

Abstract
Previously described Lewis superacids are moisture sensitive and predominantly hard in character—features that severely limit their widespread use in orbital‐controlled reactions and under non‐inert conditions. Described here are adducts of bis(perchlorocatecholato)germane, the first hard and soft Lewis superacid based on germanium. Remarkably, the synthesis of this compound is performed in water, and the obtained H2O adduct constitutes a strong Brønsted acid. If applied as an adduct with aprotic donors, it displays excellent activity in a diverse set of Lewis acid catalyzed transformations, covering hydrosilylation, hydrodefluorination, transfer hydrogenation, and carbonyl–olefin metathesis. Given the very straightforward synthetic access from two commercially available precursors, the unlimited water stability and the soft Lewis acidic character, it promotes the transfer of Lewis superacidity into organic synthesis and materials science.
Keywords: germanium, Lewis acids, structure elucidation, superacids, water
Bis(perchlorocatecholato)germane is a hard and soft Lewis superacid with unlimited water stability. Its utility in catalysis as both a Lewis and Brønsted acid was demonstrated by application to a variety of catalytic transformations, such as hydrodefluorination, hydrosilylation, transfer hydrogenation, and carbonyl–olefin metathesis.
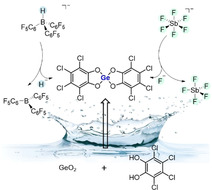
Lewis acids play a vital role in all domains of chemistry. [1] For the most efficient and broad applicability of an ideal Lewis acid, two features are critical to be upheld simultaneously: 1) high affinity toward a full range of hard and soft Lewis basic substrates and 2) high stability against external stress such as temperature, moisture, or redox events. [2] The most comprehensive value to scale the hard Lewis acidity is the fluoride ion affinity (FIA). [3] Lewis acids that are exceeding the FIA of SbF5 are considered Lewis superacids (LSA).[ 2 , 4 ] A value that reflects the soft Lewis acidity is the hydride ion affinity (HIA), and Lewis acids that are exceeding the HIA of B(C6F5)3 have been classed as soft Lewis superacids (sLSA). [2] Overall, Lewis superacids promise the extra portion of strength but have been used only to a limited extend. [2] Their hesitant use is directly connected to the second feature: stability. In this regard, significant improvements have been made during the last decade. The daunting oxidizing and corrosive power of the prototypical SbF5 has been tamed by Krossing's hallmark Lewis acid Al(OC(CF3)3)3, among others.[ 4 , 5 ] The limited thermal stability of aluminum‐based Lewis acids has been tackled by an evolved ligand design in Al(OC(C6F5)3)3. [6] Still, most LSAs suffer from high oxophilicity and the lability of M−O bonds, making them highly prone to hydrolysis. Hence, not only the requirement for strictly inert conditions but also the incompatibility with released water during Lewis acid‐mediated condensation reactions and poor functional group tolerance are still major obstacles. Indeed, an entirely water‐stable neutral LSA is unknown, but to the best of our knowledge, only approximated with the air‐stable Sb(C6F5)4 +. [7]
Although some water‐compatible metal triflates also reach considerable Lewis acidities, this class suffers from poorly defined compositions. [8] Moreover, the limitation to one type of ligand (OTf in this case) prevents any steric or electronic tuning. We recently emphasized the influence of perhalogenated catecholate ligands on the Lewis acidity of silicon and introduced the first neutral silicon LSA. [9] However, like virtually all other LSA, bis(catecholato)silanes are highly sensitive towards moisture. In stark contrast, germanium catecholates have been known to be stable against moisture since their first isolation in 1954, but were never considered as Lewis acids. [10] In general, germanium Lewis acids are only little explored, and a germanium LSA is unknown. [11] Herein, we describe the first germanium LSA, and the first LSA which offers unlimited water stability (Figure 1). Importantly, the compound qualifies not only as hard LSA (FIA > SbF5) but also as soft LSA (HIA > B(C6F5)3).
Figure 1.

a) Previous limitations for the broad applicability of Lewis superacids (LSA). b) The herein described bis(perchlorocatecholato)germane 1.
Heating two equivalents of perchlorocatechol (CatClH2) with GeO2 in water leads to the dissolution of all insoluble starting materials (Figure 2 a). Reaction monitoring via 1H NMR spectroscopy indicated the disappearance of the OH signals of CatClH2 and the simultaneous formation of a characteristic 13C NMR signal set of a new catecholate species. The product Ge(catCl)2‐(H2O)4 (1‐(H2O)4) was isolated upon vacuum removal of the solvent and washing with dichloromethane in 94 % yield at 5 g scale. [12] The isolated compound was recrystallized from water and analyzed by single‐crystal X‐ray diffraction (SCXRD, Figure 2 b). The unit cell contains the bis‐aqua adduct of 1 with four additional hydrogen‐bonded water molecules. The coordination geometry around Ge is almost ideally octahedral, with the two catecholates lying coplanar and the two (H2O)3 clusters in trans orientation.
Figure 2.

a) Synthesis of selected adducts of 1, together with the molecular structures of b) 1‐(H2O)6 (only two water molecules of the second coordination sphere are depicted, Ge‐O1 184.5(3) pm, Ge‐O3 195.7(3) pm) and c) [NEt4]2[trans‐1‐Cl2] (cation omitted for clarity, Ge‐O1 187.6(4) pm, Ge‐Cl 235.3(2) pm). [28]
Interestingly, the structure of the nonhalogenated derivative, trans‐Ge(catH)2‐(H2O)2, was derived by vibrational spectroscopy in 1970, [13] but an SCXRD confirmation remained elusive. 1‐(H2O)n not only represents a rare LSA‐water adduct (vide infra for proof), [14] but a LSA which does not reveal any signs of decomposition over months under bench‐top conditions.
Storing 1‐(H2O)4 in a mixture of CH3CN/CH2Cl2 or acetone in the presence of molecular sieves (3 Å) leads to the replacement of bound H2O, and the isolation of 1‐(CH3CN)2 or 1‐(acetone)2 in 95 % or 91 % yield, respectively. The composition of the two adducts was confirmed by elemental analysis and NMR spectroscopy. The binding of CH3CN was corroborated by the blue shifting of the C=N stretching mode (2324 cm−1, cf. 2253 cm−1 in free CH3CN). 1‐(acetone)2 possesses decent solubility in nonpolar solvents such as CH2Cl2 or toluene. The addition of other neutral Lewis bases such as DMSO, Et3PO, or (n‐BuO)3PO to 1‐(CH3CN)2 or 1‐(H2O)6 resulted in the formation of the respective Lewis adducts. All compounds were fully characterized, including SCXRD (see the Supporting Information (SI)). The addition of one equiv of KF/18‐crown‐6 to 1‐(CH3CN)2 furnished the mono‐fluorido adduct [K@18‐crown‐6][1‐F], constituting a square‐pyramidal structure with the two catecholates in the basal positions. [15] Interestingly, also crystals of the octahedral mixed fluoride‐aqua adduct [K@18‐crown‐6][trans‐H2O‐1‐F] were obtained and analyzed crystallographically (see SI). Upon reaction with two equivalents of NEt4Cl, the formation of octahedral [NEt4]2[trans‐1‐Cl2] readily occurred (Figure 2 c). The two chloride substituents are arranged in trans‐fashion, with Ge‐Cl bond lengths of 235 pm, slightly longer compared to GeCl6 2− or GeCl5 − (229–232 pm). [16] It represents, beyond GeCl6 2−, only the second dianionic chlorido germanate and, to the best of our knowledge, the first dianionic GeO4Cl2 structural motif. [17] Next, we sought after a donor‐free variant of Lewis acid 1. Spontaneous hydroboration of both acetones in 1‐(acetone)2 with 9‐BBN granted access, but a very poor solubility prevented NMR spectroscopy in nondonor solvents or crystallization of 1. Yet, IR spectroscopy, elemental analysis, and a clean NMR spectrum after dissolving the powder in [D6]DMSO supported its pristine composition as donorless 1.
Having various forms of 1 in hand, we considered its Lewis acidity and its hydrolytic stability by quantum chemical methods. The fluoride ion affinity (FIA) and the hydride ion affinity (HIA) were computed first and compared with those of other literature‐known germanium and silicon Lewis acids (Figure 3 a). Notably, it exceeds the FIA and HIA of all previously reported germanium‐based Lewis acids by more than 100 kJ mol−1. The FIA of 1 is very close to the value determined for its silicon counterpart and exceeds that of SbF5, thus meeting the criteria for hard Lewis superacidity. Excitingly, the HIA of 1 sharply exceeded the HIA of Si(catCl)2 and that of B(C6F5)3, rendering this new compound also a soft Lewis superacid. The COSMO‐RS corrected FIAsolv revealed the expected decrease in affinities by solvation damping.[ 2 , 3b ] The hydrolytic stability was considered by comparing the computed enthalpies of the hydrolysis steps of 1 with those of the moisture‐sensitive Si(catCl)2 (Figure 3 b). Indeed, the hydrolytic opening of the first M‐O‐C‐C‐O five‐membered ring is thermodynamically more favorable by 43 kJ mol−1 for silicon as the central atom. Similar trends exist for the remaining steps, making the total hydrolysis of Si(catCl)2 more exothermic by about 70 kJ mol−1. This difference can be explained as the result of diminished ring strain in the five‐membered ring of 1 in comparison to Si(catCl)2 (Figure 3 c), exemplified by comparison of the molecular structures of Si(catCl)2‐(OPEt3)2 [9b] with the one of Ge(catCl)2‐(OPEt3)2 (see SI for full comparison). The longer Ge−O bonds (186 pm) in comparison to Si‐O (176 pm) cause less deviation from the preferred 120° O‐C‐C valence angle in the parent catecholate moiety (see SI, section 4).
Figure 3.

Computational evaluation of a) the ion affinities of 1 in comparison to other strong Lewis acids (DLPNO‐CCSD(T)/aug‐cc‐pVQZ//PW6B95‐D3(BJ)/def2‐TZVPP and COSMO‐RS) and b) the reaction enthalpies in kJ mol−1 of hydrolysis in comparison to Si(catCl)2 (PW6B95‐D3(BJ)/def2‐TZVPP). c) Comparison of relevant bond lengths and angles in Si(catCl)2‐(OPEt3)2 and 1‐(OPEt3)2.
Subjecting 1‐(CH3CN)2 in CD2Cl2 to Et3PO provided an estimate of Lewis acidity according to the Gutmann‐Beckett method. [18] As pentacoordination is rather uncommon for neutral germanium complexes, [19] the formation of the respective bis‐adduct 1‐(OPEt3)2 was strongly favored. [20] Interestingly, both the cis and trans bis‐adducts were observed by NMR spectroscopy in a 1:1.3 ratio. The chirality in the C2‐symmetric cis adducts leads to strong diastereotopic splitting of the CH2‐proton signals. Hence, the preferential coordination state in 1‐(OPEt3)2 differs in solution and the solid state, for which SCXRD illustrated only the trans adduct. The induced 31P NMR chemical shift upon binding of the Lewis acid (δ(31P)=75.1 and 70.6 ppm, cis and trans) is significantly more downfield shifted compared to that of GeF4‐(OPEt3)2 (60.1 ppm), [21] and in the range of the respective silicon bis‐adduct Si(catCl)2‐(OPEt3)2 (73.1 ppm). [9b]
While 1 does not exist in the condensed phase as soluble “free” Lewis acid, the adduct with acetonitrile still exhibits Lewis superacidic behavior. In the reaction of 1‐(CH3CN)2 with [PPh4][SbF6], rapid formation of the mono‐fluorido adduct [1‐F]− was detected by 13C and 19F NMR spectroscopy, as well as ESI mass spectrometry. No defined reaction product was isolated due to follow‐up reactions initiated by the strong oxidant SbF5. Treating a solution of the hydridoborate salt [tBu3PH][HB(C6F5)3] with 1‐(CH3CN)2 led to hydride abstraction and formation of the adduct CH3CN‐B(C6F5)3, experimentally supporting also the soft Lewis superacidic character of 1.
With an established picture of the features of 1‐(CH3CN)2 concerning Lewis acidity and stability, the utility in Lewis acid catalysis was probed. First, the hydrosilylation of aldehydes was tested with 0.5–5 mol % of 1‐(CH3CN)2 as the catalyst. Aliphatic, electron‐rich, and electron‐poor aromatic aldehydes were cleanly hydrosilylated with Et3SiH as reducing agent (Figure 4 a). The feasibility of this process for electron‐rich aromatic aldehydes outperforms the catalytic aptitude of Si(catX)2 or electrophilic phosphonium cations.[ 9a , 22 ] Those harder Lewis acids tolerate only electron‐poor aromatic aldehydes as substrates but tend to deoxygenate electron‐rich aromatic aldehydes in stoichiometric fashion.
Figure 4.

Catalytic applications of 1‐(donor) in a) the hydrosilylation of aldehydes, b) the hydrodefluorination of 1‐adamantylfluoride, c) the transfer hydrogenation of DPE, d) Friedel–Crafts dimerization of 1,1‐DPE, and e) the intramolecular carbonyl–olefin metathesis. The conversion was determined by 1H NMR integration against an internal standard.
The catalytic hydrodefluorination of an aliphatic C−F bond was successfully achieved for 1‐adamantyl fluoride in CD2Cl2 (Figure 4 b). An exceptionally high TON of >1900 (0.05 mol % cat. loading) was determined, underpinning the robustness of the catalyst towards eventual trace impurities or moisture. The intermediate detection of a signal corresponding to the mono‐fluorido adduct [1‐F]− indicated C−F bond activation over Si−H bond activation as a more likely pathway for the reaction. Despite the outstanding activity with 1‐fluoroadamantane as a substrate, no reaction could be observed for other C−F bonds. This lack of reactivity is attributed to the relatively strong binding of acetonitrile to 1‐(CH3CN)2 and the inability of weaker donor substrates to displace the ligand for subsequent activation of the substrate by the Lewis acid. The significant computed hydride ion affinity of 1 tempted us to probe the catalytic activity in transfer hydrogenation reactions. Using 1,4‐cyclohexadiene as hydrogen surrogate, the hydrogenation of 1,1‐diphenylethylene (1,1‐DPE) proceeded smoothly under full conversion at room temperature employing 10 mol % catalyst (Figure 4 c). In the absence of a reductant, 1‐(CH3CN)2 catalyzes the Friedel–Crafts dimerization of 1,1‐DPE in typical Lewis acid fashion (Figure 4 d). After heating at 50 °C for 24 h, 48 % conversion to the dimer was observed, very similar to the value previously reported for B(C6F5)3. [23] Interestingly, 1‐(CH3CN)2 was also found to catalyze the intramolecular carbonyl‐olefin metathesis of the β‐ketoester A, which presents a powerful method for the formation of carbon–carbon bonds. [24] The reaction proceeds with 5 mol % catalyst at room temperature, where full conversion of the substrate is achieved within 20 h. The formation of the byproduct acetone did not preclude catalytic turnover. Contrastingly, direct use of the acetone adduct 1‐(acetone)2 does not lead to any visible reaction even after prolonged heating. This observation emphasizes the critical role of displacement of the first Lewis base in a bis‐adduct of 1. Several tests on the potential involvement of hidden proton catalysis were performed for the mentioned reactions (see SI). The outcome of those tests corroborated true Lewis acid catalysis of 1‐(CH3CN)2 under the above‐described conditions. Although, 1‐(H2O)n was either inactive or considerably slower, a background reactivity by Brønsted acid catalysis was indicated in some reactions. Hence, the catalytic activity of the water adducts 1‐(H2O)n itself was inspected. With 10 mol % of 1‐(H2O)6 and Et3SiH as the hydride source, clean transfer hydrogenation of 1,1‐diphenylethylene was achieved (Figure 4 c).
Control experiments using 1‐(CH3CN)2 and D2O verified that the transferred deuterium originates from bound water. Transfer hydrogenation combining water as a proton source and Et3SiH as a hydride source was reported only recently for the first time, using an iridium catalyst. [25] Yet, it is unprecedented for a main‐group catalyst. This reactivity renders aprotic adducts of 1 not only active in Lewis‐acid catalysis but qualifies protic adducts of 1 as easily accessible Brønsted acids for proton‐driven catalysis. A quantitative description of the Brønsted acidity of 1‐(H2O)2 was obtained by the computed gas‐phase acidity (GA=standard Gibbs energy of deprotonation in the gas phase). For 1‐(H2O)2, a GA value of 1168 kJ mol−1 was calculated at the BP86/def2‐TZVP level of theory. [26] This acidity exceeds the computed values of HSO3F (1233 kJ mol−1) or H2SO4 (1272 kJ mol−1), [27] and approaches the strength of the Lewis superacid—water adduct H2O‐Al(OC(CF3)3)3, for which a GA of 1148 kJ mol−1 was calculated at the same level of theory. [14c] Indeed, the Brønsted acidity of a 0.1 mM solution of 1 in water was assessed qualitatively, in which the measured pH value of ≈4 corresponds to complete dissociation. It is worth reiterating that this Brønsted superacidic system is obtained simply by heating commercially available perchlorochatechol and GeO2 in water.
In conclusion, adducts of bis(perchlorocatecholato)germane 1 were prepared as the first neutral Lewis superacid that is entirely stable under aqueous conditions, compellingly emphasized by its preparation in water. The Lewis superacidity of 1 and the hydrolytic stability was proven and rationalized by theory and experiment. If 1 is used as an adduct of aprotic donors, it serves as a versatile Lewis acid catalyst. As complex with protic donors, it promises applications in the field of Brønsted superacids. The robustness allows handling and catalytic transformations under standard bench‐top conditions. The soft Lewis superacidic features of 1 might enable high activity in orbital controlled processes. Ultimately, it will allow to merge 1) the activity expected from a Lewis superacid, 2) the selectivity achievable with a Lewis acid that is easy to derivatize, and 3) the robustness of an entirely water‐stable species. Given those desirable features paired with its straightforward synthesis, we foresee many potential applications in organic synthesis and materials science.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Prof. H.‐J. Himmel for his constant support, the DFG (GR5007/2‐1), and FCI for financial support. The federal state Baden‐Würtemberg is acknowledged for providing computational resources (BWFor/BWUni). Till Kalkuhl is acknowledged for experimental support. Open access funding enabled and organized by Projekt DEAL.
D. Roth, H. Wadepohl, L. Greb, Angew. Chem. Int. Ed. 2020, 59, 20930.
References
- 1.
- 1a. Corma A., García H., Chem. Rev. 2003, 103, 4307–4366; [DOI] [PubMed] [Google Scholar]
- 1b. Yamamoto H., Ishihara K., Acid catalysis in modern organic synthesis, Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 2. Greb L., Chem. Eur. J. 2018, 24, 17881–17896. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Christe K. O., Dixon D. A., McLemore D., Wilson W. W., Sheehy J. A., Boatz J. A., J. Fluorine Chem. 2000, 101, 151–153; [Google Scholar]
- 3b. Erdmann P., Leitner J., Schwarz J., Greb L., ChemPhysChem 2020, 21, 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Müller L. O., Himmel D., Stauffer J., Steinfeld G., Slattery J., Santiso-Quiñones G., Brecht V., Krossing I., Angew. Chem. Int. Ed. 2008, 47, 7659–7663; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 7772–7776. [Google Scholar]
- 5.
- 5a. Wiesner A., Gries T. W., Steinhauer S., Beckers H., Riedel S., Angew. Chem. Int. Ed. 2017, 56, 8263–8266; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8375–8378; [Google Scholar]
- 5b. Körte L. A., Schwabedissen J., Soffner M., Blomeyer S., Reuter C. G., Vishnevskiy Y. V., Neumann B., Stammler H.-G., Mitzel N. W., Angew. Chem. Int. Ed. 2017, 56, 8578–8582; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8701–8705. [Google Scholar]
- 6. Kögel J. F., Timoshkin A. Y., Schröder A., Lork E., Beckmann J., Chem. Sci. 2018, 9, 8178–8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pan B., Gabbaï F. P., J. Am. Chem. Soc. 2014, 136, 9564–9567. [DOI] [PubMed] [Google Scholar]
- 8. Kobayashi S., Ogawa C., Chem. Eur. J. 2006, 12, 5954–5960. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Liberman-Martin A. L., Bergman R. G., Tilley T. D., J. Am. Chem. Soc. 2015, 137, 5328–5331; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Maskey R., Schädler M., Legler C., Greb L., Angew. Chem. Int. Ed. 2018, 57, 1717–1720; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1733–1736; [Google Scholar]
- 9c. Hartmann D., Schädler M., Greb L., Chem. Sci. 2019, 10, 7379–7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bevillard P., Bull. Soc. Chim. Fr. 1954, 21, 296–303. [Google Scholar]
- 11.
- 11a. Clark H. C., Willis C. J., J. Am. Chem. Soc. 1962, 84, 898–900; [Google Scholar]
- 11b. Graddon D. P., Rana B. A., J. Organomet. Chem. 1979, 165, 157–161; [Google Scholar]
- 11c. Brauer D. J., Bürger H., Eujen R., Angew. Chem. Int. Ed. Engl. 1980, 19, 836–837; [Google Scholar]; Angew. Chem. 1980, 92, 859–860; [Google Scholar]
- 11d. Denmark S. E., Jacobs R. T., Dai-Ho G., Wilson S., Organometallics 1990, 9, 3015–3019; [Google Scholar]
- 11e. Pelzer S., Neumann B., Stammler H.-G., Ignat′ev N., Hoge B., Chem. Eur. J. 2016, 22, 3327–3332; [DOI] [PubMed] [Google Scholar]
- 11f. Pelzer S., Neumann B., Stammler H.-G., Ignat'ev N., Hoge B., Chem. Eur. J. 2016, 22, 16460–16466; [DOI] [PubMed] [Google Scholar]
- 11g. Kinder T. A., Pior R., Blomeyer S., Neumann B., Stammler H.-G., Mitzel N. W., Chem. Eur. J. 2019, 25, 5899–5903. [DOI] [PubMed] [Google Scholar]
- 12.The amount of bound water per formula unit depends on work-up and can vary between 4 and 6.
- 13. Kurnevich G. I., Vishnevskii V. B., J. Appl. Spectrosc. 1970, 13, 1201–1204. [Google Scholar]
- 14.
- 14a. Malkov A. A., Romm I. P., Guryanova E. N., Noskov Y. G., Petrov E. S., Polyhedron 1997, 16, 4081–4086; [Google Scholar]
- 14b. Chakraborty D., Chen E. Y. X., Organometallics 2003, 22, 207–210; [Google Scholar]
- 14c. Kraft A., Possart J., Scherer H., Beck J., Himmel D., Krossing I., Eur. J. Inorg. Chem. 2013, 3054–3062; [Google Scholar]
- 14d. Niemann M., Neumann B., Stammler H. G., Hoge B., Angew. Chem. Int. Ed. 2019, 58, 8938–8942; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9033–9038. [Google Scholar]
- 15. Sau A. C., Day R. O., Holmes R. R., J. Am. Chem. Soc. 1980, 102, 7972–7973. [Google Scholar]
- 16.
- 16a. Gruber H., Müller U., Z. Kristallogr. New Cryst. Struct. 1997, 212, 497; [Google Scholar]
- 16b. Carmalt C. J., Lomeli V., Chem. Commun. 1997, 2095–2096. [Google Scholar]
- 17. Baukov Y. I., Tandura S. N., PATAI′S Chemistry of Functional Groups, Wiley, Hoboken, 2009. [Google Scholar]
- 18.
- 18a. Mayer U., Gutmann V., Gerger W., Monatsh. Chem. 1975, 106, 1235–1257; [Google Scholar]
- 18b. Beckett M. A., Strickland G. C., Holland J. R., Sukumar Varma K., Polymer 1996, 37, 4629–4631. [Google Scholar]
- 19. Parr J. in Comprehensive Coordination Chemistry II (Eds.: McCleverty J. A., Meyer T. J.), Pergamon, Oxford, 2003, pp. 545–608. [Google Scholar]
- 20.By using a very small ratio of OPEt3/1, the formation of a putative monoadduct was observed, which was in rapid exchange with the bis-adduct, thus not serving as accurate probe for the GB-examination. DFT-computations on the formation of 1-(OPEt3) and 1-(OPEt3)2 revealed that the association of the second OPEt3 is thermodynamically more favorable than the first association. Interestingly, for Si(catCl)2 the inverse is the case. See the Supporting Information.
- 21. Cheng F., Davis M. F., Hector A. L., Levason W., Reid G., Webster M., Zhang W., Eur. J. Inorg. Chem. 2007, 2488–2495. [Google Scholar]
- 22. Süsse L., LaFortune J. H. W., Stephan D. W., Oestreich M., Organometallics 2019. , 38, 712–721. [Google Scholar]
- 23. Mehta M., Goicoechea J. M., Angew. Chem. Int. Ed. 2020, 59, 2715–2719; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 2737–2741. [Google Scholar]
- 24. Ludwig J. R., Zimmerman P. M., Gianino J. B., Schindler C. S., Nature 2016, 533, 374–379. [DOI] [PubMed] [Google Scholar]
- 25. Wang D.-W., Wang D.-S., Chen Q.-A., Zhou Y.-G., Chem. Eur. J. 2010, 16, 1133–1136. [DOI] [PubMed] [Google Scholar]
- 26.After deprotonation, the second water molecule is not coordinated via its oxygen atom, but through hydrogen bonding to catechol oxygens according to the gas phase DFT geometry.
- 27. Kraft A., Beck J., Steinfeld G., Scherer H., Himmel D., Krossing I., Organometallics 2012, 31, 7485–7491. [Google Scholar]
- 28.CCDC 2015935–2015941 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary