

Abstract
A concise synthetic route towards a new family of phosphorus‐containing polycyclic aromatic hydrocarbons starting from the versatile acridophosphine has been established. The structural and optoelectronic properties of these compounds were efficiently modulated through derivatization of the phosphorus center. X‐ray crystallographic analysis, UV/Vis spectroscopic, and electrochemical studies supported by DFT calculations identified the considerable potential of these scaffolds for the development of organophosphorus functional materials with tailored properties upon further functionalization.
Keywords: olefination, phosphorus heterocycles, photochemistry, polycyclic aromatic hydrocarbons
Phosphorus in place: A new family of phosphorus‐containing polycyclic scaffolds was realized through a concise synthetic route. The impact of the phosphorus functionalization on the structural, electrochemical, and photophysical properties was investigated both in experiment and theory to reveal the highly tunable nature of these compounds.
Polycyclic aromatic hydrocarbons (PAHs) are of great interest for the development of functional materials for diverse applications such as organic solar cells, field‐effect transistors, and light emitting diodes.1 However, the crucial step towards these applications is the ability to tailor the properties of PAHs by means of synthetic organic chemistry. Controlling the size and shape of the π‐conjugated system allows to adjust the energy levels of frontier molecular orbitals (FMOs) and consequently to modulate the UV/Vis absorption and emission characteristics.2 A particularly potent strategy to tune the properties of PAHs involves the incorporation of different heteroatoms into the sp2 carbon framework.3, 4, 5
In this context, the highly tunable electronic nature of trivalent phosphorus renders phosphorus‐containing PAHs promising candidates for novel functional materials.6, 7 Hence, the oxidation of trivalent phosphorus with its Lewis basic lone pair affords electron‐withdrawing pentavalent phosphoryl species. In these systems π back donation from the filled p orbital at the oxygen into the antibonding σ* orbital at the phosphorus, called negative hyperconjugation, results in a highly polarized phosphorus–oxygen bond. This effect concomitantly increases the relative electronegativity of the phosphorus center, turning it into an electron acceptor.6b, 8 In this regard, acridophosphines with their bridging carbonyl moiety represent an appealing compound family for molecular engineering.9, 10 Hence, the reactive carbonyl group provides for the introduction of functional groups in the periphery through carbonyl chemistry, while the derivatization of the phosphorus center enables fine tuning of the resulting compounds in terms of their optoelectronic properties.11 Despite their indisputable synthetic versatility, acridophosphines remained largely dormant until we recently recognized their potential to realize a series of dicyanomethylene‐bridged scaffolds with tunable electron acceptor properties.11 The ability of the bridging carbonyl to undergo a Knoevenagel condensation with malononitrile stimulated us to explore its reactivity towards π expansion in order to achieve a new type of phosphorus‐containing PAHs.
Herein, we report the first, to the best of our knowledge, Barton–Kellogg olefination of acridophosphine, which was followed by Mallory photocyclization to achieve unprecedented phosphorus‐containing PAHs of general formula A (Figure 1). Subsequent derivatization of the phosphorus center provided for fine‐tuning of the FMO energy levels and the photophysical properties of the newly synthesized phosphorus‐containing PAHs. In fact, the newly synthesized PAHs represent organophosphorus analogues of the hitherto elusive dibenzonaphthanthrene (8H‐benzo[gh]naphtho[1,2,3,4‐pqr]tetraphene).12 Boron‐containing counterparts B of this interesting PAH were recently realized as exceptionally stable redox‐active luminophores by Wagner and co‐workers.13
Figure 1.
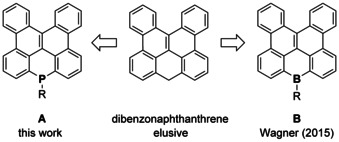
The relation of the heteroatom‐containing PAHs to their hitherto elusive all‐carbon congeners.
Parent compound 1 was synthesized by carbanion‐mediated cyclization of a carbamoyl‐substituted triphenylphosphine oxide precursor according to Snieckus and co‐workers (for synthetic details, see the Supporting Information).9, 11 Reaction of compound 1 with Lawesson's reagent afforded thioketone 2 as a dark blue crystalline solid (Scheme 1). Thioketone 2 was subjected to a Barton–Kellogg olefination14 reaction with diphenyldiazomethane (S2) followed by reduction of the resulting intermediate thiirane with copper to afford diphenylvinylidene‐bridged 3 as a white crystalline solid in an overall yield of 90 %. In accordance with the recently reported successful oxidative photocyclizations of π‐expanded phosphole sulfides by Hissler and co‐workers,15 Mallory reaction involving 3 afforded a mixture of the monocyclized compound 4 (52 % yield) and the fully cyclized counterpart 5 (29 % yield), both as pale‐yellow solids. Increasing the reaction time did not improve the yield of 5. Both compounds are air stable and can be readily purified by column chromatography on SiO2. It should be emphasized that the attempted syntheses of 5 (and 4) by Lewis acid‐mediated oxidative cyclodehydrogenation (Scholl reaction) from 3 as well as from the corresponding phosphine oxide and the trivalent phosphine failed,16 most likely due to incompatibility of the reactive phosphorus center with the required reaction conditions.
Scheme 1.
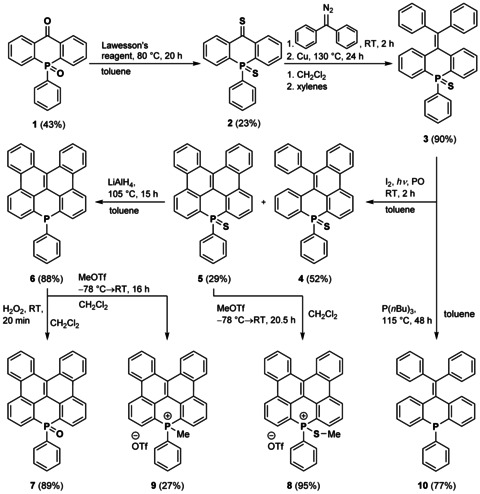
Synthesis of phosphorus‐containing PAHs 3–10. PO: propylene oxide.
To explore the impact of phosphorus functionalization on the properties of the dibenzonaphthanthrene scaffold, the thiophosphoryl unit in 5 was reduced with P(nBu)3 to afford trivalent phosphine 6 in 88 % yield as a white solid. Phosphine 6 is fairly stable in the solid state but oxidizes readily in solution under ambient conditions in air towards phosphine oxide 7. On a preparative scale, phosphine oxide 7 was accessible in 89 % yield from oxidation of 6 with H2O2. Methylation of 5 and 6 with methyl triflate furnished the phosphonium salts 8 and 9, respectively. In addition, phosphine 10 was synthesized upon reduction of precursor 3 (72 % yield) as a model compound in order to evaluate the impact of cyclization on the structural and optoelectronic properties (Scheme 1). The 31P NMR resonances of compounds 3–5 and 7–9 (25.7, 22.8, 21.1, 7.3, 29.2 and 0.0 ppm, respectively) in CD2Cl2 at room temperature appear low‐field shifted in comparison to the trivalent phosphines 6 and 10 (6: −37.0 ppm, 10: −17.7 and −25.2 ppm); this is in accordance with their increased electron‐deficient nature. Whereas only sharp signals are observed in the room temperature 1H NMR spectrum of the twofold cyclized phosphine 6, the model compound 10 shows broadened signals under the same conditions, thus indicating the occurrence of a conformational process.
This process is most likely analogous to the wing‐flip motion of the butterfly‐shaped phosphaanthracenyl fragment, as already described for related acridophosphines (Figure 2).11, 17 From the variable‐temperature (VT) 31P NMR line‐shape analysis (see the Supporting Information), a Gibbs free activation energy ΔG ≠ of 15.6 kcal mol−1 was obtained for the proposed wing‐flip in 10, with ΔH ≠=16.1 kcal mol−1 and ΔS ≠=1.8 cal mol−1 K−1. Considering the increased steric demand of the phenyl moieties, the value of ΔG ≠ is slightly higher as that reported previously for the dicyanovinylidene‐bridged system with ΔG ≠=14.3 kcal mol−1.11
Figure 2.
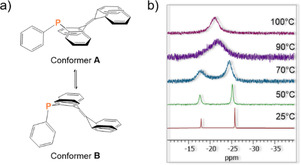
a) Schematic representation of the conformational equilibrium and b) selected VT 31P NMR spectra (121 MHz) of phosphine 10 in 1,1,2,2‐[D2]tetrachloroethane.
Single crystals of compounds 3–5 and 7 suitable for X‐ray crystallographic analysis were obtained by slow evaporation of the compound solutions in CH2Cl2/n‐hexane mixture (Figure 3).18 The noncyclized phosphine sulfide 3 exhibits a characteristic distorted butterfly‐shaped conformation caused by the spatial demands of the phosphorus center in combination with the repulsion of the diphenylvinylidene moiety and the adjacent hydrogens at the phosphaanthracenyl fragment. Each cyclization step enforces a more planar polycyclic scaffold. The fully cyclized phosphine sulfide 5 and phosphine oxide 7 comprise a [4]helicene‐like subunit embedded within the organophosphorus π system. The helical pitch as expressed by the distance “ortho–ortho” carbons differs only slightly between compounds 5 (2.96 Å) and 7 (2.95 Å), and is in good agreement with the value reported for 4‐methyl‐[4]helicene (3.00 Å; for a definition of the structural parameters, see the Supporting Information).19 In contrast, the dihedral angle of the [4]helicene fragment is much larger for the phosphine sulfide 5 (28.2°) and the phosphine oxide 7 (34.3°) in comparison to 4‐methyl‐[4]helicene (13.0°). In this context it is worth mentioning that our attempts to separate the enantiomers of the twofold cyclized compounds by chiral HPLC were not successful, most likely due to the low energy barrier of racemization comparable to unsubstituted [4]helicene.19 All studied compounds show pyramidalization around the phosphorus atom ranging from Σ( C‐P‐C) 312.2(3)° for the monocyclized phosphine sulfide 4 to 314.6(6)° for phosphine oxide 7. These values are in range of those observed for the dicyanovinyl‐substituted acridophosphines previously.11
Figure 3.
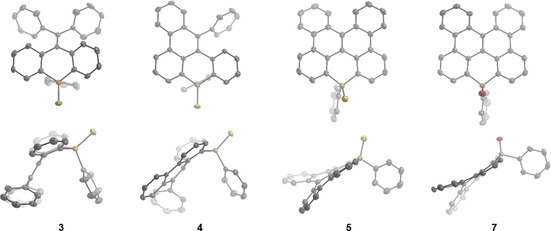
X‐ray crystal structures of 3, 4, 5, and 7 at the 50 % probability level (hydrogens are omitted for clarity).
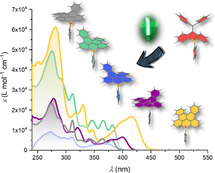
The impact of planarization along with the phosphorus functionalization on the optoelectronic properties of the compounds was investigated by UV/Vis absorption spectroscopy in CH2Cl2 at room temperature (Figure 4 and Table 1). Solutions of the noncyclized phosphine sulfide 3 and phosphine 10 are colorless and consequently show absorptions exclusively in the UV region with lowest‐energy absorption maxima at 303 and 332 nm, respectively. For compounds 4–9 a significant bathochromic shift with the longest‐wavelength absorption maxima (λ max) ranging from 371 nm for phosphine 6 to 416 nm for phosphonium salt 8 are observed, which is mostly due to the enlarged π system, see below. Interestingly, the consecutive planarization of the π system when going from noncyclized 3 via 4 to twofold cyclized 5 leads to considerably increased extinction coefficients with the concomitant appearance of the vibronic fine structure for the dibenzonaphthanthrene scaffolds 5 and 7. Compounds 3 and 5 do not feature any emission, which is presumably a consequence of the heavy atom effect of the sulfur promoting spin‐orbit coupling in the excited state.20 However, the partially cyclized compound 4 is weakly fluorescent with a quantum yield (Φ) of 0.02 although it comprises a thiophosphoryl unit. The twofold cyclized compounds 6 (Φ=0.14), 7 (Φ=0.16), 8 (Φ=0.02), and 9 (Φ=0.29) are emissive with Stokes shifts ranging from 1861 cm−1 for phosphonium salt 9 to 2300 cm−1 for phosphonium salt 8.
Figure 4.
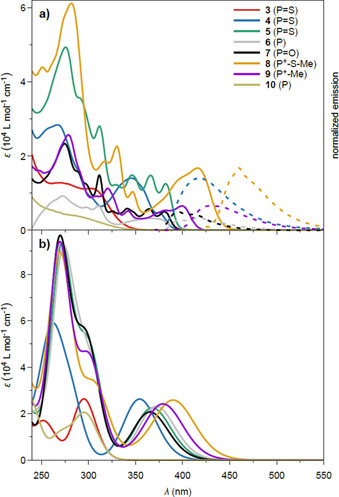
a) UV/Vis absorption (solid lines) and scaled emission spectra (dashed lines; 4: λ ex=384 nm, 6: λ ex=371 nm, 7: λ ex=395 nm, 8: λ ex=416 nm, 9: λ ex=360 nm) recorded in CH2Cl2 at room temperature; b) Calculated UV/Vis absorption spectra at the COSMO‐BHLYP/TZVP level of theory. The theoretical spectra are red‐shifted by 0.3 eV to account for the systematic overestimation of excitation energies of the BHLYP density functional.
Table 1.
Photophysical and electrochemical data of the phosphorus‐containing PAHs.
|
Compound |
λ abs [nm][a] (ϵ [m −1 cm−1]) |
E gap [eV][b] |
λ em [nm][a]
|
Stokes shift [cm−1] |
E ox [V] |
|---|---|---|---|---|---|
|
3 |
303 (11 300) |
3.64 |
– |
– |
– |
|
4 |
374 (4 700) |
3.17 |
417 |
2 757 |
+0.98 |
|
5 |
382 (12 600) |
3.13 |
– |
– |
+0.88 |
|
6 |
371 (3 100) |
3.12 |
416 |
2 295 |
+0.57 |
|
7 |
381 (5 000) |
3.15 |
411 |
1 916 |
– |
|
8 |
416 (16 800) |
2.86 |
460 |
2 300 |
– |
|
9 |
399 (6 600) |
2.98 |
431 |
1 861 |
– |
|
10 |
332 (4 800) |
3.66 |
– |
– |
– |
[a] In CH2Cl2. [b] Optical bandgap calculated from the end absorption wavelength.
The redox properties of 4–8 were investigated by cyclic voltammetry in CH2Cl2 containing nBu4NPF6 as the supporting electrolyte. The potentials were referenced against the ferrocenium/ferrocene (Fc+/Fc) redox couple. The cyclic voltammograms of compounds 4–6 exhibit a single irreversible oxidation event at +0.98, +0.88, and +0.57 V, respectively (see the Supporting Information). The cyclic voltammograms of phosphine sulfide 3 and phosphine oxide 7 show no redox events within the electrochemical window of CH2Cl2 (between ca. −2.0 to +2.0 V vs. Fc+/Fc). Phosphonium salt 8 features one irreversible reduction event at −1.55 V. These findings not only illustrate the effect of different substituents at the phosphorus center but also highlight the impact of cyclization on the electronic nature of these organophosphorus polycyclic scaffolds.
Further insight into the electronic character of the compounds was obtained by density functional (DFT) calculations. Molecular geometries were optimized by using the B3LYP density functional21 including the Grimme D3 correction for dispersion interactions.22 Solvent effects were captured by the conductor‐like screening model23 (COSMO) with ϵ r=8.93 mimicking CH2Cl2. To expand the MOs, we used the def2‐TZVP basis24 from the Turbomole basis set library. UV/Vis absorption spectra and frontier molecular orbitals (FMOs) were calculated adopting the BHLYP density functional,25 which provides a more adequate description of charge‐transfer excitations than B3LYP; otherwise, the computational setup of the ground‐state calculations was kept. Further computational details can be found in the Supporting Information.
For all molecules, the energetically lowest absorption band corresponds to the HOMO→LUMO excitation, which can be characterized as a π–π transition. The frontier orbitals of compounds 3–6 are shown in Figure 5, illustrating the effect of the cyclization: as the cyclization proceeds, the conjugation in the polycyclic scaffold enhances and the HOMO–LUMO gap decreases leading to the observed red‐shift of the lowest absorption band with cyclization, Figure 4 b). The effect of the substituent at the phosphorus atom is apparently smaller: there is some interaction to the MOs on the polycyclic scaffold, but this does not change their principal shape, see for example, FMOs of 5 and 6 in Figure 5 (FMOs of 5–9 in the Supporting Information). Depending on the substituent, the HOMO–LUMO gap of the fully cyclized molecules varies in a range of 0.3 eV leading to the different lowest absorption energies of compounds 5–9, however without changing the overall appearance of the spectra (Figure 4).
Figure 5.
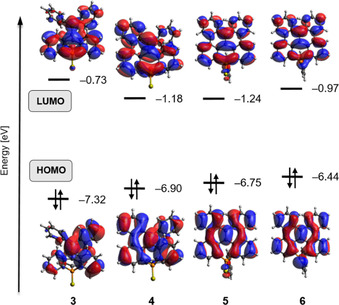
Frontier molecular orbitals (FMOs) of compounds 3–6 (COSMO‐BHLYP/TZVP level of theory).
In summary, starting from readily accessible acridophosphine we have established a concise synthetic strategy towards a new family of phosphorus‐containing PAHs. The polycyclic scaffolds can be regarded as organophosphorus congeners of the hitherto elusive PAH dibenzonaphthanthrene. Our synthetic approach relies on a sequence of Barton–Kellogg olefination followed by Mallory photocyclization as the key steps. Based on UV/Vis spectroscopic and electrochemical studies supported by DFT calculations, we have revealed the role of the substituent at the phosphorus center together with the size and shape of the polycyclic π system on its electronic nature. Taking into account the versatility and robustness of our synthetic approach, we are convinced that our findings are of considerable potential for the development of organophosphorus π‐conjugated materials with tailored properties and functions.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors gratefully acknowledge the funding by the Deutsche Forschungsgemeinschaft (DFG)—projects no. 182849149–SFB 953 and 401247651–KI 1662/3‐1. J.D.R.A. thanks the Evangelisches Studienwerk Villigst e.V. for generous financial support. Open access funding enabled and organized by Projekt DEAL.
J. D. R. Ascherl, C. Neiß, A. Vogel, J. Graf, F. Rominger, T. Oeser, F. Hampel, A. Görling, M. Kivala, Chem. Eur. J. 2020, 26, 13157.
References
- 1.
- 1a. Watson M. D., Fechtenkotter A., Müllen K., Chem. Rev. 2001, 101, 1267–1300; [DOI] [PubMed] [Google Scholar]
- 1b. Wu J. S., Pisula W., Müllen K., Chem. Rev. 2007, 107, 718–747; [DOI] [PubMed] [Google Scholar]
- 1c. Pisula W., Feng X., Müllen K., Chem. Mater. 2011, 23, 554–567; [Google Scholar]
- 1d. Nielsen C. B., Holliday S., Chen H.-Y., Cryer S. J., McCulloch I., Acc. Chem. Res. 2015, 48, 2803–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Pisula W., Feng X., Müllen K., Adv. Mater. 2010, 22, 3634–3649; [DOI] [PubMed] [Google Scholar]
- 2b. Rieger R., Müllen K., J. Phys. Org. Chem. 2010, 23, 315–325; [Google Scholar]
- 2c. Yan X., Li L.-S., J. Mater. Chem. 2011, 21, 3295–3300. [Google Scholar]
- 3. Dral P. O., Kivala M., Clark T., J. Org. Chem. 2013, 78, 1894–1902. [DOI] [PubMed] [Google Scholar]
- 4.For selected reviews on heteroatom-containing PAHs, see:
- 4a. Stępień M., Gońka E., Żyła M., Sprutta N., Chem. Rev. 2017, 117, 3479–3716; [DOI] [PubMed] [Google Scholar]
- 4b. Hirai M., Tanaka N., Sakai M., Yamaguchi S., Chem. Rev. 2019, 119, 8291–8331; [DOI] [PubMed] [Google Scholar]
- 4c. Schaub T. A., Padberg K., Kivala M., J. Phys. Org. Chem. 2020, 33, e4022. [Google Scholar]
- 5.For selected recent, illustrative examples of heteroatom-containing PAHs, see:
- 5a. Saito S., Matsuo K., Yamaguchi S., J. Am. Chem. Soc. 2012, 134, 9130–9133; [DOI] [PubMed] [Google Scholar]
- 5b. Tan Y.-Z., Osella S., Liu Y., Yang B., Beljonne D., Feng X., Müllen K., Angew. Chem. Int. Ed. 2015, 54, 2927–2931; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2970–2974; [Google Scholar]
- 5c. Dong R., Pfeffermann M., Skidin D., Wang F., Fu Y., Narita A., Tommasini M., Moresco F., Cuniberti G., Berger R., Müllen K., Feng X., J. Am. Chem. Soc. 2017, 139, 2168–2171; [DOI] [PubMed] [Google Scholar]
- 5d. Oki K., Takase M., Mori S., Shiotari A., Sugimoto Y., Ohara K., Okujima T., Uno H., J. Am. Chem. Soc. 2018, 140, 10430–10434; [DOI] [PubMed] [Google Scholar]
- 5e. Berezin A., Biot N., Battisti T., Bonifazi D., Angew. Chem. Int. Ed. 2018, 57, 8942–8946; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9080–9084; [Google Scholar]
- 5f. Navakouski M., Zhylitskaya H., Chmielewski P. J., Lis T., Cybińska J., Stępień M., Angew. Chem. Int. Ed. 2019, 58, 4929–4933; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4983–4987; [Google Scholar]
- 5g. Ðorđević L., Valentini C., Demitri N., Mézière C., Allain M., Sallé M., Folli A., Murphy D., Mañas-Valero S., Coronado E., Bonifazi D., Angew. Chem. Int. Ed. 2020, 59, 4106–4114; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 4135–4143. [Google Scholar]
- 6.For recent reviews on phosphorus-containing PAHs, see:
- 6a. Baumgartner T., Réau R., Chem. Rev. 2006, 106, 4681–4727; [DOI] [PubMed] [Google Scholar]
- 6b. Baumgartner T., Acc. Chem. Res. 2014, 47, 1613–1622; [DOI] [PubMed] [Google Scholar]
- 6c. Hindenburg P., Romero-Nieto C., Synlett 2016, 27, 2293–2300; [Google Scholar]
- 6d. Szűcs R., Bouit P.-A., Nyulászi L., Hissler M., ChemPhysChem 2017, 18, 2618–2630; [DOI] [PubMed] [Google Scholar]
- 6e. Schaub T. A., Padberg K., Kivala M., Chem. Lett. 2019, 48, 1358–1367. [Google Scholar]
- 7.For selected examples of P-containing PAHs, see:
- 7a. Hellwinkel D., Weil A., Sattler G., Nuber B., Angew. Chem. Int. Ed. Engl. 1990, 29, 689–692; [Google Scholar]; Angew. Chem. 1990, 102, 677–680; [Google Scholar]
- 7b. Durben S., Baumgartner T., Angew. Chem. Int. Ed. 2011, 50, 7948–7952; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8096–8100; [Google Scholar]
- 7c. Reus C., Stolar M., Vanderkley J., Nebauer J., Baumgartner T., J. Am. Chem. Soc. 2015, 137, 11710–11717; [DOI] [PubMed] [Google Scholar]
- 7d. Greulich T. W., Suzuki N., Daniliuc C. G., Fukazawa A., Yamaguchi E., Studer A., Yamaguchi S., Chem. Commun. 2016, 52, 2374–2377; [DOI] [PubMed] [Google Scholar]
- 7e. Hindenberg P., Busch M., Paul A., Bernhardt M., Gemessy P., Rominger F., Romero-Nieto C., Angew. Chem. Int. Ed. 2018, 57, 15157–15161; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15377–15381; [Google Scholar]; Padberg K., Ascherl J. D. R., Hampel F., Kivala M., Chem. Eur. J. 2020, 26, 3474–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Reed A. E., Schleyer P. v. R., J. Am. Chem. Soc. 1990, 112, 1434–1445; [Google Scholar]
- 8b. Leyssens T., Peeters D., J. Org. Chem. 2008, 73, 2725–2730. [DOI] [PubMed] [Google Scholar]
- 9. Gray M., Chapell B. J., Taylor N. J., Snieckus V., Angew. Chem. Int. Ed. Engl. 1996, 35, 1558–1560; [Google Scholar]; Angew. Chem. 1996, 108, 1609–1611. [Google Scholar]
- 10. Segall Y., Granoth I., Kalir A., Chem. Commun. 1974, 501b–502. [Google Scholar]
- 11. Schaub T. A., Brülls S. M., Dral P. O., Hampel F., Maid H., Kivala M., Chem. Eur. J. 2017, 23, 6988–6992. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Clar E., Polycyclic Hydrocarbons. Volume I and II, Springer, Berlin, 1964. [Google Scholar]
- 13.
- 13a. Hertz V. M., Bolte M., Lerner H.-W., Wagner M., Angew. Chem. Int. Ed. 2015, 54, 8800–8804; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8924–8928; [Google Scholar]
- 13b. Hertz V. M., Massoth J. G., Bolte M., Lerner H.-W., Wagner M., Chem. Eur. J. 2016, 22, 13181–13188. [DOI] [PubMed] [Google Scholar]
- 14. Hammer N., Shubina T. E., Gisselbrecht J.-P., Hampel F., Kivala M., J. Org. Chem. 2015, 80, 2418–2424. [DOI] [PubMed] [Google Scholar]
- 15. Bouit P.-A., Escande A., Szűcs R., Szieberth D., Lescop C., Nyulászi L., Hissler M., Réau R., J. Am. Chem. Soc. 2012, 134, 6524–6527. [DOI] [PubMed] [Google Scholar]
- 16. Grzybowski M., Skonieczny K., Butenschön H., Gryko D. T., Angew. Chem. Int. Ed. 2013, 52, 9900–9930; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10084–10115. [Google Scholar]
- 17. Chen K.-C., Ealick S. E., van der Helm D., Barycki J., Berlin K. D., J. Org. Chem. 1977, 42, 1170–1177. [Google Scholar]
- 18.Deposition numbers 1981909, 1982210, 1982209, and 1981910 (3, 4, 5, and 7) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 19.
- 19a. Grimme S., Peyerimhoff S. D., Chem. Phys. 1996, 204, 411–417; [Google Scholar]
- 19b. Wolstenholme D. J., Matta C. F., Cameron T. S., J. Phys. Chem. A 2007, 111, 8803–8813. [DOI] [PubMed] [Google Scholar]
- 20. Lakowicz J. R., Principles of Fluorescence Spectroscopy, Springer, Heidelberg, 2006. [Google Scholar]
- 21. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- 22. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 23. Klamt A., Schüürmann G., J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar]
- 24. Weigend F., Ahlrichs R., Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 25. Becke A. D., J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary