

Abstract
Introduction
New HIV prevention strategies are urgently required. The discovery of broadly neutralising antibodies (bNAbs) has provided the opportunity to evaluate passive immunisation as a potential prevention strategy and facilitate vaccine development. Since 2014, several bNAbs have been isolated from a clade C-infected South African donor, CAPRISA 256. One particular bNAb, CAP256-VRC26.25, was found to be extremely potent, with good coverage against clade C viruses, the dominant HIV clade in sub-Saharan Africa. Challenge studies in non-human primates demonstrated that this antibody was fully protective even at extremely low doses. This bNAb was subsequently structurally engineered and the clinical variant is now referred to as CAP256V2LS.
Methods and analysis
CAPRISA 012B is the second of three trials in the CAPRISA 012 bNAb trial programme. It is a first-in-human, phase I study to assess the safety and pharmacokinetics of CAP256V2LS. The study is divided into four groups. Group 1 is a dose escalation of CAP256V2LS administered intravenously to HIV-negative and HIV-positive women. Group 2 is a dose escalation of CAP256V2LS administered subcutaneously (SC), with and without the dispersing agent recombinant human hyaluronidase (rHuPH20) as single or repeat doses in HIV-negative women. Groups 3 and 4 are randomised placebo controlled to assess two (CAP256V2LS+VRC07-523LS; CAP256V2LS+PGT121) and three (CAP256V2LS+VRC07-523LS+PGT121) bNAb combinations administered SC to HIV-negative women. Safety will be assessed by the frequency of reactogenicity and adverse events related to the study product. Pharmacokinetic disposition of CAP256V2LS alone and in combination with VRC07-523LS and PGT121 will be assessed via dose subgroups and route of administration.
Ethics and dissemination
The University of KwaZulu-Natal Biomedical Research Ethics Committee (BREC) and the South African Health Products Regulatory Authority (SAHPRA) have granted regulatory approval (trial reference numbers: BREC00000857/2019 and SAHPRA 20200123). Trial results will be disseminated through conference presentations, peer-reviewed publications and the clinical trial registry.
Trial registration number
PACTR202003767867253; Pre-results.
Keywords: infectious diseases, hiv & aids, microbiology, public health, epidemiology
Strengths and limitations of this study.
This is the first-in-human trial to assess the safety and pharmacokinetics of the monoclonal antibody CAP256V2LS.
The trial investigates the administration of CAP256V2LS in combination with two potent antibodies, VRC07-523LS and PGT121.
The trial assesses the subcutaneous administration of monoclonal antibodies for HIV prevention.
The study evaluates the use of a dispersing agent, recombinant human hyaluronidase (rHuPH20), together with antibodies against HIV.
The study is not powered to show the efficacy of CAP256V2LS against HIV.
Introduction
Despite extensive prevention and treatment efforts, South Africa remains the country worst affected by the HIV-AIDS pandemic.1 Here, young women carry a disproportionately high burden of the disease with a persistently high HIV incidence.2 3 Insights from universal testing and treatment trials demonstrate that early treatment alone is not sufficient to reduce the number of new infections and achieve epidemic control, but that effective HIV prevention methods are also needed.4 In African women, clinical trials evaluating daily oral tenofovir disoproxil fumarate (TDF) and emtricitabine (TDF/FTC) for pre-exposure prophylaxis demonstrated inconsistent results, most likely owing to varying adherence levels.5 While an effective vaccine remains a major challenge, new HIV prevention strategies are urgently required.6
The discovery of broadly neutralising antibodies (bNAbs) has allowed scientists to evaluate passive immunisation as a potential HIV prevention strategy.7 8 These antibodies are generally recovered from the memory B cells of chronically HIV-infected individuals and effectively neutralise diverse strains of HIV-1 indicating their breadth of response. Preclinical studies have demonstrated that passive immunisation using bNAbs protects rhesus macaques from simian-human immunodeficiency virus (SHIV) infection.9–12 However, there are currently no clinical trial data that show the ability of bNAbs to prevent HIV-1 infection in humans.13
In 2014 and subsequently, several bNAbs targeting the V2 region of the HIV-1 envelope glycoprotein were isolated from a South African donor participating in the Centre for the AIDS Programme of Research in South Africa (CAPRISA) 002 Acute Infection study.14 15 This study was established in 2004 in KwaZulu-Natal, South Africa and followed HIV-negative participants for identification of subsequent HIV seroconversion. This participant was infected with a clade C virus and superinfected with a different clade C virus, 15 weeks later.16 One particular bNAb, referred to as CAP256-VRC26.25, was isolated and found to be 10 times more potent than the previously published members of this lineage. Its overall potency (IC50=0.001 μg/mL) was comparable with or better than that of existing bNAbs.17
The exceptional potency of this antibody may be related to the reduced dependence on the N160 glycan, the unique long heavy-chain complementarity-determining region 3 (CDRH3) conformation or other structural features that have yet to be identified.18–21 Further research using site-directed mutagenesis allowed for the manufacturing of an improved LS version of CAP256-VRC26.25. This mutation increases the binding affinity for the neonatal Fc-receptor, resulting in an increased recirculation of functional immunoglobulin G (IgG), thereby increasing plasma half-life.22 In vivo studies demonstrated that CAP256-VRC26.25LS was fully protective against a SHIV challenge in monkeys, even at the lowest dose of 0.08 mg/kg, with protection achieved at serum antibody concentrations of <0.75 µg/mL.23 This was the lowest dose of any bNAb to show protection in monkeys.24
Furthermore, when tested against a panel of 200 acute infection clade C pseudoviruses, CAP256-VRC26.25 emerged as the most potent member of bNAbs targeting the V2 loop.10 17 The neutralisation profile of CAP256-VRC26.25LS was particularly well suited as a complementary bNAb in combinations with bNAbs targeting other epitopes.17
CAP256-VRC26.25LS was subsequently engineered to prevent proteolytic clipping of the heavy chain through mutation of the lysine at position 100 to alanine.25 This single amino acid change, K127A, was made in the CDRH3 region to improve manufacturability without altering neutralisation potency or breadth. This non-clipped variant of the antibody is referred to as CAP256V2LS.
Preclinical studies in rhesus macaques demonstrated that CAP256V2LS has a half-life of 14.3 days when administered via the intravenous (IV) route and 9.9 days when administered via the subcutaneous (SC) route. Toxicology reports showed that CAP256V2LS displayed low polyspecific autoreactivity to HEp-2 cells and cardiolipin.26 In non-human primate (NHP) studies, non-pathological implications such as anticardiolipin activity were associated with the administration of anti-Env monoclonal antibodies (mAbs). Although these mAbs have polyspecific reactivities to host antigens,27 the immune response of NHPs to therapeutic mAbs is not considered to be predictive of the human response. This is due to the differences at the species level. Thus, the ability to compare relative immunogenicity of mAbs in NHPs and humans is low.28 Its high potency and good breadth against clade C HIV viruses and long half-life make CAP256V2LS an excellent candidate for further clinical development. This is particularly important for southern Africa, where clade C is the dominant circulating virus.
The research and development pathway of the CAP256V2LS bNAb led to the CAPRISA 012 clinical trial programme that consists of three trials conducted in South Africa (figure 1). This programme aims to evaluate the concept of bNAbs as long-acting pre-exposure prophylaxis with two to three SC doses per year to reduce HIV incidence among young women in three trials: CAPRISA 012A, a phase I trial of VRC07-523LS and PGT121 in HIV-negative women, previously described in this journal,29 CAPRISA 012B, described here, and CAPRISA 012C, a phase II combination bNAb trial in young women.
Figure 1.
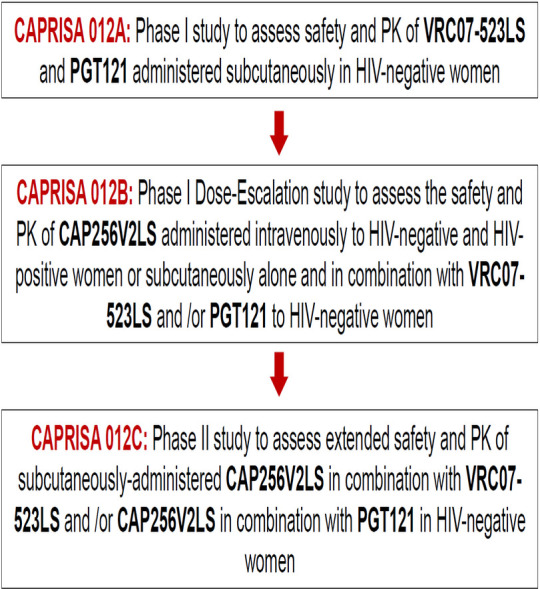
Caprisa 012 clinical trial programme.
In CAPRISA 012B, CAP256V2LS will be assessed alone and in combination with VRC07-523LS and/or PGT121. VRC07-523LS targets the HIV-1 Envelope CD4 binding site and PGT121 targets the V3 glycan-dependent epitope region of the HIV Envelope protein. Given the vast genetic diversity of HIV-1, the use of multiple bNAbs may be required to ensure adequate coverage of circulating strains. Recent clinical trial data demonstrate that VRC07-523LS is safe with a half-life of 38 days after IV administration and 33 days after SC administration.30 Preliminary clinical trial data of PGT121 also demonstrate safety with a half-life of 22 days.31
This trial is also one of the first to assess the use of a recombinant human hyaluronidase (rHuPH20) together with an anti-HIV monoclonal antibody.32 rHuPH20 is the active ingredient of the investigational ENHANZE Drug Product (EDP) and optimises the SC delivery of coadministered therapeutics by depolymerising hyaluronan in the extracellular matrix of the SC space that normally serves to restrict increased flow volumes. EDP allows the comixing of antibodies with rHuPH20 at the clinical site. Clinical trials conducted in oncology have demonstrated safety and favourable results with this product.33
SC administration of rHuPH20 was well tolerated in healthy participants, participants with diabetes, rheumatoid arthritis, cancer and dehydration. SC administrations of rHuPH20 alone or in combination with morphine, ceftriaxone, ondansetron, insulin, adalimumab, IgG and hydration fluids were also well tolerated.34 35 Most adverse events (AEs) reported were mild, transient injection site reactions, including erythema, pruritus, tenderness, induration and paraesthesia. Moderate injection site reactions, occurring less frequently, include burning, erythema, pain and numbness. Mild-to-moderate headache was also reported.33 Local tissue changes induced by rHuPH20 are reversible within 24–48 hours after administration, without inflammatory or histological changes. Coadministration demonstrated beneficial effects such as improved absorption, increased bioavailability and decreased pharmacokinetics (PK) variability.33 36 rHuPH20 is currently coformulated with two approved anticancer therapies, trastuzumab and rituximab.
Methods and analysis
Patient and public involvement
The CAPRISA Community Advisory Board (CAB) plays a central role in study planning and recruitment of participants. The CAB includes local community leaders, traditional leaders, leadership of local HIV/AIDS organisations, local health service provider representatives, previous study participants and HIV-positive local community members. During trial preparation and prior to study start, the study concept is presented to the CAB members for their feedback. Recruitment will include community events, local clinics, street recruitment and use of snowballing techniques. A recruitment and retention plan will be drawn up at the study start and will be reviewed and updated regularly.
Study setting
The CAPRISA 012B trial will be conducted at the CAPRISA eThekwini Clinical Research Site. This site is based in a busy commuter area in the city centre of Durban, KwaZulu-Natal, South Africa.
Study population selection
The study population will consist of 66 women, 52 HIV-negative and 14 HIV-positive women who have not yet started antiretroviral therapy (ART). All eligibility criteria must be met, and HIV-positive women will be recruited based on additional inclusion criteria (table 1).
Table 1.
Eligibility criteria
| Inclusion criteria | Exclusion criteria | Additional inclusion criteria* |
|
|
|
*Additional eligibility criteria for HIV-positive participants in groups 1c and 1d.
FDA, food and drug administration.
Study design
CAPRISA 012B is a first-in-human, phase I study to assess the safety, tolerability and PK of CAP256V2LS. The study is divided into four groups (table 2). Group 1 is a dose escalation of CAP256V2LS administered IV to HIV-negative and HIV-positive women. Group 2 is a dose escalation of CAP256V2LS administered SC with and without rHuPH20 at a single or repeat dose to HIV-negative women. Groups 3 and 4 are randomised placebo controlled to assess two (CAP256V2LS+VRC07-523LS; CAP256V2LS+PGT121) and three (CAP256V2LS+VRC07-523LS+PGT121) bNAb combinations administered SC to HIV-negative women. Participants will be followed-up for 24 weeks after the administration of the last dose of study product/s. HIV-positive participants in groups 1 c and 1d will have two additional follow-up visits after 12 and 24 months.
Table 2.
CAPRISA 012B group, dose and arm allocation
| Group | Participants | Regimen | SA site (N=66) | Dose (mg/kg) |
| Group 1: dose escalation of IV administration of CAP256V2LS | ||||
| 1a | HIV negative | CAP256V2LS | 4 | 5 mg/kg IV one dose |
| 1b | HIV negative | CAP256V2LS | 4 | 10 mg/kg IV one dose |
| 1c | HIV positive | CAP256V2LS | 4/2§ | 20 mg/kg IV one dose |
| 1d | HIV positive | CAP256V2LS | 4/4§ | 20 mg/kg IV one dose |
| Group 2: dose escalation of SC administration of CAP256V2LS | ||||
| 2a | HIV negative | CAP256V2LS | 4 | 5 mg/kg SC one dose |
| 2b | HIV negative | CAP256V2LS* | 4 | 5 mg/kg SC one dose |
| 2c | HIV negative | CAP256V2LS* | 4 | 10 mg/kg SC one dose |
| 2d | HIV negative | CAP256V2LS* | 4 | 10 mg/kg SC with one repeat dose at 16/24 weeks† |
| 2e | HIV negative | CAP256V2LS* | 4 | 20 mg/kg SC one dose |
| 2f | HIV negative | CAP256V2LS* | 4 | 20 mg/kg SC with one repeat dose at 16/24 weeks† |
| Group 3: dose escalation of the two antibody combinations | ||||
| 3a | HIV negative | CAP256V2LS*+VRC07-523.LS* | 4/1§ | 10 mg/kg SC/10 mg/kg SC one dose |
| 3b | HIV negative | CAP256V2LS*+VRC07-523.LS* | 4/1§ | 20 mg/kg SC/20 mg/kg SC one dose |
| 3c | HIV negative | CAP256V2LS*+PGT121‡ | 4/1§ | 20 mg/kg SC/5 mg/kg SC one dose |
| Group 4: three antibody combination | ||||
| 4a | HIV negative | CAP256V2LS*+PGT121‡+ VRC07-523.LS* |
4/1§ | 20 mg/kg SC/5 mg/kg SC/20 mg/kg SC one dose |
*Antibody will be injected with hyaluronidase, so that the antibody dose can be administered as a single SC injection.
†First two participants will receive two doses 24 weeks apart and the next two participants will receive two doses 16 weeks apart.
‡PGT121 may be replaced with PGT121LS at a higher dose, based on its availability at the time of study initiation.
§Placebo allocation.
SC, subcutaneous.
Study objectives
Primary objectives
-
To evaluate the safety and tolerability of CAP256V2LS administered.
SC in HIV-negative women.
IV in HIV-negative and HIV-positive women.
SC in combination with VRC07-523LS and/or PGT121 in HIV-negative women.
Secondary objectives
To characterise the PK profile of CAP256V2LS administered SC as a single dose or as two doses 16 or 24 weeks apart.
To characterise the PK profile of CAP256V2LS administered SC in combination with VRC07-523LS and/or PGT121.
To characterise the PK profile of CAP256V2LS administered IV as a single dose in HIV-negative and HIV-positive women.
To evaluate the antiviral activity of CAP256V2LS administered IV to HIV-positive women not on ART.
To evaluate the concentrations and functional activity of CAP256V2LS in plasma and genital samples following SC and IV administration.
To determine whether administration of CAP256V2LS induces anti-monoclonal antibody responses.
To assess the acceptability of SC administration of monoclonal antibodies among participants.
Primary endpoints
Proportion of participants with mild, moderate and severe reactogenicity events within the first 3 days after IV or SC administration of CAP256V2LS.
Proportion of participants with mild, moderate and severe AEs as well as serious adverse events (SAEs) related to the IV or SC administration of CAP256V2LS.
Secondary endpoints
The difference in the elimination half-life, clearance, volume of distribution and area under the concentration decay curve of CAP256V2LS mAb among study groups.
Change in plasma HIV-1 RNA levels from baseline (only for groups 1 c and 1d).
Changes in the concentration of serum anti-CAP256V2LS titres from baseline.
The difference in the functional activity including IgG/IgA binding responses, cellular immune responses and antibody function in plasma and mucosal surfaces compared with baseline.
Proportion of participants reporting CAP256V2LS injections to be acceptable as per study questionnaire.
Sample size calculation
The main objective of this study is to assess the safety, tolerability and PK of CAP256V2LS; hence, the ability of the study to detect SAEs is key. The probability of detecting no SAE, at least one, or at least two SAEs at a specified true event rate will be calculated. These probabilities highlight the likelihood of the study to detect either rare or common AEs. In addition, the 95% CI for the true event rate was calculated. Currently, limited safety data are available to guide the estimation of the true event rate that might be observed in the study. In the absence of available AE rates for CAP256V2LS alone or in combination with other bNAbs, a range of hypothesised event rates to calculate the probability of observing no events, at least one event, or at least two events were used. Among the four participants receiving the active product in each of the groups, there is a 34% chance of observing at least one event if the true event rate is 10%. When the true event rate is two or three times higher, this probability rises to 59% and 76% (table 3).
Table 3.
Probability of observing no events, at least one event or at least two events, for a range of hypothetical true event rates
| True eventrate (%) | No. of participants | 0 events | 1+ events | 2+ events |
| 1 | 4 | 0.96 | 0.04 | <0.01 |
| 8 | 0.92 | 0.08 | <0.01 | |
| 12 | 0.89 | 0.11 | 0.01 | |
| 16 | 0.85 | 0.15 | 0.01 | |
| 24 | 0.79 | 0.21 | 0.02 | |
| 56 | 0.57 | 0.43 | 0.11 | |
| 5 | 4 | 0.81 | 0.19 | 0.01 |
| 8 | 0.66 | 0.34 | 0.06 | |
| 12 | 0.54 | 0.46 | 0.12 | |
| 16 | 0.44 | 0.56 | 0.19 | |
| 24 | 0.29 | 0.71 | 0.34 | |
| 56 | 0.06 | 0.94 | 0.78 | |
| 10 | 4 | 0.66 | 0.34 | 0.05 |
| 8 | 0.43 | 0.57 | 0.19 | |
| 12 | 0.28 | 0.72 | 0.34 | |
| 16 | 0.19 | 0.81 | 0.49 | |
| 24 | 0.08 | 0.92 | 0.71 | |
| 56 | <0.01 | >0.99 | 0.98 | |
| 20 | 4 | 0.41 | 0.59 | 0.18 |
| 8 | 0.17 | 0.83 | 0.50 | |
| 12 | 0.07 | 0.93 | 0.73 | |
| 16 | 0.03 | 0.97 | 0.86 | |
| 24 | <0.01 | >0.99 | >0.99 | |
| 56 | <0.01 | >0.99 | >0.99 | |
| 30 | 4 | 0.24 | 0.76 | 0.35 |
| 8 | 0.06 | 0.94 | 0.74 | |
| 12 | 0.01 | 0.99 | 0.91 | |
| 16 | <0.01 | >0.99 | 0.97 | |
| 24 | <0.01 | >0.99 | >0.99 | |
| 56 | <0.01 | >0.99 | >0.99 |
Since the phase I assessment of the safety of CAP256V2LS administered SC includes eight participants receiving a combination of CAP256V2LS and VRC07-523.LS (ie, groups 3a and 3b combined), there is an 8% chance of observing at least one event, if the true event rate is 1%, but it is as high as 83%, if the true event is 20%. These probabilities are also applicable to the groups that will receive repeat doses (group 2d and 2 f). For the 16 participants receiving CAP256V2LS administered IV in group 1, there is an 85% chance of observing no events if the event rate is 1%, and less than 1% chance, if the event rate is 30-fold higher. However, if all 56 women receiving CAP256V2LS active product at enrolment are combined, the former probability changes to 57% for no events and remains very low (<1%) given the event rates of 10%, 20% and 30%. As expected, an increase in sample size increases the likelihood of detecting rare events.
Study procedures
Informed consent
Prior to screening and enrolment, informed consent will be obtained from every participant in accordance with the South African Good Clinical Practice guidelines. The informed consent procedure will be conducted in either English or isiZulu as per participant preference. If a participant is illiterate, an impartial witness will be present throughout the informed consent procedure to ensure that all questions are answered to the satisfaction of the potential participant. For the purposes of this study, consent for all study-related procedures, pharmacogenetic studies and storage of specimens will be obtained.
Screening
All potential participants will complete the informed consent procedure for screening and provide relevant identification documents. In order to rule out coenrolment in other intervention trials, potential participants’ details will be checked on the Biometric Co-Enrolment Prevention System. The study team will also evaluate and review proof of contraceptive use, such as family planning records. HIV precounselling will be performed, and if the participant meets eligibility, the sociodemographic and behavioural questionnaires will be administered. Post-test counselling is provided after disclosure of all HIV results and if the participant is ineligible to enrol into the study, referral to one of several HIV/AIDS care programmes will be facilitated. HIV testing in this study will be conducted by following a study HIV algorithm. At the screening, HIV testing will be performed using two rapid antibody testing kits. Any participant with discordant results will be regarded as ineligible and referred to medical care.
A comprehensive medical history and physical examination will be conducted to determine eligibility. All pre-existing conditions and concomitant medication information will be documented. Screening laboratory tests will be conducted as per the schedule of evaluations (SOE) to determine further eligibility. These tests include urinalysis, haematology, blood chemistry tests, liver function tests, testing for sexually transmitted infections including syphilis serology and hepatitis B virus assays. Serum and plasma specimens will also be stored, for further analyses. In addition, a genital specimen using a menstrual cup device will be obtained and stored.37
Randomisation
An unblinded statistician who is not involved in study conduct will be responsible for generating the randomisation sequence (for groups 1 c, 1d, 3a to 3 c and 4a) using SAS V.9.4 software or latest. Participants will be assigned to unique participant identification numbers stratified by HIV status and group number. Sequentially numbered, sealed, opaque envelopes containing the group number and envelope number or treatment code (for use by the unblinded pharmacist only) will be provided to the study coordinator, to be opened once a participant has been deemed eligible and is ready to be enrolled into the study.
The pharmacist will store envelopes in a secured location within the research pharmacy with access restricted to delegated study pharmacists only. The study pharmacist will also receive a randomisation list consisting of the unique three-digit envelope number and study group with the corresponding study drug or placebo and relevant dosage from the unblinded statistician. The envelope number will enable the unblinded pharmacist to assign the correct treatment to the correct participant.
Enrolment
Enrolment will take place within 56 days of screening. After the informed consent is obtained and eligibility criteria are met, the participant will be allocated to a study group (table 2). At this visit, vital signs will be recorded, a targeted physical examination will be conducted and all laboratory results from the screening visit will be reviewed. For women of childbearing potential, a negative pregnancy test on that day must be obtained prior to product administration. Prior to the infusion/injection, the study team must ensure that the participant is eligible to receive study product. All study product administrations will be completed according to the assigned group and may be via an IV infusion or SC injection. Once safety has been established in the first participants, enrolment into the next groups as well as dose escalation will take place in a sequential manner, following review at each step of safety data from the preceding groups. For the bNAb combination groups, two separate injections, each containing a single bNAb, will be administered. After receiving the study product, all participants will be observed for a minimum of 1 hour after the first product and any repeat product administrations. PK analysis on blood draws will be conducted for both the IV and SC administration groups at the timelines outlined in the SOE.
Management of HIV-positive participants enrolled in group 1c and 1D
All potential volunteers who meet the eligibility criteria for enrolment will receive extensive ART counselling on entering the study. The benefits of early ART initiation, including the universal treatment policy, will be explained. Only participants who, after appropriate counselling, are willing to defer ART initiation will be eligible to enrol. At each study visit, the participant’s decision to defer ART initiation will be reviewed, and participants are allowed to change their mind and start treatment at any stage during the study. During the study, HIV-positive participants will be monitored closely with regular clinical and safety assessments. HIV viral load monitoring and CD4 count measurements will take place as per SOE. Participants will be counselled on reducing the risk of HIV transmission to their sexual partners. Furthermore, a multidisciplinary approach to HIV transmission risk mitigation will be followed as per guidance from previous publications.38
Specific criteria to initiate ART within 8 weeks of product administration are listed below and have been outlined as per guidance previously described for studies designed for ART interruption.39 Once any of these criteria are met, the participant will receive ART counselling and will be initiated on ART as per South African guidelines. Criteria to start ART within 8 weeks of product administration include:
Two consecutive viral loads >10 000 copies/mL after 2 weeks after product administration.
CD4 count <350 cells/mL.
Pregnancy.
If ART is deemed medically necessary.
If requested by the participant at any stage during the study.
Safety monitoring
Reactogenicity assessments
Reactogenicity events are 12 common infusion/injection-related signs and symptoms. These are a subset of AEs and have specific reporting requirements. Reactogenicity signs and symptoms are solicited from the start of the infusion/injection through the 3-day post-infusion/injection reactogenicity period. Reactogenicity events may be infusion/injection site reactions (infusion/injection-related erythema/redness or induration/swelling), local symptoms (pain, tenderness) or systemic signs or symptoms (increased body temperature, malaise and/or fatigue, myalgia, headache, chills, arthralgia, nausea and vomiting). After study product administration, the participant will be seen at the clinic for safety assessments on the day of product administration (day 0), as well as day 1, day 2 and day 3 (4 days in total). Clinicians and nurses will assess the product administration site(s) for local reactogenicity on the day of product administration and during the scheduled follow-up visits for all groups. Participants will keep a daily diary of local and systemic symptoms and record their temperature for 3 days after each product administration. In the event of a missed reactogenicity clinic visit, study staff will review the diary together with the participant once they present to the clinic and will determine the severity of the reactions. For any reactogenicity symptoms that are not resolved within 3 days, clinicians will follow and collect resolution information.
Safety monitoring
Safety monitoring includes internal monitoring by the study team, the protocol safety review team (PSRT) and the data safety monitoring board (DSMB), as well as external monitoring including audits. Safety reporting of SAEs, AEs and other important reportable events will be the responsibility of the entire study team. In addition, the study statistician will prepare routine study safety progress reports, which include reports of AEs experienced by study participants (blinded to treatment assignment), for review by the PSRT. The PSRT will review the clinical safety data on a weekly basis via electronic distribution of reports and will have face-to-face meetings as required. The PSRT will be responsible for decisions related to participant safety. In addition, an independent DSMB will meet in-person and/or via teleconference semiannually and review the study data and study conduct. The DSMB could recommend that the study should proceed as designed, should proceed with design modifications or should be discontinued. Furthermore, the Principal Investigator will permit authorised representatives such as external monitors and auditors to inspect the site facilities and records relevant to the study.
Follow-up visits
All enrolled participants in both intervention and control arms will have follow-up visits as specified in the SOE. At these visits, behavioural questionnaires will be administered together with HIV risk-reduction counselling. Contraception counselling and provision will also take place. Targeted physical examinations will be conducted and all reactogenicity and AEs will be recorded. Laboratory investigations specified in the SOE will be conducted by the CAPRISA Research Laboratories. Where required, sample processing and storage of specimens for potential future testing (blood and vaginal specimens) will also be undertaken. An accredited contract laboratory will perform all safety blood testing and provide a backup laboratory service when required.
Statistical analysis
Baseline characteristics including demographics and laboratory measurements will be summarised using descriptive statistics either by group or overall. Summaries of the number and percentage of participants experiencing any AE or reactogenicity will be analysed. AEs and SAEs will be coded into the Medical Dictionary for Regulatory Activities preferred terms. The number and percentages of participants experiencing each specific AE will be tabulated by severity and relationship to study product. For the calculations in these tables, each participant’s AE will be counted once under the maximum severity or strongest recorded causal relationship to study product. A complete listing of AEs for each participant will provide details including severity, relationship to study product, onset, duration and outcome. Tolerability evaluation will be mostly descriptive and consist of solicited AEs that occur within 1 hour following study product administration and reasons for any withdrawal or discontinuation based on participant discomfort. This early assessment of tolerability of the monoclonal antibodies will inform which parameters should be solicited or routinely assessed to further characterise the tolerability profile in a larger number of participants. Where appropriate, some of the data will be presented graphically. The analysis will be carried out using either SAS V.9.4 or higher or R.
PK analysis
PK disposition of CAP256V2LS alone and in combination with VRC07-523LS and PGT121 will be evaluated in this study and PK will be assessed via dose subgroups and route of administration. Sixteen participants will receive CAP256V2LS by the IV route. The two lower dose levels will be administered to HIV-negative participants and the highest dose level will be administered to HIV-positive participants. These PK data will serve to characterise dose effects on CAP256V2LS clearance, volumes of distribution and elimination half-life. The PK data across group 1 will also enable a preliminary evaluation of the potential impact of HIV status on CAP256V2LS PK. The PK results from the IV administration groups will be compared with the data from the SC administration groups in order to estimate bioavailability.
A total of 24 participants will receive CAP256V2LS by the SC route, either as a single dose or a repeat dose. A subset of participants will receive CAP256V2LS with SC hyaluronidase to allow the larger doses of antibody to be administered SC. These data will allow concentration profiles, bioavailability and absorption patterns to be established. The impact of repeat CAP256V2LS SC administration on PK will also be determined. The PK data from the combination groups (groups 3 and 4) will be compared with those from group 2 to determine if coadministration of VRC07-523LS or PGT121 affects CAP256V2LS PK. In addition, the VRC07-523LS and PGT121 PK results will be compared with findings from PK studies in CAPRISA 012A and reported in the literature to establish whether CAP256V2LS impacts the PK of VRC07-523LS and PGT121.
Data management
Data for the CAPRISA 012B study will be collected on case report forms (CRFs) designed specifically to address the protocol requirements. Data will be managed by the CAPRISA Data Management department, using DFdiscover (DF/Net Research, Inc), software specifically designed for clinical trial data management. The site will record data on paper CRFs that will be directly captured onto the DFdiscover system and validated by the data management staff. All source documents will be kept in the participants’ study files and medical charts at the clinical research site. All original CRFs and study-related documents will be securely stored at the site, during the study and after study completion.
Ethics and dissemination
Regulatory approval has been granted by the University of KwaZulu-Natal (UKZN) Biomedical Research Ethics Committee and by the South African Health Products Regulatory Authority (trial reference numbers: BREC00000857/2019 and SAHPRA 20200123). The study team will disseminate the trial results by sharing with the scientific community at international conferences, through peer-reviewed journal publications and presentations to the wider community. Trial results will be uploaded onto the UKZN repository and the Pan-African Clinical Trial Registry.
Supplementary Material
Footnotes
Contributors: SAK and QAK conceived the trial. SAK, QAK, SM and NG designed the trial. SM and NG wrote the study protocol. EC will conduct the PK simulations and analysis. NYZ performed sample size calculations and the statistical analysis strategy. KC, NDR, PM, JM and LM contributed to antibody development. CB, TG, DA, NS, CW, DHB, JM, JL, CH, BP and PEF contributed to the planning and conduct of the trial. All authors reviewed the manuscript and consented to publication.
Funding: This study is supported by the European and Developing Countries Clinical Trials Partnership (EDCTP Grant number: RIA2017S) and the South African Medical Research Council (SAMRC), Special Initiative on HIV Prevention Technology.
Competing interests: None declared.
Patient and public involvement: Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement
Data are available upon reasonable request. The study team will disseminate the trial results as broadly as possible. The research team will share findings from the study with the scientific community at international conferences, through peer-reviewed journal publications and through presentations to the wider community. Trial results will be uploaded onto the University of KwaZulu-Natal (UKZN) repository and the Pan-African Clinical Trial Registry (PACTR).
Ethics statements
Patient consent for publication
Not required.
References
- 1.UNAIDS . HIV/AIDS JUNPo. Geneva: UNAIDS, 2018. [Google Scholar]
- 2.de Oliveira T, Kharsany ABM, Gräf T, et al. Transmission networks and risk of HIV infection in KwaZulu-Natal, South Africa: a community-wide phylogenetic study. Lancet HIV 2017;4:e41–50. 10.1016/S2352-3018(16)30186-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dellar RC, Dlamini S, Karim QA. Adolescent girls and young women: key populations for HIV epidemic control. J Int AIDS Soc 2015;18:19408. 10.7448/IAS.18.2.19408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdool Karim SS. HIV-1 epidemic control-insights from test-and-treat trials. N Engl J Med 2019;381:286–8. 10.1056/NEJMe1907279 [DOI] [PubMed] [Google Scholar]
- 5.Fang W, Mphoyi BI, Motake DR, et al. Efficacy, adherence and side effects of PreP for HIV-1 prevention. Int J Biol 2019;11:80. 10.5539/ijb.v11n4p80 [DOI] [Google Scholar]
- 6.Fauci AS, Marston HD. Ending the HIV-AIDS pandemic—follow the science. N Engl J Med 2015;373:2197–9. 10.1056/NEJMp1502020 [DOI] [PubMed] [Google Scholar]
- 7.Anderson DJ, Politch JA, Vaca GB, et al. Use of monoclonal antibodies to prevent the sexual transmission of human immunodeficiency virus type 1. Curr Immunol Rev 2019;15:123–30. 10.2174/1573395514666180605091240 [DOI] [Google Scholar]
- 8.Pegu A, Hessell AJ, Mascola JR, et al. Use of broadly neutralizing antibodies for HIV-1 prevention. Immunol Rev 2017;275:296–312. 10.1111/imr.12511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hessell AJ, Poignard P, Hunter M, et al. Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat Med 2009;15:951–4. 10.1038/nm.1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker LM, Huber M, Doores KJ, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 2011;477:466–70. 10.1038/nature10373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moldt B, Rakasz EG, Schultz N, et al. Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc Natl Acad Sci U S A 2012;109:18921–5. 10.1073/pnas.1214785109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barouch DH, Whitney JB, Moldt B, et al. Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature 2013;503:224–8. 10.1038/nature12744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sok D, Burton DR. Recent progress in broadly neutralizing antibodies to HIV. Nat Immunol 2018;19:1179–88. 10.1038/s41590-018-0235-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Loggerenberg F, Mlisana K, Williamson C, et al. Establishing a cohort at high risk of HIV infection in South Africa: challenges and experiences of the CAPRISA 002 acute infection study. PLoS One 2008;3:e1954. 10.1371/journal.pone.0001954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doria-Rose NA, Schramm CA, Gorman J, et al. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 2014;509:55–62. 10.1038/nature13036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheward DJ, Marais J, Bekker V, et al. Hiv superinfection drives de novo antibody responses and not neutralization breadth. Cell Host Microbe 2018;24:593–9. 10.1016/j.chom.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagh K, Bhattacharya T, Williamson C, et al. Optimal combinations of broadly neutralizing antibodies for prevention and treatment of HIV-1 clade C infection. PLoS Pathog 2016;12:e1005520. 10.1371/journal.ppat.1005520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore PL, Gray ES, Sheward D, et al. Potent and broad neutralization of HIV-1 subtype C by plasma antibodies targeting a quaternary epitope including residues in the V2 loop. J Virol 2011;85:3128–41. 10.1128/JVI.02658-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhiman JN, Anthony C, Doria-Rose NA, et al. Viral variants that initiate and drive maturation of V1V2-directed HIV-1 broadly neutralizing antibodies. Nat Med 2015;21:1332–6. 10.1038/nm.3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doria-Rose NA, Bhiman JN, Roark RS, et al. New member of the V1V2-directed CAP256-VRC26 lineage that shows increased breadth and exceptional potency. J Virol 2016;90:76–91. 10.1128/JVI.01791-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gorman J, Chuang G-Y, Lai Y-T, et al. Structure of super-potent antibody CAP256-VRC26.25 in complex with HIV-1 envelope reveals a combined mode of Trimer-Apex recognition. Cell Rep 2020;31:107488. 10.1016/j.celrep.2020.03.052 [DOI] [PubMed] [Google Scholar]
- 22.Gaudinski MR, Coates EE, Houser KV, et al. Safety and pharmacokinetics of the Fc-modified HIV-1 human monoclonal antibody VRC01LS: a phase 1 open-label clinical trial in healthy adults. PLoS Med 2018;15:e1002493. 10.1371/journal.pmed.1002493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Julg B, Tartaglia LJ, Keele BF, et al. Broadly neutralizing antibodies targeting the HIV-1 envelope V2 apex confer protection against a clade C SHIV challenge. Sci Transl Med 2017;9:eaal1321. 10.1126/scitranslmed.aal1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pegu A, Borate B, Huang Y, et al. A meta-analysis of passive immunization studies shows that serum-neutralizing antibody titer associates with protection against SHIV challenge. Cell Host Microbe 2019;26:336–46. 10.1016/j.chom.2019.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivleva VB, Schneck NA, Gollapudi D, et al. Investigation of sequence clipping and structural heterogeneity of an HIV broadly neutralizing antibody by a comprehensive LC-MS analysis. J Am Soc Mass Spectrom 2018;29:1512–23. 10.1007/s13361-018-1968-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaccine Research Centre . Investigators brochure CAP256V2LS (VRC-HIVMAB 0102-00-AB), version 10, 2020. [Accessed 12 Feb 2020].
- 27.Haynes BF, Moody MA, Verkoczy L, et al. Antibody polyspecificity and neutralization of HIV-1: a hypothesis. Hum Antibodies 2005;14:59–67. 10.3233/HAB-2005-143-402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Meer PJK, Kooijman M, Brinks V, et al. Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. MAbs 2013;5:810–6. 10.4161/mabs.25234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mahomed S, Garrett N, Capparelli E, et al. Assessing the safety and pharmacokinetics of the monoclonal antibodies, VRC07-523LS and PGT121 in HIV negative women in South Africa: study protocol for the CAPRISA 012A randomised controlled phase I trial. BMJ Open 2019;9:e030283. 10.1136/bmjopen-2019-030283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaudinski MR, Houser KV, Doria-Rose NA, et al. Safety and pharmacokinetics of broadly neutralising human monoclonal antibody VRC07-523LS in healthy adults: a phase 1 dose-escalation clinical trial. Lancet HIV 2019;6:e667–79. 10.1016/S2352-3018(19)30181-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stephenson K, Julg B, Ansel J, et al. Therapeutic activity of PGT121 monoclonal antibody in HIV-infected adults. Conference on retroviruses and opportunistic infections; Seattle, Washington 2019.
- 32.Mahomed S, Garrett N, Baxter C, et al. Clinical trials of broadly neutralizing monoclonal antibodies for HIV prevention: a review. J Infect Dis 2020. doi: 10.1093/infdis/jiaa377. [Epub ahead of print: 30 Jun 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Locke KW, Maneval DC, LaBarre MJ. ENHANZE® drug delivery technology: a novel approach to subcutaneous administration using recombinant human hyaluronidase PH20. Drug Deliv 2019;26:98–106. 10.1080/10717544.2018.1551442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morcos PN, Zhang X, McIntyre C, et al. Pharmacokinetics and pharmacodynamics of single subcutaneous doses of tocilizumab administered with or without rHuPH20. Int J Clin Pharmacol Ther 2013;51:537–48. 10.5414/CP201847 [DOI] [PubMed] [Google Scholar]
- 35.Morrow L, Muchmore DB, Ludington EA, et al. Reduction in intrasubject variability in the pharmacokinetic response to insulin after subcutaneous co-administration with recombinant human hyaluronidase in healthy volunteers. Diabetes Technol Ther 2011;13:1039–45. 10.1089/dia.2011.0115 [DOI] [PubMed] [Google Scholar]
- 36.Shpilberg O, Jackisch C. Subcutaneous administration of rituximab (MabThera) and trastuzumab (Herceptin) using hyaluronidase. Br J Cancer 2013;109:1556–61. 10.1038/bjc.2013.371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Archary D, Liebenberg LJ, Werner L, et al. Randomized cross-sectional study to compare HIV-1 specific antibody and cytokine concentrations in female genital secretions obtained by menstrual cup and cervicovaginal lavage. PLoS One 2015;10:e0131906. 10.1371/journal.pone.0131906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peluso MJ, Dee L, Campbell D, et al. A collaborative, multidisciplinary approach to HIV transmission risk mitigation during analytic treatment interruption. J Virus Erad 2020;6:34–7. 10.1016/S2055-6640(20)30009-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Julg B, Dee L, Ananworanich J, et al. Recommendations for analytical antiretroviral treatment interruptions in HIV research trials-report of a consensus meeting. Lancet HIV 2019;6:e259–68. 10.1016/S2352-3018(19)30052-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon reasonable request. The study team will disseminate the trial results as broadly as possible. The research team will share findings from the study with the scientific community at international conferences, through peer-reviewed journal publications and through presentations to the wider community. Trial results will be uploaded onto the University of KwaZulu-Natal (UKZN) repository and the Pan-African Clinical Trial Registry (PACTR).