

Abstract
Supramolecular chemistry has enabled the design of tunable biomaterials that mimic the dynamic and viscoelastic characteristics of the extracellular matrix. However, the noncovalent nature of supramolecular bonds renders them inherently weak, limiting their applicability to many biomedical applications. To address this, we formulated double network (DN) hydrogels through a combination of supramolecular and covalent networks to tailor hydrogel viscoelastic properties. Specifically, DN hydrogels were formed through the combination of supramolecular guest-host (GH) hyaluronic acid (HA) networks with covalent networks from the photocrosslinking of acrylated poly(ethylene glycol) modified fibrinogen (PEG-fibrinogen) and PEG diacrylate. DN hydrogels exhibited higher compressive moduli, increased failure stresses, and increased toughness when compared to purely covalent networks. While GH concentration had little influence on the compressive moduli across DN hydrogels, an increase in the GH concentration resulted in more viscous behavior of DN hydrogels. High viability of encapsulated bovine mesenchymal stromal cells (MSCs) was observed across groups with enhanced spreading and proliferation in DN hydrogels with increased GH concentration. This combination of supramolecular and covalent chemistries enables the formation of dynamic hydrogels with tunable properties that can be customized towards repair of viscoelastic tissues.
Introduction
Synthetic hydrogels offer a versatile spectrum of mechanical and chemical properties to replicate aspects of native tissues or to systematically investigate their influence on biological processes.1,2 As such, hydrogels play central roles in approaches to the engineering of tissues, as well as three-dimensional (3D) culture systems to understand cell behavior.3 However, many of the synthetic hydrogels used in these approaches exhibit static and elastic characteristics, which do not capture the dynamic complexity of the native extracellular matrix (ECM). To address this, hydrogels have been engineered to change over time through network erosion processes, including those sensitive to proteolytic4–6 or hydrolytic7,8 degradation. More recently, hydrogels have been designed to improve their viscoelastic behavior, including through control over stress-relaxation9–11 or by introducing reversible crosslinking,12,13 to better emulate ECM dynamics.
Despite these advances in the design of viscoelastic hydrogels, there are limitations to such approaches, particularly as the use of non-covalent crosslinking can decrease the overall mechanical resilience and stability of hydrogels and limit their applications in the repair of viscoelastic tissues. To address this, hydrogels based on double network (DN) structures have evolved as promising materials for tissue repair strategies, owing to their high toughness and high water content.14 DN hydrogels represent a subset of interpenetrating polymer networks (IPNs) and are defined by a primary network that is highly crosslinked and typically brittle, and a secondary ductile but weak network.15 The asymmetric nature of these two entangled networks results in hydrogels with improved strength and toughness primarily through chain entanglement and energy dissipation mechanisms.16 Indeed, the natural ECM consists of networks of numerous proteins and sugars, which contribute to the complex mechanical properties of tissues.
Critical for the design of these tough DN hydrogels is the ability of the secondary network to self-heal, which is often achieved by dynamic bond rupture and reforming upon deformation. In particular, physical networks, including those based on ionic,17–19 hydrogen,20 and supramolecular21,22 bonding, have enabled the formation of DN hydrogels with self-healing network properties that protect the primary network from failure. While some of these systems have been optimized towards viable cell encapsulation,17,18,23 the static and elastic nature of the primary network and the resulting DN may restrict cell functions, such as morphological changes that are critical towards cell proliferation and new tissue formation.
Here, we developed a DN hydrogel system that incorporates high strength and toughness, while still maintaining encapsulated cell activity, and applied this system to elucidate the role of viscoelasticity in cell behavior. To accomplish this, the primary network was composed of fibrinogen modified with acrylated poly(ethylene)glycol (PEG) and optional PEG-diacrylate, which was photocrosslinked and conserved the bioactivity and degradability of the fibrinogen backbone.24–26 For the secondary network, we employed supramolecular guest-host (GH) interactions to assemble the network with supramolecular bonds through a complex of β-cyclodextrin (CD, host) and adamantane (Ad, guest), which were separately coupled to hyaluronic acid (HA).27,28
The combination of covalent PEG-fibrinogen and supramolecular GH networks enabled the formation of DN hydrogels with independent control over the viscous and elastic properties, which holds promise for repairing native tissues and as 3D culture systems in vitro.
Experimental
Material synthesis and characterization
All chemicals were purchased from MilliporeSigma unless otherwise indicated.
Hyaluronic acid (HA, 75 kDa; Lifecore Biomedical) was converted to the tetrabutylammonium salt (HA-TBA) by ion exchange against Dowex 50Wx8 hydrogen form and neutralized with aqueous tetrabutylammonium hydroxide (0.4 M).8 HA-TBA was modified with 1-adamantane acetic acid (Ad) to form Ad-HA (Fig. S1, ESI†) or 6-(6-aminohexyl)amino-6-deoxy-b-cyclodextrin (CD) to form CD-HA (Fig. S2, ESI†) via an anhydrous reaction in DMSO, according to our previously published methods.27 Briefly, coupling of adamantane (3.0 equiv.) to HA (1 equiv. disaccharides) was performed through an esterification reaction with di-tert-butyl dicarbonate (Boc2O, 0.54 equiv.) and 4-dimethylaminopyridine (DMAP, 1.5 equiv.). Coupling of cyclodextrin (0.6 equiv.) to HA (1 equiv.) was performed via a reaction in the presence of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP, 0.6 equiv.). For all HA derivatives, purification was performed by dialysis and lyophilization, and functionalization of the polymers was quantified by 1H NMR (Bruker 360 MHz) as previously described.27
Synthesis of PEG-fibrinogen was performed from linear PEG as previously described (10 kDa).24 Briefly, PEG-diacrylate (PEG-DA, Fig. S3, ESI†) was obtained by reacting PEG-OH (1 equiv.) under argon with acryloyl chloride (1.5 equiv.) and triethylamine (1.5 equiv.) in dichloromethane. Bovine fibrinogen (1 equiv.) was covalently coupled to PEG-DA (145 equiv.) in an 8 M urea solution in the presence of tris (2-carboxyethyl) phosphine hydrochloride (TCEP HCl, 68 equiv.). The PEG-fibrinogen product was precipitated in acetone and dialyzed.
Hydrogel formation
Ad-HA and CD-HA of 29% and 25% modification, respectively, were used for all experiments and the GH concentration (0, 3, 5%) denotes the combined polymer weight percent, while maintaining a 1:1 ratio of adamantane and β-cyclodextrin. DN hydrogels were prepared from separate solutions of Ad-HA and CD-HA dissolved in PEG-fibrinogen to obtain a final concentration of 8.5 mg ml−1 PEG-fibrinogen in PBS containing 0.05% 2-hydroxy-4’-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959) photoinitiator. The concentration of PEG-DA (1, 2, 3%) indicates the weight percent of additional PEG-DA added. The two-component solution was manually mixed and briefly centrifuged to remove entrapped air. Hydrogels were cast into ca. 300 μm thick films between two coverslips and photocrosslinked with ultraviolet (UV) light (EXFO OmniCure Series 1500, 320–390 nm filter, 5 mW cm−2, 5 min).
Mechanical characterization
Shear rheology.
Hydrogels were formed as described and rheological properties were examined using an AR2000 stress-controlled rheometer (TA Instruments) fitted with a 20 mm diameter cone and plate geometry and 27 μm gap. Rheological properties were measured by oscillatory frequency sweeps (0.01–100 Hz; 1% strain), oscillatory time sweeps (0.1 Hz, 1% strain) and oscillatory strain sweeps (0.01–500% strain).
Dynamic mechanical analysis.
Compressive moduli were examined by dynamic mechanical analysis (TA Instruments, Q800, 0.5 N min−1). Hydrogels were cast into 5 mm diameter cylinders, secured via a preload (0.01 N), and compressed (0.5 N min−1) to determine the Young’s moduli (slope from 10–20% strain), failure strains and failure stresses.
Tensile testing.
Hydrogels were cast into dog-bone shaped samples using polydimethylsiloxane (PDMS) molds (3.0 mm thick, 5.0 mm width at center). Samples were secured using custom clamps with pre-tension (0.01 N) and then extended at 5.0 mm s−1 (Instron 5848, 5 N load cell). Engineering stress-strain curves were employed to measure tensile moduli (slope from 40–50% strain), failure strains and failure stresses. Toughness was determined by integration of the area under stress-strain profiles.
Cell encapsulation, viability and immunofluorescence
Bovine mesenchymal stromal cells (MSCs) were isolated from bone marrow of calves (4–6 months old) obtained from Research 87 Inc (Boylston, MA, USA), as previously described.29 MSCs were passaged once in high glucose Dulbecco’s modified Eagle’s Medium (DMEM, 10% fetal bovine serum, 1% penicillin–streptomycin) and encapsulated at a density of 5 × 106 cells mL−1. Hydrogels were prepared as described and cultured for one day (24 hours) or three days.
Viability was assessed by fluorescence staining with calcein AM (2 μM) and ethidium homodimer-1 (4 μM) for 30 min, imaging with an Olympus epifluorescent microscope, and quantifying with ImageJ software.
For immunofluorescence staining, hydrogels were fixed with 10% buffered formaldehyde in PBS at RT for 30 min, permeabilized with 1% Triton X100 (2 hours, 4 °C) and stained with rhodamine-conjugated phalloidin (1: 100 in 1% bovine serum albumin; Invitrogen R415) for 2 hours at RT, followed by incubation in 5 mg ml−1 Hoechst 33342 for 30 min. Z-stack images were acquired on a Nikon A1R Confocal Microscope at 20 × 0.75 NA and 40 × 0.95 NA.
MSC proliferation was assessed using 5-ethynyl-2’-deoxyuridine (EdU) incorporation over three days in culture. Upon fixing as described, EdU was visualized with AlexaFlour 488 azide using a Click-iT EdU kit according to the manufacturer’s instructions (Thermofisher Scientific). Cell nuclei were counterstained with Hoechst before confocal imaging and the fraction of proliferating cells was quantified as the fraction of nuclei stained positive for EdU.
Statistical analysis
All experiments were performed with three replicates, and statistical significance was assessed using GraphPad Prism 7 software. Comparisons among groups were made using one-way ANOVA with Bonferroni post hoc testing.
Results and discussion
Single networks have tunable properties
The supramolecular network is based on reversible GH complexes between Ad and CD moieties coupled to HA and a GH hydrogel is formed immediately upon mixing of the separate Ad-HA and CD-HA polymer solutions (Fig. 1A). Oscillatory shear rheology confirmed the expected frequency dependence of the storage and loss moduli (G′ and G″, Fig. 1B), due to the dynamic bonding of Ad and CD. As expected, the GH hydrogel moduli increased with increasing concentration of GH polymers. To examine the response of the GH network to increased strains and subsequent recovery, such as upon deformation or loading, hydrogels were subjected to increasing oscillatory strains (0.05–500%) followed by low strain (1%). Strain sweeps indicated a decrease in moduli with increasing strains, with yielding at high strains (~90% strain at yield) (Fig. 1C). The network exhibited a rapid recovery to the initial modulus (G′ = 0.37 ± 0.14 kPa, 5% GH) within seconds of the transition back to low strain (Fig. 1C). Again, network properties were altered through the concentration of GH polymer, where lower concentrations resulted in an overall reduction in modulus (G′ = 0.23 ± 0.21 kPa, 3% GH), but still exhibited yielding and self-healing behaviors (Fig. 1C). Thus, such a GH system exhibits the desired self-healing and tunable properties of a dynamic network.
Fig. 1.
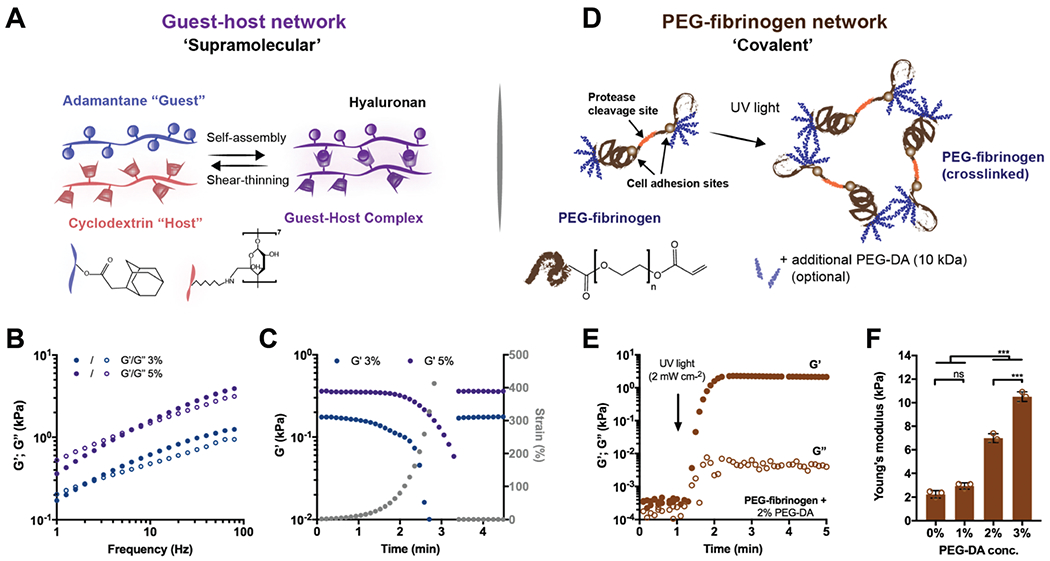
Double network (DN) hydrogels consist of two independent polymer networks with controlled properties. (A) Schematic illustrating adamantane (Ad-HA, blue) and β-cyclodextrin (CD-HA, red) modified hyaluronic acid (HA) supramolecular assembly through guest–host (GH, purple) bond formation. Representative (B) frequency sweeps (1.0–100 Hz, 0.5% strain) and (C) strain sweeps (1.0 Hz, 1–500% strain, then recovery to 1% strain) of storage (G′, filled symbols) and loss (G″, empty symbols) moduli of GH hydrogels at concentrations of 3% (blue) and 5% (purple). (D) Schematic illustrating PEG-fibrinogen that contains natural protease cleavage and cell adhesion sites and is functionalized with acrylated poly(ethylene glycol) (PEG), through a reaction with PEG-diacrylate (PEG-DA). To form hydrogels, unreacted acrylate groups on PEG-fibrinogen and optional additional PEG-DA are polymerized with ultraviolet light in the presence of a photoinitiator (I2959). (E) Representative time sweep (1.0 Hz, 0.5% strain) of the crosslinking of PEG-fibrinogen hydrogels (8.5 mg mL−1) with 2% PEG-DA. (F) Young’s moduli of PEG-fibrinogen (8.5 mg mL−1) with varying concentrations of PEG-DA (mean ± SD, ***p ≤ 0.001, ns = no significant difference by one-way ANOVA with Bonferroni post hoc).
The covalent network is composed of a fibrinogen backbone with reactive end groups (acrylated PEG-fibrinogen) and PEG-DA, and is formed through a photocrosslinking mechanism (Fig. 1D). Photopolymerization, in the presence of UV light and a radical generating photoinitiator (Irgacure 2959), resulted in the formation of PEG-fibrinogen hydrogels, which exhibited primarily elastic properties (tan delta (tan(δ)) < 0.01) due to the covalent crosslinking (Fig. 1E). The elasticity of the network was altered by adjusting the PEG-DA content, resulting in variable moduli (Young’s modulus: 2.23 ± 0.35 to 10.51 ± 0.41 kPa), which was independent of the fibrinogen concentration (constant at 8.5 mg ml−1, Fig. 1F). The results confirm that the single network hydrogels have distinct properties from each other, based on their respective mode of crosslinking (i.e., supramolecular versus covalent bonds).
DN hydrogels exhibit viscoelastic properties
Given the precise control over the properties of either network, we expected that the combination of supramolecular and covalent networks would enable facile tuning of the elasticity and viscosity of DN hydrogels (Fig. 2A). When GH and PEG-fibrinogen/PEG-DA polymers were mixed in solution, hydrogels were initially soft due to the rapid self-healing of the GH bonds, but the elastic and viscous moduli then increased when exposed to UV light due to covalent PEG-DA (2%) crosslinking (G′: 2.48 ± 0.13 kPa, G″: 0.82 ± 0.12 kPa, Fig. 2B). GH bonds contributed largely to the properties of the DN hydrogels, as network entanglement and supramolecular bond formation resulted in a viscous modulus two orders of magnitude greater than that of the covalent only crosslinked PEG-fibrinogen/PEG-DA alone (0% GH, Fig. 1E). Next, the GH polymer concentration was increased to 5% while maintaining the PEG-DA concentration at 2% (Fig. 2C). Although this resulted in a minimal change in the elasticity (G′: 2.59 ± 0.38 kPa), the viscous modulus increased similarly to the trend observed within single GH networks (G″: 1.34 ± 0.23 kPa).
Fig. 2.
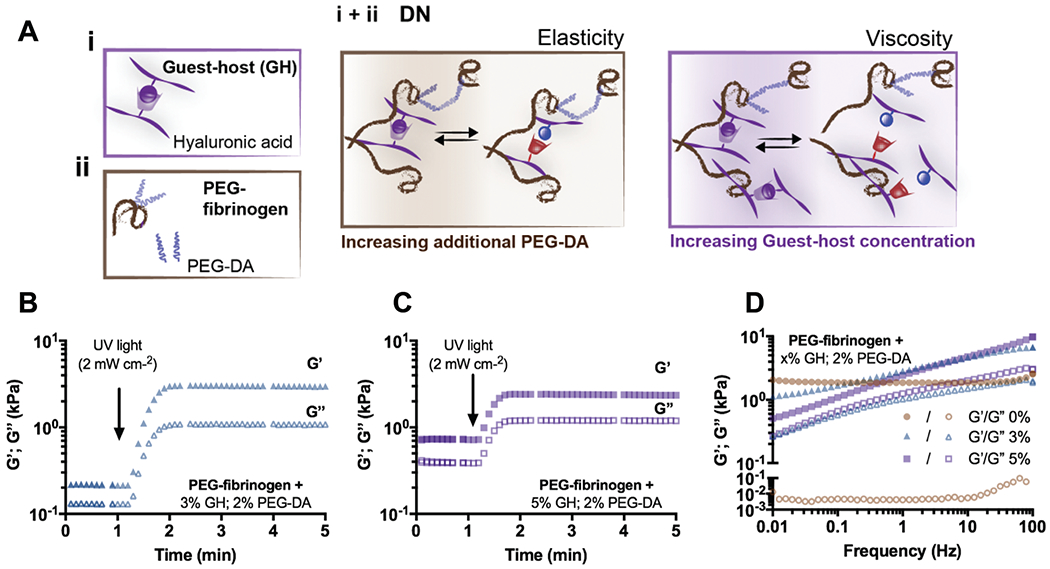
DN hydrogels exhibit viscoelastic properties. (A) Schematic illustrating DN network formation through the combination of (i) a guest-host (GH) network and (ii) covalently crosslinked PEG-fibrinogen (8.5 mg mL−1) with or without additional PEG-DA. Schematics of network tunability where the viscoelasticity of DN hydrogels is controlled through the amount of additional PEG-DA (elasticity) and the GH concentration (viscosity). Representative time sweeps (1.0 Hz, 0.5% strain) of storage (G′, filled symbols) and loss (G″, empty symbols) moduli of DN hydrogels (PEG-fibrinogen (8.5 mg mL−1) plus 2% PEG-DA) containing either (B) 3% or (C) 5% GH concentration. (D) Representative frequency sweeps (0.01–100 Hz, 0.5% strain) of DN hydrogels (PEG-fibrinogen (8.5 mg mL−1) plus 2% PEG-DA) without (0%) or with (3% or 5%) GH of different concentrations.
The frequency response of the hydrogels was investigated and further illustrated the variability in the network structure for the DN hydrogels based on the composition (Fig. 2D). Specifically, while 0% GH hydrogels resulted in steady elastic and viscous moduli across the frequency range (G′: 2.14 ± 0.16 kPa, G″: 0.005 ± 0.001 kPa), DN hydrogels exhibited frequency dependent moduli, as indicated by reductions of G′ and G″ at low frequencies (Fig. 2D). In both 3% and 5% DNs, material properties were dominated by the elastic moduli, which was attributed to the covalent network structures. These results suggest that supramolecular bonds are conserved within DNs, enabling viscoelastic behavior dependent on the polymer concentration of the GH network.
Viscous and elastic properties of DN hydrogels are tuned independently
Noting that DN hydrogels displayed dynamic properties, the influence of the DN composition on the viscoelasticity of the system was investigated. These inputs (e.g., polymer concentration, ratio of supramolecular to covalent crosslinking) govern the configurations in which the networks may assemble and could influence DN crosslink densities, structural inhomogeneities and entanglements, which may impact energy dissipation mechanisms.30 Therefore, the concentration of individual polymers, either GH or PEG-DA, was altered and DN hydrogel properties were systematically investigated by rheology.
To evaluate how the hydrogel composition influenced elastic properties, storage moduli of various DN compositions were examined. PEG-DA concentration was found to exhibit a greater influence on the elastic modulus than the GH concentration across formulations (Fig. 3A). For instance, an increase in PEG-DA concentration from 1 to 3% resulted in a 47–77% increase in the elastic modulus, whereas an increase in the GH concentration from 0 to 3% resulted in negligible changes. While these findings demonstrate the impact on DN hydrogel elasticity, the utilization of DNs to control viscous behavior has rarely been investigated.18 To evaluate the viscosity of the system, tan(δ) was examined. Tan(δ) is a measure of the dampening in the material and is the ratio of the loss (G″) and the storage (G′) modulus. A reduction of tan(δ) was observed with increased covalent crosslinking (e.g., greater PEG-DA concentration); however, higher GH polymer concentrations resulted in a pronounced increase in tan(δ) (Fig. 3B). Moreover, tan(δ) was highly tunable through modulation of the GH to PEG-DA molar ratio (Fig. 3C). Specifically, an increase in the relative GH concentration resulted in more viscous behavior of the DN hydrogels as indicated by the increase of tan(δ). Taken together, the findings demonstrate a system where the viscous and elastic properties can be modulated independently. Although altering the elasticity of DN hydrogels with varying PEG-fibrinogen amounts is possible, we chose a constant concentration of 8.5 mg ml−1, which has been shown to support homogeneous hydrogel formation and cell compatibility.26,31 Importantly, DN mechanical properties cover the range of viscoelasticity measured in a number of soft tissues, including cardiac and skeletal muscle as well as lung tissue.32 While other DN hydrogel systems have demonstrated similar elastic properties,18,21–23 they often lack the tunable viscous aspects that may be needed to mimic the viscoelastic properties of many soft tissues and ECM.32,33
Fig. 3.
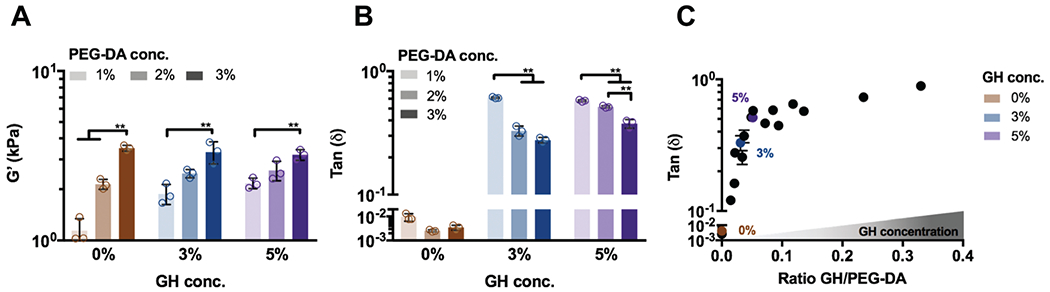
Viscoelastic properties of DN hydrogels are tuned through the amount of independent networks. Rheological measurements (1.0 Hz, 0.5% strain) of (A) storage modulus (G′ and (B) tan(δ) for DN hydrogels with PEG-fibrinogen (8.5 mg mL−1) and varied GH (0, 3, 5%) and PEG-DA (1, 2, 3%) concentrations (conc., n = 3 replicates per group, mean ± SD, **p ≤ 0.01 by one-way ANOVA with Bonferroni post hoc). (C) Tan(δ) of DN hydrogels with various molar ratios of GH/PEG-DA (n = 3 replicates per group, mean ± SD). Colored symbols represent examples of DN hydrogels (PEG-fibrinogen (8.5 mg mL−1) plus 2% PEG-DA) with varied GH concentrations.
DNs exhibit high mechanical strength and toughness
In addition to capturing the viscoelastic behavior of native ECM, high mechanical strength and toughness of hydrogels are often critical towards their applications in repair and augmentation of tissues. Since a high degree of tunability was observed for DN hydrogels with 2% PEG-DA, this covalent crosslink density was used in subsequent studies (unless otherwise noted) and the GH concentration was varied at 0%, 3%, and 5%. When tested in compression, PEG-fibrinogen hydrogels with only covalent crosslinking (0% GH) and DNs with 3% GH exhibited recovery following compression to 90% strain, whereas ductile and unrecoverable failure was observed for 5% DN hydrogels (Fig. 4A and Video S1, ESI†), indicating that the ratio of covalent to supramolecular crosslinks is critical to DN hydrogel mechanics. Compressive stress-strain relationships demonstrated increased failure stresses for DN hydrogels compared with covalent-only hydrogels, but little changes in the moduli when compared with 0% GH (inset, Fig. 4B). Further, the dependence of the Young’s modulus on PEG-DA concentration and only minimally on GH content was similar to observations by rheology, confirming the tunability of the system (Fig. 4C).
Fig. 4.
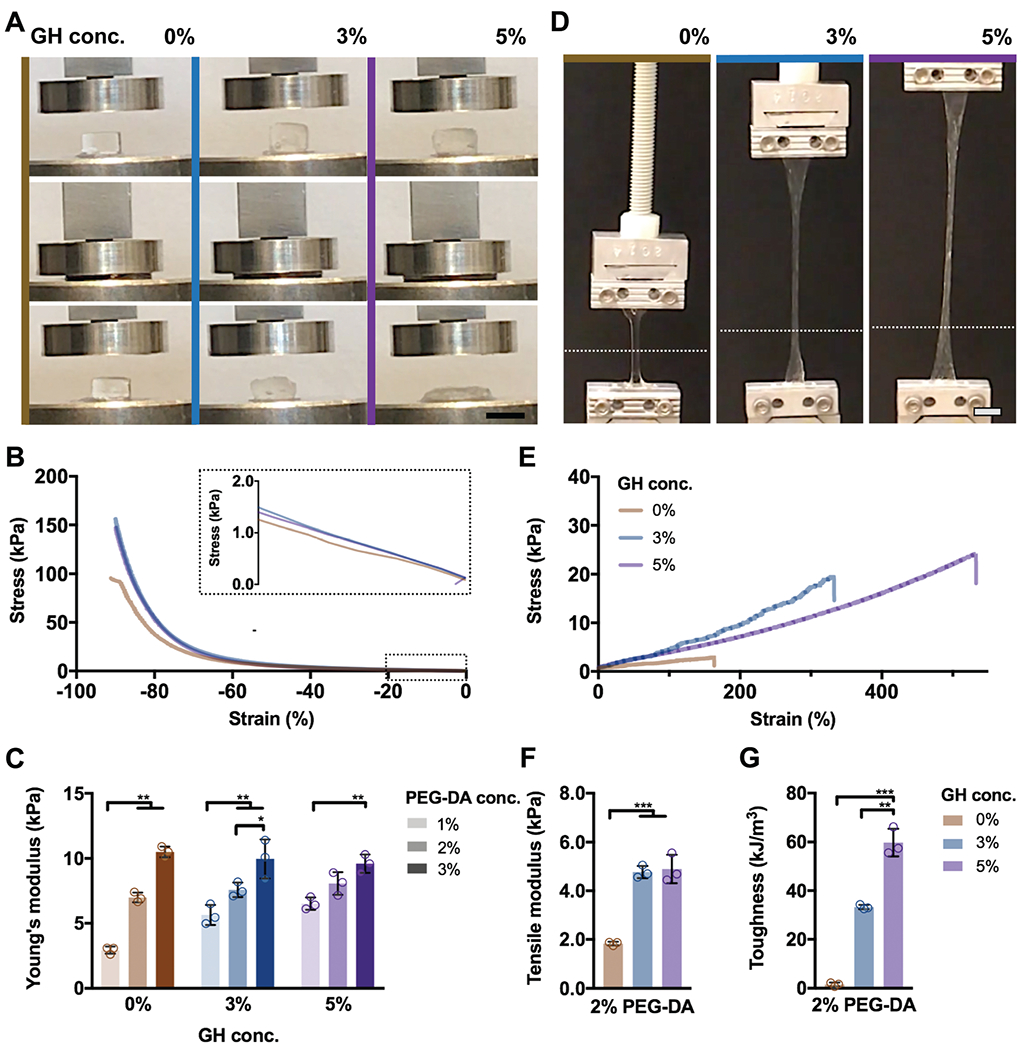
DN hydrogels exhibit high mechanical strength and toughness. (A–C) Compressive and (D–G) tensile testing of DN hydrogels (PEG-fibrinogen (8.5 mg mL−1) plus 2% PEG-DA) without (0%) or with (3%, 5%) GH of different concentrations. (A) Images of DN hydrogel compressive testing (scale bars 5 mm) and corresponding (B) stress–strain profiles (0.5 N min−1) and (C) Young’s moduli (n = 3 replicates per group, mean ± SD, **p ≤ 0.01) for DN hydrogels. (D) Images of DN hydrogel tensile testing, where the starting position of the top grid is indicated (dotted line, scale bars 5 mm) and corresponding (E) stress-strain profiles (5 mm s−1), (F) tensile moduli, and (G) toughness for DN hydrogels (n = 3 replicates per group, mean ± SD, **p ≤ 0.01, ***p ≤ 0.001).
When subjected to tensile loading, elongation was observed for all hydrogels (Fig. 4D and Video S2, ESI†). DN hydrogels exhibited approximately eight-fold and ten-fold increases in failure stresses at 3% and 5% GH concentrations, respectively, when compared to covalent-only hydrogels with similar improvements in failure strains (Fig. 4E and Fig. S4, ESI†). Along with these changes in tensile properties, increased moduli were observed for DNs compared with 0% GH hydrogels (Fig. 4F). Despite similar tensile moduli for both 3% and 5% DNs, increasing GH concentrations further enhanced the toughness of DN hydrogels up to 57.4 ± 8.8 kJ m−3 (Fig. 4G).
Since the GH hydrogel assembly is reversible, DNs retained the ability to self-heal and undergo repeated mechanical loading. Cut hydrogel fragments exhibited rapid healing due to GH interactions, enabling resistance to separation (Video S3, ESI†). Thus, supramolecular interactions endowed DNs with enhanced compressive and tensile strengths as well as the ability to withstand repeated loading, properties that may be very useful depending on their application.
DNs enable control over cell behavior in 3D hydrogels
In addition to providing mechanical support and resilience, cytocompatibility and matrix remodeling are critical towards functional tissue repair; however, these cellular processes are limited in many hydrogels, including many DN systems. Using the hydrogel system developed, we investigated the influence of DN hydrogel properties – the same elasticity (G′ 2.4 ± 0.26 kPa) but altered viscosity (G″ 0.00–1.34 kPa, Fig. 5A) – on cell morphology and activity. Encapsulated MSCs exhibited high viability (>85%) under all conditions throughout three days in culture (Fig. S5, ESI†). To examine cell morphologies in these hydrogels, cytoskeletal organization was visualized using F-Actin staining (Fig. 5A). After one day of culture in growth media, encapsulated MSCs exhibited generally rounded morphologies with some protrusions across all hydrogels. However, after three days, cell spreading was greatly enhanced in DN hydrogels (3%, 5%) – cells under both conditions adopted spindle-like morphologies with thin and elongated protrusions.
Fig. 5.
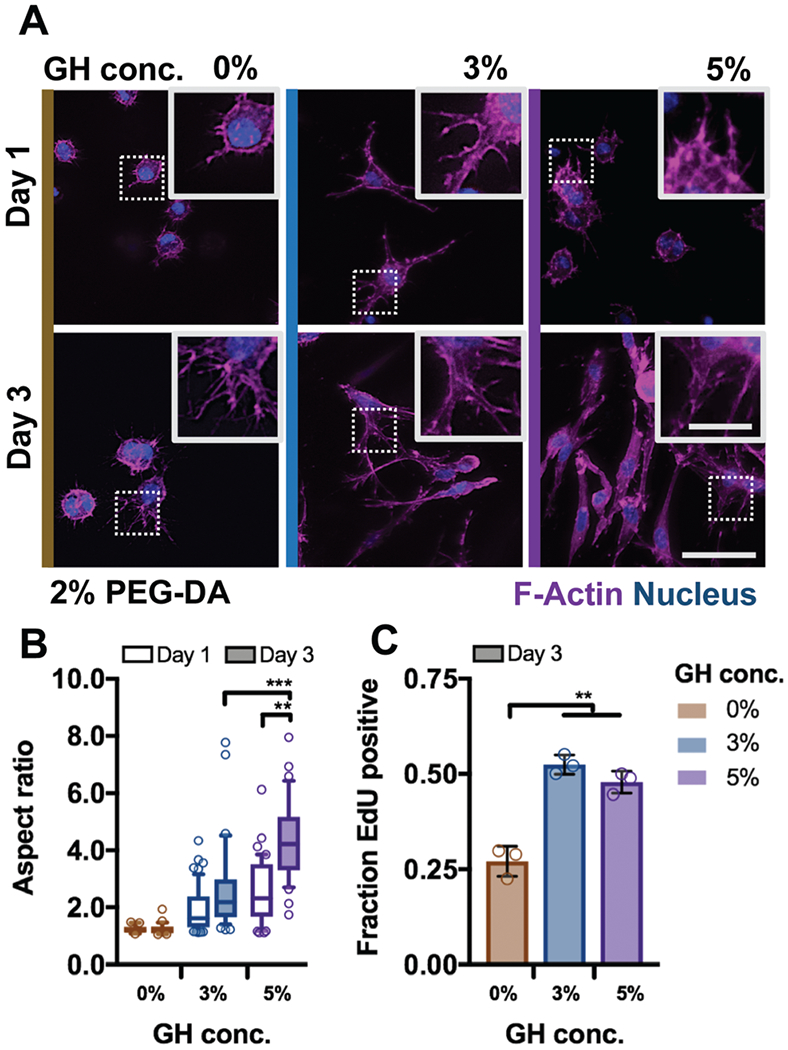
DN hydrogels are cytocompatible and enable control over cell spreading. (A) Representative images of F-Actin immunofluorescence of bovine MSCs cultured for one and three days in DN hydrogels (PEG-fibrinogen (8.5 mg mL−1) plus 2% PEG-DA) without (0%) or with (3% or 5%) GH of different concentrations (scale bar 50 μm, inset 20 μm). Quantification of (B) cell aspect ratio (n ≥ 50 cells per group, box plots show 25/50/75th percentiles, whiskers show 10/90th percentiles, **p ≤ 0.01, ***p ≤ 0.001 by one-way ANOVA with Bonferroni post hoc) after one and three days of culture and (C) fraction of cells after three days of culture with nuclei positively stained for 5-ethynyl-2’-deoxyuridine (EdU, n = 3 replicates per group, mean ± SD, **p ≤ 0.01 by one-way ANOVA with Bonferroni post hoc).
When comparing temporal profiles of cell spreading with increasing GH concentrations, cell aspect ratios (a measure of spreading) increased as a function of viscosity and time (Fig. 5B). No significant differences in aspect ratios were observed for cells in covalent only (0% GH) hydrogels following three days of culture. Functional outcomes of cell spreading were also altered; MSC proliferation in DNs, as assayed by EdU incorporation (Fig. S6, ESI†), was enhanced by ~25% of values in 0% GH hydrogels (Fig. 5C). It should be noted that the fibrinogen backbone of these hydrogels is susceptible to degradation by proteinases (Fig. S7, ESI†). Although protrusions in 0% GH hydrogel matrices indicated that cells have started to proteolytically degrade their matrix environment within three days, remodeling of such dense polymer networks likely necessitates longer culture times.25,26
Taken together, these findings suggest that cellular remodeling of DN hydrogels allows for cell spreading and proliferation when compared to purely covalently crosslinked hydrogels. Moreover, cell behavior is regulated by cell-mediated rearrangement of the dynamic GH bonds, and emphasizes, consistent with previous reports,9,11 that cells respond to the increasing viscosity (i.e. higher GH concentration) of the hydrogel microenvironment. Notably, we found that network rearrangements occurred quickly (within three days) through cellular remodeling to influence cell spreading and proliferation. This link between hydrogel remodeling and cell response suggests that the rapid dynamics of GH bonds not only enable encapsulated MSCs to rearrange their microenvironment, but also influence cell activity and function. Although the molecular mechanisms remain to be elucidated, an increase in ligand density and integrin clustering has been found to be critical for activating signaling pathways that mediate cell spreading in viscoelastic hydrogels.9,10,34 Other dynamic systems (e.g., ionic,9 dynamic covalent bonds35) often require several days for cellular remodeling; thus, this DN hydrogel may add a valuable strategy for capturing ECM dynamics at varying time scales.36,37 Beyond studying cell behavior in vitro, the identified network structural parameters may be harnessed to accommodate the dynamic needs during tissue healing and can be further engineered to enhance endogenous repair (e.g., through release of growth factors,38,39 chemoattractants,39 cytokines40). A recent work demonstrating enhanced cell invasion and tissue formation through introducing viscosity to implanted hydrogels illustrated this potential.41
Conclusions
Supramolecular and covalent interactions were used to form DN hydrogels that are viscoelastic to create dynamic matrices for cell encapsulation. Network entanglement resulted in the desired hydrogel properties with enhanced mechanical strength and toughness when compared to single-network hydrogels. Owing to rapid association of supramolecular bonds, internal self-healing of DN hydrogels resulted in recoverable primary networks, enabling repetitive loading. Furthermore, the viscoelastic properties were controlled independently through alterations of the concentration of either network. Using this tunability, an increase in network viscosity (e.g. through higher supramolecular polymer concentration) influenced cell behavior, enhancing cell spreading and proliferation. The ability of this system to not only enable cellular remodeling, but also recapitulate the mechanical resilience of many tissues may provide new avenues towards functional tissue repair.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was financially supported by the National Science Foundation (JAB: DMR Award 1610525, JHG: Graduate Research Fellowship), the Swiss National Science Foundation (CL), the USA–Israel Binational Science Foundation (DS, AA, OK, HSY: Award 2015697), and the Israel Science Foundation (DS, OK: Award 1245/14). The authors thank the Penn Center for Musculoskeletal Disorders for tensile testing.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb02593b
Conflicts of interest
There are no conflicts to declare.
References
- 1.Seliktar D, Science, 2012, 336, 1124–1128. [DOI] [PubMed] [Google Scholar]
- 2.Guvendiren M and Burdick JA, Curr. Opin. Biotechnol, 2013, 24, 841–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caliari SR and Burdick JA, Nat. Methods, 2016, 13, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS and Burdick JA, Nat. Mater, 2013, 12, 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schultz KM, Kyburz KA and Anseth KS, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, E3757–E3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson SB, Lin CC, Kuntzler DV and Anseth KS, Biomaterials, 2011, 32, 3564–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bryant SJ and Anseth KS, J. Biomed. Mater. Res., Part A, 2003, 64, 70–79. [DOI] [PubMed] [Google Scholar]
- 8.Sahoo S, Chung C, Khetan S and Burdick JA, Biomacromolecules, 2008, 9, 1088–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee HP, Lippens E, Duda GN and Mooney DJ, Nat. Mater, 2016, 15, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lou J, Stowers R, Nam S, Xia Y and Chaudhuri O, Biomaterials, 2018, 154, 213–222. [DOI] [PubMed] [Google Scholar]
- 11.Cameron AR, Frith JE and Cooper-White JJ, Biomaterials, 2011, 32, 5979–5993. [DOI] [PubMed] [Google Scholar]
- 12.Shih H and Lin C-C, J. Mater. Chem. B, 2016, 4, 4969–4974. [DOI] [PubMed] [Google Scholar]
- 13.Rosales AM, Vega SL, DelRio FW, Burdick JA and Anseth KS, Angew. Chem., Int. Ed, 2017, 56, 12132–12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong JP, Katsuyama Y, Kurokawa T and Osada Y, Adv. Mater, 2003, 15, 1155–1158. [Google Scholar]
- 15.Haque MA, Kurokawa T and Gong JP, Polymer, 2012, 53, 1805–1822. [Google Scholar]
- 16.Zhao X, Soft Matter, 2014, 10, 672–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeon O, Shin J-Y, Marks R, Hopkins M, Kim T-H, Park H-H and Alsberg E, Chem. Mater, 2017, 29, 8425–8432. [Google Scholar]
- 18.Liang Z, Liu C, Li L, Xu P, Luo G, Ding M and Liang Q, Sci. Rep, 2016, 6, 33462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun J-Y, Zhao X, Illeperuma WRK, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ and Suo Z, Nature, 2012, 489, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo M, Pitet LM, Wyss HM, Vos M, Dankers PYW and Meijer EW, J. Am. Chem. Soc, 2014, 136, 6969–6977. [DOI] [PubMed] [Google Scholar]
- 21.Rodell CB, Dusaj NN, Highley CB and Burdick JA, Adv. Mater, 2016, 28, 8419–8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li C, Rowland MJ, Shao Y, Cao T, Chen C, Jia H, Zhou X, Yang Z, Scherman OA and Liu D, Adv. Mater, 2015, 27, 3298–3304. [DOI] [PubMed] [Google Scholar]
- 23.Yan Y, Li M, Yang D, Wang Q, Liang F, Qu X, Qiu D and Yang Z, Biomacromolecules, 2017, 18, 2128–2138. [DOI] [PubMed] [Google Scholar]
- 24.Almany L and Seliktar D, Biomaterials, 2005, 26, 2467–2477. [DOI] [PubMed] [Google Scholar]
- 25.Dikovsky D, Bianco-Peled H and Seliktar D, Biophys. J, 2008, 94, 2914–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dikovsky D, Bianco-Peled H and Seliktar D, Biomaterials, 2006, 27, 1496–1506. [DOI] [PubMed] [Google Scholar]
- 27.Loebel C, Rodell CB, Chen MH and Burdick JA, Nat. Protoc, 2017, 12, 1521–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodell CB, Kaminski AL and Burdick JA, Biomacromolecules, 2013, 14, 4125–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loebel C, Szczesny SE, Cosgrove BD, Alini M, Zenobi-Wong M, Mauck RL and Eglin D, Biomacromolecules, 2017, 18, 855–864. [DOI] [PubMed] [Google Scholar]
- 30.Costa AMS and Mano JF, Eur. Polym. J, 2015, 72, 344–364. [Google Scholar]
- 31.Mironi-Harpaz I, Wang DY, Venkatraman S and Seliktar D, Acta Biomater, 2012, 8, 1838–1848. [DOI] [PubMed] [Google Scholar]
- 32.Levental I, Georges PC and Janmey PA, Soft Matter, 2007, 3, 299–306. [DOI] [PubMed] [Google Scholar]
- 33.Chaudhuri O, Biomater. Sci, 2017, 5, 1480–1490. [DOI] [PubMed] [Google Scholar]
- 34.Maheshwari G, Brown G, Lauffenburger DA, Wells A and Griffith LG, J. Cell Sci, 2000, 113, 1677–1686. [DOI] [PubMed] [Google Scholar]
- 35.McKinnon DD, Domaille DW, Cha JN and Anseth KS, Adv. Mater. , 2014, 26, 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H and Heilshorn SC, Adv. Mater, 2015, 27, 3717–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Eyckmans J and Chen CS, Nat. Mater, 2017, 16, 1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shekaran A, Garcia JR, Clark AY, Kavanaugh TE, Lin AS, Guldberg RE and Garcia AJ, Biomaterials, 2014, 35, 5453–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee CH, Lee FY, Tarafder S, Kao K, Jun Y, Yang G and Mao JJ, J. Clin. Invest, 2015, 125, 2690–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spiller KL, Nassiri S, Witherel CE, Anfang RR, Ng J, Nakazawa KR, Yu T and Vunjak-Novakovic G, Biomaterials, 2015, 37, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Darnell M, Young S, Gu L, Shah N, Lippens E, Weaver J, Duda G and Mooney D, Adv. Healthcare Mater, 2017, 6, DOI: 10.1002/adhm.201601185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.