

Abstract
Diabetic skin has impaired wound healing properties following injury. We have further shown that diabetic skin has weakened biomechanical properties at baseline. We hypothesize that the biomechanical properties of diabetic skin decline during the progression of the diabetic phenotype, and that this decline is due to the dysregulation of miR-29a, resulting in decreased collagen content. We further hypothesize that treatment with mesenchymal stem cells (MSCs) may improve diabetic wound healing by correction of the dysregulated miR-29a expression. We analyzed the biomechanical properties, collagen gene expression, collagen protein production, and miR-29a levels in skin harvested from 6 to 18 week old mice during the development of the diabetic phenotype. We also examined the correction of these impairments by both MSC treatment and the inhibition of miR-29a. Diabetic skin demonstrated a progressive impairment of biomechanical properties, decreased collagen content, and increased miR-29a levels during the development of the diabetic phenotype. MSC treatment decreased miR-29a levels, increased collagen content, and corrected the impaired biomechanical properties of diabetic skin. Additionally, direct inhibition of miR-29a also increased collagen content in diabetic skin. This decline in the biomechanical properties of diabetic skin during the progression of diabetes may increase the susceptibility of diabetic skin to injury and miR-29a appears to play a key role in this process.
Diabetes mellitus is a worldwide pandemic. An estimated 29 million Americans suffer from diabetes, which results in substantial morbidity and mortality as well as annual health care expenditures in excess of $176 billion (Data from the 2014 National diabetes fact sheet; available at http://www.cdc.gov/diabetes/data/statistics/2014Statistics-Report.html). This significant healthcare burden is the result of a progressive disease process and associated complications, particularly the development of chronic wounds and infections.1 Prolonged, uncontrolled diabetes can lead to impaired skin integrity, neuropathy, and increased susceptibility to bacterial infection, all of which predispose the skin to injury.2-5 Chronic wounds are the leading cause for hospital admission in diabetic patients and are the number one cause of nontraumatic lower extremity amputations.6 Current clinical therapies focus on foot hygiene and preventive measures, as 85% of all amputations in diabetes are thought to be preventable. Preventive strategies have been proven effective; however, they often fail secondary to poor patient compliance and the persistent pathophysiologic derangements of diabetic wounds.7,8 Given the projected increase in the burden of disease attributable to chronic diabetic wounds, innovative strategies are needed for the both prevention and treatment of this growing clinical problem.
Numerous studies have shown that the biomechanical properties of diabetic skin are impaired after injury.9 However, few studies have assessed the biomechanical properties of diabetic skin at baseline. Previously, we have shown that both murine and human diabetic skin are biomechanically inferior, with both lower elasticity and decreased maximum stress relative to nondiabetic skin at baseline.2 We have further demonstrated that protein levels of type I collagen—the main component of the skin’s extracellular matrix responsible for its structural integrity—are lower in diabetic skin, despite their elevated gene expression.2 This discrepancy suggests a possible defect in the regulation of collagen protein synthesis at the posttranscriptional level, as would be observed due to the effect of microRNAs on collagen synthesis.
MicroRNAs are a class of small, noncoding RNA molecules that inhibit gene expression at the posttranscriptional level.10 Complementary binding of microRNAs to the 3′-untranslated region of their target messenger RNA (mRNA) results in posttranscriptional repression and/or mRNA degradation.11 In particular, microRNA-29a (miR-29a) has been shown to play an important role in the posttranscriptional regulation of collagen production and is inversely associated with collagen content.12,13
Mesenchymal stem cells (MSCs) are multipotent cells that play an important role in tissue repair by promoting the production of growth factors, cytokines, collagens, and the correction of dysregulated miRNAs expression, while also maintaining the ability to differentiate into multiple cell lineages; these characteristics form the foundation for the therapeutic role of MSCs in wound healing.14-18 In mice, MSCs have been shown to correct impaired diabetic wound healing; however, there has not yet been an evaluation of the impact of MSCs on the biomechanical properties of diabetic skin.19,20
Thus, we hypothesize that the biomechanical properties of diabetic skin decline with the development and progression of the diabetic phenotype and that treatment with MSCs improves the impaired biomechanical properties. Furthermore, we hypothesize that treatment with MSCs improves the biomechanical properties of diabetic skin via the correction of abnormal miR-29a expression. To test these hypotheses, we characterized the biomechanical properties of diabetic and nondiabetic human skin, and the biomechanical properties of diabetic and nondiabetic murine skin before and after treatment with MSCs in 6–18 week old mice; furthermore, we defined the expression profile of miR-29a and its target gene in human and murine, diabetic and nondiabetic skin and evaluated the expression of miR-29a before and after treatment with either MSCs or a lentivirus expressing miR-29a inhibitor in diabetic and nondiabetic murine skin.
METHODS
Animals
All experimental protocols were approved by the Institutional Animal Care and Use Committee at University of Colorado Denver—Anschutz Medical Campus and followed the guidelines described in the NIH Guide for the Care and Use of Laboratory Animals. Age-matched, female, genetically diabetic C57BKS.Cg-m/Leprdb/J mice and heterozygous, nondiabetic, female controls were used in these experiments. In our study population, the development of the diabetic phenotype was monitored by serial measurements of weight and serum blood glucose, with blood glucose >200 mg/dL considered confirmatory for a diagnosis of diabetes.
Murine skin biomechanical testing
To evaluate the biomechanical characteristics of skin during the development of the diabetic phenotype, biomechanical testing was performed on skin from 4-, 8-, 12-, 16-, and 18-weeks-old diabetic and nondiabetic female mice (n = 5 per group). Biomechanical testing was also done 28 days after either MSC or PBS (vehicle or control) treatment of skin from 12-week-old diabetic and nondiabetic mice. Skin samples immediately underwent testing after harvest; all samples had their subcutaneous tissue removed, and a uniform, dumbbell-shaped testing unit was stamped out using well-established, previously described techniques.21 Cranial-caudal orientation was preserved. Two Verhoeff stain lines were placed on either end of the dumbbell shape, demarcating the gauge length of the testing area. We measured the cross-sectional area of each sample first, by using a custom laser-based device that translates the tissue underneath a charged, coupled device laser into a measurement of thickness. Position data using two linear variable differential transformers were used in conjunction with the thickness data to calculate an average cross-sectional area within the gauge length. Thus, the mean cross-sectional area of the sample tested was calculated as precisely as possible to ensure that an accurate modulus and maximum stress were calculated. The ends of the testing unit were fixed between sandpaper using a cyanoacrylate adhesive to prevent slipping. The testing unit was mounted in custom designed fixtures and tested in tension using an Instron 5543 test frame (Instron, Norwood, MA). Each testing unit underwent a previously established protocol.22 In short, the testing unit was submerged in a 37 °C PBS bath, preloaded to 0.005 Newtons (N), held for 120 seconds, and then subjected to a constant increase in force until the sample failed (as evidenced by tearing of the skin) and was no longer able to hold tension. Using the stain lines, local tissue strain was measured optically and analyzed using Matlab software (The MathWorks, Natick, MA).23 Maximum stress to failure was calculated using linear regression from the linear region of the stress-strain curve. The modulus of elasticity was calculated as the slope of the stress-strain curve within the elastic region of the ramp to failure.
MSC treatment of diabetic and nondiabetic skin
MSCs were isolated from bone marrow of transgenic adult mice expressing green fluorescent protein as previously described.24 MSC identities were confirmed by staining for cell surface markers, as previously described.25 An 8 mm2 skin area on the back of 12 weeks old diabetic and nondiabetic female mice was treated with 40 μL of an intradermal injection of either 1 × 106 MSC or PBS alone (n = 5 per group). The site was marked with India ink for later identification. Twenty-eight days after injection, mice were euthanized with inhaled CO2 prior to cervical dislocation and the skin was harvested for analysis.
MiR-29a inhibition in vivo
An 8 mm2 area on the back of 12 weeks old diabetic and nondiabetic female mice was treated with an intradermal injection of 107 PFU/mL lentivirus expressing miR-29a inhibitor (miR-29ai) or a lentivirus expressing a control miR (miR-CTL) in a total volume of 40 μL. The site was marked with India ink for later identification. Seven days after injection, mice were euthanized and skin harvested for analysis.
Extraction of collagen and Western blot for collagens I and III
Collagen was extracted using previously published methods.26,27 Skin samples were cut into 1-mm pieces and homogenized in a 0.5 M acetic acid solution containing 1× protease inhibitor cocktail and 5 mmol/L EDTA. Homogenates were then centrifuged for 4 hours at 4 °C at 16.1 × 103g. The lipid layer was aspirated and the process repeated. The protein concentration of the supernatant, which contains total collagen, was quantified and standardized to 1 μg/2 μL using a sample buffer and was subsequently boiled at 95 0C for 5 minutes. Samples were run with standardized quantities (positive control) of collagen I and collagen III (Abcam 7533 and Abcam 7535, respectively, Abcam, Cambridge, United Kingdom). Tris-acetate gels (3–8%) were run at 150 V for 1 hour and then transferred at 30 V overnight at 4 °C. The membranes were blocked 1:1 Odyssey blocking buffer-1X-PBS for 1 hour at room temperature then incubated with collagen I or III antibodies (Abcam, Cambridge, MA) at a 1:2000 and 1:1000 dilution, respectively, for 1 hour at room temperature, washed with 1×-PBS-0.1%Tween, and subsequently incubated with Li-Cor secondary antibody (anti-rabbit IgG; LiCor, Lincolin, NE) at 1:10,000 for 1 hour at room temperature. The immunoreactive bands were quantified using the Li-COR Odyssey infrared imaging system. Collagen I and III protein expression was normalized to the expression of the respective positive control.
Histology
Skin samples were immediately fixed in 10% neutral buffered formalin (Sigma-Aldrich, St. Louis, MO). The tissue was then processed by using a histoprocessor (Leica TP1050). Paraffin sections (4 μm thick) were obtained and mounted on slides (Fisher Scientific, Pittsburgh, PA) and incubated overnight at 11.1 °C. Slides were deparaffinized in xylene (3 × 10 min) followed by graded ethanol rehydration (2 × 100%, 95%, 75%) to distilled water. Masson’s trichrome staining was then performed. Two blinded observers analyzed collagen deposition. A total of 10 high-power fields per slide were examined.
Real time PCR analysis
Total RNA from samples of skin from diabetic and non-diabetic mice and humans was extracted and purified after homogenization in TRIzol (Invitrogen, Life Technologies, Carlsbad, CA) and following the manufacturer’s instructions. RNA was converted into cDNA using the SuperScript First-Strand Synthesis System (Invitrogen, Life Technologies). The miRNA Reverse transcriptase kit was used to prepare cDNA for miRNA. Real time quantitative PCR was performed with the BioRad CFX-960 thermal cycler (BioRad, Hercules, CA). Primers for collagen Iα2 (Col1α2), collagen IIIα1 (Col3α1), and miR-29a were amplified using the TaqMan gene expression assay (Applied Biosystems, Foster City, CA). Internal normalization was achieved by using the housekeeping gene (18s for gene expression and U6 for miRNA expression). Samples (n = 5 per group) were amplified in triplicate and results were averaged for each individual sample. Results are reported as mean ± SEM.
Acquisition of human skin
All research was approved through the institutional review board at the University of Colorado—Anschutz Medical Campus. Samples were obtained through National Disease Research Interchange. Human skin samples were collected from the anterior portion of the lower extremity of individuals both with and without Type 2 diabetes (n = 5 per group); these samples were immediately flash frozen in liquid nitrogen. Samples were obtained from patients who were 55–75 years of age and were without known comorbid malignancy or history of either radiation or chemotherapy. Clinical data was not available regarding the gender or duration of diabetes in these individuals.
Statistical analysis
Results are expressed as mean ± SEM. Statistical significance involving single comparisons was determined by a Student’s t test. For multiple comparisons, ANOVA followed by an appropriate post hoc test as noted was performed. A p < 0.05 was taken as significant. All statistics were performed using Graphpad Prism Software (LaJolla, CA).
