statistically significant at a value of less than 0.05.
Results
Spheroids in ALS
PAS-stained sections exhibited distinct PAS-positive spheroids greater than 9.07 µm in diameter in the RNFL in 9 of 10 patients with ALS and 5 of 10 age-matched controls. Spheroids were most commonly observed in the peripheral RNFL (Fig. 1B) and peripapillary RNFL (Figs. 2B and 2C) and, rarely, in the central RNFL in patients with ALS, compared with corresponding areas in controls (Figs. 1A and 2A). The density of PAS-positive spheroids was significantly greater in patients with ALS than in controls (median, 2.6 spheroids per mm2 [interquartile range (IQR), 1.8 spheroids per mm2] vs. median, 0.3 spheroids per mm2 [IQR, 1.8 spheroids per mm2]; P = 0.027) (Fig. 3A, Table 2). The density of PAS-positive spheroids in the peripapillary RNFL was significantly greater in patients with ALS than in controls (median, 4.6 spheroids per mm2 [IQR, 7.1 spheroids per mm2] vs. 0.0 spheroids per mm2 [IQR, 0.0 spheroids per mm2]; P = 0.047) (Fig. 3B, Table 2). The PAS-positive spheroid density in the peripheral and central RNFL did not differ significantly between patients with ALS and controls (median, 2.1 spheroids per mm2 [IQR, 4.4 spheroids per mm2] vs. median, 0.0 spheroids per mm2 [IQR, 2.9 spheroids per mm2]; P > 0.05 and median, 0.0 spheroids per mm2 [IQR, 0.0 spheroids per mm2] vs. median, 0.0 spheroids per mm2 [IQR, 0.0 spheroids per mm2]; P > 0.05, respectively) (Figs. 3C and 3D, Table 2).
Figure 1.

Spheroids identified in the peripheral retina of patients with ALS. PAS-stained sections of the peripheral retina from (A) control (case 8) and (B) ALS (case 4). Several spheroids stained in deep purple are visible in the peripheral RNFL of the ALS patient (B, arrows). Abbreviations: RNFL = retinal nerve fiber layer; GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer; ONL = outer nuclear layer. Scale bar = 100 µm.
Figure 2.

Spheroids identified in the peripapillary retina of patients with ALS. PAS-stained sections of the peripapillary retina from (A) control (case 7) and (B) ALS (case 2). The boxed region in (B) is expanded in (C). Spheroids stained in deep purple were observed in the peripapillary RNFL of the ALS patient (C; arrow). Abbreviations: RNFL = retinal nerve fiber layer; LC = lamina cribrosa. (A, B) Scale bar = 100 µm; (C) scale bar = 50 µm.
Figure 3.

Increased spheroid density in the retinas of patients with ALS. Boxplots show the distribution of the density of PAS-positive spheroids in the RNFL, overall (A), and in peripapillary (B), peripheral (C), and central (D) regions. The density of spheroids is significantly greater in patients with ALS than in controls across the retina (A) and in the peripapillary region (B). No significant difference in spheroid density was observed in the peripheral (C) or central retina (D). For each boxplot, the center line represents the median. The upper and lower limits of each box represent the 75th and 25th percentiles, respectively. Individual data points are displayed as open circles for controls and open diamonds for patients with ALS.
Table 2.
Retinal Spheroids and Axon Pathology in Patients With ALS Compared With Controls
| Exact Wilcoxon Matched-Pairs Signed-Rank Test | ||||
|---|---|---|---|---|
| Median | IQR | S | P Value | |
| PAS-positive spheroids per mm2: Total RNFL | ||||
| Control | 0.3 | 1.8 | ||
| ALS | 2.6 | 1.8 | 21.5 | .0273 |
| PAS-positive spheroids per mm2: Peripapillary RNFL | ||||
| Control | 0.0 | 0.0 | ||
| ALS | 4.6 | 7.1 | 12.0 | .0469 |
| PAS-positive spheroids per mm2: Peripheral RNFL | ||||
| Control | 0.0 | 2.9 | ||
| ALS | 2.1 | 4.4 | 6.5 | >0.05 |
| PAS-positive spheroids per mm2: Central RNFL | ||||
| Control | 0.0 | 0.0 | ||
| ALS | 0.0 | 0.0 | 0.5 | >0.05 |
| P-NF–positive spheroids per mm2 | ||||
| Control | 0.0 | 0.0 | ||
| ALS | 1.3 | 0.8 | 22.5 | .0039 |
| NP-NF–positive spheroids per mm2 | ||||
| Control | 0.0 | 0.0 | ||
| ALS | 0.0 | 0.0 | 1.5 | >0.05 |
| Mean pixel intensity of P-NF (AU) | ||||
| Control | 7328.9 | 2711.5 | ||
| ALS | 22365.4 | 5154.7 | 27.5 | .0020 |
P-NF in ALS
P-NF immunofluorescence revealed P-NF–positive spheroids ranging from 8 to 15 µm in diameter in the RNFL in 9 of 10 patients with ALS and none of 10 controls. These spheroids were observed in the peripheral (Fig. 4B) and peripapillary RNFL (Figs. 5B and 5C) in patients with ALS, compared with corresponding controls (Figs. 4A and 5A, respectively). P-NF–positive spheroids were not observed in the central RNFL in patients with ALS (Fig. 4D) nor in controls (Fig. 4C). The density of P-NF–positive spheroids was significantly greater in patients with ALS than in controls (median, 1.3 spheroids per mm2 [IQR, 0.8 spheroids per mm2] vs. median, 0.0 spheroids per mm2 [IQR, 0.0 spheroids per mm2]; P = 0.004) (Fig. 6, Table 2).
Figure 4.
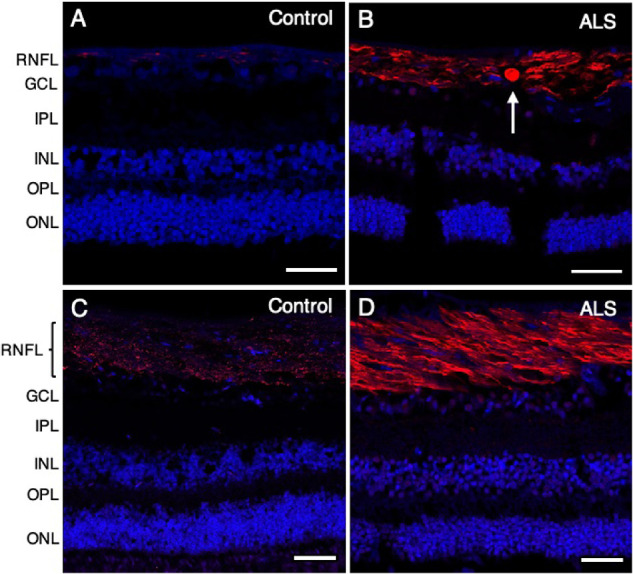
Increased phosphorylated neurofilament in the peripheral and central retina of patients with ALS. Phosphorylated neurofilament is shown in red using SMI 31, with nuclei counterstained with DAPI in blue. P-NF–positive spheroids ranging from 8 to 15 µm in diameter were observed in the peripheral retina (B, arrow) but not in the central retina in patients with ALS (D). Compared with controls (A, C), a stronger P-NF signal was observed in patients with ALS (B, D) in both peripheral (B) and central (D) retinal regions. (A) Control (case 9); (B) ALS (case 7); (C) control (case 3); (D) ALS (case 5). Abbreviations: RNFL = retinal nerve fiber layer; GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer; ONL = outer nuclear layer. Scale bar = 50 µm.
Figure 5.

Increased phosphorylated neurofilament in the peripapillary retina of patients with ALS. Phosphorylated neurofilament is shown in red using SMI 31, with nuclei counterstained with DAPI in blue. Compared with controls (A), a significant increase in P-NF immunoreactivity in the peripapillary region of the RNFL was observed in patients with ALS (B). The boxed region in (B) is expanded in (C). P-NF–positive spheroids ranging from 8 to 15 µm in diameter were observed in all 10 ALS cases (n = 10) (C; arrows), and in none of the 10 control cases. (A) Control (case 8); (B, C) ALS (case 4). Abbreviations: RNFL = retinal nerve fiber layer; LC = lamina cribrosa. (A, B) Scale bar = 100 µm; (C) Scale bar = 25 µm.
Figure 6.
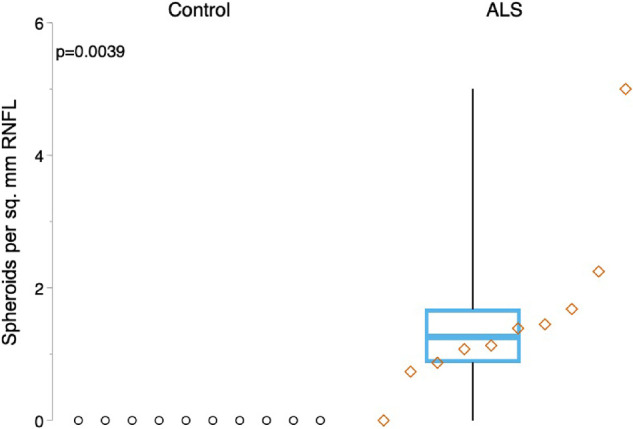
Increased density of P-NF–positive spheroids in the retinas of patients with ALS. Boxplots show the density distribution of P-NF–positive spheroids in the RNFL. Spheroid density is significantly increased in patients with ALS (right) compared with controls (P = 0.004). For each boxplot, the center line represents the median. The upper and lower limits of each box represent the 75th and 25th percentiles, respectively. Individual data points are displayed as open circles for controls and open diamonds for patients with ALS.
Patients with ALS showed stronger P-NF signal in the RNFL in the peripheral (Fig. 4B), central (Fig. 4D), and peripapillary regions (Fig. 5B) than in corresponding regions in controls (Figs. 4A, 4C, and 5A, respectively). The P-NF signal intensity in the RNFL was significantly greater in patients with ALS than in controls (median, 22364.4 AU [IQR, 5154.7 AU] vs. median, 7328.9 AU [IQR, 2711.5 AU]; P = 0.002) (Fig. 7, Table 2).
Figure 7.
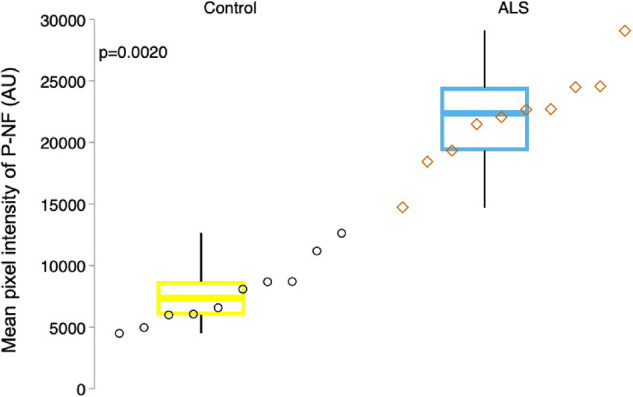
Increased phosphorylated neurofilament in the retina of patients with ALS. Boxplots show the intensity distribution of P-NF immunofluorescence in the RNFL in all retinal regions. P-NF intensity is significantly increased in patients with ALS (right) compared with controls (P = 0.002). For each boxplot, the center line represents the median. The upper and lower limits of each box represent the 75th and 25th percentiles, AU = arbitrary units. Individual data points are displayed as open circles for controls and open diamonds for patients with ALS.
NP-NF in ALS
NP-NF immunofluorescence revealed distinct spheroids ranging from 7 to 10 µm in diameter in the RNFL in 2 of 10 patients with ALS (Fig. 8B) and none of 10 controls (Fig. 8A). The density of NP-NF spheroids did not differ significantly between patients with ALS and controls (median, 0.0 spheroids per mm2 [IQR, 0.0 spheroids per mm2] vs. median, 0.0 spheroids per mm2 [IQR, 0.0 spheroids per mm2]; P > 0.05) (Table 2). In 8 of 10 patients with ALS, the NP-NF signal was increased in the RNFL and inner plexiform layer (Fig. 8B) compared with controls (Fig. 8A).
Figure 8.
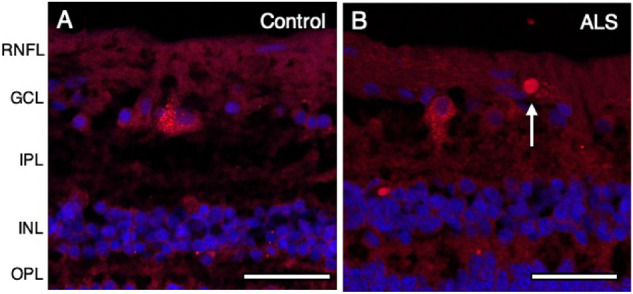
NP-NF in the peripheral retina of patients with ALS. NP-NF is shown in red using SMI 32, with nuclei counterstained in blue with DAPI. NP-NF-positive spheroids ranging from 7 to 10 µm were observed in the RNFL in the ALS group (B) but not in controls, where the NP-NF signal was confined to the RGC cell body (A). (A) Control (case 3); (B) ALS (case 5). Abbreviations: RNFL = retinal nerve fiber layer; GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer. Scale bar = 50 µm.
ALS Clinical Profile and Correlation of Retinal Spheroids and Neurofilament
The clinical characteristics of patients with ALS were assessed in relation to retinal spheroids and neurofilament findings. There was no significant association between densities of PAS-positive spheroids (overall, peripapillary, peripheral, and central retina), the density of P-NF–positive or of NP-NF–positive spheroids, and clinical characteristics of age at death, gender, disease duration, mode of disease onset (bulbar vs. nonbulbar), average bulbar score on the revised ALS functional rating scale, and rate of disease progression (slow, moderate, or fast) (P > 0.05). No significant correlation was observed between P-NF signal intensity in the RNFL and any of the clinical characteristics discussed in this article (P > 0.05).
Discussion
In this study of a rare collection of retinas collected from patients with ALS shortly after death, we identified spheroids and axon pathology in the RNFL. The phenotypic findings of PAS-positive spheroids and P-NF–positive spheroids located in the RNFL are consistent with reported findings in motor neurons of patients with ALS.42–47 Transgenic mice with dysfunctional microtubule-associated motor proteins also display such findings.57–59 The axons of RGCs in patients with ALS also showed significantly increased P-NF immunoreactivity compared with controls. In our study, 8 of 10 patients with ALS showed P-NF–positive spheroids, although only two patients with ALS showed rare NP-NF–positive spheroids. Because NF-H phosphorylation is a regulator of neurofilament transport,60 and increased phosphorylation of NF-H has been associated with slower axonal transport rates,61 the increased frequency of P-NF–positive spheroids and increased P-NF immunoreactivity in the RNFL in patients with ALS may be a biomarker of axon pathology in ALS.
The presence of NP-NF in the RNFL of patients with ALS suggests axonal transport alteration, because NP-NF is not typically detected in healthy axons.41 However, only 2 of 10 patients with ALS showed NP-NF–positive spheroids. Furthermore, the pattern of increased NP-NF in the inner plexiform layer in ALS cases is indicative of increased NP-NF in RGC dendrites. This finding suggests that, in ALS, altered transport in RGCs may not be limited to axons. Further investigation is warranted to assess the distribution of other cellular components within RGCs of patients with ALS, such as mitochondria and autophagosomes, which have been shown to accumulate in affected motor neurons owing to dysfunctional transport.62,63 Retinal immunostaining for other known molecular markers of ALS, such as TDP-43,64 FUS,65 OPTN,66 UBQLN2,67 and p62,68 may shed further insights.
The pathologic phenotypes reported in this article did not correlate significantly with the patients’ clinical characteristics (age at death, gender, disease duration, mode of disease onset, clinical severity of disease, or rate of disease progression). Although it is known that most nonbulbar onset cases have better prognosis compared with those with bulbar onset,9 it is of note that ALS (case 3) with nonbulbar onset had a fast rate of disease progression. Studies with larger sample sizes with various disease stages in addition to end-stage ALS are needed to determine whether retinal findings and clinical aspects of the disease are correlated.
Because the retina and optic nerve are considered part of the CNS, knowledge obtained from studying and visualizing the eye has in many cases been used as a means to gain insight into CNS processes, as well as to assist in the diagnosis and monitoring of CNS pathologies. Conventional retinal imaging techniques, such as OCT and confocal scanning laser ophthalmoscopy, have uncovered ocular manifestations of various CNS disorders, including stroke,69 multiple sclerosis,70 Alzheimer's disease,71 and Parkinson's disease.72 Moreover, ocular manifestations often precede symptoms in the brain; thus, investigations of the eye may serve as a means for early diagnosis.73 OCT has previously been used to assess retinal pathology in ALS, and marked thinning of the RNFL has been reported in patients with ALS compared with controls.74–77 A recent study by Rojas et al.78 reported macular thickening in patients with ALS compared with controls, and, on follow-up 6 months later, reported significant peripapillary RNFL thinning in patients with ALS compared with baseline, indicating possible progression of disease. The authors also reported a moderate correlation between OCT RNFL parameters and revised ALS functional rating scale score.78 In our study, retinal spheroids were increased in the peripapillary RNFL of patients with ALS. These preliminary observations suggest that prospective studies with a large sample size and detailed sectoral analysis may be helpful.
Patients with ALS show elevated neurofilament levels in the cerebrospinal fluid and serum at early symptom onset relative to healthy subjects.79 Thus, detection of neurofilament has been proposed as a sensitive marker of axon damage and neurodegeneration, tightly linked to the symptomatic phase of ALS. Here we present evidence for spheroids in the RNFL—distinct retinal lesions associated with ALS. These lesions, representing axon pathology in ALS, may be a promising new biomarker for ALS detectable in vivo. We propose these newly identified spheroids and axon pathology in ALS as a novel retinal phenotype. Future clinical studies are needed to determine whether these retinal findings can be used as a biomarker to detect and monitor ALS disease progression by noninvasive retinal imaging.
Acknowledgments
Supported by the Henry Farrugia Research Fund, the Dan Sullivan Research Fund, and the Human Eye Biobank for Research. The authors thank Eileen Girard (MLT) for her excellent technical support.
Grant Support: Henry Farrugia Research Fund, Dan Sullivan Research Fund.
Disclosure: K. Sharma, None; M. Amin Mohammed Amin, None; N. Gupta, None; L. Zinman, None; X. Zhou, None; H. Irving, None; Y. Yücel, None
References
- 1. Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017; 377(2): 162–172. [DOI] [PubMed] [Google Scholar]
- 2. Strong MJ. Revisiting the concept of amyotrophic lateral sclerosis as a multisystems disorder of limited phenotypic expression. Curr Opin Neurol. 2017; 30(6): 599–607. [DOI] [PubMed] [Google Scholar]
- 3. Moura MC, Novaes MR, Eduardo EJ, Zago YS, Freitas Rdel N, Casulari LA. Prognostic factors in amyotrophic lateral sclerosis: a population based study. PLoS One. 2015; 10(10): e0141500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Logroscino G, Traynor BJ, Hardiman O, et al.. Incidence of amyotrophic lateral sclerosis in Europe. J Neurol Neurosurg Psychiatr. 2010; 81(4): 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnston CA, Stanton BR, Turner MR, et al.. Amyotrophic lateral sclerosis in an urban setting: a population based study of inner city London. J Neurol. 2006; 253(12): 1642–1643. [DOI] [PubMed] [Google Scholar]
- 6. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994; 330(9): 585–591. [DOI] [PubMed] [Google Scholar]
- 7. Jaiswal MK. Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. 2019; 39(2): 733–748. [DOI] [PubMed] [Google Scholar]
- 8. Radunovic A, Mitsumoto H, Leigh PN. Clinical care of patients with amyotrophic lateral sclerosis. Lancet Neurol. 2007; 6(10): 913–925. [DOI] [PubMed] [Google Scholar]
- 9. Chio A, Logroscino G, Hardiman O, et al.. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. 2009; 10(5-6): 310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elamin M, Bede P, Byrne S, et al.. Cognitive changes predict functional decline in ALS: a population-based longitudinal study. Neurology. 2013; 80(17): 1590–1597. [DOI] [PubMed] [Google Scholar]
- 11. Strong MJ, Abrahams S, Goldstein LH, et al.. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017; 18(3-4): 153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Phukan J, Elamin M, Bede P, et al.. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatr. 2012; 83(1): 102–108. [DOI] [PubMed] [Google Scholar]
- 13. Al-Chalabi A, Hardiman O, Kiernan MC, Chio A, Rix-Brooks B, van den Berg LH. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 2016; 15(11): 1182–1194. [DOI] [PubMed] [Google Scholar]
- 14. Turner MR. Progress and new frontiers in biomarkers for amyotrophic lateral sclerosis. Biomark Med. 2018; 12(7): 693–696. [DOI] [PubMed] [Google Scholar]
- 15. Mulder DW, Kurland LT, Offord KP, Beard CM. Familial adult motor neuron disease: amyotrophic lateral sclerosis. Neurology. 1986; 36(4): 511–517. [DOI] [PubMed] [Google Scholar]
- 16. Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014; 17(1): 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013; 14(3): 161–176. [DOI] [PubMed] [Google Scholar]
- 18. Ragagnin AMG, Shadfar S, Vidal M, Jamali MS, Atkin JD. Motor neuron susceptibility in ALS/FTD. Front Neurosci. 2019; 13: 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Figlewicz DA, Krizus A, Martinoli MG, et al.. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet. 1994; 3(10): 1757–1761. [DOI] [PubMed] [Google Scholar]
- 20. Al-Chalabi A, Andersen PM, Nilsson P, et al.. Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum Mol Genet. 1999; 8(2): 157–164. [DOI] [PubMed] [Google Scholar]
- 21. Nicolas A, Kenna KP, Renton AE, et al.. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron. 2018; 97(6): 1268–1283.e1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brenner D, Yilmaz R, Muller K, et al.. Hot-spot KIF5A mutations cause familial ALS. Brain. 2018; 141(3): 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Puls I, Jonnakuty C, LaMonte BH, et al.. Mutant dynactin in motor neuron disease. Nat Genet. 2003; 33(4): 455–456. [DOI] [PubMed] [Google Scholar]
- 24. Munch C, Sedlmeier R, Meyer T, et al.. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004; 63(4): 724–726. [DOI] [PubMed] [Google Scholar]
- 25. Smith BN, Ticozzi N, Fallini C, et al.. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014; 84(2): 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu CH, Fallini C, Ticozzi N, et al.. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012; 488(7412): 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smith BN, Vance C, Scotter EL, et al.. Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol Aging. 2015; 36(3): 1602.e17–1602.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alami NH, Smith RB, Carrasco MA, et al.. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014; 81(3): 536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baldwin KR, Godena VK, Hewitt VL, Whitworth AJ.. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum Mol Genet. 2016; 25(12): 2378–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G. Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci USA. 2010; 107(47): 20523–20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moller A, Bauer CS, Cohen RN, Webster CP, De Vos KJ. Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum Mol Genet. 2017; 26(23): 4668–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hirano A, Nakano I, Kurland LT, Mulder DW, Holley PW, Saccomanno G. Fine structural study of neurofibrillary changes in a family with amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1984; 43(5): 471–480. [DOI] [PubMed] [Google Scholar]
- 33. Corbo M, Hays AP.. Peripherin and neurofilament protein coexist in spinal spheroids of motor neuron disease. J Neuropathol Exp Neurol. 1992; 51(5): 531–537. [DOI] [PubMed] [Google Scholar]
- 34. Rouleau GA, Clark AW, Rooke K, et al.. SOD1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann Neurol. 1996; 39(1): 128–131. [DOI] [PubMed] [Google Scholar]
- 35. Sasaki S, Iwata M.. Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis. Neurology. 1996; 47(2): 535–540. [DOI] [PubMed] [Google Scholar]
- 36. Zhang B, Tu P, Abtahian F, Trojanowski JQ, Lee VM. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol. 1997; 139(5): 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ligon LA, LaMonte BH, Wallace KE, Weber N, Kalb RG, Holzbaur EL. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport. 2005; 16(6): 533–536. [DOI] [PubMed] [Google Scholar]
- 38. Williamson TL, Cleveland DW.. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999; 2(1): 50–56. [DOI] [PubMed] [Google Scholar]
- 39. Pant HC, Veeranna Grant P. Regulation of axonal neurofilament phosphorylation. Curr Top Cell Regul. 2000; 36: 133–150. [DOI] [PubMed] [Google Scholar]
- 40. Tsang YM, Chiong F, Kuznetsov D, Kasarskis E, Geula C. Motor neurons are rich in non-phosphorylated neurofilaments: cross-species comparison and alterations in ALS. Brain Res. 2000; 861(1): 45–58. [DOI] [PubMed] [Google Scholar]
- 41. Sternberger LA, Sternberger NH.. Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc Natl Acad Sci USA. 1983; 80(19): 6126–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Delisle MB, Carpenter S. Neurofibrillary axonal swellings and amyotrophic lateral sclerosis. J Neurol Sci. 1984; 63(2): 241–250. [DOI] [PubMed] [Google Scholar]
- 43. Julien JP. A role for neurofilaments in the pathogenesis of amyotrophic lateral sclerosis. Biochem Cell Biol. 1995; 73(9-10): 593–597. [DOI] [PubMed] [Google Scholar]
- 44. Carpenter S. Proximal axonal enlargement in motor neuron disease. Neurology. 1968; 18(9): 841–851. [DOI] [PubMed] [Google Scholar]
- 45. Munoz DG, Greene C, Perl DP, Selkoe DJ. Accumulation of phosphorylated neurofilaments in anterior horn motoneurons of amyotrophic lateral sclerosis patients. J Neuropathol Exp Neurol. 1988; 47(1): 9–18. [DOI] [PubMed] [Google Scholar]
- 46. Leigh PN, Dodson A, Swash M, Brion JP, Anderton BH. Cytoskeletal abnormalities in motor neuron disease. An immunocytochemical study. Brain. 1989; 112(Pt 2): 521–535. [DOI] [PubMed] [Google Scholar]
- 47. Murayama S, Bouldin TW, Suzuki K. Immunocytochemical and ultrastructural studies of upper motor neurons in amyotrophic lateral sclerosis. Acta Neuropathol. 1992; 83(5): 518–524. [DOI] [PubMed] [Google Scholar]
- 48. Yucel Y, Gupta N.. Glaucoma of the brain: a disease model for the study of transsynaptic neural degeneration. Prog Brain Res. 2008; 173: 465–478. [DOI] [PubMed] [Google Scholar]
- 49. Yu DY, Cringle SJ, Balaratnasingam C, Morgan WH, Yu PK, Su EN. Retinal ganglion cells: energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog Retin Eye Res. 2013; 36: 217–246. [DOI] [PubMed] [Google Scholar]
- 50. Bagheri N, Bell BA, Bonilha VL, Hollyfield JG. Imaging human postmortem eyes with SLO and OCT. Adv Exp Med Biol. 2012; 723: 479–488. [DOI] [PubMed] [Google Scholar]
- 51. Cedarbaum JM, Stambler N, Malta E, et al.. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999; 169(1-2): 13–21. [DOI] [PubMed] [Google Scholar]
- 52. Krenacs L, Krenacs T, Stelkovics E, Raffeld M. Heat-induced antigen retrieval for immunohistochemical reactions in routinely processed paraffin sections. In: Oliver C, Jamur MC, eds. Immunocytochemical Methods and Protocols, Methods in Molecular Biology, Vol. 588 Totowa, NJ: Humana Press; 2010;103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Magaki S, Hojat SA, Wei B, So A, Yong WH. An introduction to the performance of immunohistochemistry. Methods Mol Biol. 2019; 1897: 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sternberger LA, Harwell LW, Sternberger NH. Neurotypy: regional individuality in rat brain detected by immunocytochemistry with monoclonal antibodies. Proc Natl Acad Sci USA. 1982; 79(4): 1326–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Choi YJ, Di Nardo A, Kramvis I, et al.. Tuberous sclerosis complex proteins control axon formation. Genes Dev. 2008; 22(18): 2485–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mizusawa H, Matsumoto S, Yen SH, Hirano A, Rojas-Corona RR, Donnenfeld H. Focal accumulation of phosphorylated neurofilaments within anterior horn cell in familial amyotrophic lateral sclerosis. Acta Neuropathol. 1989; 79(1): 37–43. [DOI] [PubMed] [Google Scholar]
- 57. LaMonte BH, Wallace KE, Holloway BA, et al.. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002; 34(5): 715–727. [DOI] [PubMed] [Google Scholar]
- 58. Laird FM, Farah MH, Ackerley S, et al.. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci. 2008; 28(9): 1997–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xia CH, Roberts EA, Her LS, et al.. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J Cell Biol. 2003; 161(1): 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ackerley S, Thornhill P, Grierson AJ, et al.. Neurofilament heavy chain side arm phosphorylation regulates axonal transport of neurofilaments. J Cell Biol. 2003; 161(3): 489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ackerley S, Grierson AJ, Brownlees J, et al.. Glutamate slows axonal transport of neurofilaments in transfected neurons. J Cell Biol. 2000; 150(1): 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Magrane J, Manfredi, G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antiox Redox Signal. 2009; 11(7): 1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sasaki S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2011; 70(5): 349–359. [DOI] [PubMed] [Google Scholar]
- 64. Neumann M, Sampathu DM, Kwong LK, et al.. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006; 314(5796): 130–133. [DOI] [PubMed] [Google Scholar]
- 65. Deng HX, Zhai H, Bigio EH, et al.. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol. 2010; 67(6): 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Deng HX, Bigio EH, Zhai H, et al.. Differential involvement of optineurin in amyotrophic lateral sclerosis with or without SOD1 mutations. Arch Neurol. 2011; 68(8): 1057–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Williams KL, Warraich ST, Yang S, et al.. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol Aging. 2012; 33(10): 2527.e3–10. [DOI] [PubMed] [Google Scholar]
- 68. Keller BA, Volkening K, Droppelmann CA, Ang LC, Rademakers R, Strong MJ. Co-aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathol. 2012; 124(5): 733–747. [DOI] [PubMed] [Google Scholar]
- 69. Wong TY, Klein R, Couper DJ, et al.. Retinal microvascular abnormalities and incident stroke: the Atherosclerosis Risk in Communities Study. Lancet. 2001; 358(9288): 1134–1140. [DOI] [PubMed] [Google Scholar]
- 70. Fisher JB, Jacobs DA, Markowitz CE, et al.. Relation of visual function to retinal nerve fiber layer thickness in multiple sclerosis. Ophthalmology. 2006; 113(2): 324–332. [DOI] [PubMed] [Google Scholar]
- 71. Guo L, Duggan J, Cordeiro MF. Alzheimer's disease and retinal neurodegeneration. Curr Alzheimer Res. 2010; 7(1): 3–14. [DOI] [PubMed] [Google Scholar]
- 72. Archibald NK, Clarke MP, Mosimann UP, Burn DJ. The retina in Parkinson's disease. Brain. 2009; 132(Pt 5): 1128–1145. [DOI] [PubMed] [Google Scholar]
- 73. London A, Benhar I, Schwartz M. The retina as a window to the brain-from eye research to CNS disorders. Nat Rev Neurol. 2013; 9(1): 44–53.. [DOI] [PubMed] [Google Scholar]
- 74. Ringelstein M, Albrecht P, Sudmeyer M, et al.. Subtle retinal pathology in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2014; 1(4): 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rohani M, Meysamie A, Zamani B, Sowlat MM, Akhoundi FH. Reduced retinal nerve fiber layer (RNFL) thickness in ALS patients: a window to disease progression. J Neurol. 2018; 265(7): 1557–1562. [DOI] [PubMed] [Google Scholar]
- 76. Simonett JM, Huang R, Siddique N, et al.. Macular sub-layer thinning and association with pulmonary function tests in amyotrophic lateral sclerosis. Sci Rep. 2016; 6: 29187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Volpe NJ, Simonett J, Fawzi AA, Siddique T. Ophthalmic manifestations of amyotrophic lateral sclerosis (An American Ophthalmological Society thesis). Trans Am Ophthalmol Soc. 2015; 113: T121–T1215. [PMC free article] [PubMed] [Google Scholar]
- 78. Rojas P, de Hoz R, Ramirez AI, et al.. Changes in retinal OCT and their correlations with neurological disability in early ALS patients, a follow-up study. Brain Sci. 2019; 9(12): 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Weydt P, Oeckl P, Huss A, et al.. Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann Neurol. 2016; 79(1): 152–158. [DOI] [PubMed] [Google Scholar]