

Abstract
Key points
Heart failure (HF), the leading cause of death in developed countries, occurs in the setting of reduced (HFrEF) or preserved (HFpEF) ejection fraction. Unlike HFrEF, there are no effective treatments for HFpEF, which accounts for ∼50% of heart failure.
Abnormal intracellular calcium dynamics in cardiomyocytes have major implications for contractility and rhythm, but compared to HFrEF, very little is known about calcium cycling in HFpEF.
We used rat models of HFpEF and HFrEF to reveal distinct differences in intracellular calcium regulation and excitation‐contraction (EC) coupling.
While HFrEF is characterized by defective EC coupling at baseline, HFpEF exhibits enhanced coupling fidelity, further aggravated by a reduction in β‐adrenergic sensitivity.
These differences in EC coupling and β‐adrenergic sensitivity may help explain why therapies that work in HFrEF are ineffective in HFpEF.
Abstract
Heart failure with reduced or preserved ejection fraction (respectively, HFrEF and HFpEF) is the leading cause of death in developed countries. Although numerous therapies improve outcomes in HFrEF, there are no effective treatments for HFpEF. We studied phenotypically verified rat models of HFrEF and HFpEF to compare excitation‐contraction (EC) coupling and protein expression in these two forms of heart failure. Dahl salt‐sensitive rats were fed a high‐salt diet (8% NaCl) from 7 weeks of age to induce HFpEF. Impaired diastolic relaxation and preserved ejection fraction were confirmed in each animal echocardiographically, and clinical signs of heart failure were documented. To generate HFrEF, Sprague‐Dawley (SD) rats underwent permanent left anterior descending coronary artery ligation which, 8–10 weeks later, led to systolic dysfunction (verified echocardiographically) and clinical signs of heart failure. Calcium (Ca2+) transients were measured in isolated cardiomyocytes under field stimulation or patch clamp. Ultra‐high‐speed laser scanning confocal imaging captured Ca2+ sparks evoked by voltage steps. Western blotting and PCR were used to assay changes in EC coupling protein and RNA expression. Cardiomyocytes from rats with HFrEF exhibited impaired EC coupling, including decreased Ca2+ transient (CaT) amplitude and defective couplon recruitment, associated with transverse (t)‐tubule disruption. In stark contrast, HFpEF cardiomyocytes showed saturated EC coupling (increased I Ca, high probability of couplon recruitment with greater Ca2+ release synchrony, increased CaT) and preserved t‐tubule integrity. β‐Adrenergic stimulation of HFpEF myocytes with isoprenaline (isoproterenol) failed to elicit robust increases in I Ca or CaT and relaxation kinetics. Fundamental differences in EC coupling distinguish HFrEF from HFpEF.
Keywords: β‐adrenergic stimulation, calcium, excitation‐contraction coupling, HFpEF
Key points
Heart failure (HF), the leading cause of death in developed countries, occurs in the setting of reduced (HFrEF) or preserved (HFpEF) ejection fraction. Unlike HFrEF, there are no effective treatments for HFpEF, which accounts for ∼50% of heart failure.
Abnormal intracellular calcium dynamics in cardiomyocytes have major implications for contractility and rhythm, but compared to HFrEF, very little is known about calcium cycling in HFpEF.
We used rat models of HFpEF and HFrEF to reveal distinct differences in intracellular calcium regulation and excitation‐contraction (EC) coupling.
While HFrEF is characterized by defective EC coupling at baseline, HFpEF exhibits enhanced coupling fidelity, further aggravated by a reduction in β‐adrenergic sensitivity.
These differences in EC coupling and β‐adrenergic sensitivity may help explain why therapies that work in HFrEF are ineffective in HFpEF.
Introduction
Heart failure – a syndrome marked by pulmonary congestion and exercise intolerance – is the leading cause of hospitalization and death in developed countries (Shah et al. 2017; Lam et al. 2018). Two entirely different pathophysiological entities produce heart failure. The better‐understood entity is heart failure with reduced ejection fraction (HFrEF), in which the heart is dilated and contracts poorly; the second, heart failure with preserved ejection fraction (HFpEF), is characterized by impaired relaxation without dilatation. Patients exhibit similar signs and symptoms regardless of the cause (Pieske et al. 2019). Although numerous drugs and devices have been proven effective in HFrEF, there are no effective treatments for HFpEF, which accounts for ∼50% of heart failure (Dunlay et al. 2017). It is well established that abnormal Ca2+ regulation and defective excitation‐contraction (EC) coupling underlie abnormal contractility in HFrEF. Common features in human and animal models of HFrEF include reduction in the Ca2+ transient (CaT) amplitude, slowing of CaT decay, increased sarcoplasmic reticulum (SR) Ca2+ leak through phosphorylated RyRs, increased Ca2+ efflux by Na+/Ca2+ exchange (NCX) and depleted SR Ca2+ (Zima et al. 2014). Much less is known about Ca2+ regulation in HFpEF, but an unproven assumption is that slowed SR Ca2+ uptake is the dominant cause of diastolic dysfunction, the signature feature of HFpEF haemodynamics (Cain et al. 1998; Ouzounian et al. 2008; Zile & Gaasch, 2011). Here we used phenotypically verified rat models of HFpEF and HFrEF to uncover distinct differences in EC coupling and Ca2+ regulation between these two forms of heart failure. We found that differences in the subcellular mechanism of EC coupling and β‐adrenergic sensitivity distinguish systolic abnormalities in HFrEF from those present in HFpEF, and help explain why HFrEF‐targeted interventions to improve contractile function have largely failed when applied to HFpEF patients.
Methods
Ethical approval
All animal care and experiments conform to the principles and regulations as described in the editorial by Grundy (2015), were in accordance with institutional guidelines and approved by the Cedars‐Sinai Institutional Animal Care and Use Committee (IACUC005280), and adhered to the Guide for the Care and Use of Laboratory Animals prepared by the Committee for the Update of the Guide for the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Research, National Research Council (US).
Animal models of HFpEF and HFrEF
Animals were housed by the Cedars‐Sinai Department of Comparative Medicine, which is fully accredited by the AAALAC International (Association for the Assessment and Accreditation of Laboratory Animal Care), under a 10 h:14 h dark/light cycle. Lights were switched on at 06.00 h and off at 20.00 h. The ambient room temperature was maintained at 20–25°C. Humidity was not controlled.
We implemented the Dahl salt‐sensitive (DS) rat model of HFpEF as described previously (Fig. 1 A) (Gallet et al. 2016). Briefly, 7‐week‐old male DS rats (Charles River Laboratories, MA, USA) underwent baseline transthoracic echocardiography under 2.5% isoflurane anaesthesia and were then randomly assigned for 11 weeks to either a high‐salt diet (AIN‐76A plus 8% NaCl, Research Diets, NJ, USA) to induce hypertension or a normal‐salt diet (AIN‐76A plus 0.3% NaCl) to serve as controls. Chow and water were available ad libitum. At endpoint (after 11 weeks), only high‐salt‐fed rats with preserved EF (above 65%), diastolic dysfunction confirmed by either elevated E/e′ ratio and/or abnormal E/A ratio (reduced E/A or E/A pseudonormalization) and a clinical heart failure score >2 (Cho et al. 2018 b), were selected for experimentation. In our previous studies, we found that mortality of high‐salt‐fed rats over the high‐salt feeding period was ∼50% (Gallet et al. 2016; Cho et al. 2018 b). The vast majority (∼90%) of surviving rats qualified for use based upon the above criteria. As we reported (Cho et al. 2018 a), about 17% of mortality is attributable to sudden cardiac death, while the rest is due to non‐cardiac causes (seizures, anorexia). There was no evidence of stroke in rats used in our study by gross necropsy.
Figure 1. Generation and characteristics of rat models of HFpEF and HFrEF.
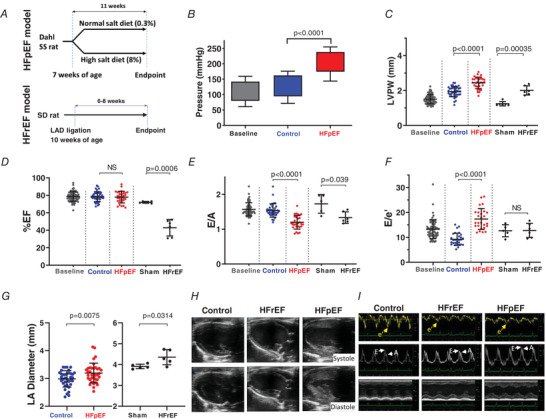
A, HFpEF model: Dahl salt‐sensitive (SS) rats were fed a high‐salt diet (8% NaCl) or a normal‐salt diet (0.3% NaCl) starting at 7 weeks of age. We obtained echocardiograms at baseline and 11 weeks later at endpoint immediately before use in experiments. HFrEF model: Sprague‐Dawley (SD) rats underwent permanent ligation of the left anterior descending (LAD) coronary artery or sham procedure. Echocardiograms were performed at baseline and endpoint. B, systolic and diastolic blood pressures measured using tail cuff photoplethysmography. Top of filled bar indicates mean systolic pressure (+ SD), bottom of bar indicates mean diastolic pressure (− SD). C–G, echo‐derived measures including left ventricular (LV) posterior wall thickness at diastole (LVPW), ejection fraction (%EF), E wave to A wave (E/A) ratio, E wave to e′ (E/e′) ratio, and left atrial (LA) diameter in control, sham, HFrEF and HFpEF. H, representative B‐mode echo images showing left ventricular end diastole and end systole in control, HFrEF and HFpEF. I, representative tissue Doppler, pulse‐wave Doppler at mitral annulus and M‐mode echo recordings in control, HFrEF and HFpEF. Results are shown as means ± SD. Statistics are calculated by one‐way ANOVA (B–F) and Student's t test (G). [Color figure can be viewed at wileyonlinelibrary.com]
The rat post‐infarct model of HFrEF was generated as described (Pfeffer et al. 1979; Terrovitis et al. 2009; Wang et al. 2016) in animals aged 9–10 weeks. Briefly, male Sprague‐Dawley (SD) rats were anaesthetized under 4% isoflurane and the heart was exposed by thoracotomy. The left anterior descending coronary artery was permanently ligated to induce myocardial infarction, the chest was closed and animals were allowed to recover. We administered carprofen (5 mg kg−1) subcutaneously after anaesthesia induction to provide post‐operative analgesia. Chow (PicoLab Rodent Diet 20; LabDiet, St Louis, MO, USA) and water were available ad libitum. Following 8–10 weeks of chronic adaptation to the infarct, reduced EF (EF% < 50) was confirmed by echocardiography. Mortality was 14% over 10 weeks. For echocardiographic comparison, a separate group of animals underwent a sham procedure.
Transthoracic echocardiography
We used Doppler/echocardiography to measure systolic and diastolic function as described (Cho et al. 2017). Rats were rapidly anaesthetized under 5% isoflurane and maintained under 2.5% isoflurane for transthoracic echocardiography (Vevo 3100, VisualSonics, Toronto, Canada) on a heated platform with paw electrodes to measure surface ECG (electrocardiogram). Systolic function was assessed by calculating EF from M‐mode images obtained in the parasternal short axis view. LA diameter was measured from B‐mode images obtained in the parasternal long axis view. Diastolic function was assessed by calculating E/A and E/e′ ratios from an apical 4‐chamber view. To measure E wave (representing early, passive ventricular filling) and A wave (representing late, atrial‐assisted ventricular filling) velocities, we used pulse‐wave Doppler mode, placed at the tip of the mitral valve, to record mitral inflow velocity. To assess e′ wave velocity, tissue Doppler mode was used at the septal aspect of the mitral annulus. Each measurement was repeated 3 times and analysed offline.
Cardiomyocyte isolation
We enzymatically isolated adult rat left ventricular cardiomyocytes using a modification of our atrial myocyte isolation protocol (Yue et al. 2017). Following intraperitoneal injection with heparin sodium (1000 IU), rats were anaesthetized under 5% isoflurane, the heart was quickly excised by thoracotomy, placed in ice‐cold phosphate buffered saline (PBS) and transferred to a Langendorff system. After cannulation, the aorta was retrogradely perfused at a constant flow rate of 9 ml min−1 with oxygen‐bubbled, Ca2+‐free Tyrode solution containing (in mm): 136 NaCl, 5.4 KCl, 0.33 NaH2PO4, 1 MgCl2, 10 HEPES, 10 dextrose, pH 7.4, maintained at 37°C using a heat exchange coil and water bath. After 3 min of perfusion to flush the coronary vasculature of blood, the solution was switched to Tyrode solution containing 0.1 mg ml−1 Liberase TH (MilliporeSigma, St Louis, MO, USA) and 15 μm CaCl2. The heart was perfused with this enzyme solution for 12 min, removed from the apparatus, and the left ventricular (LV) free wall was dissected and removed. The LV free wall was then cut with a razor into approximately 2 mm3 pieces and placed into a 25 ml flask containing warmed enzyme solution diluted 1:1 with Ca2+‐free Tyrode solution and gently shaken at 37°C for secondary digestion. Following approximately 5 min of shaking, tissue was gently triturated using a flame‐polished wide‐mouthed Pasteur pipette to liberate cardiomyocytes. The suspension of dissociated cells was collected (while minimizing the collection of undigested tissue chunks) using a serological pipette and filtered through a 140 μm mesh into a 15 ml tube containing Tyrode solution supplemented with 0.2 mm CaCl2 and 1% BSA. Concurrently, 5 ml of fresh, diluted enzyme solution was added back to the 50 ml flask containing the remaining partially digested tissue and was returned to the heated shaker for another 5 min. The 15 ml tube containing the suspension of dissociated cells was gently centrifuged (20 g, 2 min), the supernatant was removed, and the pellet was resuspended in Tyrode solution containing 0.4 mm CaCl2. Cells were allowed to settle by gravity for ∼10 min, and then similarly resuspended in 0.6, 0.8 and finally 1.0 mm CaCl2 Tyrode solution. The above procedure was repeated for each of the cell suspensions collected at 5‐min intervals from the secondary digestion, typically a total of 5 times. The fraction(s) with the greatest proportion of healthy myocytes (as estimated by observing shape and striations using phase‐contrast light microscopy) were used for experiments. Isolations routinely yielded >75–80% calcium‐tolerant rod‐shaped cardiomyocytes with clear striations. Cells were stored at room temperature for up to 6 h in 1 mm CaCl2 Tyrode solution.
Single cell patch clamp
We recorded membrane currents from isolated left ventricular myocytes using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) in whole‐cell configuration, connected to a computer by a Digidata 1400a A‐D converter (Molecular Devices), and controlled by pClamp software (version 10.4; Molecular Devices) at 20–22°C. Patch electrodes were pulled from borosilicate glass (TW150F‐3, World Precision Instruments, Sarasota, FL, USA) using a Flaming‐Brown horizontal micropipette puller (P‐97, Sutter Instruments, Novato, CA, USA). Pipettes had an electrical resistance of 0.75–1.2 MΩ when filled with internal solution containing (in mm): 130 CsCl, 10 NaCl, 0.33 MgCl2, 20 tetraethylammonium chloride, 10 HEPES, 0.05 cAMP, 4 mgATP (pH adjusted to 7.2 with CsOH). In experiments measuring the I Ca,L response to isoprenaline, cAMP was omitted from the internal solution. For experiments involving simultaneous recording of membrane currents and intracellular Ca2+ (described below), isolated cardiomyocytes were incubated in 10 μm fluo‐4 AM (ThermoFisher Scientific, Waltham, MA, USA) with 0.02% Pluronic F‐127 (ThermoFisher) at 20–22°C for 30 min and then allowed to de‐esterify by washing 3 times for 5 min in normal Tyrode solution. Fluo‐4 AM‐loaded cells were stored in the dark and used up to 2 h following loading. Cells were pipetted into a low volume, laminar flow chamber mounted on an inverted microscope and superfused at a flow rate of 1–2 ml min−1 with external solution appropriate to each experiment. Cells were first bathed in 1 mm Ca2+ Tyrode solution. Upon obtaining a gigaseal and low access resistance breakthrough, the bath was rapidly replaced with a solution designed to isolate I Ca,L containing (in mm): 136 NaCl, 20 CsCl, 1 mgCl2, 10 HEPES, 10 dextrose, 1 CaCl2, 0.01 tetrodotoxin, pH 7.4. Series resistance was electronically compensated ∼ 75%. Cells were held at −80 mV between recordings and I Ca,L was elicited by applying a 1 s prepulse to −50 mV (to inactivate residual I Na), followed by a 200 ms depolarizing step to a test potential of 0 mV. Upon return to −80 mV, SR Ca2+ load and I NaCa were simultaneously assessed by applying a 5 s pulse of 10 mm caffeine (in Tyrode solution) to the cell using a rapid perfusion system (Groenke et al. 2013).
Ultra‐rapid laser scanning confocal microscopy of subcellular calcium
To record Ca2+ release from individual couplons evoked by depolarization, we used the ultra‐rapid line‐scan mode (12 μs/line with two‐line averaging for an effective temporal resolution of 24 μs/line) of the Leica SP5 resonant scanning laser confocal microscope system (Leica Instruments, Wetzlar, Germany) featuring an inverted microscope with a 63×/1.2NA water‐immersion objective. Image acquisition was synchronized with whole‐cell patch clamp depolarization and triggered using digital outputs from the Digidata 1400a. Spatial resolution was 0.19 μm/pixel. To reduce photobleaching of the Ca2+ indicator and to minimize phototoxicity, the laser was shuttered and triggered to open only during the 100 ms acquisition period. All line‐scan images, including those described below for field‐stimulated and voltage clamp‐evoked CaTs, were acquired by placing the scanning line parallel to the long axis of the cell, taking care to avoid intersection with the nucleus. Images of fluo‐4 transients were converted to TIFF format and saved to hard disk.
We analysed the ultra‐rapid line scan images using custom software (Chantawansri et al. 2008) programmed in IDL 6.1 (Harris Geospatial Solutions, Broomfield, CO, USA). Briefly, we identified the exact scan line where the voltage clamp was triggered by the patch clamp amplifier, allowing submillisecond determination of Ca2+ release latency at each pixel along the scan line. Ca2+ release site latency (which we refer to as spark latency) was defined as the time interval between triggered depolarization and the earliest time point where fluorescence at a Ca2+ release site increased 10% above resting value. We identified individual couplons releasing Ca2+ by identifying the pixel locations along the scan line where Ca2+ release from single couplons originated, using a spark location strategy based on the work of Cheng et al. (1999), as we described previously (Chantawansri et al. 2008). We used the following simplifying assumptions to calculate spark probability (P spark), the probability of Ca2+ release from individual couplons upon depolarization (Inoue & Bridge, 2003). Couplons are ideally spaced evenly every 1.8 μm. Thus, the maximum number of couplons activated along a 180 μm line would be 100 (180 μm/1.8 μm per couplon). If all 100 couplons were activated, we would assign a P spark of 1.0. The synchronicity of Ca2+ release was calculated as the standard deviation of Ca2+ release latency at all couplons actively releasing Ca2+ along the scan line (σspark). Local Ca2+ uptake rates (T ½) were similarly determined at each of these locations (but with conventional line scan speeds, e.g. 2.5 ms per line) by measuring the time in milliseconds for the CaT relaxation to fall 50% from peak to baseline. The variability in Ca2+ uptake was calculated as the standard deviation of uptake at the locations measured (σT ½).
Field‐stimulated Ca2+ transients
Cardiomyocytes were loaded with fluo‐4 AM as above, or with fura‐2 AM (2 μm with 0.02% Pluronic F‐127 for 15 min at room temperature, followed by three washes of 5 min to allow de‐esterification) and placed into a low volume perfusion chamber with platinum field‐stimulation electrodes (RC‐27, Warner Instruments, Hamden, CT, USA). The bath was perfused at 1–2 ml min−1. Cells were excited using a MyoPacer Field Stimulator at 10–30 V, 3 ms pulsewidth (IonOptix, Westwood, MA, USA). Before recording CaTs by line scanning (fluo‐4) or ratiometric photometry (fura‐2), we paced cells for 10 s to equilibrate SR Ca2+ content. Evoked CaTs were recorded for 10 s, and the final two CaTs used for analysis. For fura‐2 experiments, we used a custom‐built system where cells were excited by LED at wavelengths of 360 and 390 nm on a Leica DMI3000B inverted microscope fitted with a 40×/1.3NA oil‐immersion objective. We used a photomultiplier (IONOPTIX) to measure epifluorescence at 510 nm during alternate excitation (500 Hz) at the above wavelengths. A mask was placed in the light path so epifluorescence from only a well‐focused portion of one cell was recorded. We used the F 360/F 390 ratio (after background subtraction) as a measure of relative changes in free cytosolic Ca. In some experiments, the bath Tyrode solution was then exchanged by perfusion with Tyrode solution containing isoprenaline (ISO, 1 μm) and cells were incubated for 3 min, after which CaTs were recorded again as above.
Simultaneous patch clamp and fluo‐4 fluorescence measurements
For some experiments, fluo‐4 fluorescence was recorded with a photodiode during simultaneous voltage clamp (as described above) using a custom‐designed photometric epifluorescence detection system attached to a Zeiss Axiovert TV100 inverted microscope with a 40×/1.2NA water immersion lens. A 485 nm LED light source was used for excitation and a longpass filter used to restrict emission to >510 nm. Fluorescence was normalized to baseline (F/F 0) after background subtraction.
Baseline correction for diastolic calcium
To correct for increased baseline fluo‐4 fluorescence in HFrEF and HFpEF compared to control (consistent with elevated diastolic Ca2+ as found in the fura‐2 experiments described in Results), we employed the following correction (F/F 0c):
where F/F 0c is the corrected fluo‐4 fluorescence and R dias model/R dias control was the measured ratio of fura‐2 diastolic fluorescence in either the HFrEF model or the HFpEF model compared to the control model. The ratios used were 1.125 for HFrEF and 1.25 for HFpEF.
Protein isolation, electrophoresis and immunoblotting
Tissue was homogenized in RIPA buffer (Thermo Fisher) using twice the manufacturer‐recommended amounts of protease and phosphatase inhibitor cocktails (Halt, Thermo Fisher). Protein quantification was performed by Pierce BCA protein assay (Thermo Fisher). Electrophoresis was performed using NuPage protein gels of 4–12%. Proteins were transferred to PVDF membranes using a Trans‐Blot Turbo transfer system (BioRad). Membranes were blocked for 1 h in tris‐buffered saline + 0.1% Tween‐20 (TBS‐T) solution containing 5% bovine serum albumin. Antibodies used were Cav1.2 (Alomone ACC‐003, 1:600), pSer1928 Cav1.2 (Badrilla, A010‐70 1:1000), SERCA2 (Invitrogen, MA3‐919, 1:1000), PLN (Thermofisher, PA5 78410, 1:1000), pThr17 PLN (Badrilla, A010‐13AP, 1:1000), pSer16 PLN (Bardilla, A010‐12AP, 1:5000), NCX (R3F1 (Porzig et al. 1993) 1:500) and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (3683S, Cell Signalling, 1:3000). Immunoreactivity was detected by enhanced chemiluminescence (SuperSignal West Pico Plus, Thermo Fisher) using a ChemiDoc Imager (BioRad).
Immunoprecipitation
Tissue was homogenized in homogenization buffer containing (in mm): HEPES 20, NaCl 150, EDTA 5, KF 25, sodium orthovanadate 1 with glycerol 20%, triton X‐100 0.5% and protease inhibitor cocktail (Roche, cOmplete), pH 6.8. Anti‐RyR antibody (Abcam, ab2868) was incubated with Dynabeads Protein G (Invitrogen) for 40 min at room temperature (1 μl antibody + 11 μl beads per sample). The beads were washed with PBS‐0.05% Tween‐20 twice and homogenization buffer once. A 400 μl aliquot of lysate (total protein concentration of 1 mg ml−1) was incubated at 4°C overnight with antibody‐bead in homogenization buffer. After washing, electrophoresis was performed using protein gels of 8%. Proteins were transferred to PVDF membranes using the Trans‐Blot semi‐dry transfer system (BioRad). Membranes were blocked for 1 h in PBS‐0.05% Tween‐20 solution containing 5% dried skimmed milk. Antibodies used were RyR2 (Abcam, ab2868, 1:5000) and pSer2808 RyR2 (Badrilla, A010‐30AP, 1:1000). Data were normalized to RyR in the immunoprecipitated sample.
Reverse transcription quantitative polymerase chain reaction
Following isolation and reacclimating to 1 mm Ca2+ in Tyrode solution, cardiomyocytes were pelleted by centrifugation and flash frozen. Total RNA was isolated from myocyte samples using Rneasy Mini Kit (Qiagen). RNA quality was determined using a spectrophotometer and was reverse transcribed using a cDNA conversion kit from 2 μg RNA with the High Capacity RNA‐to‐cDNA Kit (Applied Biosystems). cDNA in combination with RT² SYBR Green qPCR Mastermix (Cat. No. 330529) was used with RT2 qPCR Assays. Primers were designed to detect target transcripts as follows:
GAPDH (AF106860.2), 5′‐AGTTCAACGGCACAGTCAAG‐3′ (forward), 5′‐TACTCAGCACCAGCA TCACC‐3′ (reverse)
rat a2d3 (NM_175595), 5′‐TCCGAACGCACCATCAAG‐3′ (forward), 5′‐ACTGTCCACCACCACCAT‐3′ (reverse)
rat a2d2 (NM_175592), 5′‐CAGTGGTGGGTGTCAAAC‐3′ (forward), 5′‐TACCTCGCAGTCCATCTC‐3′ (reverse)
rat a2d1 (NM_012919), 5′‐AGCCTATGTGCCATCAATTAC‐3′ (forward), 5′‐AGTCATCCTCTTCCATT TCAAC‐3′ (reverse)
CaVB1 (NM_017346.1), 5′‐GCGAGCACCTGGCGGAGTAC‐3′ (forward), 5′‐GCGGTAGCCATGGTG CGGTT‐3′ (reverse)
CaVB2 (NM_053851.1), 5′‐AGTGGGACAGGTCGAGGCCT‐3′ (forward), 5′‐CACGGTGTGGAACA TAGCGGTC‐3′ (reverse)
CaVB3 (NM_012828.2), 5′‐TTCACCCCTGGAGCGGGACA‐3′ (forward), 5′‐ACGGTGAGGCTGG TACAGGTC‐3′ (reverse)
CaVB4 (NM_012828.2), 5′‐ACCTGGAGGCATATTGGCGTGC‐3′ (forward), 5′‐TGGTTGCTATGCCT CATCCGCT‐3′ (reverse)
CaVB2 splice variants:
CaVB2a: 5′‐ATGCAGTGCTGCGGGCTGGTAC‐3′ (forward)
CaVB2b: 5′‐ATGCTTGACAGGCAGTTGGTG‐3′ (forward)
CaVB2c: 5′‐ATGGACCAGGCGAGTGGACTGG‐3′ (forward)
CaVB2d: 5′‐AAAGCGCGGCTCCCG‐3′ (forward)
Common CaVB2 antisense primer: 5′‐TCCCGGTCCTCTTCCAAAGACA‐3′ (forward)
CT values were exported to an Excel file to create a table of CT values. This table was then uploaded on to the data analysis web portal at http://www.qiagen.com/geneglobe. Each cycle threshold (CT) value was normalized to the CT value of GAPDH. Fold change was calculated with 2^(−ddCT) compared with the control group. The P values were calculated based on a Student's t test of the replicate 2^ (−ΔCT) values for each gene in the control group and HFpEF groups.
Statistics
Data are expressed as means ± SD. Statistical analysis was performed using Prism 8.0 (GraphPad Software, Inc.; La Jolla, CA, USA) or Microsoft Excel. The Shapiro‐Wilk statistic was used to test for normality. One‐way ANOVA with Holm‐Sidak post hoc correction for multiple comparisons was used for statistical comparison of measures among three groups (control, HFrEF, HFpEF). Two‐way ANOVA with Holm‐Sidak post hoc correction for multiple comparisons was used when appropriate. For measures comparing only HFpEF to control, Student's unpaired or paired two‐tailed t test (normal distribution) or Mann‐Whitney rank test (non‐normal distribution) was used to compared group means. P < 0.05 was considered statistically significant.
Results
Echocardiographic and morphometric assessment of HFrEF and HFpEF
As we have shown previously using invasive haemodynamic monitoring and echocardiography, when compared to DS rats fed low‐salt diets (control), DS rats fed high salt develop profound hypertension (mean blood pressure 238 ± 16/175 ± 31 mmHg; N = 10; P < 0.0001 vs. control; Fig. 1 B) and posterior wall thickening (in mm: HFpEF 2.4 ± 0.33 (N = 30) vs. control 1.9 ± 0.32 (N = 32), P < 0.0001; Fig. 1 C), ultimately exhibiting the cardinal haemodynamic and clinical features of human HFpEF (Ogihara et al. 2002; Gallet et al. 2016; Cho et al. 2017). Notably, systolic function is preserved (EF > 65%, Fig. 1 D and H), diastolic function is impaired (reduced E/A ratio and increased E/e′ ratio; Fig. 1 E, F and I), with objective signs of heart failure: pulmonary congestion as manifested by increased lung to body weight ratio (HFpEF 4.9 ± 0.5 (N = 27) vs. control 3.8 ± 0.3 (N = 22); P < 0.0001) and increased left atrial size (Fig. 1 G), consistent with adaptation to elevated ventricular filling pressure. We also found increased LV mass by echo (HFpEF 1154 ± 204 mg (N = 17) vs. control 855 ± 138 mg (N = 29); P < 0.001) as well as increased single myocyte capacitance (HFpEF 317 ± 70 pF (N = 29) vs. control 250 ± 45 pF (N = 17); P < 0.002), indicative of hypertrophy. Other features of this model, including the high mortality (∼50%), are summarized in our previous study (Cho et al. 2017).
To investigate HFrEF, we created a left anterior descending artery (LAD) coronary ligation model and used pre‐infarct SD or age‐matched low‐salt DS rats as controls. Six to eight weeks following LAD ligation, SD rats developed HFrEF characterized by reduced ejection fraction (EF; HFrEF 42.9 ± 9.5% (N = 6) vs. sham 72.0 ± 1.1% (N = 6); P < 0.0001) as described previously (Pfeffer et al. 1979; Wang et al. 2016; Fig. 1 D and H). HFrEF rats also showed evidence of elevated filling pressures with increased left atrial diameter (Fig. 1 G), and pulmonary congestion with increased lung /body weight ratio (HFrEF 3.83 ± 0.22 mg g−1 (N = 4) vs. sham 3.42 ± 0.11 mg g−1 (N = 4); P < 0.02) and increased LV mass (HFrEF 1054 ± 62 (N = 6) vs. sham 777 ± 197 (N = 6); P < 0.020). Cell capacitance was not significantly increased in HFrEF (control 244 ± 65 pF (N = 25); HFrEF 271 ± 72 pF (N = 31); p > 0.05 (NS)).
Excitation‐contraction coupling is impaired in HFrEF but enhanced in HFpEF
To probe EC coupling, we recorded CaTs at 1 Hz using the ratiometric Ca2+ indicator fura‐2 in single ventricular cardiomyocytes isolated from control, HFrEF and HFpEF animals. As is evident in representative signals (Fig. 2 A), CaT amplitude was reduced in HFrEF myocytes (ΔF 360/F 390 = 0.92 ± 0.41; P < 0.05), but increased in HFpEF (ΔF 360/F 390 = 1.71 ± 0.53; P < 0.001) versus control (ΔF 360/F 390 = 1.21 ± 0.46) (Fig. 2 B). While diastolic Ca2+ was higher than control in both HF models (F 360/F 390: control 2.18 ± 0.53; HFrEF 2.45 ± 49, P < 0.05; HFpEF 2.72 ± 0.52, P < 0.001) (Fig. 2 C), systolic Ca2+ was increased only in HFpEF (F 360/F 390: control 3.38 ± 0.80 vs. HFpEF 4.43 ± 0.78, P < 0.001) (Fig. 2 D). In fluo‐4 loaded HFpEF cells, increased pacing frequency did not cause further increase in diastolic Ca2+ (Fig. 7 C).
Figure 2. Calcium transients are reduced in HFrEF but enhanced in HFpEF.
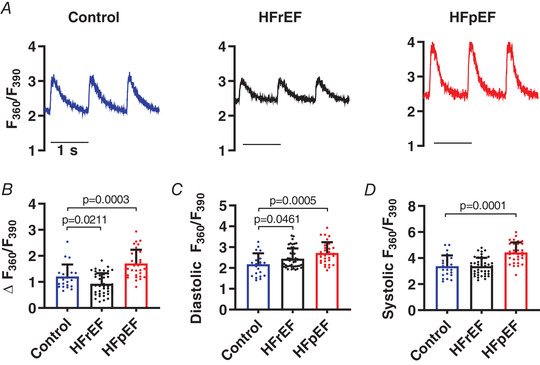
A, representative fura‐2 Ca2+ transients (F 360/F 390) elicited by field stimulation at 1 Hz in cardiomyocytes isolated from control, HFrEF and HFpEF animals. B–D, pooled data analysing the amplitude (Δ), diastolic and systolic fura‐2 ratios (n = 23, 44 and 32 cells from 3 control, 5 HFrEF and 5 HFpEF rats). Results are shown as means + SD. Statistics are calculated by one‐way ANOVA. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 7. Defective local uptake of calcium in HFpEF cells.
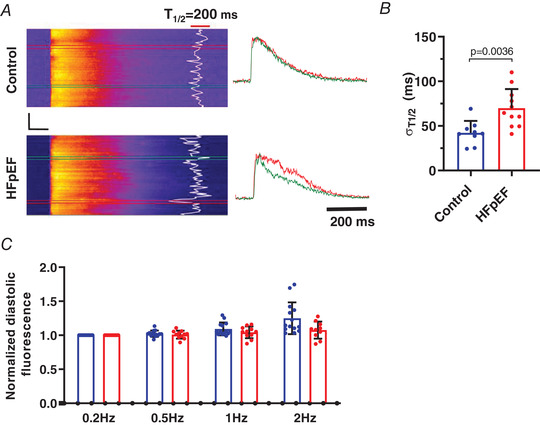
A, representative confocal line scan (2.5 ms/line) CaTs elicited by field stimulation in cardiomyocytes isolated from control and HFpEF animals; fluorescence transients obtained at colour‐coded boxed locations are plotted to the right. CaT relaxation half‐time (T ½) at individual scan locations is overlaid in white. Black scale bars: vertical = 20 μm, horizontal = 100 ms. B, mean (+ SD) uptake variability, calculated as the standard deviation of localized T ½ at each spatial location along the scan line, n = 9 cells from 3 control rats and 11 cells from three HFpEF rats. C, normalized diastolic fluo‐4 fluorescence measured during action potentials recorded in current clamp at increasing frequencies in 14 cells from 3 control rats (blue bars) and 10 cells from 3 HFpEF rats (red bars). There was no statistically significant elevation in diastolic Ca2+ fluorescence in the HFpEF group compared to 0.2 Hz pacing rate. Statistics are calculated as Student's two‐tailed unpaired t test (B) and two‐way ANOVA (C). [Color figure can be viewed at wileyonlinelibrary.com]
Further dissection using simultaneous patch clamp and fluo‐4 fluorescence photometry (Fig. 3 A) revealed unchanged L‐type Ca2+ current (I Ca,L; Fig. 3 B) and sarcoplasmic reticulum (SR) Ca2+ load as assessed by caffeine (Fig. 3 E) in HFrEF cells. NCX current in response to caffeine was modestly increased (pA pF−1: control −0.91 ± 0.33 (N = 27); HFrEF −1.05 ± 0.21 (N = 52); P < 0.05). However, EC coupling gain (expressed as amplitude of the CaT (ΔF/F 0c) divided by the peak amplitude of the eliciting Ca2+ current (I Ca,L) at 0 mV (pA pF−1)) was reduced in HFrEF (control −0.71 ± 0.28; HFrEF −0.46 ± 0.22; P < 0.05), a hallmark of defective EC coupling (Fig. 3 D).
Figure 3. Excitation‐contraction coupling is defective in HFrEF and enhanced in HFpEF.
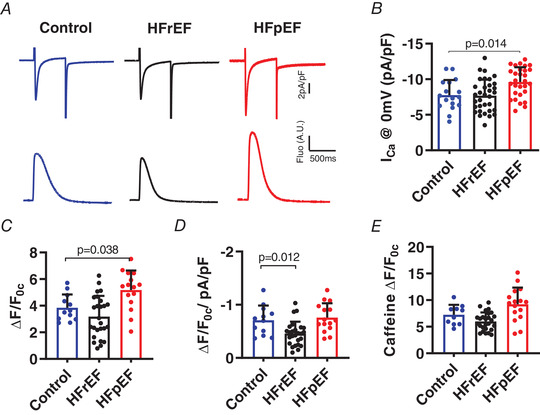
A, simultaneous recordings of I Ca,L (upper) and fluo‐4 CaTs (lower) upon depolarisation to 0 mV. B and C, pooled results for I Ca,L amplitude from 7 control, 6 HFrEF and 9 HFpEF rats (B), and triggered CaT amplitude (ΔF/F 0c) (C). D, EC coupling gain expressed as amplitude of the CaT (ΔF/F 0c) divided by peak I Ca (pA pF−1) at 0 mV. E, summary plots of CaT (ΔF/F 0c) induced by caffeine (10 mm) to assess SR Ca2+ stores. Results are shown as means + SD. Statistics are calculated by one‐way analysis of variance (ANOVA). [Color figure can be viewed at wileyonlinelibrary.com]
In contrast, I Ca,L (Fig. 3 B) was increased in HFpEF (pA pF−1: control −7.77 ± 2.11; HFpEF −9.61 ± 2.09; P < 0.02) but without a significant change in EC coupling gain (Fig. 3 D), SR Ca2+ content (Fig. 3 E), or NCX current in response to caffeine (pA pF−1: control −1.03 ± 0.46 (N = 9); HFpEF −1.04 ± 0.29 (N = 17), p > 0.05 (NS)). Cav1.2 expression, but not phosphorylation, was enhanced in HFpEF (Fig. 8 A, P < 0.05). The lack of phosphorylation differs from HFrEF (Bryant et al. 2015), consistent with the observed differences in I Ca,L. qPCR showed no significant changes in the abundance of Cav1.2‐interacting subunit transcripts (Table 1). Thus, in HFpEF, an increase in I Ca,L due to enhanced protein expression (with an increase in the number of L‐type Ca2+ channels per couplon) could account for the increased CaT amplitude, while in HFrEF, impaired coupling between Ca2+ entry and Ca2+ release must be invoked (given preserved I Ca,L and SR Ca2+ load) to account for the decrease in CaTs, echoing the conclusions of extensive previous work on HFrEF myocytes (Gomez et al. 2001; Zima et al. 2014).
Figure 8. Immunoblots of major Ca2+‐handling proteins in HFpEF.
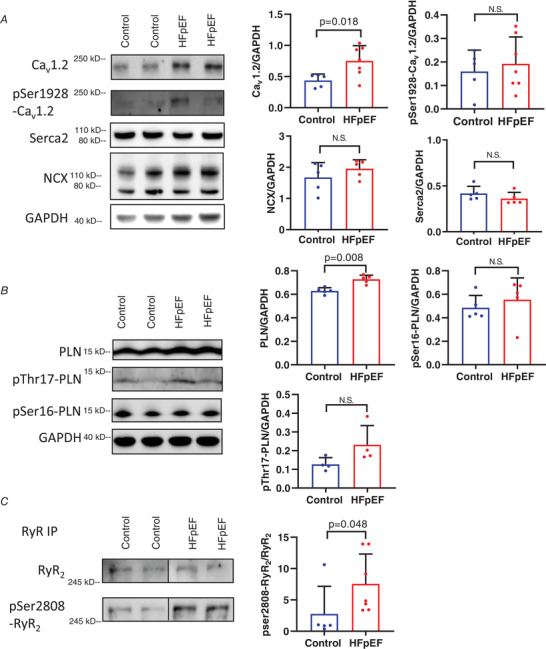
A and B, representative western blots from ventricular cardiomyocytes (left) and pooled data (right) showing mean density relative to GAPDH for major Ca2+‐handling proteins (Cav1.2, SERCA2, NCX and phospholamban (PLN) and assessment of phosphorylation at known sites (pSer1928 Cav1.2, pThr17‐PLN and pSer16‐PLN). C, immunoprecipitation studies of RyR2 and pSer2808 RyR2 showing significant increase in phosphorylation of RyR2 relative to expression of RyR2. Data were normalized to RyR2 in the immunoprecipitated sample. Data expressed as means + SD. Statistics are calculated as Mann‐Whitney rank test. [Color figure can be viewed at wileyonlinelibrary.com]
Table 1.
PCR analysis of LTCC‐interacting subunits
| AVG Ct | ΔCt vs. GAPDH | Standard deviation | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Control | HFpEF | Control | HFpEF | Control | HFpEF | Fold change | 95% CI | P value | |
| GAPDH | 16.49 | 16.24 | 0 | 0 | 0 | 0 | 1 | (1.00, 1.00) | 0 |
| a2D‐3 | 33.73 | 32.3 | 17.238 | 16.053 | 0.679 | 2.414 | 2.272 | (0.00001, 5.57) | 0.326 |
| a2D‐2 | 24.33 | 24 | 7.839 | 7.755 | 0.535 | 1.575 | 1.060 | (0.41, 1.71) | 0.893 |
| a2D‐1 | 24.27 | 23.56 | 7.778 | 7.321 | 1.089 | 1.936 | 1.373 | (0.81, 1.94) | 0.148 |
| CaVB2d | 25.92 | 25.41 | 9.426 | 9.162 | 0.685 | 1.346 | 1.200 | (0.60, 1.80) | 0.331 |
| CaVB2c | 28.66 | 28.18 | 12.172 | 11.936 | 0.854 | 1.442 | 1.178 | (0.69, 1.66) | 0.328 |
| CaVB2b | 22.36 | 21.98 | 5.868 | 5.738 | 0.519 | 1.520 | 1.094 | (0.44, 1.75) | 0.903 |
| CaVB2a | 35 | 35 | 18.509 | 18.756 | 0.000 | 0.000 | 0.843 | (0.00001, 1.74) | 0.946 |
| CaVB4 | 32.46 | 31.91 | 15.970 | 15.664 | 1.691 | 1.784 | 1.237 | (0.00001, 3.27) | 0.384 |
| CaVB3 | 26.54 | 25.36 | 10.051 | 9.117 | 1.799 | 1.085 | 1.911 | (0.00001, 4.45) | 0.406 |
| CaVB2 | 26.11 | 26.14 | 9.618 | 9.896 | 1.615 | 1.488 | 0.825 | (0.26, 1.39) | 0.454 |
| CaVB1 | 31.16 | 30.06 | 14.670 | 13.820 | 0.973 | 1.181 | 1.802 | (0.61, 2.99) | 0.102 |
Calcium spark recruitment is compromised in HFrEF but not in HFpEF
Ultra‐fast laser scanning confocal microscopy can be used to record microscopic aspects of EC coupling and local Ca‐induced Ca2+ release (CICR) including probability of Ca2+ release from individual couplons (spark probability (P spark)), synchronicity of Ca2+ release from all couplons along the scan line (spark variance (σspark)), and latency of Ca2+ release (spark latency) in response to triggering by I Ca,L (Inoue & Bridge, 2003; Chantawansri et al. 2008). These values allow quantification of subcellular changes in couplon recruitment that might rationalize differences in EC coupling efficiency between HFrEF and HFpEF. In patch‐clamped HFrEF myocytes (Fig. 4), P spark was lower than control (P spark: control 0.63 ± 0.10 (N = 26) vs. HFrEF 0.48 ± 0.06 (N = 9); P < 0.001, Fig. 4 C), with fewer couplons along the scan line releasing Ca2+ upon triggering by a voltage clamp to 0 mV, despite unchanged I Ca,L and SR Ca2+ load. Furthermore, the timing of Ca2+ release at each couplon along the scan line was highly dyssynchronous in HFrEF when expressed as variance (σspark in ms: HFrEF 1.68 ± 0.36 (N = 13) vs. control 1.28 ± 0.38 (N = 14); P < 0.001; Fig 4 D), similar to prior descriptions (Litwin et al. 2000; Chantawansri et al. 2008; Heinzel et al. 2008). In contrast, two‐thirds of the couplons along the scan line in HFpEF released Ca2+ in response to I Ca (i.e. P spark = 0.66 ± 0.10) with high synchronicity (σspark = 0.88 ± 0.17 ms) compared to HFrEF (P < 0.001 vs. HFpEF) (Fig. 3 B–D). Likewise, Ca2+ release latency (i.e. the time from depolarization to Ca2+ release) was shorter in HFpEF (5.42 ± 0.63 ms) than HFrEF (7.19 ± 1.42 ms, P < 0.001 vs. HFpEF) (Fig. 4 B and E), consistent with more effective ryanodine receptor (RyR) recruitment by I Ca in HFpEF myocytes. These results pinpoint fundamental differences in the behaviour and interaction of L‐type Ca2+ channels (LCCs) and RyRs in HFpEF versus HFrEF.
Figure 4. Microscopic EC coupling is compromised in HFrEF but enhanced in HFpEF.
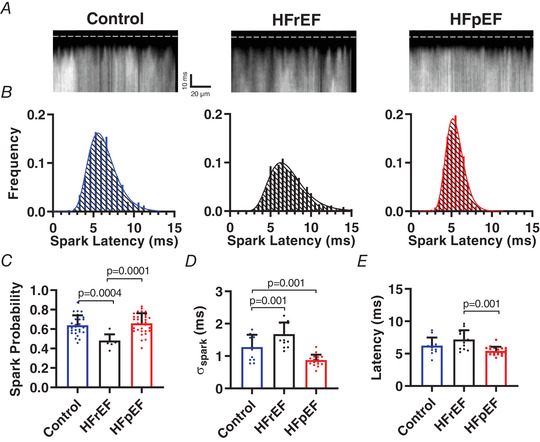
A, individual Ca2+ sparks triggered by depolarisation to 0 mV resolved using ultra‐high‐speed resonant confocal line scanning in fluo‐4‐loaded patch‐clamped cardiomyocytes. Dashed line indicates onset of depolarisation. B, histograms of spark latencies in control (blue), HFrEF (black) and HFpEF (red). C–E, mean (+ SD) spark probability, spark latency variability (plotted as standard deviation of latency), and spark latency (n = 13, 14 and 19 cells from 6, 3 and 5 control, HFrEF and HFpEF rats, respectively). Statistics are calculated by one‐way ANOVA. [Color figure can be viewed at wileyonlinelibrary.com]
The EC coupling machinery is concentrated within the transverse‐axial tubule (TAT) system (Kockskamper et al. 2001; Yue et al. 2017), which is disorganized and depleted in HFrEF (Louch et al. 2006; Song et al. 2006). Conversely, little is known about remodelling of the TAT system in HFpEF, although it is reportedly intact in human HFpEF tissue (Frisk et al. 2018). In freshly isolated cardiomyocytes, TAT architecture (visualized with Di‐4 Anepps (Fig. 5 A) and quantified as previously described (Guo & Song, 2014)) was overtly disrupted in HFrEF (TTdensity = 3.73 ± 1.01% in HFrEF vs. 5.90 ± 0.85% in control; P < 0.001; Fig. 5 B; TTintegrity = 1.65 ± 0.52 in HFrEF vs. 2.59 ± 0.69 in control; P < 0.001; Fig. 5 C), but remained well‐organized in HFpEF (TTdensity = 5.60 ± 0.80%; TTintegrity = 2.63 ± 0.82; both p > 0.05 (NS) vs. control; Fig. 5 B and C). Axial tubule density was similar between groups (Fig. 5 D). Loss of t‐tubule integrity probably contributes to spark dyssynchrony in HFrEF, while preserved TAT architecture supports effective spark recruitment in HFpEF by enabling direct engagement of Ca2+ release channels by Ca2+ entering the dyadic cleft through LCCs.
Figure 5. Transverse‐tubules are depleted in HFrEF but intact in HFpEF.
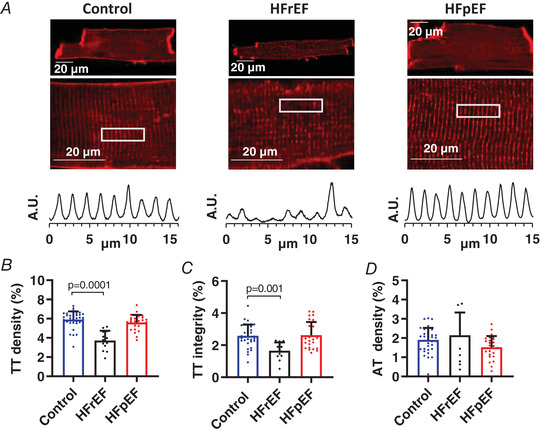
A, representative t‐tubule staining with Di‐4‐ANEPPS (upper) and intensity profiles (lower) assessing t‐tubule regularity in the regions outlined by white box. B–D, mean (+ SD) t‐tubule (TT) density, t‐tubule (TT) integrity and axial tubule (AT) density in Di‐4‐Anepps‐loaded cells (n = 36, 13 and 26 cells from three, two and four control, HFrEF and HFpEF animals, respectively). Statistics are calculated by one‐way analysis of variance (ANOVA). [Color figure can be viewed at wileyonlinelibrary.com]
β‐Adrenergic responses of I Ca, Ca2+ transients, contractility and relaxation are impaired in HFpEF
Given that EC coupling abnormalities in HFrEF are well understood, and our findings there largely confirmatory, we focus more exclusively on HFpEF as we continue. Symptoms in HFpEF patients are most evident upon exertion (Borlaug et al. 2010), reflecting reduced inotropic and lusitropic reserves (Norman et al. 2011). The β‐agonist isoprenaline (ISO) activates protein kinase A (PKA) signalling and, through downstream phosphorylation of key EC coupling proteins, increases contractility and accelerates relaxation. We found that while including ISO (1 μm) in the bath solution increased I Ca,L (at 0 mV) in control cells by 96%, it was much less effective in HFpEF, increasing I Ca,L (which has a greater amplitude at baseline compared to control) by only 38% (P < 0.001; Fig. 6 A and B). Similarly, ISO‐induced increases in CaT amplitude and decay kinetics were severely blunted in HFpEF myocytes compared to control (Fig. 6 C–F), without further elevating diastolic Ca2+ (data not shown). Thus, key aspects of EC coupling in HFpEF myocytes are resistant to β‐stimulation, with potential implications for both systolic and diastolic function in HFpEF during exercise.
Figure 6. Impaired β‐adrenergic response of I Ca,L and Ca2+ transient kinetics in HFpEF.
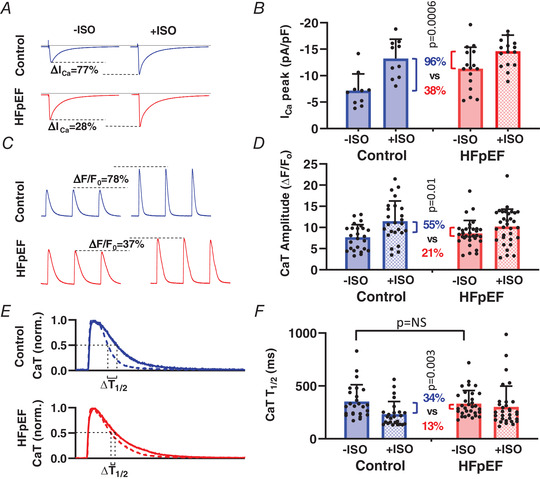
A and B, representative (A) and mean + SD I Ca,L (B) upon depolarisation to 0 mV, at baseline (−ISO) and after isoprenaline (+ISO), in 10 cells from three control rats and 16 cells from three HFpEF rats. C and D, representative (C) and mean CaT amplitudes (D) elicited by field stimulation before (−ISO) and after (+ISO) isoprenaline in 25 cells from 4 control rats and 32 cells from 5 HFpEF rats. E, normalized CaT before (continuous line) and after (dashed line) ISO indicating measurement of CaT relaxation half‐time (T ½). F, mean (+ SD) Ca2+ uptake rate (T ½) before and after ISO in 25 cells from 4 control rats and 32 cells from 5 HFpEF rats. Statistics are calculated using paired Student's t test to compare the effect of ISO on relative change in I Ca, CaT amplitude and CaT T ½, as shown by the colour‐coded percentage changes accompanied by P values. [Color figure can be viewed at wileyonlinelibrary.com]
Abnormal relaxation is a key characteristic of HFpEF hearts. Speculation (Loffredo et al. 2014), yet untested in a phenotype‐verified model, posits that reduced or defective SR Ca2+ reuptake contributes to impaired relaxation in HFpEF. Although our HFpEF rat model exhibits profound diastolic dysfunction (Fig. 1 E, F and I; see also Gallet et al. 2016), CaT decay was comparable in control and HFpEF cells at baseline (CaT T ½ = 352 ± 158 ms in control vs. 332 ± 124 ms in HFpEF, p > 0.05 (NS); Fig. 6 F). Only upon closer examination using confocal imaging (Fig. 7 A) did we observe significant subcellular spatiotemporal inhomogeneities of Ca2+ uptake in HFpEF (σT ½ = 69.9 ± 21.5 ms) compared to control cells (σT ½ = 42.3 ± 13.4 ms; P < 0.01) (Fig. 7 B). In other disease models, notably HFrEF, high local variability in Ca2+ uptake has been associated with abnormal relaxation and arrhythmia (Hohendanner et al. 2013).
Except for an increase in phospholamban (PLN), we found no significant changes in expression of the major proteins responsible for removal of cytosolic Ca2+, namely the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA), and the sodium‐calcium exchanger (NCX) (Fig. 8 A). Likewise, there was no significant increase in phosphorylation of PLN at either the Ser16 or Thr17 phosphorylation sites (Fig. 8 B).These findings contrast with the literature in HFrEF, where reduced SERCA expression and hypophosphorylation of PLN are well documented (Eisner et al. 2013; Marks, 2013; Hulot et al. 2017). We did find a significant increase in RyR phosphorylation Ser2808 in HFpEF (Fig. 8 C), which is also reported in HFrEF (Wehrens et al. 2006), but this was not associated with depletion of SR Ca2+ (Fig. 3 E). Thus, post‐translational regulation, not expression, of EC coupling proteins appears to be responsible for the blunted β‐adrenergic response in HFpEF rat myocytes.
Discussion
While both HFrEF and HFpEF patients have the clinical syndrome of heart failure, key differences in cardiac function point to distinct underlying pathological mechanisms. Defective EC coupling is a central feature of HFrEF, and mechanistically links cellular pathophysiological and cytoarchitectural changes to organ level systolic dysfunction. We find that HFpEF differs qualitatively from HFrEF in terms of Ca2+ cycling. That conclusion is based on investigations of EC coupling in the DS rat model of HFpEF, which exhibits the major haemodynamic and clinical features of the human syndrome, including hypertension, diastolic dysfunction, pulmonary congestion, cachexia, lethargy and exercise intolerance (Guazzi et al. 2001; Gallet et al. 2016; Cho et al. 2018 b; Pieske et al. 2019), and relevant co‐morbidities such as insulin resistance (Ogihara et al. 2002). In stark contrast to the defective CICR mechanism that typifies HFrEF, EC coupling is actually enhanced at baseline in HFpEF rats, with increased I Ca, increased CaT amplitude, a high probability of couplon recruitment (P spark), improved Ca2+ release synchrony (decreased σspark) and shorter Ca2+ release latency compared to control rats. These physiological enhancements in EC coupling, the opposite of changes in HFrEF, are likely to be adaptations that allow the heart to maintain effective contractile function in the face of increased ventricular wall stiffness produced by the known increased collagen deposition/fibrosis of this model (Gallet et al. 2016) and possibly titin hypophosphorylation (Hamdani et al. 2013), a factor underlying diastolic dysfunction in other rat models of HFpEF.
For EC coupling to occur with high fidelity, two things are required. First, LCCs and RyRs must maintain close proximity. The ordered network of t‐tubules ensures this juxtaposition by placing sarcolemmal LCCs a short distance away (across the diadic cleft) from RyRs on the junctional SR. Second, a sufficient number of LCCs in each couplon must open with a high enough probability, open time and short enough latency to trigger RyRs synchronously (Inoue & Bridge, 2003). Reduced Ca2+ spark probability and synchrony have been identified in post‐infarct HFrEF in rabbit (Litwin et al. 2000) as well as a metabolic stress model of ischaemia featuring reduced LCC open probability, increased latency and shorter open times (Chantawansri et al. 2008). Disruption of t‐tubules in HFrEF has been directly associated with the systolic impairments characteristic of the disease, including reduced efficiency and synchrony of CICR (Louch et al. 2006; Heinzel et al. 2008; Frisk et al. 2016; Hong & Shaw, 2017). As t‐tubules remodel, the spatially dependent communication between LCCs and RYRs is disrupted (Song et al. 2006). This produces a pool of ‘orphaned’ RyRs, which exhibit delayed Ca2+ release, while their hyperphosphorylation in HF contributes to SR leakiness and reductions in SR Ca2+ content. In sum, these factors, including t‐tubule disruption (Fig. 5), contribute prominently to the impaired EC coupling we observe in HFrEF and probably contribute to reduced contractility (Fig. 1 D and H).
In marked contrast, HFpEF rats exhibit an enhanced CICR mechanism versus HFrEF, characterized by increased I Ca, high P spark featuring high release synchronicity, and short Ca2+ release site latency, yielding increased CaT amplitude. This high efficiency EC coupling could be helpful in overcoming the fibrosis‐induced stiffness associated with HFpEF (Gallet et al. 2016), but also predicts a lack of contractile reserve, as described in humans (Borlaug et al. 2010). The increased CaT with insignificant increase in SR Ca2+ also suggests an increase in fractional release of SR Ca. Since the t‐tubule network in HFpEF appears similar to control, the enhanced CICR in HFpEF must be related to physiological changes in the behaviour and interactions of EC coupling proteins, e.g. LCCs and RyRs. Consistent with our findings, t‐tubule disruption is not a prominent feature in HFpEF human heart tissue, unlike in HFrEF (Frisk et al. 2018).
Enhanced systolic function has been described in other models of HFpEF, including aged sheep (Dibb et al. 2004) and inbred hypertrophic heart rat (Curl et al. 2018). Similar changes in the amplitude and relaxation kinetics of the CaT have also been reported in rodent models of hypertrophy that lack the key features of HFpEF (specifically clinical signs and symptoms of severe heart failure associated with high mortality), including the spontaneously hypertensive rat (SHR) and a pressure overload rat model created by aortic banding (Delbridge et al. 1997; Shorofsky et al. 1999). Notably, increased I Ca or blunted response to isoprenaline have not been reported in the SHR or aortic banding model, though increased efficacy of SR Ca2+ release triggered by I Ca was noted in the SHR model (Shorofsky et al. 1999).
While we have focused this study on abnormal systolic function of HFpEF, the role of intracellular Ca2+ dynamics in the diastolic dysfunction of HFpEF requires discussion. Prior speculation (Loffredo et al. 2014) has posited that slowed Ca2+ reuptake into the SR contributes to impaired relaxation in HFpEF. Here we found no changes in global CaT decay rates in HFpEF using two different Ca2+ indicators in both patch‐clamped and field stimulated cells. Furthermore, we did not observe any changes in Ca2+ efflux proteins or SERCA/PLB function. However, we did observe an increase in resting Ca2+, an increased heterogeneity of Ca2+ uptake rates, and perhaps most importantly a pronounced deficit in global Ca2+ uptake that only became apparent upon stimulation with β‐agonists. The increase in resting Ca2+ could contribute to increased resting stiffness based on binding kinetics to myofilaments. The reason for increased resting Ca2+ in HFpEF is not clear. Since we observed no change in NCX activity/expression or SERCA/PLB expression or phosphorylation (Fig. 8), we speculate that increased Ca2+ influx through LCCs together with increased Ca2+ leak by phosphorylated RyRs (Fig. 8) may be the cause (Wehrens et al. 2006; Dridi et al. 2020; Ke et al. 2020). Since we used the same Na+ concentration in our internal pipette solution for control and HFpEF studies, it seems unlikely that changes in intracellular Na+ can be invoked as a cause of reduced Ca2+ efflux by NCX. We also did not assess the activity or expression of the alternative Ca2+ efflux protein, the plasma membrane Ca2+ ATPase. Dissection of the mechanism(s) of elevated diastolic Ca2+ will require further study.
A lack of β‐adrenergic reserve was similarly seen in the diminished enhancement of I Ca and CaT amplitude in response to ISO. In normal cardiomyocytes, the PKA signalling cascade extensively alters cardiac function, both acutely through the phosphorylation of ion‐handling proteins, and chronically through altered gene expression. In EC coupling, activation of PKA results in increased phosphorylation of a multitude of ion channels, transporters and intimately associated regulators (LCC, RYR2, PLN, PDEs, AKAPs, Ahnak) (Boularan & Gales, 2015) with the composite function of enhancing SR Ca2+ release and uptake during the cardiac cycle. We found no increase in baseline phosphorylation of Cav1.2 (Fig. 8 A) that might blunt further increase in current with ISO. Prior studies in the DS rat HFpEF model found no sarcolemmal downregulation of the β1‐adrenergic receptor or enhanced translocation of βARK1/GRK2 in the heart (Nishio et al. 2008). This starkly contrasts with HFrEF, in which β1 receptor downregulation is a key feature of the disease (Lohse et al. 2003). These differences suggest a novel mode of regulation of the β‐adrenergic/PKA signalling pathway in HFpEF, which we will address in future studies.
Limitations
Both our HFpEF and HFrEF models display the key features of heart failure: increased left atrial size, pulmonary congestion (i.e. increased lung/body weight ratio) and decreased exercise tolerance (Borlaug & Redfield, 2011). While our rat model of HFpEF possesses a phenotype generally consistent with the human disease, extrapolation of any animal model findings to the human condition must be approached with caution. Certain aspects of Ca2+ handling and electrophysiological mechanisms are different in rats compared to larger animals, especially humans. For example, the action potential duration in the rat is far shorter than in humans, providing an abbreviated time for Ca2+ to enter the cell through Ca2+ channels. Nevertheless, key aspects of EC coupling remain conserved across species, including the CICR mechanism and its dependence upon the close association between t‐tubules and RyRs on SR membranes (Eisner et al. 2017). Decreased t‐tubule density and disorganization have been observed in HFrEF across many models and species (Swift et al. 2008; Heinzel et al. 2008; Lyon et al. 2009), but not in HFpEF (Frisk et al. 2018).
We chose the DS rat model because it has the virtue of having two comorbid features commonly found in human HFpEF: hypertension and insulin resistance. Models encompassing even greater multi‐system maladaptation have been developed recently (Schiattarella et al. 2019), though none in which calcium handling and EC coupling have been rigorously examined as in the present work. Expanding our findings into other HFpEF models, particularly humans, should be a priority in future studies.
We chose the SD rat for generation of HFrEF via infarct because SD rats are the parent strain of DS rats (Rapp & Dene, 1985). This also had the advantage of allowing us to use DS rats fed a low‐salt diet as controls for cellular studies comparing HFrEF to HFpEF. Notably, sham surgery control SD rats had nearly identical morphometric parameters to low‐salt fed DS rats. Thus, the latter served as control for most of the cellular studies we performed in both HFpEF and HFrEF. Although our rat infarct model of HFrEF displayed reduced systolic function, we found incomplete echocardiographic evidence of the diastolic dysfunction (decreased E/A but no change in E/e′; Fig. 1 E, F and I) reported by other groups (Litwin et al. 1994; Prunier et al. 2002). Since our goal was to use the infarct model to compare CICR as it relates to systolic function in HFpEF with HFrEF, the lack of demonstrable diastolic dysfunction does not detract from our key results: systolic function in HFrEF is characterized by defective EC coupling while systolic function in HFpEF is characterized by enhanced EC coupling. The lack of pacing‐induced increases of diastolic Ca2+ in isolated HFpEF myocytes (Fig. 7 C) may be explained by the relatively low pacing rates, bath (Ca2+) and temperature, conditions that were used to preserve cell viability during the long experiments.
Using the gold standard ratiometric Ca2+ indicator fura‐2, we detected a Ca2+ handling signature in HFrEF myocytes consistent with the literature (decreased CaT amplitude, increased diastolic Ca2+ concentration). Analysis of raw fluo‐4 Ca2+ measurements revealed a significant decrease in caffeine‐induced SR Ca2+ release in HFrEF, similar to what has been reported by others using fluo‐4 (Gomez et al. 2001). However, we decided to be extraordinarily conservative about this non‐ratiometric data and corrected it for the increased diastolic Ca2+ we had observed in the separate fura‐2 experiments, as the ensuing ΔF/F 0 calculations are highly sensitive to resting fluorescence values. This unique approach is uncommon because it is rare to have the availability of diastolic information from parallel experiments using fura‐2. Consequently, we found there was only a trend for reduction in caffeine‐induced Ca2+ release (e.g. SR Ca2+ load; p = 0.145) in HFrEF. We conclude from our experiments that t‐tubule remodelling (and presumably orphan RyR2s) is the primary contributor to dyssynchronous Ca2+ release in the rat infarct HFrEF model, which is already well established (Song et al. 2006). If the reduction in SR load had been significant, it would have further reduced the extent of Ca2+ release and EC coupling gain, exaggerating the already‐dramatic difference we observe in EC coupling between HFpEF and HFrEF. Notably, in the metabolic inhibition model of ischaemia, SR content is preserved but EC coupling fidelity is significantly reduced (Chantawansri et al. 2008).
Conclusions
Our results highlight fundamental differences in EC coupling, cytoarchitecture, Ca2+ handling and integrative pump function between HFpEF and HFrEF. Systolic function in HFpEF is normal or enhanced at baseline, reflecting increased I Ca, a maximum degree of coupling fidelity, intact t‐tubules and preserved global SR Ca2+ uptake. Relaxation in HFpEF may be impaired by the marked spatiotemporal inhomogeneity of Ca2+ decay throughout the myocyte in combination with non‐Ca2+‐dependent mechanisms such as increased collagen deposition and altered titin phosphorylation. Beyond these baseline abnormalities, the blunted cellular response to β‐adrenergic stimulation is an important feature in this HFpEF model, which helps explain why common therapies to improve cardiac performance in HFrEF lack clinical efficacy in HFpEF.
Additional information
Competing interests
None.
Author contributions
P.J.K., E.M. and J.I.G. designed the protocols. P.J.K., S.L., R.Z., S.A. and R.E.S. conducted the experiments. P.J.K., S.L., R.Z., X.Y., S.A. and R.E.S. analysed the data and prepared the figures. P.J.K., E.C., E.M. and J.I.G. wrote and edited the article. All authors approved the final version of the article and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This research was supported by National Institutes of Health grants T32 HL116273 to J.I.G. and E.M., HL048509 to J.I.G., R01 HL147570 to J.I.G. and E.C. and Department of Defense grant PR150620 to E.M.
Supporting information
Statistical Summary Document
Biography
Peter Kilfoil undertook predoctoral training in ion channel physiology at the University of Louisville, following which he came to the Smidt Heart Institute at Cedars‐Sinai Medical Center in Los Angeles to investigate the electrophysiological changes that occur in HFpEF. With an initial hypothesis that he would see predominantly diastolic changes, he soon uncovered unanticipated changes in systolic calcium handling as well, some of which can be explained by altered β‐adrenergic responses. Further investigating the signalling changes responsible for this altered systolic function is a future goal and may provide important insights into this disease.

Edited by: Don Bers & Robert Harvey
Linked articles: This article is highlighted in a Perspectives article by Louch and a Journal Club article by Durland. To read these articles, visit https://doi.org/10.1113/JP280691 and https://doi.org/10.1113/JP280739.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Borlaug BA, Olson TP, Lam CS, Flood KS, Lerman A, Johnson BD & Redfield MM (2010). Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J Am Coll Cardiol 56, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlaug BA & Redfield MM (2011). Diastolic and systolic heart failure are distinct phenotypes within the heart failure spectrum. Circulation 123, 2006‐2014; discussion 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boularan C & Gales C (2015). Cardiac cAMP: production, hydrolysis, modulation and detection. Front Pharmacol 6, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant SM, Kong CH, Watson J, Cannell MB, James AF & Orchard CH (2015). Altered distribution of I Ca impairs Ca release at the t‐tubules of ventricular myocytes from failing hearts. J Mol Cell Cardiol 86, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain BS, Meldrum DR, Joo KS, Wang JF, Meng X, Cleveland JC Jr, Banerjee A & Harken AH (1998). Human SERCA2a levels correlate inversely with age in senescent human myocardium. J Am Coll Cardiol 32, 458–467. [DOI] [PubMed] [Google Scholar]
- Chantawansri C, Huynh N, Yamanaka J, Garfinkel A, Lamp ST, Inoue M, Bridge JH & Goldhaber JI (2008). Effect of metabolic inhibition on couplon behavior in rabbit ventricular myocytes. Biophys J 94, 1656–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Song LS, Shirokova N, Gonzalez A, Lakatta EG, Rios E & Stern MD (1999). Amplitude distribution of calcium sparks in confocal images: theory and studies with an automatic detection method. Biophys J 76, 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Kilfoil PJ, Zhang R, Solymani RE, Bresee C, Kang EM, Luther K, Rogers RG, de Couto G, Goldhaber JI, Marban E & Cingolani E (2018a). Reverse electrical remodeling in rats with heart failure and preserved ejection fraction. JCI Insight 3, e121123. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cho JH, Zhang R, Aynaszyan S, Holm K, Goldhaber JI, Marban E & Cingolani E (2018b). Ventricular arrhythmias underlie sudden death in rats with heart failure and preserved ejection fraction. Circ Arrhythm Electrophysiol 11, e006452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Zhang R, Kilfoil PJ, Gallet R, de Couto G, Bresee C, Goldhaber JI, Marban E & Cingolani E (2017). Delayed repolarization underlies ventricular arrhythmias in rats with heart failure and preserved ejection fraction. Circulation 136, 2037–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curl CL, Danes VR, Bell JR, Raaijmakers AJA, Ip WTK, Chandramouli C, Harding TW, Porrello ER, Erickson JR, Charchar FJ, Kompa AR, Edgley AJ, Crossman DJ, Soeller C, Mellor KM, Kalman JM, Harrap SB & Delbridge LMD (2018). Cardiomyocyte functional etiology in heart failure with preserved ejection fraction is distinctive‐a new preclinical model. J Am Heart Assoc 7, e007451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbridge LM, Satoh H, Yuan W, Bassani JW, Qi M, Ginsburg KS, Samarel AM & Bers DM (1997). Cardiac myocyte volume, Ca2+ fluxes, and sarcoplasmic reticulum loading in pressure‐overload hypertrophy. Am J Physiol Heart Circ Physiol 272, H2425–H2435. [DOI] [PubMed] [Google Scholar]
- Dibb KM, Rueckschloss U, Eisner DA, Isenberg G & Trafford AW (2004). Mechanisms underlying enhanced cardiac excitation contraction coupling observed in the senescent sheep myocardium. J Mol Cell Cardiol 37, 1171–1181. [DOI] [PubMed] [Google Scholar]
- Dridi H, Kushnir A, Zalk R, Yuan Q, Melville Z & Marks AR (2020). Intracellular calcium leak in heart failure and atrial fibrillation: a unifying mechanism and therapeutic target. Nat Rev Cardiol (in press; 10.1038/s41569-020-0394-8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlay SM, Roger VL & Redfield MM (2017). Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol 14, 591–602. [DOI] [PubMed] [Google Scholar]
- Eisner D, Caldwell J & Trafford A (2013). Sarcoplasmic reticulum Ca‐ATPase and heart failure 20 years later. Circ Res 113, 958–961. [DOI] [PubMed] [Google Scholar]
- Eisner DA, Caldwell JL, Kistamas K & Trafford AW (2017). Calcium and excitation‐contraction coupling in the heart. Circ Res 121, 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisk M, Le C, Dahl CP, Lunde IG, Almaas VM, Gullestad L, Aakhus S, Sejersted OM, Tonnessen T & Louch WE (2018). T‐tubule loss is a prominent feature of HFrEF but not HFpEF. Biophys J 114, 618A–618A. [Google Scholar]
- Frisk M, Ruud M, Espe EK, Aronsen JM, Roe AT, Zhang L, Norseng PA, Sejersted OM, Christensen GA, Sjaastad I & Louch WE (2016). Elevated ventricular wall stress disrupts cardiomyocyte t‐tubule structure and calcium homeostasis. Cardiovasc Res 112, 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallet R, de Couto G, Simsolo E, Valle J, Sun B, Liu W, Tseliou E, Zile MR & Marban E (2016). Cardiosphere‐derived cells reverse heart failure with preserved ejection fraction (HFpEF) in rats by decreasing fibrosis and inflammation. JACC Basic Transl Sci 1, 14–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez AM, Guatimosim S, Dilly KW, Vassort G & Lederer WJ (2001). Heart failure after myocardial infarction: altered excitation‐contraction coupling. Circulation 104, 688–693. [DOI] [PubMed] [Google Scholar]
- Groenke S, Larson ED, Alber S, Zhang R, Lamp ST, Ren X, Nakano H, Jordan MC, Karagueuzian HS, Roos KP, Nakano A, Proenza C, Philipson KD & Goldhaber JI (2013). Complete atrial‐specific knockout of sodium‐calcium exchange eliminates sinoatrial node pacemaker activity. PLoS One 8, e81633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guazzi M, Brenner DA, Apstein CS & Saupe KW (2001). Exercise intolerance in rats with hypertensive heart disease is associated with impaired diastolic relaxation. Hypertension 37, 204–208. [DOI] [PubMed] [Google Scholar]
- Guo A & Song LS (2014). AutoTT: automated detection and analysis of T‐tubule architecture in cardiomyocytes. Biophys J 106, 2729–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdani N, Franssen C, Lourenco A, Falcao‐Pires I, Fontoura D, Leite S, Plettig L, Lopez B, Ottenheijm CA, Becher PM, Gonzalez A, Tschope C, Diez J, Linke WA, Leite‐Moreira AF & Paulus WJ (2013). Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail 6, 1239–1249. [DOI] [PubMed] [Google Scholar]
- Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, Claus P, Dymarkowski S, Maes F, Bogaert J, Rademakers F, D'Hooge J & Sipido K (2008). Remodeling of T‐tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res 102, 338–346. [DOI] [PubMed] [Google Scholar]
- Hohendanner F, Ljubojevic S, MacQuaide N, Sacherer M, Sedej S, Biesmans L, Wakula P, Platzer D, Sokolow S, Herchuelz A, Antoons G, Sipido K, Pieske B & Heinzel FR (2013). Intracellular dyssynchrony of diastolic cytosolic (Ca2+) decay in ventricular cardiomyocytes in cardiac remodeling and human heart failure. Circ Res 113, 527–538. [DOI] [PubMed] [Google Scholar]
- Hong T & Shaw RM (2017). Cardiac t‐tubule microanatomy and function. Physiol Rev 97, 227–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulot JS, Salem JE, Redheuil A, Collet JP, Varnous S, Jourdain P, Logeart D, Gandjbakhch E, Bernard C, Hatem SN, Isnard R, Cluzel P, Le Feuvre C, Leprince P, Hammoudi N, Lemoine FM, Klatzmann D, Vicaut E, Komajda M, Montalescot G, Lompre AM, Hajjar RJ & Investigators A‐H (2017). Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: results from the AGENT‐HF randomized phase 2 trial. Eur J Heart Fail 19, 1534–1541. [DOI] [PubMed] [Google Scholar]
- Inoue M & Bridge JH (2003). Ca2+ sparks in rabbit ventricular myocytes evoked by action potentials: involvement of clusters of L‐type Ca2+ channels. Circ Res 92, 532–538. [DOI] [PubMed] [Google Scholar]
- Ke HY, Yang HY, Francis AJ, Collins TP, Surendran H, Alvarez‐Laviada A, Firth JM & MacLeod KT (2020). Changes in cellular Ca2+ and Na+ regulation during the progression towards heart failure in the guinea pig. J Physiol 598, 1339–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockskamper J, Sheehan KA, Bare DJ, Lipsius SL, Mignery GA & Blatter LA (2001). Activation and propagation of Ca2+ release during excitation‐contraction coupling in atrial myocytes. Biophys J 81, 2590–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam CSP, Gamble GD, Ling LH, Sim D, Leong KTG, Yeo PSD, Ong HY, Jaufeerally F, Ng TP, Cameron VA, Poppe K, Lund M, Devlin G, Troughton R, Richards AM & Doughty RN (2018). Mortality associated with heart failure with preserved vs. reduced ejection fraction in a prospective international multi‐ethnic cohort study. Eur Heart J 39, 1770–1780. [DOI] [PubMed] [Google Scholar]
- Litwin SE, Katz SE, Morgan JP & Douglas PS (1994). Serial echocardiographic assessment of left ventricular geometry and function after large myocardial infarction in the rat. Circulation 89, 345–354. [DOI] [PubMed] [Google Scholar]
- Litwin SE, Zhang D & Bridge JH (2000). Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ Res 87, 1040–1047. [DOI] [PubMed] [Google Scholar]
- Loffredo FS, Nikolova AP, Pancoast JR & Lee RT (2014). Heart failure with preserved ejection fraction: molecular pathways of the aging myocardium. Circ Res 115, 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Engelhardt S & Eschenhagen T (2003). What is the role of beta‐adrenergic signaling in heart failure? Circ Res 93, 896–906. [DOI] [PubMed] [Google Scholar]
- Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I & Sejersted OM (2006). T‐tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol 574, 519–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE & Gorelik J (2009). Loss of T‐tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A 106, 6854–6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks AR (2013). Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest 123, 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M, Sakata Y, Mano T, Ohtani T, Takeda Y, Miwa T, Hori M, Masuyama T, Kondo T & Yamamoto K (2008). Beneficial effects of bisoprolol on the survival of hypertensive diastolic heart failure model rats. Eur J Heart Fail 10, 446–453. [DOI] [PubMed] [Google Scholar]
- Norman HS, Oujiri J, Larue SJ, Chapman CB, Margulies KB & Sweitzer NK (2011). Decreased cardiac functional reserve in heart failure with preserved systolic function. J Card Fail 17, 301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogihara T, Asano T, Ando K, Sakoda H, Anai M, Shojima N, Ono H, Onishi Y, Fujishiro M, Abe M, Fukushima Y, Kikuchi M & Fujita T (2002). High‐salt diet enhances insulin signaling and induces insulin resistance in Dahl salt‐sensitive rats. Hypertension 40, 83–89. [DOI] [PubMed] [Google Scholar]
- Ouzounian M, Lee DS & Liu PP (2008). Diastolic heart failure: mechanisms and controversies. Nat Clin Pract Cardiovasc Med 5, 375–386. [DOI] [PubMed] [Google Scholar]
- Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA & Braunwald E (1979). Myocardial infarct size and ventricular function in rats. Circ Res 44, 503–512. [DOI] [PubMed] [Google Scholar]
- Pieske B, Tschope C, de Boer RA, Fraser AG, Anker SD, Donal E, Edelmann F, Fu M, Guazzi M, Lam CSP, Lancellotti P, Melenovsky V, Morris DA, Nagel E, Pieske‐Kraigher E, Ponikowski P, Solomon SD, Vasan RS, Rutten FH, Voors AA, Ruschitzka F, Paulus WJ, Seferovic P & Filippatos G (2019). How to diagnose heart failure with preserved ejection fraction: the HFA‐PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur Heart J 40, 3297–3317. [DOI] [PubMed] [Google Scholar]
- Porzig H, Li Z, Nicoll DA & Philipson KD (1993). Mapping of the cardiac sodium‐calcium exchanger with monoclonal antibodies. Am J Physiol Cell Physiol 265, C748–C756. [DOI] [PubMed] [Google Scholar]
- Prunier F, Gaertner R, Louedec L, Michel JB, Mercadier JJ & Escoubet B (2002). Doppler echocardiographic estimation of left ventricular end‐diastolic pressure after MI in rats. Am J Physiol Heart Circ Physiol 283, H346–H352. [DOI] [PubMed] [Google Scholar]
- Rapp JP & Dene H (1985). Development and characteristics of inbred strains of Dahl salt‐sensitive and salt‐resistant rats. Hypertension 7, 340–349. [PubMed] [Google Scholar]
- Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, Hill TM, Mammen PPA, Huang J, Lee DI, Hahn VS, Sharma K, Kass DA, Lavandero S, Gillette TG & Hill JA (2019). Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah KS, Xu H, Matsouaka RA, Bhatt DL, Heidenreich PA, Hernandez AF, Devore AD, Yancy CW & Fonarow GC (2017). Heart failure with preserved, borderline, and reduced ejection fraction: 5‐year outcomes. J Am Coll Cardiol 70, 2476–2486. [DOI] [PubMed] [Google Scholar]
- Shorofsky SR, Aggarwal R, Corretti M, Baffa JM, Strum JM, Al‐Seikhan BA, Kobayashi YM, Jones LR, Wier WG & Balke CW (1999). Cellular mechanisms of altered contractility in the hypertrophied heart: big hearts, big sparks. Circ Res 84, 424–434. [DOI] [PubMed] [Google Scholar]
- Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW & Cheng H (2006). Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A 103, 4305–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift F, Birkeland JA, Tovsrud N, Enger UH, Aronsen JM, Louch WE, Sjaastad I & Sejersted OM (2008). Altered Na+/Ca2+‐exchanger activity due to downregulation of Na+/K+‐ATPase alpha2‐isoform in heart failure. Cardiovasc Res 78, 71–78. [DOI] [PubMed] [Google Scholar]
- Terrovitis J, Lautamaki R, Bonios M, Fox J, Engles JM, Yu J, Leppo MK, Pomper MG, Wahl RL, Seidel J, Tsui BM, Bengel FM, Abraham MR & Marban E (2009). Noninvasive quantification and optimization of acute cell retention by in vivo positron emission tomography after intramyocardial cardiac‐derived stem cell delivery. J Am Coll Cardiol 54, 1619–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Liu T, Shi S, Li R, Shan Y, Huang Y, Hu D & Huang C (2016). Chronic administration of catestatin improves autonomic function and exerts cardioprotective effects in myocardial infarction rats. J Cardiovasc Pharmacol Ther 21, 526–535. [DOI] [PubMed] [Google Scholar]
- Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A & Marks AR (2006). Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A 103, 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X, Zhang R, Kim B, Ma A, Philipson KD & Goldhaber JI (2017). Heterogeneity of transverse‐axial tubule system in mouse atria: Remodeling in atrial‐specific Na+‐Ca2+ exchanger knockout mice. J Mol Cell Cardiol 108, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zile MR & Gaasch WH (2011). Abnormal calcium homeostasis: one mechanism in diastolic heart failure. J Am Coll Cardiol 58, 155–157. [DOI] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W & Terentyev D (2014). Ca handling during excitation‐contraction coupling in heart failure. Pflugers Arch 466, 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistical Summary Document
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.