RESULTS
The onset of diabetes starts at 6 weeks of age in diabetic mice
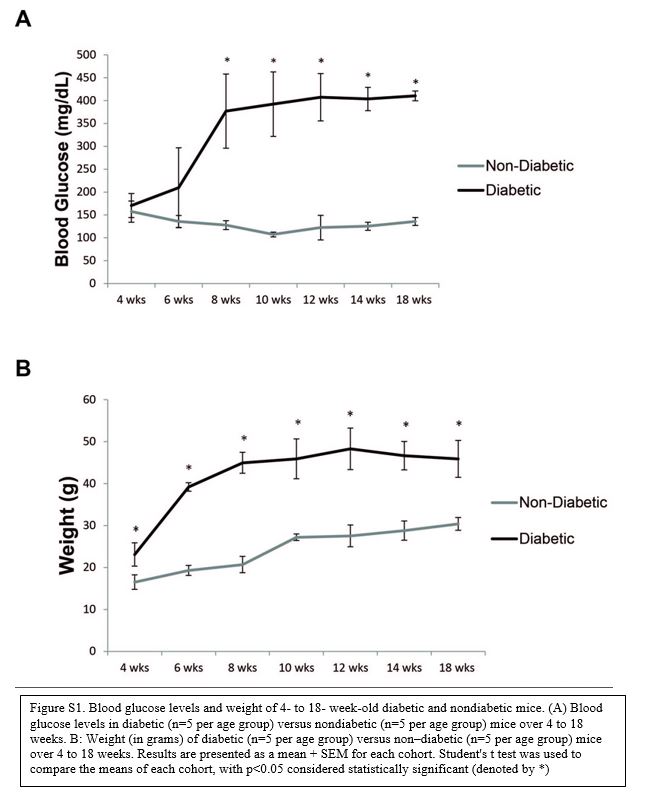
Weight and blood glucose levels measurement in 4-, 6-, 8-, 10-, 12-, 16-, and 18-weeks-old diabetic and nondiabetic mice showed a progressive increase in the weight and glucose levels in diabetic mice with a significant difference at each time point compared with nondiabetic mice starting at week 6 (Supporting Information Figure S1), with diabetic mice developing average serum glucose levels >200 mg/dL after 6 weeks of age. The increase in these levels reflects the development and progression of the diabetic phenotype in the diabetic mice.
The biomechanical properties of diabetic skin decline with the development of the diabetic phenotype
Dorsal skin from these mice was exposed to mechanical stress (n = 5 per age group). The biomechanical analysis was initially performed in mice 4–18 weeks of age; no significant difference in the elasticity, strength, or maximum load was noted when comparing diabetic and nondiabetic mice at 4 weeks of age, suggesting that the diabetic phenotype is not established enough to impact the components of the extracellular matrix in this age group. Beyond 8 weeks of age, diabetic murine skin demonstrated a consistently decreased ability to resist mechanical stress when compared with nondiabetic skin. There was a trend for increased maximum stress to failure in diabetic skin compared to nondiabetic skin at 4 weeks, prior to development of the diabetic phenotype; however, from week 8 to week 18, diabetic skin showed significantly lower maximum stress to failure, when compared with nondiabetic skin (Figure 1A). The elastic modulus was significantly higher in diabetic skin at 4 weeks of age (9.14 ± 4.84 vs. 2.72 ± 1.7 MPa, p = 0.01); however, by 8 weeks (3.82 ± 0.3 vs. 7.14 ± 2.46 MPa, p = 0.06) and up to 18 weeks (6.0 ± 1.8 vs. 7.36 ± 1.2 MPa, p = 0.03), nondiabetic skin demonstrated superior biomechanical strength (Figure 1B).
Figure 1.
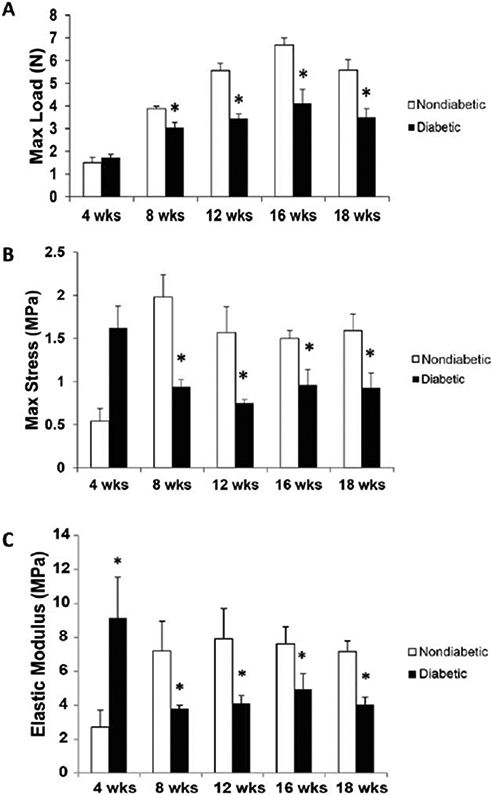
Biomechanical properties of diabetic and nondiabetic murine skin at 4, 8, 12, 16, and 18 weeks of age. (A) The maximum load sustained prior to failure (N) in diabetic versus nondiabetic skin samples over 4–18 weeks of age. (B) The maximum stress to failure (MPa) in diabetic versus nondiabetic skin samples over 4–18 weeks of age. (C) The elastic modulus (MPa) measured in diabetic versus nondiabetic skin samples over 4–18 weeks of age. Data is presented as a mean + standard error of the mean (SEM) for each cohort. Student’s t test was used to compare non-diabetic skin vs. diabetic skin at each time point, with *p < 0.05. Abbreviation: Max = Maximum.
Collagen protein expression is decreased in diabetic skin
Real time PCR analysis of collagen gene expression demonstrated significant differences between diabetic and non-diabetic murine skin. Col1α2 and Col3α1 gene expression were higher in the diabetic murine skin at all age-points when compared to nondiabetic murine skin (Figure 2A and B). However, the protein expression of Col-I was lower in diabetic skin at 18 weeks when compared with nondiabetic skin at 18 weeks or diabetic skin at 4 weeks (Figure 2C, D). Col-III protein levels (upper band; marked with black arrows; 130 KDa), however, were much higher in both diabetic and nondiabetic murine skin at 18 weeks when compared with the 4 week time point. In addition, we detected the presence of a lower molecular weight (MW) band of Col-III (~120 KDa). Interestingly, this lower MW form of Col-III seems to be predominant in both diabetic and nondiabetic skin at a younger age.
Figure 2.
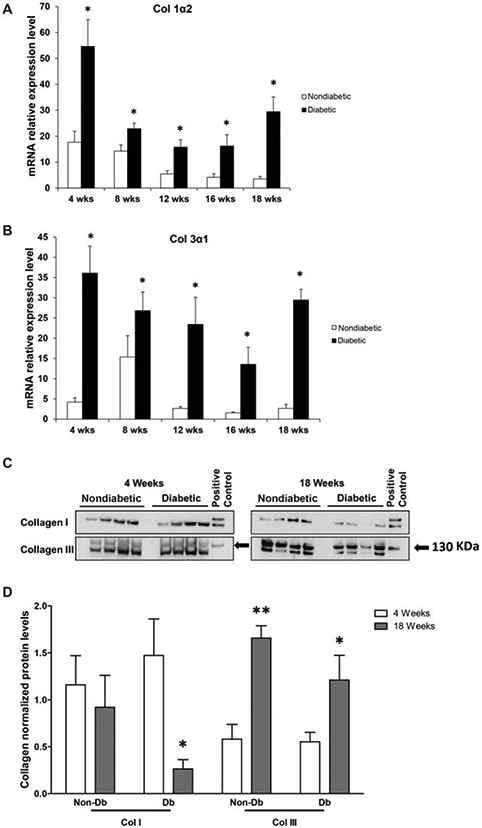
Collagen gene and protein expression in diabetic and nondiabetic murine skin. (A) Relative gene expression for collagen Iα2 in skin samples from diabetic (n = 5) versus nondiabetic (n = 5) mice at 4 and 18 weeks of age. (B) Relative gene expression for collagen IIIα1 as measured in skin samples from diabetic (n = 5) versus nondiabetic (n = 5) mice at 4 and 18 weeks of age. (C) Collagen I and III (upper band; black arrows) levels as demonstrated by Western blots obtained from skin samples from age-matched, nondiabetic, and diabetic mice at 4 and 18 weeks of age. (D) Collagen I and III protein levels as quantified by Western blot. These findings are representative of five independent experiments. Data is presented as a mean + SEM for each cohort. Student’s t test was used to compare nondiabetic skin to diabetic skin at each time point, with *p < 0.05; **p < 0.001.
MiR-29a expression is increased in diabetic skin
Real-time PCR analysis showed increased miR-29a expression levels in murine diabetic skin with the development and progression of the diabetic phenotype (p < 0.001; Figure 3A). MiR-29a expression was significantly higher in the diabetic skin at both 12 weeks (36.6 ± 4.0 vs. 5.7 ± 1.3, p < 0.001) and 18 weeks (54.7 ± 5.2 vs. 29.1 ± 3.3, p < 0.001) compared with both nondiabetic skin and diabetic skin from earlier age points. Furthermore, Figure 3B demonstrates that miR-29a expression in human diabetic skin is significantly up-regulated compared with miR-29a levels in nondiabetic human skin.
Figure 3.
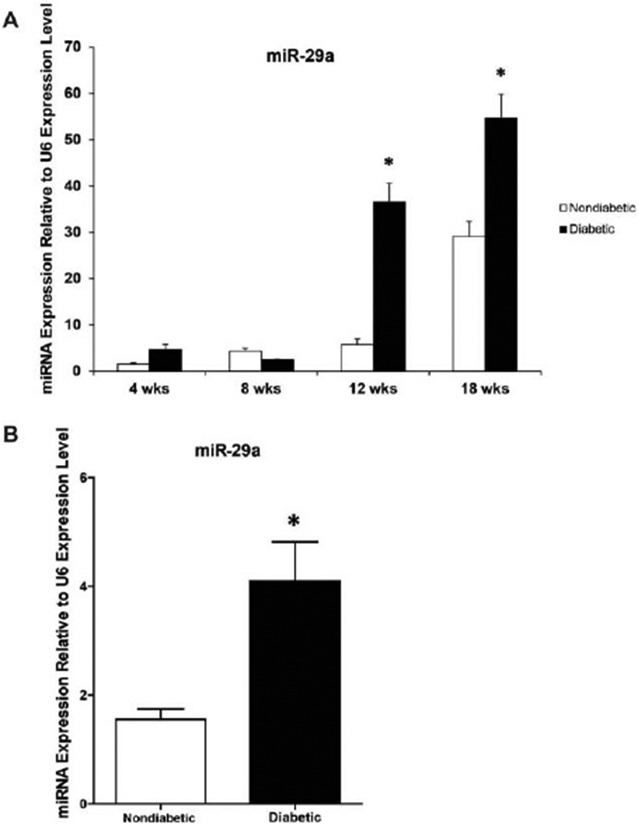
MiR-29a gene expression in diabetic and non-diabetic murine (A) and human (B) skin. (A) Real-time quantitative PCR analysis of miR-29a levels in murine diabetic and nondiabetic skin at different age points. (B) Real-time quantitative PCR analysis of miR-29a levels in human diabetic and nondiabetic skin. MiR-29a gene expression was calculated after normalizing with U6. Results are presented as a mean- + SEM for each cohort. Student’s t test was used to compare nondiabetic skin to diabetic skin at each time point, with *p < 0.05.
MSC treatment corrected the impaired biomechanical properties of diabetic skin
While there was no significant difference in the maximum load tolerated by murine diabetic skin, treatment with MSCs demonstrated a significant improvement in elasticity and strength, as shown by the increased modulus (10.01 ± 2.06 vs. 1.82 ± 0.39) and maximum stress to failure (1.57 ± 0.21 vs. 0.62 ± 0.06) compared with PBS-treated skin (Figure 4A). Treatment of nondiabetic skin with MSC did not affect the maximum stress or elastic modulus, however it did improve the maximum load (Figure 4B).
Figure 4.
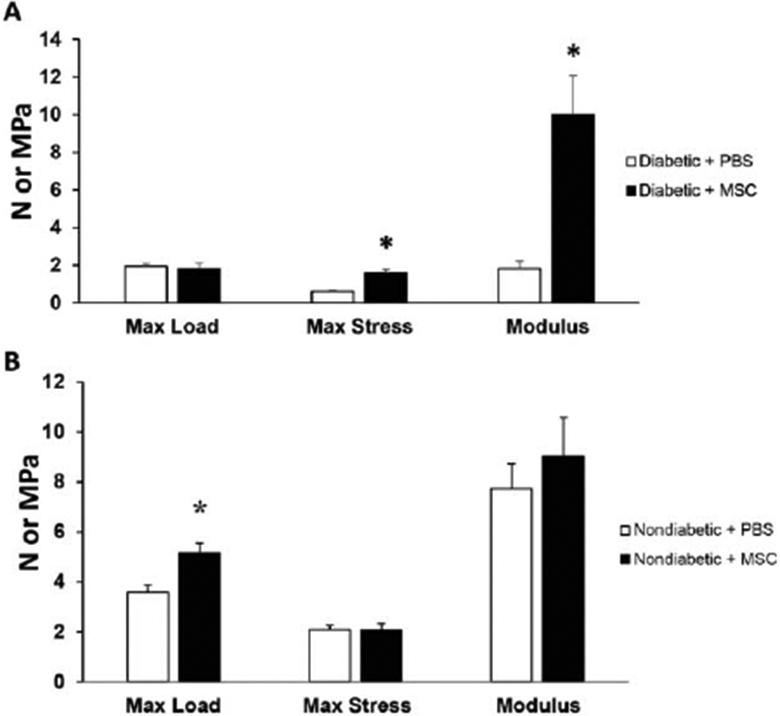
Effect of MSC treatment on the biomechanical properties of diabetic and nondiabetic murine skin. (A) The maximum load (N), maximum stress (MPa), and elastic modulus (MPa) measured in skin samples from 12 weeks old diabetic mice previously treated with 1 × 106 MSC (black bars) or PBS (white bars) for 28 days. (B) The maximum load (N), maximum stress (MPa), and elastic modulus (MPa) measured in skin samples from 12-week-old nondiabetic mice treated with 1 × 106 MSC (black bars) or PBS (white bars) for 28 days. Results are presented as a mean + SEM for each cohort. Student’s t test was used to compare the maximum load, maximum stress, and elastic modulus for nondiabetic skin treated with PBS vs. nondiabetic skin treated with MSC or diabetic skin treated with PBS vs. diabetic skin treated with MSC, with *p < 0.05. Max, Maximum.
MSC increased collagen levels in diabetic skin
Lower Col-I protein expression was observed in diabetic murine skin treated with PBS when compared to nondiabetic skin treated with PBS. However, in diabetic skin treated with MSCs, Col-I protein levels increased to the levels observed in nondiabetic murine skin treated with PBS (Figure 5A and B). Col-III protein levels were increased in the diabetic skin treated with MSC (Figure 5A and B). Masson’s Trichrome staining showed that collagen levels in diabetic skin (PBS) are significantly lower than in nondiabetic skin (PBS). 28 days following MSC treatment, collagen levels where increased in diabetic skin compared with the diabetic skin treated with PBS. There was no significant difference in nondiabetic skin treated with MSC compared with PBS (Figure 5C).
Figure 5.

Effect of MSC treatment on collagen and miR-29a levels in diabetic and nondiabetic murine skin. (A) Western blot depicting collagen I and III protein levels in 16-week-old nondiabetic and diabetic murine skin treated with MSCs or PBS for 28 days. (B) Quantification of collagen I and III protein by Western blot. C: Masson’s trichrome stain of representative sections of diabetic and nondiabetic skin after 28 days of treatment with 1 × 106 MSC (n = 5) or PBS (n = 5) and graphical representation of collagen (blue stain) content quantification from masson’s trichrome staining. (D) Relative expression of miR-29a in diabetic and nondiabetic murine skin treated with MSC or PBS for 28 days. Data was analyzed by one-way ANOVA followed by Bonferroni’s post hoc test (*p < 0.05; **p < 0.001; ***p < 0.0001; ns = nonsignificant).
Mir-29a levels were decreased with MSC treatment
Real time PCR analysis showed decreased miR-29a expression in the MSC-treated diabetic skin compared with the PBS-treated group 28 days following treatment. A similar decrease in miR-29a expression was seen in the nondiabetic skin treated with MSC (Figure 5D).
MiR-29a inhibition increases collagen expression in diabetic skin
Following injection with lenti-miR-29ai (miR-29a inhibitor), levels of miR-29a in the diabetic skin (Figure 6A) were decreased to the levels observed in nondiabetic skin. Collagen protein expression was increased in the lenti-miR-29a inhibitor treated group compared with the control group (Figure 6B). Masson’s trichrome staining showed that miR-29a inhibition increased collagen levels in diabetic skin compared with control (Figure 6C).
Figure 6.

Effect of miR-29a inhibition on miR-29a levels and collagen protein expression in diabetic and nondiabetic murine skin. (A) Relative expression of miR-29a in diabetic or nondiabetic skin 7 days following treatment with either lenti-miR29a inhibitor (miR-29a-i) or lenti-miR-control (miR-CTL). (B) Western blot analysis and quantification of collagen I protein expression in diabetic or nondiabetic murine skin after 7 days of treatment with miR-29a-i or miR-CTL. Results are presented as a mean + SEM for each cohort. (C) Masson’s trichrome stain of representative sections of diabetic and nondiabetic skin after treatment with lenti-miR-29a inhibitor (n = 5) or lenti-miR-CTL (n = 5) for 7 days and graphical representation of collagen (blue stain) content quantification from Masson’s trichrome staining. Data was analyzed by one-way ANOVA followed by Bonferroni’s post hoc test (*p < 0.05; **p < 0.001; ***p < 0.0001; ns=nonsignificant).
DISCUSSION
We have demonstrated that the biomechanical properties of diabetic skin decline significantly with the development and progression of the diabetic phenotype, and we have further shown that this decline in the biomechanical properties of diabetic skin is associated with decreased levels of collagen I. This decrease in collagen I protein content is despite significant increases in collagen I gene expression, which we have shown may be attributed to increasing levels of miR-29a inhibiting translation at the posttranscriptional level. We also show that inhibition of miR-29a in diabetic skin decreases miR-29a expression to the levels observed in nondiabetic skin at baseline, while subsequently increasing collagen levels. Results further demonstrate that treatment of murine diabetic skin with MSCs corrects the impaired biomechanical properties, the elevated levels of miR-29a, and the decreased collagen levels observed in diabetic skin at baseline. These results suggest that the correction of the diabetic phenotype observed in skin treated with MSCs is mediated by inhibition of miR-29a. In addition to our previous work describing the impaired biomechanical properties of human diabetic skin,2 we now show that human diabetic skin demonstrates elevated miR-29a expression, as was observed in murine diabetic skin. To our knowledge, this is the first evidence that the biomechanical properties of diabetic skin decline with the progression of the diabetic phenotype and that up-regulated expression of miR-29a may be partly responsible for the low collagen levels observed in diabetic skin. Furthermore, this is the first evaluation of miR-29a expression in human skin.
Factors that place diabetic skin at an increased risk of injury may play a significant role in the pathogenesis of chronic diabetic wounds. Normal wound healing requires that the skin surrounding the wound be capable of providing structural support for the formation of granulation tissue. However, diabetic skin demonstrates impaired biomechanical properties, disorganized extracellular matrix, and chronic inflammation following injury.28 The impaired wound healing response observed in diabetic patients has been attributed to alterations in growth factor production, cellular recruitment, angiogenesis, and extracellular matrix production and degradation.28 However, little research has focused on baseline impairments in diabetic skin that may predispose to injury. Very few studies have sought to characterize the baseline biomechanical properties of intact diabetic skin; instead, the majority of studies have examined it in the wounded state. The limited number of studies investigating baseline biomechanical properties of diabetic skin describe increased stiffness and decreased elasticity.29,30 In this study, we not only examine the baseline biomechanical characteristic of diabetic skin, confirming the decreased tensile strength of diabetic skin, but also demonstrate this decrease in tensile strength is associated with decreased collagen deposition attributed to posttranscriptional modification of collagen expression, likely due to increased levels of miR-29a. The decreased elasticity, tensile strength, and collagen deposition in baseline diabetic skin may contribute to an increased predisposition to injury and the subsequent poor wound healing observed in diabetic skin.
Collagen is the main component of the skin’s extracellular matrix and plays an important role in its integrity. The maintenance of collagen protein levels is regulated by a complex balance between synthesis and degradation, including the activity of matrix metalloproteinases and posttranscriptional regulation by microRNAs. Collagen is a validated target of miR-29a.31,32 As an inhibitor of collagen expression, miR-29a expression is typically reduced in fibrosis-associated disease, and has been shown to play a key role in cardiac,33 pulmonary,32 and kidney fibrosis,34,35 and fibroblast cell lines from systemic sclerosis also tend to display reduced miR-29 expression.36 Forced expression of miR-29 in such cell lines significantly decreased the levels of mRNA and protein for type I and type III collagen.36 Moreover, systemic delivery of miR-29a can attenuate liver fibrosis in carbon tetrachloride (CCl4)-induced mice34,37 and renal fibrosis by decreasing collagen expression.35 In patients with hypertrophic cardiomyopathy, circulating miR-29a is proposed to be the only biomarker for both hypertrophy and fibrosis.38 Together, these findings support miR-29a is the key regulator of collagen expression. We have sought to examine one aspect of this complex balance, demonstrating that miR-29a expression is increased in the diabetic phenotype, with a concomitant decrease in collagen protein content. Conversely, miR-29a inhibition was successful in decreasing miR-29a expression in diabetic skin and was further associated with an increase in collagen protein content.
Previous studies have shown that MSCs can accelerate healing in both nondiabetic and diabetic mouse models,25,39,40 and have also been shown to improve healing in nondiabetic human skin.39 This correction with MSC therapy involves multiple aspects of the wound healing response, including reepithelialization, granulation tissue formation, and neovascularization.25,40 Many of the effects of MSC treatment are thought to be due to the release of soluble factors that regulate the local cellular response, affecting multiple signaling pathways.41-43 One of the novel findings of this study is that diabetic skin treated with MSCs also show significantly improved biomechanical properties. This improvement is associated with a down-regulation in miR-29a expression and up-regulation in the protein levels of its target gene, collagen. These data provide evidence that treatment with MSCs enhances the ability of diabetic skin to resist stress by reversing the impaired miR-29a and collagen levels seen in baseline diabetic skin.
These results indicate promising directions for pursuit of novel therapies for prevention of diabetic wounds; however, our study design does have several limitations. While in vivo analysis using human tissues would be optimal, there are insurmountable ethical considerations regarding application of a novel therapy to a human subject without prior evaluation using animal models. As such, our study is limited by use of an animal model of diabetes; however, our model used diabetic C57BKS.Cg-m/Leprdb/J mice, which have been recommended as the strain of choice for evaluation of similar diabetic complications, including diabetic neuropathy.44 Finally, we acknowledge that the dermal extracellular matrix is composed of multiple elements, with collagen being only one component. However, a comprehensive analysis of the impact of the diabetic phenotype on all components of the dermal matrix is beyond the scope of this analysis.
Our results suggest that therapies directed at miR-29a regulation may improve the impaired biomechanical properties of diabetic skin, with the goal of preventing the initial injuries leading to chronic diabetic wounds. As the global burden of diabetes reaches pandemic proportions, novel therapies are needed to improve preventative strategies to minimize the burden of disease attributable to chronic diabetic wounds.
Supplementary Material
Figure S1. Blood glucose levels and weight of 4- to 18- week-old diabetic and nondiabetic mice. (A) Blood glucose levels in diabetic (n=5 per age group) versus nondiabetic (n=5 per age group) mice over 4 to 18 weeks. B: Weight (in grams) of diabetic (n=5 per age group) versus non–diabetic (n=5 per age group) mice over 4 to 18 weeks. Results are presented as a mean + SEM for each cohort. Student’s t test was used to compare the means of each cohort, with p<0.05 considered statistically significant (denoted by *).
{kind=link}
ACKNOWLEDGMENTS
Source of Funding: The research presented in this article was supported by National Institutes of Health Diabetes Pathfinder Grant 7DP2 DK-083085-01 to K.W.L.
Glossary
- Col
Collagen
- MSC
Mesenchymal stem cells
- MiR
MicroRNA
Footnotes
Conflict of Interest: The authors have nothing to disclose and declare no conflict of interest.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- 1.Faries PL, Teodorescu VJ, Morrissey NJ, Hollier LH, Marin ML. The role of surgical revascularization in the management of diabetic foot wounds. Am J Surg 2004; 187(5A): 34S–7S. [DOI] [PubMed] [Google Scholar]
- 2.Bermudez DM, Herdrich BJ, Xu J, Lind R, Beason DP, Mitchell ME, et al. Impaired biomechanical properties of diabetic skin implications in pathogenesis of diabetic wound complications. Am J Pathol 2011; 178: 2215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Britland ST, Young RJ, Sharma AK, Clarke BF. Association of painful and painless diabetic polyneuropathy with different patterns of nerve fiber degeneration and regeneration. Diabetes 1990; 39: 898–908. [DOI] [PubMed] [Google Scholar]
- 4.Cheng HT, Dauch JR, Hayes JM, Hong Y, Feldman EL. Nerve growth factor mediates mechanical allodynia in a mouse model of type 2 diabetes. J Neuropathol Exp Neurol 2009; 68: 1229–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falanga V Wound healing and its impairment in the diabetic foot. Lancet 2005; 366: 1736–43. [DOI] [PubMed] [Google Scholar]
- 6.Reiber GE, Vileikyte L, Boyko EJ, del Aguila M, Smith DG, Lavery LA, et al. Causal pathways for incident lower-extremity ulcers in patients with diabetes from two settings. Diabetes care 1999; 22: 157–62. [DOI] [PubMed] [Google Scholar]
- 7.Stokes J 3rd, Kannel WB, Wolf PA, Cupples LA, D’Agostino RB. The relative importance of selected risk factors for various manifestations of cardiovascular disease among men and women from 35 to 64 years old: 30 years of follow-up in the Framingham Study. Circulation 1987; 75(6 Pt 2): V65–73. [PubMed] [Google Scholar]
- 8.Londahl M, Katzman P, Nilsson A, Hammarlund C. Hyperbaric oxygen therapy facilitates healing of chronic foot ulcers in patients with diabetes. Diabetes Care 2010; 33: 998–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andreassen TT, Oxlund H. The influence of experimental diabetes and insulin treatments on the biochemical properties of rat skin incisional wounds. Acta Chir Scand 1987; 153: 405–9. [PubMed] [Google Scholar]
- 10.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Natarajan R, Putta S, Kato M. MicroRNAs and diabetic complications. J Cardiovasc Trans Res 2012; 5: 413–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Men LL, Xu JY, Du JL, Song GR, Xu L, Yang Y, et al. [Relationship of amyloid-beta with vascular endothelial cell damage induced by diabetic serum]. Zhonghua Yi Xue Za Zhi 2012; 92: 2406–9. [PubMed] [Google Scholar]
- 13.Maurer B, Stanczyk J, Jungel A, Akhmetshina A, Trenkmann M, Brock M, et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis and rheumatism 2010;62(6):1733–43. [DOI] [PubMed] [Google Scholar]
- 14.Seitz O, Schurmann C, Hermes N, Muller E, Pfeilschifter J, Frank S, et al. Wound healing in mice with high-fat diet- or ob gene-induced diabetes-obesity syndromes: a comparative study. Experimental diabetes research 2010;2010:476969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wetzler C, Kampfer H, Stallmeyer B, Pfeilschifter J, Frank S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: prolonged persistence of neutrophils and macrophages during the late phase of repair. J Invest Dermatol 2000; 115: 245–53. [DOI] [PubMed] [Google Scholar]
- 16.Xu J, Wu W, Zhang L, Dorset-Martin W, Morris MW, Mitchell ME, et al. The role of microRNA-146a in the pathogenesis of the diabetic wound-healing impairment: correction with mesenchymal stem cell treatment. Diabetes 2012; 61: 2906–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barry FP. Biology and clinical applications of mesenchymal stem cells. Birth Defects Res Part C Embryo Today: Rev 2003; 69: 250–6. [DOI] [PubMed] [Google Scholar]
- 18.Parikka V, Vaananen A, Risteli J, Salo T, Sorsa T, Vaananen HK, et al. Human mesenchymal stem cell derived osteoblasts degrade organic bone matrix in vitro by matrix metalloproteinases. Matrix Biol 2005; 24: 438–47. [DOI] [PubMed] [Google Scholar]
- 19.Crevensten G, Walsh AJ, Ananthakrishnan D, Page P, Wahba GM, Lotz JC, et al. Intervertebral disc cell therapy for regeneration: mesenchymal stem cell implantation in rat intervertebral discs. Ann Biomed Eng 2004; 32: 430–4. [DOI] [PubMed] [Google Scholar]
- 20.Sasaki M, Abe R, Fujita Y, Ando S, Inokuma D, Shimizu H. Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. J Immunol 2008; 180: 2581–7. [DOI] [PubMed] [Google Scholar]
- 21.Soslowsky LJ, Thomopoulos S, Tun S, Flanagan CL, Keefer CC, Mastaw J, et al. Neer Award 1999. Overuse activity injures the supraspinatus tendon in an animal model: a histologic and biomechanical study. J Shoulder Elbow Surg 2000; 9: 79–84. [PubMed] [Google Scholar]
- 22.Christner PJ, Gentiletti J, Peters J, Ball ST, Yamauchi M, Atsawasuwan P, et al. Collagen dysregulation in the dermis of the Sagg/+ mouse: a loose skin model. J Invest Dermatol 2006; 126: 595–602. [DOI] [PubMed] [Google Scholar]
- 23.Derwin KA, Soslowsky LJ, Green WD, Elder SH. A new optical system for the determination of deformations and strains: calibration characteristics and experimental results. J Biomech 1994; 27: 1277–85. [DOI] [PubMed] [Google Scholar]
- 24.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 2006; 103: 12481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Badillo AT, Redden RA, Zhang L, Doolin EJ, Liechty KW. Treatment of diabetic wounds with fetal murine mesenchymal stromal cells enhances wound closure. Cell Tissue Res 2007; 329: 301–11. [DOI] [PubMed] [Google Scholar]
- 26.Miller EJ, Rhodes RK. Preparation and characterization of the different types of collagen. Methods Enzymol 1982; 82PtA: 33–64. [DOI] [PubMed] [Google Scholar]
- 27.Schram K, Wong MM, Palanivel R, No EK, Dixon IM, Sweeney G. Increased expression and cell surface localization of MT1-MMP plays a role in stimulation of MMP-2 activity by leptin in neonatal rat cardiac myofibroblasts. J Mol Cell Cardiol 2008; 44: 874–81. [DOI] [PubMed] [Google Scholar]
- 28.Blakytny R, Jude E. The molecular biology of chronic wounds and delayed healing in diabetes. Diabet Med 2006; 23: 594–608. [DOI] [PubMed] [Google Scholar]
- 29.Nikkels-Tassoudji N, Henry F, Letawe C, Pierard-Franchimont C, Lefebvre P, Pierard GE. Mechanical properties of the diabetic waxy skin. Dermatology 1996; 192: 19–22. [DOI] [PubMed] [Google Scholar]
- 30.Reihsner R, Melling M, Pfeiler W, Menzel EJ. Alterations of biochemical and two-dimensional biomechanical properties of human skin in diabetes mellitus as compared to effects of in vitro non-enzymatic glycation. Clin Biomech 2000; 15: 379–86. [DOI] [PubMed] [Google Scholar]
- 31.Cushing L, Kuang P, Lu J. The role of miR-29 in pulmonary fibrosis. Biochem Cell Biol 2015; 93: 109–18. [DOI] [PubMed] [Google Scholar]
- 32.Pandit KV, Milosevic J, Kaminski N. MicroRNAs in idiopathic pulmonary fibrosis. Transl Res 2011; 157: 191–9. [DOI] [PubMed] [Google Scholar]
- 33.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A 2008; 105: 13027–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qin W, Chung AC, Huang XR, Meng XM, Hui DS, Yu CM, et al. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J Am Soc Nephrol 2011; 22: 1462–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang B, Komers R, Carew R, Winbanks CE, Xu B, Herman-Edelstein M, et al. Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol 2012; 23: 252–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maurer B, Stanczyk J, Jüngel A, Akhmetshina A, Trenkmann M, Brock M, et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum 2010; 62: 1733–43. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Wu L, Wang Y, Zhang M, Li L, Zhu D, et al. Protective role of estrogen-induced miRNA-29 expression in carbon tetrachloride-induced mouse liver injury. J Biol Chem 2012; 287: 14851–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roncarati R, Viviani Anselmi C, Losi MA, Papa L, Cavarretta E, Da Costa Martins P, et al. Circulating miR-29a, among other up-regulated microRNAs, is the only biomarker for both hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2014; 63: 920–7. [DOI] [PubMed] [Google Scholar]
- 39.Falanga V, Iwamoto S, Chartier M, Yufit T, Butmarc J, Kouttab N, et al. Autologous bone marrow-derived cultured mesenchymal stem cells delivered in a fibrin spray accelerate healing in murine and human cutaneous wounds. Tissue Eng 2007; 13: 1299–312. [DOI] [PubMed] [Google Scholar]
- 40.Javazon EH, Keswani SG, Badillo AT, Crombleholme TM, Zoltick PW, Radu AP, et al. Enhanced epithelial gap closure and increased angiogenesis in wounds of diabetic mice treated with adult murine bone marrow stromal progenitor cells. Wound Repair Regen 2007; 15: 350–9. [DOI] [PubMed] [Google Scholar]
- 41.Chen L, Tredget EE, Wu PY, Wu Y. Paracrine factors of mesenchymal stem cells recruit macrophages and endothelial lineage cells and enhance wound healing. PloS One 2008; 3: e1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim WS, Park BS, Sung JH, Yang JM, Park SB, Kwak SJ, et al. Wound healing effect of adipose-derived stem cells: a critical role of secretory factors on human dermal fibroblasts. J Dermatol Sci 2007; 48: 15–24. [DOI] [PubMed] [Google Scholar]
- 43.Lee EY, Xia Y, Kim WS, Kim MH, Kim TH, Kim KJ, et al. Hypoxia-enhanced wound-healing function of adipose-derived stem cells: increase in stem cell proliferation and up-regulation of VEGF and bFGF. Wound Repair Regen 2009; 17: 540–7. [DOI] [PubMed] [Google Scholar]
- 44.Sullivan KA, Hayes JM, Wiggin TD, Backus C, Su Oh S, Lentz SI, et al. Mouse models of diabetic neuropathy. Neurobiol Dis 2007; 28: 276–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Blood glucose levels and weight of 4- to 18- week-old diabetic and nondiabetic mice. (A) Blood glucose levels in diabetic (n=5 per age group) versus nondiabetic (n=5 per age group) mice over 4 to 18 weeks. B: Weight (in grams) of diabetic (n=5 per age group) versus non–diabetic (n=5 per age group) mice over 4 to 18 weeks. Results are presented as a mean + SEM for each cohort. Student’s t test was used to compare the means of each cohort, with p<0.05 considered statistically significant (denoted by *).