

Abstract
Clinical trials are fundamental for advances in cancer treatment. The traditional framework of phase 1 to 3 trials is designed for incremental advances between regimens. However, our ability to understand and treat cancer has evolved with the increase in drugs targeting an expanding array of therapeutic targets, the development of progressively comprehensive data sets, and emerging computational analytics, all of which are reshaping our treatment strategies. A more robust linkage between drugs and underlying cancer biology is blurring historical lines that define trials on the basis of cancer type. The complexity of the molecular basis of cancer, coupled with manifold variations in clinical status, is driving the individually tailored use of combinations of precision targeted drugs. This approach is spawning a new era of clinical trial types. Although most care is delivered in a community setting, large centers support real‐time multi‐omic analytics and their integrated interpretation by using machine learning in the context of real‐world data sets. Coupling the analytic capabilities of large centers to the tailored delivery of therapy in the community is forging a paradigm that is optimizing service for patients. Understanding the importance of these evolving trends across the health care spectrum will affect our treatment of cancer in the future and is the focus of this review.
Keywords: big data, clinical trial, clinical trial protocol, precision medicine
Short abstract
With advances in cancer biology, precision therapeutics, and big data, clinical trial designs are evolving. They are transforming cancer care and research across the biomedical enterprise.
Introduction
The goal of clinical trials in cancer drug development is to improve clinically meaningful outcomes for patients. The result of a clinical trial has the potential to shape the care of many future patients, to alter our understanding of human biology, and to produce long‐lasting financial ramifications for health care and industry. With such wide‐ranging impacts, the nature of its design is paramount.
The traditional model of progressing from phase 1 through phase 3 is considered the standard paradigm for drug development. However, there is a high rate of phase 3 failure, 1 and this signifies that early‐phase trials have poor specificity for predicting benefits. The high failure rate results in a large number of participants being exposed to ineffective therapies and wasted resources. Even with an effective experimental drug, this traditional paradigm is inefficient, with the time from pipeline to market averaging 12 years. 2 Because of the importance of clinical trials, their designs are constantly being evaluated and modified.
Sequencing of the human genome and advances in cancer biology have yielded an expanding number of biological targets and associated therapeutics. Our ability to probe increasingly complex data‐intensive aspects of cancer biology at the single‐patient level is progressively guiding the prospective application of targeted therapeutics and spawning rapid change in clinical trial design.
Herein, we provide a review of cancer clinical trial designs; we begin with a historical perspective, which is followed by a discussion of new directions. Emphasis is placed on precision therapeutics and emerging concepts and technologies in oncology and how they are transforming the clinical trial landscape. This transformation will affect the entire health care enterprise. This review aims to inform those across that enterprise about these coming changes and how they will affect oncology care.
Modern History of Chemotherapy Development
The first modern anticancer drug evolved from chemical warfare agents developed during World War II. After encouraging animal model studies, Louis Goodman and Alfred Gilman performed the first human chemotherapy experiment in 1942 by using nitrogen mustard for the treatment of patients with non‐Hodgkin lymphoma. 3 Despite not having an appropriate dose‐finding phase, it arguably met the modern definition of a clinical trial: a prospective, organized, systematic exposure of voluntary participants to an intervention. This marked the beginning of modern chemotherapy development.
In the 1950s, the US Congress invested in the formation of the National Institutes of Health and the National Cancer Institute (NCI). The National Cancer Chemotherapy Service Center was formed at the NCI and established many of the components of modern drug development and clinical trials. The National Cancer Chemotherapy Service Center evolved into the Cancer Therapy Evaluation Program. The work of Li et al 4 at the NCI led to the first remission of a solid tumor (choriocarcinoma) with methotrexate. The case series was published in 1958, and the use of gonadotropin as a biomarker of response was also reported.
As the effectiveness of chemotherapy and its combinations in particular was being recognized, clinical trial designs were maturing as well. Supported by the NCI, Frei et al 5 conducted a study comparing 2 dosing regimens of methotrexate with 6‐mercaptopurine in acute leukemia. Published in 1958, this study marked the prototype of the modern clinical trial with inclusion and exclusion criteria, preclinical studies, randomization, toxicity reporting, and endpoints.
Traditional Clinical Trial Design
Historically, during the very early stage of chemotherapeutic development, empirical testing with N of 1 trials or case series was able to demonstrate dramatic clinical responses in uniformly fatal diseases without a treatment alternative. 3 , 4 However, with a steady increase in treatment regimens, there came a need to systemically compare and quantify their efficacy. This prompted the creation of the traditional phase 1 to 3 clinical trial paradigm, which is well suited for studying stepwise incremental advances in clinical benefits.
Clinical Trial Framework
Phase 1 clinical trial designs
Preclinical models frequently fail to predict performance in humans, including metrics related to toxicity, efficacy, and pharmacology. If preclinical data are promising, an investigational new drug (IND) application can then be submitted to the Food and Drug Administration (FDA), after which a new chemical is first introduced into humans through a phase 1 trial as an experimental drug. A phase 1 trial seeks to define a drug's toxicity profile, to identify a dose suitable for phase 2 trials, and to characterize pharmacokinetics. Although efficacy is not a primary endpoint, it is monitored and often reported. For conventional cytotoxic drugs, the maximum tolerated dose (MTD) defined in the phase 1 trial is typically used in phase 2 and 3 studies because their efficacy generally correlates with dosage. With the newer and diverse classes of drugs, the dose‐response curve may plateau, and thus a minimally effective dose may be more appropriate.
An optimized phase 1 trial design is able to accurately identify the MTD while minimizing the number of subjects exposed to ineffective or overly toxic doses. A phase 1 design typically uses a dose‐escalation scheme, which can be rule‐based, model‐based, or model‐assisted. Although each offers advantages and limitations, retrospective and simulation studies have found ruled‐based designs such as 3 + 3 to be inferior to model‐based or model‐assisted designs in study duration and MTD selection accuracy. 6 , 7 Because of the unpredictable nature of a new chemical entity introduced into the human body, phase 1 trials remain a fundamental part of drug development.
Phase 2 clinical trial designs
Once a dose is determined and safety is established, a phase 2 study is conducted with the goal of evaluating a drug's efficacy and continuing to evaluate its safety. Phase 2 trials are critical in determining whether a drug offers sufficient clinical benefit to warrant a large‐scale phase 3 study. A common efficacy measure is a decrease in the cancer burden, which is quantified by the response rate. In rare circumstances, a drug that demonstrates dramatic efficacy in phase 2 may negate the need for phase 3 testing. Phase 2 design options are diverse and continue to be the subject of debate. Broadly, they can be categorized as single‐arm or randomized multiple‐arm.
Single‐arm phase 2 studies are the most common, and they evaluate whether there is sufficient efficacy, in comparison with historical controls, to justify continued development. Although the use of historical controls allows for a smaller sample size, changes in the standard of care and even the definition of a disease may confound the selection of appropriate historical controls. For trials focusing on participant subsets (eg, based on biomarkers), accurate historical benchmarks may be lacking. 8 Among single‐arm studies, a popular approach is Simon's 2‐stage design, in which the recruitment is performed in 2 stages, with the second stage performed only if a response threshold is first achieved 9 ; this minimizes participants' exposure to an ineffective treatment. Randomized phase 2 trials offer objective comparisons but require an increased sample size. Unlike a phase 3 trial, which measures definitive clinical benefit (eg, survival), randomized phase 2 trials typically evaluate activity representing a high probability that a drug will also be effective in phase 3 (eg, progression‐free survival). Numerous randomized phase 2 designs exist and include selection 10 and discontinuation 11 among other designs. At the conclusion of phases 1 and 2, sponsors and investigators may meet with the FDA to review the IND, the available data, and the viability of progressing to a phase 3 trial and its design. In some cases, FDA‐accelerated approval can take place on the basis of phase 2 data (see the Surrogate Endpoints section).
Phase 3 clinical trial designs
Phase 3 clinical trials compare a new drug or combination to the current standard of care. They remain the standard for determining whether the newer drug or combination offers improved benefits and thus a new standard of care. Comparative efficacy trials seek to determine whether a new drug has superior efficacy, with improved overall survival being a frequent primary endpoint. A phase 3 noninferiority trial seeks to determine whether a new drug is not worse than the standard of care in terms of efficacy. Noninferiority trials are used with drugs that may offer other advantages such as decreased toxicity or cost, but they are statistically complex with inherent limitations. 12 An example is the REAL‐2 trial, which established that capecitabine and oxaliplatin are noninferior to fluorouracil and cisplatin for the treatment of esophagogastric cancer. 13 Phase 3 trials are inherently complex and time‐ and resource‐intensive, and they require rigorous conduct to minimize bias and balance confounders to detect incremental improvements in treatment. There are many design considerations, which are beyond the scope of the current review. Phase 3 trials continue to serve as the foundation for FDA‐sanctioned regulatory approval. If safety and efficacy are established, a new drug application can then be submitted to the FDA for consideration of approval. After the final approval, the drug's safety and efficacy for the intended population can continue to be studied in phase 4 trials.
Surrogate Endpoints
With targeted agents and other novel therapeutics, clinical trial endpoints are evolving. Appropriate endpoints, especially for phase 2 trials, are a topic of ongoing debate. With the FDA's introduction of the accelerated approval pathway in 1992, which allows drug registration on the basis of phase 2 surrogate endpoints for life‐threatening illnesses without effective treatment, this debate has only intensified. 14
With advances in detection, several surrogate endpoints have gained popularity. Optimally, surrogate endpoints should effectively and accurately represent meaningful clinical benefits. Examples include minimal residual disease status in hematologic malignancies and pathologic complete response rates in solid tumors. Surrogate endpoints can be either validated or nonvalidated. A validated one has evidence‐based proof that it significantly predicts a clinically meaningful endpoint. 15 One example is disease‐free survival in early‐stage colon cancer, which highly correlates with overall survival. 16 This resulted from an analysis of more than 20,000 patients from 18 randomized trials, and it highlights the barriers associated with validation. The use of nonvalidated endpoints continues to be a concern. Quality measures and overall survival should be encouraged as the primary endpoints whenever possible.
Trial Design in the Era of Precision Oncology
Limitations of the Current Clinical Trial Paradigm
The current paradigm addresses the need to detect incremental clinical benefits between similar treatment regimens with comparable response kinetics. However, with advances in human genome sequencing and molecular testing and an increased understanding of cancer biology, there is a marked increase in drugs with diverse mechanisms that act on an expanding array of pharmacologic targets. The therapeutic effects of targeted drugs and other novel classes of medicine may differ in quality and timing in comparison with conventional cytotoxic therapeutics, and this may prompt additional trial design considerations. Examples include changes in the kinetics of disease growth (eg, stabilization and decreased doubling time) and the need for revised Response Evaluation Criteria in Solid Tumors criteria to accommodate the unique response kinetics of immunomodulatory drugs.
Also, the historical paradigm of trials based on cancer type is being eroded by molecular phenotyping. In a first for the FDA, in 2017, it approved pembrolizumab in a tumor‐agnostic fashion solely on the basis of microsatellite instability status; the approval of larotrectinib in 2018 for solid tumors with neurotrophic receptor tyrosine kinase (NTRK) gene fusions constitutes another example.
Furthermore, the current paradigm does not effectively address molecular heterogeneity within a histology and often relies on subanalysis. Moreover, it typically studies only a low number of regimens in 1 trial. Lastly, the fixed design and the associated systemic inability to incorporate accumulating data after initiation fail to leverage diagnostic and therapeutic advances while maintaining trial integrity. In this section, we review new trial concepts and designs that have evolved to accommodate such advances in precision medicine.
Adaptive Design
Adaptive design is defined by the FDA as “a clinical trial design that allows for prospectively planned modifications to one or more aspects of the design based on accumulating data from subjects in the trial.” 17 It allows prespecified modification to different aspects of the trial. It is fundamentally different from interim analysis or unplanned ad hoc changes. Adaption can be based on either noncomparative data (blinded analysis) or comparative data (unblinded analysis). Adaptation with noncomparative data can be applied to sample size refinement or to recruitment strategies on the basis of new prognostic data. For example, if the actual variance of the primary endpoint (eg, survival) is higher than anticipated, the sample size can be increased, and an inconclusive trial can thus be avoided. Comparative data adaptation is based on accumulating trial results. For example, I‐SPY2, an umbrella platform trial evaluating neoadjuvant treatment for early‐stage breast cancer, uses adaptive randomization with a statistical model to assign participants to treatment arms with the probability of higher efficacy by using ongoing response and biomarker data. 18 Adaption can be applied to both traditional and more novel clinical trial designs outlined later. Although adaptive design may aid in therapeutic development, specific statistical ramifications need to be considered during the design stage. For instance, if multiple endpoints are being evaluated, the overall chance of having false‐positive findings increases, and a multiplicity analysis must be incorporated into statistical considerations. 19 Recognizing these challenges, recent FDA guidance on adaptive designs recommends that methods should be used to determine appropriate statistical thresholds for interim and final analyses to ensure that the overall type I error is controlled at 2.5%. Importantly, to ensure trial integrity and mitigate the risk of bias from early unblinding, the FDA strongly recommends that prespecified endpoints be included in the trial design along with specifications for interim analysis. The latter may include the number, timing, and statistical methods as well as the limitation of access to comparative interim results to individuals independent of personnel involved in conducting or managing the trial. Overall, the added complexity of adaptive designs may warrant earlier and more extensive interactions with the FDA.
Main Protocol (Previously Known as Master Protocol)
A main protocol, previously known as a master protocol, addresses the concept that the biological heterogeneity of cancer underlies otherwise uniform‐appearing histology and is ultimately responsible for clinical outcomes (eg, responses to therapeutics and/or survival). Broadly, a main protocol is an organized clinical trial construct with a molecular screening process and the ability to evaluate multiple regimens in parallel; it is typically based on a molecular characterization of given cohorts. A screening process identifies participants with biomarkers or characteristics of interest, which then dictate allocation to specific arms within a given trial or to other trials. This design improves the screen success rate, the drug development efficiency, and potentially the therapeutic benefit for individuals. By recognizing the relationship across cancer biology, precision therapeutics, and clinical outcome, this approach uniquely aligns the interests of basic researchers, clinicians, and patients in a manner that fosters close collaborative relationships so as to improve care and advance biology. Common main protocol designs include basket, umbrella, and platform trials, and they are discussed next.
Basket and umbrella trials
Basket trials enroll different histologic cancer types (“included in the same basket”) that share a specific biomarker 20 (Fig. 1). Basket trials examine the hypothesis that the presence of a specific molecular target predicts a response to a matched targeted therapy that is agnostic of tumor histology. 21 It is ideal for evaluating targeted therapies with low prevalence targets. A prototypical example is VE‐BASKET, a histology‐independent phase 2 study of vemurafenib in BRAF V600–positive nonmelanoma cancers (NCT01524978). It enrolled 122 participants across more than 9 nonmelanoma histologies and led to the approval of vemurafenib as a treatment for patients with BRAF V600–positive Erdheim‐Chester disease; this was the first approval based on a basket study. 22 Other basket trials have led to the tissue‐agnostic approval of pembrolizumab for microsatellite instability–high/deficient mismatch repair tumors 23 and larotrectinib for cancer harboring NTRK gene fusion. 24
Figure 1.
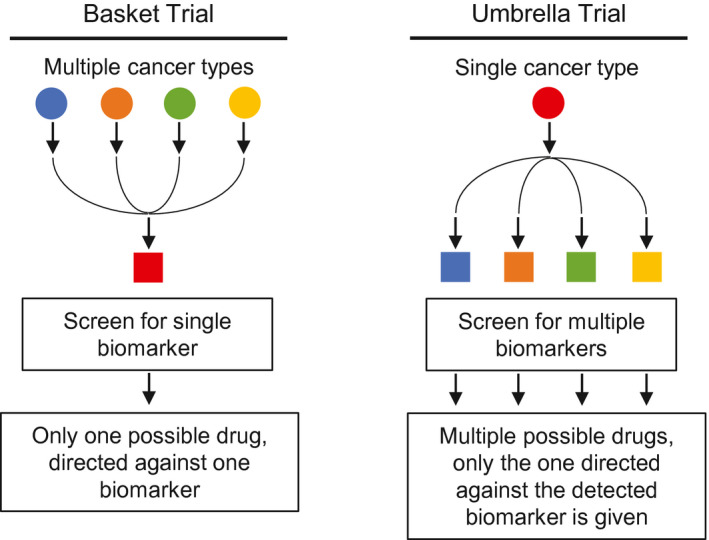
Schematic diagram of basket and umbrella trials.
Unlike basket trials, umbrella trials enroll participants with a single histologic type (“covered under the umbrella”), and on the basis of their biomarker profiles, they are shunted to different treatment arms (Fig. 1). Umbrella trials examine the notion that molecular heterogeneity underlies uniform histologies, and treatment should, therefore, be tailored accordingly. Umbrella trials are well suited to cancer types for which multiple precision therapeutic targets exist, such as adenocarcinoma of the lung. Many prominent umbrella trials also use a platform trial design as discussed next.
Platform trials
A platform trial provides an infrastructure that evaluates multiple targeted therapies for 1 or more diseases through ongoing changes in substudies and involves different therapeutics and/or target cohorts. Existing platform trials are typically randomized and contain a control arm and multiple experimental arms that undergo adaptive change as futility or efficacy is demonstrated. 25 A shared control arm increases overall allocation to experimental arms and allows a smaller sample size in comparison with traditional sequential trials. 25 The adaptive nature allows for the efficient incorporation of newly available therapeutics into an existing infrastructure. Umbrella and basket trials that allow experimental arms to enter and exit are also considered platform trials. Selected noteworthy examples include I‐SPY2, the National Cancer Institute Molecular Analysis for Therapy Choice (NCI‐MATCH) trial, the National Cancer Institute Molecular Profiling–Based Assignment of Cancer Therapy (NCI‐MPACT) trial, and the Lung Cancer Master Protocol (Lung‐MAP) trial, among others, which are discussed in detail elsewhere. 26
Challenges and considerations
These novel trial concepts offer numerous advantages but are complex in design and execution. A robust infrastructure is essential. Logistical challenges, including the potential need to work with multiple industry partners and potential competition among the substudies, need to be considered.
The emphasis on biomarkers in main protocols requires a safe and accurate screening process, which may be challenging. Testing and analytic standards for novel biomarkers may not exist. Furthermore, biomarkers may be prognostic (ie, determining outcome, regardless of treatment) instead of predictive (ie, determining responsiveness to a given therapy). Appropriate measures are required to ensure that the result is not due to intrinsic differences in cancer biology to maintain scientific validity. The focus on singular therapeutic targets also inadequately addresses tumor heterogeneity. Molecular heterogeneity can exist within a patient throughout the clinical course and among lesions. 27 Currently, there is neither a standardized measure for an individual's tumor heterogeneity nor a way to evaluate its therapeutic impact in clinical trials. Besides tumor heterogeneity, a patient may have more than 1 actionable therapeutic target, and this adds another layer of treatment and trial design challenge.
Therapeutic targets are expanding beyond genetic mutations, with the tumor microenvironment, microbiomes, and transcriptional profiling being examples of emerging therapeutic opportunities. 28 Novel trial designs are required to evaluate multidimensional biomarkers. Later, we discuss emerging concepts that are starting to advance through these boundaries.
Lastly, it is encouraging that these clinical trial designs are being increasingly recognized by regulatory agencies. The FDA has released draft guidance for industry on main/master protocols that addresses common considerations of main protocol designs (eg, each substudy within a basket trial should include specific objectives, a scientific rationale for the inclusion of each population, and a detailed statistical analysis plan of sample size justification and stopping rules for futility; in umbrella trials, appropriate dosages of each investigational drug should be established in phase 2 studies first before evaluation in a main protocol, and the common control arm is strongly recommended to be the standard of care for the target population). Because of the significant complexity, each main protocol should be submitted as a new IND and should be the only trial conducted under the IND. A pre‐IND meeting with the FDA is strongly encouraged to guide the design and conduct of the protocol.
Emerging Concepts
As appealing as the idea of identifying a tumor's Achilles' heel and applying a targeted therapy is, one study found that broad molecular testing with off‐label use of matched targeted therapies was not superior to conventional treatment. 29 Although that study has significant limitations, it highlights the complexity of tumor biology beyond a driver mutation. It also serves notice that although the targeted approach of drug development is logical and already has many examples of success, it will not be easy or straightforward. Next, we discuss emerging concepts while highlighting the proviso that although they will launch us into much higher levels of elegance and individually tailored efficacy, they will also bring along unheard‐of levels of complexity.
Systems Biology: Big Data and Multi‐Omic Trial Design
Resistance to most targeted treatments inevitably develops, 30 and this signifies that there are other factors influencing treatment outcomes beyond the driver mutation. A systems biology approach offers an avenue to refine the field of precision oncology. Systems biology evaluates the disease as a dynamic system of genes, proteins, tumor microenvironment, and other factors that interdependently interact in a networked fashion. 31 This concept is beginning to be incorporated into clinical trials using comprehensive molecular profiling beyond that of genomics. It allows for the evaluation of the mechanism of resistance in real time and provides further guidance on experimental arm selection.
Through regulator mutations and/or epigenetic changes, patients without a known driver mutation can exhibit aberrant cellular signaling similar to that of patients harboring the driver mutations. 32 The WINTHER trial compared treatment decisions based on targetable alterations as determined by DNA sequencing with those based on RNA expression profiling. 33 It demonstrated that the evaluation of gene expression reveals therapeutic targets and highlighted the need for a systems biology approach.
A consideration of the fact that cancer biology is both defined and affected by the state of the DNA, RNA, proteins, cellular microenvironment, and system (at the whole‐body level) in which it resides invariably leads to the logical conclusion that all such components should be considered together. A comprehensive translational oncology platform trial based on an integrated consideration of these multi‐omic parameters is typified in the Serial Measurements of Molecular and Architectural Responses to Therapy (SMMART) trial. 34 The SMMART platform incorporates basic concepts designed to move cancer therapeutics into the future. This platform incorporates serial, real‐time, comprehensive molecular tumor profiling; the delivery of different, prototypically unique combinations of precision targeted therapeutics; and an iterative conduct of this process to adapt therapy as the cancer and the patient adapt to the treatment (Fig. 2). For previously untested combinations of drugs, intrapatient dose escalation is built in. The platform's feasibility is established, and 4 different histologies are currently being recruited in a phase 1b study (NCT03878524). 34 After comprehensive tumor profiling, participants are assigned to a treatment according to the recommendation of a molecular tumor board (MTB). MTBs are discussed later.
Figure 2.
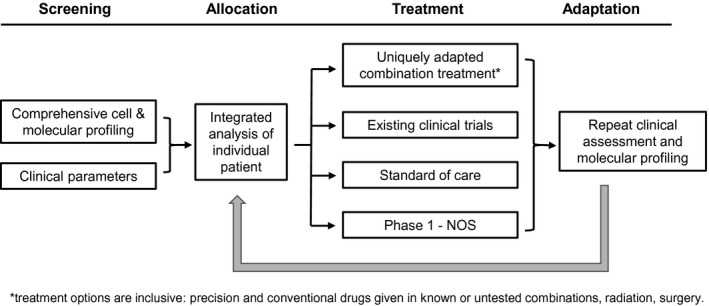
Schematic diagram of the Serial Measurements of Molecular and Architectural Responses to Therapy clinical trial platform. NOS, not otherwise specified.
Incorporating Real‐World Data
A common concern of clinical trial results is their applicability to real‐world clinical practice. Postmarketing surveillance identifies adverse events but not other key outcome metrics, focuses only on a given drug, and is not systematized. Real‐world data (RWD), defined as information generated outside a trial that advances our understanding, can fill knowledge gaps between controlled trial scenarios and real‐world patients and clinical practice. RWD has the potential to guide and refine research directions and clinical trial designs. The electronic health record has revolutionized RWD research. However, its use in precision medicine is limited by the lack of standards in dealing with complex molecular data. A systemic way to record and obtain high‐quality and comprehensive RWD on a broad scale is necessary.
The recently described master observational trial (MOT), or main observational trial, is a novel clinical trial construct that provides an avenue for collecting high‐quality and comprehensive molecular data from both clinical trials and clinical practice. 35 The MOT contains elements of a main protocol and a prospective observational trial, is geared toward precision medicine, and is focused on RWD. It differs from other prospective data registries in its modular nature and utilization of a precise standardized method of cataloging molecular data. The MOT enrolls participants with board cancer types, and the clinical outcome for each line of therapy is recorded. The diagnostic and treatment data are integrated into a main prospective observational registry. The modular nature allows other parameters to be incorporated as technologies develop. The recently announced Registry of Oncology Outcomes Associated With Testing and Treatment (ROOT) trial is one of the first examples of an MOT trial (NCT04028479). ROOT is a multicenter observational trial that collects comprehensive molecular and clinical data with the goal of building a centralized precision medicine–focused oncology database. Coupled with genome‐wide association studies and eventually multi‐omic data, RWD from an MOT can be a powerful tool for future drug development.
Molecular Tumor Board
The MTB, as a concept, is a panel of multidisciplinary experts who make treatment recommendations on the basis of clinical and molecular data. They should optimally include experts in medical and radiation oncology, pathology, bioinformatics, cancer biology, genetics, pharmacology, and other fields as needed. Recently, an evaluation by an MTB was reported to improve diagnostic decisions in comparison with standard recommendations associated with mutation analysis and to increase accrual for clinical trials. 36 The MTB is emerging as a critical component of biomarker‐driven trials. Multiple precision medicine trials have reported utilization of MTBs for the interpretation of complex molecular data and participant allocations. 33 , 34 , 37
Machine Learning
Machine learning (ML), broadly defined as computational data‐analytic techniques for building predictive models based on past data, 38 can identify patterns within large‐scale, multidimensional data sets that would otherwise be unfeasible for a human observer. Various oncology ML models have been designed to predict treatment responses. 39 , 40 One particular model was able to predict an individual's response to conventional chemotherapy with high accuracy on the basis of genomic and clinical data. 40
The integration of ML into clinical trials offers many potential applications ranging from predicting rational drug combinations to identifying new therapeutic targets. ML can also improve adaptive trial designs beyond conventional statistics‐based adaptive strategies. ML has the ability to identify a targeted therapy on the basis of a composite molecular profile and thereby refine the match between the underlying biology and the treatment regimen. It will allow for more innovative clinical trial designs. Instead of allocation based on a single biomarker, ML models can refine allocation by incorporating multidimensional data. Increased incorporation of ML into cancer treatment identification is inevitable. ML's potential for making complex, individualized, accurate predictions makes it a powerful research tool and a rapidly developing clinical tool. The FDA has published an updated regulatory framework for ML in software (as medical devices) that focuses on manufacturer transparency and real‐world performance monitoring. 41 As ML plays an increasing role in both clinical trials and clinical practice, ongoing revision of its regulation will be critical.
Individualized Dynamic Studies
It is increasingly acknowledged that tumors adapt and evolve constantly under selective treatment pressures. 42 This challenge prompted the development of the individualized dynamic model, which emphasizes longitudinal monitoring of the evolving molecular landscape and enables early discovery of emerging resistance.
Common approaches include patient‐derived xenografts (PDXs) and patient‐derived organoids (PDOs). 43 , 44 PDXs involve propagating tumor tissue in mice, whereas PDOs are patient‐derived tumor tissues embedded into a 3‐dimensional matrix culture and then grown into tumor organoids that retain the structural and functional characteristics of the source tumor. 45 Recent studies found that PDXs and PDOs for various solid tumors had a high concordance with their native tumors, and thy identified effective drugs and novel combinations that were subsequently validated in cell lines and xenograft models of the corresponding tumor type. 46 , 47 These encouraging results highlight the potential of individualized dynamic models for guiding cancer treatment and next‐generation clinical trial development. Although the FDA has guidance for industry on xenotransplantation and human cells, tissues, and their related products, regulations for PDXs and PDOs along with the potential ethical issues involved will need to be considered.
Challenges and Opportunities
Oncology is multidisciplinary in nature. However, trial designs that effectively evaluate multimodality regimens beyond simple combinations remain difficult because of various challenges, including appropriate controls and blinding. The use of biomarkers has high potential but currently lacks standardization across the expanding technologies. Radiomics, which is the method of extracting and analyzing quantitative data from imaging, is an emerging class of biomarkers with strong promise. 48 , 49 Trial designs that incorporate nontraditional biomarkers and evaluate different treatment modalities in different combinations and sequencing in the context of targeted therapy remain an unmet need. In addition to the innovation of clinical trial designs based on an improved biological understanding of cancer, similar attention should be paid to how the FDA and other regulatory bodies oversee different clinical trial designs and their associated approvals.
Current and future clinical trials require a robust and dedicated infrastructure for which large health care centers are well suited. However, the treatment of patients within their communities offers convenience and deep relationships aided by geography, and communities are where the majority of cancer care is delivered. This represents an opportunity to bridge and leverage the strengths of these 2 systems. Efforts include centralized analysis and remote MTBs with treatment delivered in the community, which can result in a collaborative relationship between health care settings.
In conclusion, the clinical trial is a fundamental part of oncology. The traditional phase 1 to 3 trial paradigm has defined the vast majority of our current standard‐of‐care treatments. However, with the rapid development of novel therapeutics and biomarkers, a different strategy is opening up before us and is being put into practice. Main protocols incorporate our current understanding of cancer biology and relevant targeted therapy. As our understanding of cancer biology continues to evolve, clinical trial designs will need to evolve as well. Trial designs that integrate emerging concepts such as comprehensive molecular profiling, ML, and RWD and established multimodality treatments will be critical to the care of patients with cancer. Lastly, clinical trial design is only 1 aspect of a trial. The same level of consideration should be applied to ensuring the integrity of clinical trial conduct, appropriate interpretation of the results, and application to clinical practice.
Funding Support
No specific funding was disclosed.
Conflict of Interest Disclosures
Raymond C. Bergan is an inventor on patents related to KBU2046 and therapeutically targeting cancer motility and is co‐owner of Third Coast Therapeutics, which has an option to license those patents. The other author made no disclosures.
Li A, Bergan RC. Clinical trial design: Past, present, and future in the context of big data and precision medicine. Cancer. 2020. 10.1002/cncr.33205
References
- 1. Harrison RK. Phase II and phase III failures: 2013‐2015. Nat Rev Drug Discov. 2016;15:817‐818. [DOI] [PubMed] [Google Scholar]
- 2. Fargen KM, Frei D, Fiorella D, et al. The FDA approval process for medical devices: an inherently flawed system or a valuable pathway for innovation? J Neurointerv Surg. 2013;5:269‐275. [DOI] [PubMed] [Google Scholar]
- 3. Goodman LS, Wintrobe MM, Dameshek W, et al. Nitrogen mustard therapy; use of methyl‐bis (beta‐chloroethyl) amine hydrochloride and tris (beta‐chloroethyl) amine hydrochloride for Hodgkin's disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J Am Med Assoc. 1946;132:126‐132. [DOI] [PubMed] [Google Scholar]
- 4. Li MC, Hertz R, Bergenstal DM. Therapy of choriocarcinoma and related trophoblastic tumors with folic acid and purine antagonists. N Engl J Med. 1958;259:66‐74. [DOI] [PubMed] [Google Scholar]
- 5. Frei E III, Holland JF, Schneiderman MA, et al. A comparative study of two regimens of combination chemotherapy in acute leukemia. Blood. 1958;13:1126‐1148. [PubMed] [Google Scholar]
- 6. Iasonos A, Wilton AS, Riedel ER, Seshan VE, Spriggs DR. A comprehensive comparison of the continual reassessment method to the standard 3 + 3 dose escalation scheme in phase I dose‐finding studies. Clin Trials. 2008;5:465‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Brummelen EM, Huitema AD, van Werkhoven E, Beijnen JH, Schellens JH. The performance of model‐based versus rule‐based phase I clinical trials in oncology: a quantitative comparison of the performance of model‐based versus rule‐based phase I trials with molecularly targeted anticancer drugs over the last 2 years. J Pharmacokinet Pharmacodyn. 2016;43:235‐242. [DOI] [PubMed] [Google Scholar]
- 8. Korn EL, Liu PY, Lee SJ, et al. Meta‐analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression‐free and overall survival benchmarks for future phase II trials. J Clin Oncol. 2008;26:527‐534. [DOI] [PubMed] [Google Scholar]
- 9. Simon R. Optimal two‐stage designs for phase II clinical trials. Control Clin Trials. 1989;10:1‐10. [DOI] [PubMed] [Google Scholar]
- 10. Simon R, Wittes RE, Ellenberg SS. Randomized phase II clinical trials. Cancer Treat Rep. 1985;69:1375‐1381. [PubMed] [Google Scholar]
- 11. Wakelee HA, Lee JW, Hanna NH, Traynor AM, Carbone DP, Schiller JH. A double‐blind randomized discontinuation phase‐II study of sorafenib (BAY 43‐9006) in previously treated non–small‐cell lung cancer patients: Eastern Cooperative Oncology Group study E2501. J Thorac Oncol. 2012;7:1574‐1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walker E, Nowacki AS. Understanding equivalence and noninferiority testing. J Gen Intern Med. 2011;26:192‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cunningham D, Starling N, Rao S, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008;358:36‐46. [DOI] [PubMed] [Google Scholar]
- 14. Hirschfeld S, Pazdur R. Oncology drug development: United States Food and Drug Administration perspective. Crit Rev Oncol Hematol. 2002;42:137‐143. [DOI] [PubMed] [Google Scholar]
- 15. Fleming TR, Powers JH. Biomarkers and surrogate endpoints in clinical trials. Stat Med. 2012;31:2973‐2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sargent DJ, Wieand HS, Haller DG, et al. Disease‐free survival versus overall survival as a primary end point for adjuvant colon cancer studies: individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. 2005;23:8664‐8670. [DOI] [PubMed] [Google Scholar]
- 17. US Food and Drug Administration . Adaptive Design Clinical Trials for Drugs and Biologics Guidance for Industry. Accessed April 10, 2020. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/adaptive‐design‐clinical‐trials‐drugs‐and‐biologics‐guidance‐industry
- 18. Barker AD, Sigman CC, Kelloff GJ, Hylton NM, Berry DA, Esserman LJ. I‐SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin Pharmacol Ther. 2009;86:97‐100. [DOI] [PubMed] [Google Scholar]
- 19. Li G, Taljaard M, Van den Heuvel ER, et al. An introduction to multiplicity issues in clinical trials: the what, why, when and how. Int J Epidemiol. 2016;46:746‐755. [DOI] [PubMed] [Google Scholar]
- 20. Hyman DM, Solit DB, Arcila ME, et al. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next‐generation sequencing enabling next‐generation targeted therapy trials. Drug Discov Today. 2015;20:1422‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Renfro LA, Sargent DJ. Statistical controversies in clinical research: basket trials, umbrella trials, and other master protocols: a review and examples. Ann Oncol. 2017;28:34‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373:726‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lemery S, Keegan P, Pazdur R. First FDA approval agnostic of cancer site—when a biomarker defines the indication. N Engl J Med. 2017;377:1409‐1412. [DOI] [PubMed] [Google Scholar]
- 24. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N Engl J Med. 2018;378:731‐739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hobbs BP, Chen N, Lee JJ. Controlled multi‐arm platform design using predictive probability. Stat Methods Med Res. 2018;27:65‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berry DA. The brave new world of clinical cancer research: adaptive biomarker–driven trials integrating clinical practice with clinical research. Mol Oncol. 2015;9:951‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613‐628. [DOI] [PubMed] [Google Scholar]
- 28. Roma‐Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20:840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Le Tourneau C, Delord JP, Goncalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open‐label, proof‐of‐concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16:1324‐1334. [DOI] [PubMed] [Google Scholar]
- 30. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR‐mutated non–small cell lung cancer. Br J Cancer. 2019;121:725‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Archer TC, Fertig EJ, Gosline SJC, et al. Systems approaches to cancer biology. Cancer Res. 2016;76:6774‐6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vaske OM, Bjork I, Salama SR, et al. Comparative tumor RNA sequencing analysis for difficult‐to‐treat pediatric and young adult patients with cancer. JAMA Netw Open. 2019;2:e1913968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodon J, Soria JC, Berger R, et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med. 2019;25:751‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mitri ZI, Parmar S, Johnson B, et al. Implementing a comprehensive translational oncology platform: from molecular testing to actionability. J Transl Med. 2018;16:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dickson D, Johnson J, Bergan R, Owens R, Subbiah V, Kurzrock R. The master observational trial: a new class of master protocol to advance precision medicine. Cell. 2020;180:9‐14. [DOI] [PubMed] [Google Scholar]
- 36. Rolfo C, Manca P, Salgado R, et al. Multidisciplinary molecular tumour board: a tool to improve clinical practice and selection accrual for clinical trials in patients with cancer. ESMO Open. 2018;3:e000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hainsworth JD, Meric‐Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open‐label, phase IIa multiple basket study. J Clin Oncol. 2018;36:536‐542. [DOI] [PubMed] [Google Scholar]
- 38. Goecks J, Jalili V, Heiser LM, Gray JW. How machine learning will transform biomedicine. Cell. 2020;181:92‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chiu YC, Chen HIH, Zhang T, et al. Predicting drug response of tumors from integrated genomic profiles by deep neural networks. BMC Med Genomics. 2019;12:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang C, Clayton EA, Matyunina LV, et al. Machine learning predicts individual cancer patient responses to therapeutic drugs with high accuracy. Sci Rep. 2018;8:16444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. US Food and Drug Administration . Artificial Intelligence and Machine Learning in Software as a Medical Device. Accessed April 10, 2020. https://www.fda.gov/medical‐devices/software‐medical‐device‐samd/artificial‐intelligence‐and‐machine‐learning‐software‐medical‐device#regulation
- 42. Aparicio S, Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med. 2013;368:842‐851. [DOI] [PubMed] [Google Scholar]
- 43. Byrne AT, Alferez DG, Amant F, et al. Interrogating open issues in cancer precision medicine with patient‐derived xenografts. Nat Rev Cancer. 2017;17:254‐268. [DOI] [PubMed] [Google Scholar]
- 44. Williams JA. Using PDX for preclinical cancer drug discovery: the evolving field. J Clin Med. 2018;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang H, Sun L, Liu M, Mao Y. Patient‐derived organoids: a promising model for personalized cancer treatment. Gastroenterol Rep (Oxf). 2018;6:243‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pauli C, Hopkins BD, Prandi D, et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017;7:462‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pauli C, Moch H, Rubin MA. Establishment of a living biobank: improved guidance of precision cancer care with in vitro and in vivo cancer models [in German]. Pathologe. 2017;38(suppl 2):160‐168. [DOI] [PubMed] [Google Scholar]
- 48. Park JE, Kim HS. Radiomics as a quantitative imaging biomarker: practical considerations and the current standpoint in neuro‐oncologic studies. Nucl Med Mol Imaging. 2018;52:99‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Forghani R, Savadjiev P, Chatterjee A, Muthukrishnan N, Reinhold C, Forghani B. Radiomics and artificial intelligence for biomarker and prediction model development in oncology. Comput Struct Biotechnol J. 2019;17:995‐1008. [DOI] [PMC free article] [PubMed] [Google Scholar]