

Abstract
Background and Aims
Telomere attrition is a major risk factor for end‐stage liver disease. Due to a lack of adequate models and intrinsic difficulties in studying telomerase in physiologically relevant cells, the molecular mechanisms responsible for liver disease in patients with telomere syndromes remain elusive. To circumvent that, we used genome editing to generate isogenic human embryonic stem cells (hESCs) harboring clinically relevant mutations in telomerase and subjected them to an in vitro, stage‐specific hepatocyte differentiation protocol that resembles hepatocyte development in vivo.
Approach and Results
Using this platform, we observed that while telomerase is highly expressed in hESCs, it is quickly silenced, specifically due to telomerase reverse transcriptase component (TERT) down‐regulation, immediately after endoderm differentiation and completely absent in in vitro–derived hepatocytes, similar to what is observed in human primary hepatocytes. While endoderm derivation is not impacted by telomere shortening, progressive telomere dysfunction impaired hepatic endoderm formation. Consequently, hepatocyte derivation, as measured by expression of specific hepatic markers as well by albumin expression and secretion, is severely compromised in telomerase mutant cells with short telomeres. Interestingly, this phenotype was not caused by cell death induction or senescence. Rather, telomere shortening prevents the up‐regulation and activation of human hepatocyte nuclear factor 4 alpha (HNF4α) in a p53‐dependent manner. Both reactivation of telomerase and silencing of p53 rescued hepatocyte formation in telomerase mutants. Likewise, the conditional expression (doxycycline‐controlled) of HNF4α, even in cells that retained short telomeres, accrued DNA damage, and exhibited p53 stabilization, successfully restored hepatocyte formation from hESCS.
Conclusions
Our data show that telomere dysfunction acts as a major regulator of HNF4α during hepatocyte development, pointing to a target in the treatment of liver disease in telomere‐syndrome patients.
Abbreviations
- AAVS1
adeno‐associated virus integration site 1
- AFP
alpha‐fetoprotein
- BMP4
bone morphogenetic protein 4
- Cas9
CRISPR‐associated 9
- CD
cluster of differentiation
- CRISPR
clustered regularly interspaced short palindromic repeats
- CXCR4
chemokine (C‐X‐C motif) receptor 4
- CYPA1
cytochrome P450 A1
- DAPI
4′,6‐diamidino‐2‐phenylindole
- DC
dyskeratosis congenita
- DKC1
dyskerin
- DOX
doxycycline
- EdU
5‐ethynyl‐2′‐deoxyuridine
- ELISA
enzyme‐linked immunosorbent assay
- EP
early passage
- FGA
fibrinogen alpha chain
- FGF
fibroblast growth factor
- FGG
fibrinogen gamma chain
- FOXA2
forkhead box A2
- gRNA
guide RNA
- H2AX
Histone H2 variant
- HCC
hepatocellular carcinoma
- HCM
hepatocyte culture medium
- hESC
human embryonic stem cell
- HGF
hepatocyte growth factor
- HNF4α
hepatocyte nuclear factor 4 alpha
- LP
late passage
- NP‐40
Nonidet P40
- OCT4
octamer‐binding transcription factor 4
- PI
propidium iodide
- RPMI
Roswell Park Memorial Institute
- sh‐
short hairpin
- SOX17
SRY (sex determining region Y)‐box 17
- TBS‐T
trishydroxymethylaminomethane‐buffered saline with 1% Tween‐20
- TERC
telomerase RNA component
- TERT
telomerase reverse transcriptase component
- TRAP
Telomere Repeat Amplification Protocol
- WT
wild type
- ZFN
zinc finger nuclease
Telomeres are repetitive DNA sequences (TTAGGG in humans) that prevent degradation and fusion of chromosomal ends.( 1 ) These structures are largely double‐stranded but end in a short single‐stranded, G‐rich 3′ overhang that spans 50‐500 nucleotides in human cells.( 1 ) Telomeres are essential structures for cellular viability as they solve two major biological hurdles, the end‐replication and the end‐repair problems.( 2 ) The end‐replication problem exists as the DNA replication machinery is unable to fully replicate DNA termini in the lagging strand, therefore resulting in the loss of DNA at chromosomal ends during each replication cycle. However, while telomeres prevent the loss of genetic information, the continuous loss of DNA during replication implies that telomeres become progressively shorter with consecutive cellular divisions. This continuous shortening of telomeres can eventually lead to a critical stage where telomere dysfunction induces cellular senescence or cell death.( 3 ) Evidence collected during the past decade has established impaired telomere maintenance as a causative effect in a number of different conditions, ranging from bone marrow failure to pulmonary fibrosis and liver disease.( 4 , 5 , 6 , 7 , 8 ) Interestingly, telomere shortening affects different tissues at different ages, and telomere length measurement in hospital settings has recently been proposed as a diagnostic tool in targeted indications, with the potential to inform treatment decisions and influence morbidity.( 9 )
Telomere shortening is prevented through the action of a specialized ribonucleoprotein enzyme termed “telomerase,” which counteracts DNA erosion by synthesizing new telomeric repeats at chromosome ends.( 10 ) The active telomerase complex is composed of TERT (the reverse transcriptase component), TERC (the telomerase RNA component), and dyskerin (DKC1), which is necessary for TERC stabilization.( 10 ) In humans, telomerase is active mostly in germ line and somatic stem cells, to facilitate their continued cellular division and long‐term homeostasis. TERT expression, and therefore telomerase activity, are quickly silenced upon cellular differentiation.( 2 ) Also bound to telomeres is shelterin, a six‐protein complex that coats telomeric DNA and prevents it from being recognized as DNA breaks,( 1 , 2 ) thereby avoiding the activation of the ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3 (ATR) pathways.( 3 )
Mutations that affect telomere integrity were initially found in dyskeratosis congenita patients, a pediatric bone marrow failure syndrome where telomere erosion prevents continued blood homeostasis. However, data from recent years indicate that adult‐onset phenotypes, such as pulmonary fibrosis and liver disease, represent the most common phenotype in patients harboring mutations that impair telomere stability.( 4 , 11 , 12 ) Interestingly, patients with liver disease usually come to clinical attention at an earlier age and with longer telomeres when compared to patients with pulmonary fibrosis,( 9 ) indicating that liver cells might be more susceptible to telomere dysfunction than the lung epithelia. Accordingly, telomerase‐deficient mice which undergo liver ablation demonstrate impaired hepatocyte regeneration and an accelerated development of liver cirrhosis after chronic liver injury.( 13 ) In addition, the promoter region of TERT is activated during liver regeneration and hepatocyte proliferation( 14 ); and, more recently, it has been described that a small population of TERT‐positive hepatocytes are responsible for hepatocyte renewal and maintenance of liver mass in mice, both in homeostasis and after critical injury.( 15 )
While these results highlight the importance of telomerase for hepatocyte regeneration and liver disease, the chain of events linking telomere shortening to hepatocyte and liver failure in humans remains elusive. In fact, while genetic mutations in telomerase have been associated with liver disease, ranging from nonalcoholic fatty liver disease and nonalcoholic steatohepatitis to fibrosis and cirrhosis, the mechanisms behind liver tissue reorganization and failure in the setting of dysfunctional telomeres have not been elucidated.( 16 , 17 , 18 , 19 ) A major hurdle to address this question has been a lack of viable models to specifically analyze the impact of exacerbated telomere shortening in human hepatocytes. For instance, it has been shown that, unlike those from human patients, hepatocytes from mice with extensive telomere damage remain viable and able to maintain and recover liver function post–acute injury.( 20 ) To overcome this limitation in the more traditional models of liver disease and telomere biology, we used the targeted differentiation of human embryonic stem cells (hESCs) into mature, hepatocyte‐like cells. We used wild‐type (WT) and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR‐associated 9 (Cas9) isogenic engineered hESCs harboring a clinically relevant mutation in the telomerase component DKC1 (DKC1_A353V mutation, which represents the most common mutation in patients with impaired telomere maintenance( 21 )). While human induced pluripotent stem cells and hESCs have been used to study telomerase biochemistry in dyskeratosis congenita (DCs)( 22 , 23 , 24 ) and mechanisms of hematopoietic failure in the setting of short telomeres,( 21 , 25 , 26 ), our data represent an initial attempt to use this technology to understand how telomere erosion leads to failure of hepatocyte development and function. We show that p53 activation following telomere erosion prevents expression of hepatocyte nuclear factor 4 alpha (HNF4α), a major transcription factor in hepatic cells, and leads to increased cellular proliferation, significantly impairing hepatocyte development and function in DC cells.
Materials and Methods
Cell Culture
H1 (WA01) hESCs were acquired from the WiCell Research Institute (Madison, WI), following all institutional guidelines determined by the Embryonic Stem Cell Research Oversight Committee at Washington University in St. Louis. hESCs were cultured in Matrigel‐coated plates (Corning, Tewksbury, MA) in mTESR1 media (Stem Cell Technologies, Vancouver, Canada) and kept in a humidified incubator at 37°C in 5% CO2 and 5% O2 levels. WT and DC mutant hESCs were maintained and passaged onto new six‐well plates every 5 days at a split ratio of 1:12.
Gene Editing
DKC1_A353V and DKC1_A353V_p53–/– were generated using CRISPR/Cas9 technology.( 21 ) CRISPR guide RNAs (gRNAs) were inserted into the MLM3636 plasmid (Addgene 43860) and cotransfected with a plasmid carrying Cas9 (Addgene 43945) using the 4D‐Nucleofector with the P4 Primary Cell 4D‐Nucleofector kit (Lonza, Allendale, NJ). Single‐stranded DNA donor oligos were cotransfected with plasmids carrying specific gRNAs and Cas9. For DKC1_A353V_p53–/–, one gRNA sequence was cotransfected with Cas9 to induce nonhomologous end‐joining, resulting in a frame shift and early termination, which was verified by targeted sequencing and protein expression analysis. Nucleofected cells were manually picked when colonies reached an appropriate size. Clones were screened and sequenced. DKC1_A353V+TERC and DKC1_A353V_shHNF4α (short hairpin HNF4α) hESCs were generated by zinc finger nuclease. Transfection targeting the adeno‐associated virus integration site 1 (AAVS1) locus was performed with X‐TremeGene 9 following the manufacturer’s instructions (Roche, Indianapolis, IN).
Hepatocyte Differentiation
Differentiation of hESCs into hepatocyte‐like cells was performed according to Mallanna and Duncan.( 27 ) Briefly, hESCs were transferred to a 100‐mm plate coated with Matrigel and incubated at 37°C in 5% CO2 and 5% O2 for 3 days or until 90%‐95% confluent. The cells were then transferred to six‐well plates previously covered with Matrigel and incubated with mTESR1 for 24 hours. On days 1 and 2 of differentiation, cells were incubated with RPMI differentiation media (RPMI 1640‐HEPES [Gibco], 1% penicillin/streptomycin [Gibco], 1% nonessential amino acids [Gibco]) supplemented with 2% B27 (minus insulin; Gibco), 100 ng/mL activin A (R&D Biosystems), 10 ng/mL bone morphogenetic protein 4 (BMP4; R&D systems), and 20 ng/mL FGF (R&D Biosystems) at ambient O2. Between days 3 and 5 of differentiation, only 2% B27 minus insulin and 100 ng/mL activin A were added to the RPMI differentiation media (changed daily). From days 6 to 10, the RPMI differentiation media was supplemented with 2% B27 (Gibco), 20 ng/mL BMP4, and 10 ng/mL FGF; and cells were incubated at 5% O2. Between days 11 and 15, cells were kept in differentiation media containing 2% B27 and 20 ng/mL HGF (Peprotech), again at 5% O2. From day 16 forward, cells were kept in HCM medium (HCM Bullet Kit, Lonza) supplemented with kit‐supplied HCM SingleQuots (excluding EGF) and 20 ng/mL Oncostatin‐M (R&D Biosystems) at 20% O2. Cells and/or media were collected on days 6 (endoderm cells), 11 (hepatic endoderm), 16 (immature hepatocytes), and 21 (mature hepatocytes).
Immunostaining
Cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X‐100, and incubated with the following antibodies: octamer‐binding transcription factor 4 (OCT4; Santa Cruz Biotechnology), TRA160 (Abcam), HNF4α (Abcam), alpha‐fetoprotein (AFP; R&D Systems), and albumin–fluorescein isothiocyanate (Dako). Cells were incubated with secondary antibodies, and nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI). For quantification of albumin‐positive cells, we counted 1,000 cells/slide on day 21.
Quantification of Albumin Secretion
Albumin secretion quantification was performed using the human albumin ELISA kit from Bethyl Laboratories (Montgomery, TX), following the manufacturers’ guidelines, and measured on a plate reader at 450 nm.
Immunoblots
Protein extraction was performed using Nonidet P40 (NP‐40) buffer supplemented with protease and phosphatase inhibitors (Roche). Quantification was performed by Bradford assay. Proteins were resolved in 10% polyacrylamide gels in 1× Tris/glycine/sodium dodecyl sulfate buffer and transferred onto nitrocellulose membranes at 400 milliamps for 1:45 hours in 1× Tris/glycine buffer with 20% methanol. Membranes were blocked in either 5% bovine serum albumin (BSA) or 5% milk in TBS buffer. Primary antibody incubation was performed overnight at 4ºC in 5% BSA in TBS buffer supplemented with 1% Tween‐20 (TBS‐T). Membranes were washed (3 times) with TBS‐T buffer and incubated in 1% milk in TBS‐T with secondary antibodies (Li‐COR, Lincoln, NE) for 1 hour. Membranes were then washed with TBS‐T and scanned using the Odyssey infrared scanner (Li‐COR). Image capture and signal analysis were done using Image Studio software (Li‐COR). Primary antibodies used in this study were histone H2 varient (H2AX; 1:1,000; Abcam, Cambridge, MA), p53 (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA), and beta‐actin (1:2,000; Sigma, St. Louis, MO).
Detection of Telomerase Activity
Telomerase activity was measured by the Telomere Repeat Amplification Protocol (TRAP). Cells were lysed in NP‐40 buffer for 20 minutes on ice, and extracts were clarified by centrifugation at 16,000g. Protein quantification was performed by Bradford assay. Telomere extension reactions were performed using 2.0 μg, 0.5 μg, and 0.125 μg of protein; and the resulting products were amplified by PCR, following a modified two‐step TRAP protocol from the manufacturer (TRAPeze; EMD Millipore, Boston, MA).
Telomere Length Analysis
Telomere length was quantified by telomere repeat fragment analysis. Isopropanol‐extracted DNA was digested (10 µg) with Rhodopseudomonas sphaeroides and Haemophilus influenzae Rf restriction enzymes (New England Biolabs, Ipswich, MA) and resolved (2.5 µg for each analysis) on a 0.8% agarose gel for 16 hours at 85 volts in TBE (Tris‐borate‐EDTA) buffer. Gel was then soaked in denaturing buffer (1.5 M NaCl and 0.5 M NaOH) for 45 minutes, followed by neutralizing buffer (1.5 M NaCl, 1 M Tris‐HCl at pH 7.4) for 1 hour. DNA was transferred to a nitrocellulose membrane by capillarity for at least 16 hours in 20× saline–sodium citrate (3 M NaCl, 0.3 M sodium citrate dehydrate at pH 7.0). After crosslinking, membrane was hybridized with a 32P‐labeled probe (CCCTAA)4 and exposed to Carestream BioMax MR film (Sigma).
Beta‐Galactosidase Staining
Senescence‐associated beta‐galactosidase staining was performed following the manufacturer’s protocol (Cell Signaling Technology, MA). Briefly, positive control (represented by senescent human fibroblasts), WT, and DKC1_A353V cells on day 21 of differentiation were stained and detached using trypsin (Gibco). Cells were collected, plated onto slides, and analyzed by microscopy. The fraction of beta‐galactosidase‐positive cells was calculated for each sample.
Quantification of Cell Death
At each time point, cell media was collected, and cells were detached with Accutase (Stem Cell Technologies) and added to the collected media. Samples were centrifuged and washed with 1× Dulbecco’s phosphate‐buffered saline (DPBS) to pool live and dead cells together. Cells were resuspended in ~100‐150 μL 1× DPBS and costained with propidium iodide (PI) solution (Invitrogen) and Hoechst 33342 staining solution (Thermo Fisher Scientific) for 2 minutes. Cell suspension was transferred to microscope slides and immediately imaged using a Leica DM6B upright digital research microscope (Leica Microsystems, Buffalo Grove, IL). Cell death was quantified by the number of PI+ nuclei relative to total nuclei. At least 200 nuclei were counted per group per time point; n = 3.
Analysis of Cellular Proliferation by 5‐ethynyl‐2′‐deoxyuridine Incorporation
The Click‐iT 5‐ethynyl‐2′‐deoxyuridine (EdU) Alexa Fluor 488 kit (Life Technologies, Carlsbad, CA) was used according to the manufacturer’s instructions. Briefly, cells were incubated with 10 μM EdU for 1 hour, then fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X‐100. Samples were incubated in the appropriate Click‐iT reaction cocktail, followed by ProLong Gold Antifade Mountant with DAPI (Invitrogen). Cells were imaged using a Leica DM6B upright digital research microscope. At least 200 nuclei were counted per group per time point; n = 2.
Flow Cytometry
Flow‐cytometric analysis was done on BD LSR Fortessa at the Department of Pathology Flow Cytometry Core. Antibodies used were the following: chemokine (C‐X‐C motif) receptor 4 (CXCR4)–phycoerythrin (BD Biosciences) and CD117‐allophycocyanin (Invitrogen).
RNA Extraction, cDNA Synthesis, and Quantitative Real‐Time PCR Analysis
RNA extraction was performed using Trizol (Invitrogen) following the manufacturer’s instructions. RNA concentrations of 500‐1,000 ng were used for cDNA synthesis using the Superscript III First Strand synthesis kit (Invitrogen) following the manufacturer’s instructions. Quantitative real‐time PCR was performed using a StepOne Plus instrument (Applied Biosystems) with 2× EvaGreen qPCR Master Mix (Lamda Biotech, St. Louis, MO). For TERC quantitative RT‐PCR analysis, Brilliant II quantitative RT‐PCR 1‐Step qPCR Master Mix (Agilent, Santa Clara, CA) was used following the manufacturer’s instructions. Gene expression data were calculated using the Delta Delta cycle threshold method. For all stages and cell lines, genes were normalized to either β‐actin or 18S ribosomal RNA (rRNA) except for comparisons with human liver RNA, which were analyzed relative to the geometric mean of 18S rRNA and β2‐microglobulin. Human liver RNA, prepared from human liver tissue, was obtained commercially (#1H21‐250; Cell Applications, San Diego, CA). All primer sequences can be found in Supporting Table S1.
Caspase Activity Measurement
Caspase activity was quantified using dedicated caspases 3, 8, and 9 colorimetric detection kits following the manufacturer’s instructions (Abcam).
Results
Telomerase is Quickly Down‐Regulated During Hepatocyte Differentiation from hESCs
Our first step to understand the consequences of telomere dysfunction on hepatocyte biology was to develop a protocol that allowed us to generate telomerase mutant, human hepatocyte‐like cells in vitro. To achieve this, we adapted a protocol( 27 ) where, starting with human pluripotent stem cells, we were able to sequentially differentiate them into different developmental stages that culminated with the formation of a hepatocyte‐like population (Fig. 1A). We initially performed this protocol in our parental hESCs (WT) and were able to successfully derive and isolate endoderm (day 6 of differentiation; CXCR4‐positive, SRY‐box transcription factor 17 [SOX17]‐positive cells), hepatic endoderm (day 11 of differentiation; HNF4α‐positive cells), immature hepatocytes (day 16 of differentiation; AFP‐positive cells), and finally mature, hepatocyte‐like cells (day 21 of differentiation) that express fibrinogen alpha chain (FGA), fibrinogen gamma chain (FGG), and cytochrome P450 A1 (CYP1A1). This progressive change in gene expression from hESCs to hepatocyte‐like cells is shown in Fig. 1B. In addition, our hepatocyte‐like cells express (Fig. 1B) and secrete (Supporting Fig. S1A) albumin. To confirm that our in vitro–derived hepatocyte‐like cells represent a physiologically relevant model to study hepatocyte biology, we compared gene expression levels of these cells against expression levels observed in human samples obtained from whole liver. There was complete silencing of pluripotency markers on our hepatocyte‐like cells (Supporting Fig. S1B). In addition, Supporting Fig. S1C shows expression of different hepatocyte markers, confirming that the hepatic signature of our in vitro–derived hepatocyte‐like cells on day 21 of differentiation resembles what is observed in whole‐liver samples.
Fig. 1.
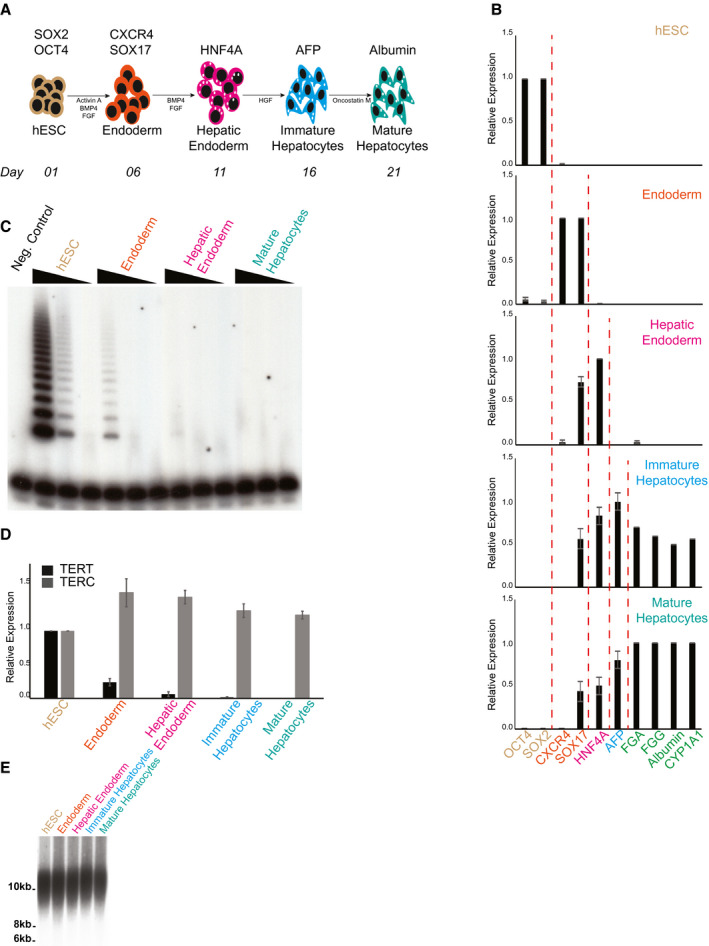
Telomerase is quickly down‐regulated during hepatocyte development from hESCs. (A) Schematic of in vitro hepatocyte differentiation. Hepatocyte differentiation is achieved through serial addition of specific cytokines and growth factors, depicted in the model. During hepatic differentiation, different transient cellular populations can be identified and isolated at specific times (indicated in the bottom line). These include endoderm, hepatic endoderm, immature hepatocytes, and mature hepatocytes. (B) Relative gene expression analysis by real‐time quantitative PCR of different markers specific for each cellular population obtained during hepatic differentiation: OCT4 and SOX2, hESCs (day 1); CXCR4 and SOX17, endoderm (day 6); HNF4α, hepatic endoderm (day 11); AFP, immature hepatocytes (day 16); FGA, FGG, albumin, and CYP1A1, mature hepatocytes (day 21). (C) Telomerase activity by TRAP during hepatic differentiation from WT hESCs. Range of concentrations represents 4‐fold serial dilutions. Negative control: NP‐40 buffer. (D) Real‐time quantitative PCR analysis of the telomerase core components TERT and TERC during hepatic differentiation from WT hESCs. (E) Telomere length analysis by telomere restriction fragment during hepatic differentiation from WT hESCs. Molecular weight (in kilobases) is shown.
It is well established that TERT is usually not expressed in human hepatocytes, which are therefore telomerase‐negative( 28 ) Accordingly, TRAP analysis shows that telomerase activity is quickly down‐regulated during the initial stages of hepatic differentiation (Fig. 1C), which can be attributable to the specific silencing of TERT expression as TERC levels remain unaltered (Fig. 1D). This pattern of expression of TERT and TERC in our in vitro–derived hepatocyte‐like cells resembles what is observed in whole‐liver samples (Supporting Fig. S1D). Importantly, despite the fast silencing of telomerase, telomeres are not significantly shortened during the 21 days of hepatic differentiation (Fig. 1E), indicating that the initial telomere length at day 1 of differentiation can be used as a reference for observed phenotypes during the entire 21‐day period of in vitro hepatocyte development.
Progressive Telomere Shortening Inhibits the Activation of HNF4α and Prevents Hepatocyte Development In Vitro
We next decided to understand whether mutations in telomerase, or telomere shortening itself, impair hepatocyte development and/or function. For that, we used isogenic hESCs where we genetically introduced (with the use of CRISPR/Cas9) a common mutation found in patients with telomeropathies, DKC1_A353V (Supporting Fig. S2A,B). These cells have normal karyotype and remain pluripotent (Supporting Fig. S2C,E). Importantly, as DKC1 is necessary for TERC stabilization,( 29 ) DKC1_A353V cells have reduced levels of TERC when compared to their unedited counterparts (Supporting Fig. S2F), mimicking the effects of this mutation in patients.( 4 ) Accordingly, the low levels of TERC observed in DKC1_A353V hESCs lead to reduced telomerase activity (Supporting Fig. S2G) and progressive telomere shortening (Supporting Fig. S2H). Combining these genetically engineered DKC1_A353V hESCs with our in vitro hepatocyte differentiation protocol, we were able to study hepatocyte development in clinically relevant telomerase mutant cells with progressively shorter telomeres. We chose two different stages, DKC1_A353V cells with longer telomeres (referred to as early passage [EP]; passage <13) and shorter telomeres (referred to as late passage [LP]; passage >30; Supporting Fig. S2H) to understand the consequences of progressive telomere shortening in hepatocyte development and function.
Mutations in telomerase or telomere length did not interfere with early endoderm development, measured both by quantification of CXCR4+CD117+ cells by flow cytometry (Fig. 2A) and by the expression of forkhead box A2 (FOXA2) and SOX17 by quantitative real‐time PCR (Fig. 2B), as both DKC1_A353V_EP and DKC1_A353V_LP cells show similar endoderm formation when compared to WT cells. However, it became clear that telomere dysfunction severely compromised hepatic development as HNF4α levels were significantly reduced, specifically in DKC1_A353V_LP cells when compared to WT and DKC1_A353V_EP cells on day 11 of differentiation (Fig. 2C). This impairment of hepatic differentiation was not caused by a failure to silence pluripotency genes (Supporting Fig. S3A) or early endoderm genes (Supporting Fig. S3B). Likewise, expression of FOXA2, a marker for hepatic endoderm, was similarly activated in DKC1_A353V_EP and DKC1_A353V_LP cells on day 11 of differentiation (Supporting Fig. S3B), indicating that HNF4α seems to be specifically inhibited in these cells at this developmental stage. As HNF4α is a major transcriptional factor in hepatic cells,( 30 , 31 ) it is not surprising that the continued hepatic development of DKC1_A353V_LP cells failed to generate a hepatocyte‐like population when compared either to WT or to DKC1_A353V_EP cells (Fig. 2D‐G). DKC1_A353V_LP cells showed significantly reduced expression levels of the hepatocyte markers AFP, FGA, FGG, and albumin (Fig. 2D); a reduced amount of albumin‐positive cells (Fig. 2E‐F); and reduced levels of albumin secretion (Fig. 2G) on day 21 of differentiation.
Fig. 2.
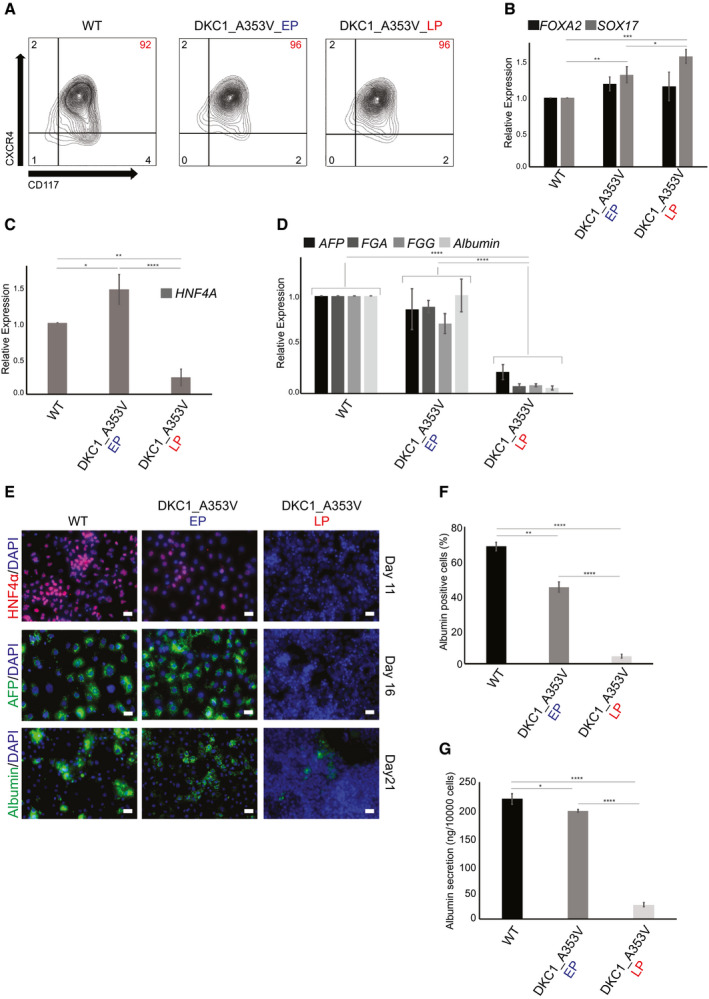
Telomere shortening impairs hepatocyte development in DKC1_A353V hESCS. (A,B) Endoderm generation from WT and DKC1_A353V hESCs at early (EP) and late (LP) passages, as assessed by (A) formation of a CXCR4+CD117+ population by flow cytometry (percent of population of interest indicated in red, top right) and (B) relative expression (analyzed by real‐time quantitative PCR) of the endoderm markers FOXA2 and SOX17. (C) Generation of a hepatic endoderm population quantified by the relative gene expression of HNF4α (by real‐time quantitative PCR) in WT and DKC1_A353V cells at different passages. (D) Relative expression of hepatocyte markers (by real‐time quantitative PCR) after 21 days of differentiation in WT and DKC1_A353V cells at different passages. (E) Immunocytochemistry showing expression of different markers (indicated in the figure) during differentiation of WT and DKC1_A353V cells in early or late passages. The specific day during differentiation is indicated on the right. Scale bars, 50 μM. (F) Quantification of albumin‐positive cells by immunofluorescence after 21 days of differentiation in WT and DKC1_A353V cells at different passages. A total of 1,000 cells/slide was counted. (G) Quantification of albumin secretion (by ELISA) after 21 days of differentiation in WT and DKC1_A353V cells at different passages. n = 3, mean ± SEM, *P ≤ 0.05; **P ≤ 0.0025; ***P ≤ 0.001; ****P ≤ 0.0001. Statistical analysis was performed using one‐way ANOVA followed by Tukey’s post hoc test.
We next decided to determine if reactivation of telomerase, and therefore the reelongation of telomeres, would be able to rescue hepatocyte development in DKC1_A353V cells. To achieve that, we introduced TERC into the AAVS1 safe harbor locus( 32 ) of DKC1_A353V_LP cells (referred to hereafter as DKC1_A353V_TERC cells; Fig. 3A). DKC1_A353V_TERC cells quickly regained TERC expression (Fig. 3B). Forced expression of TERC was able to successfully rescue hepatic endoderm development, with DKC1_A353V_TERC cells expressing high levels of HNF4α on day 11 of differentiation (Fig. 3C). This culminated with normal hepatocyte development where the expression of specific hepatocyte markers in DKC1_A353V_TERC cells on day 21 was comparable to that observed in WT (Fig. 3D). Likewise, the number of albumin‐positive cells (Fig. 3E) and albumin secretion (Fig. 3F) in DKC1_A353V_TERC cells was also rescued to levels similar to those observed in WT cells.
Fig. 3.
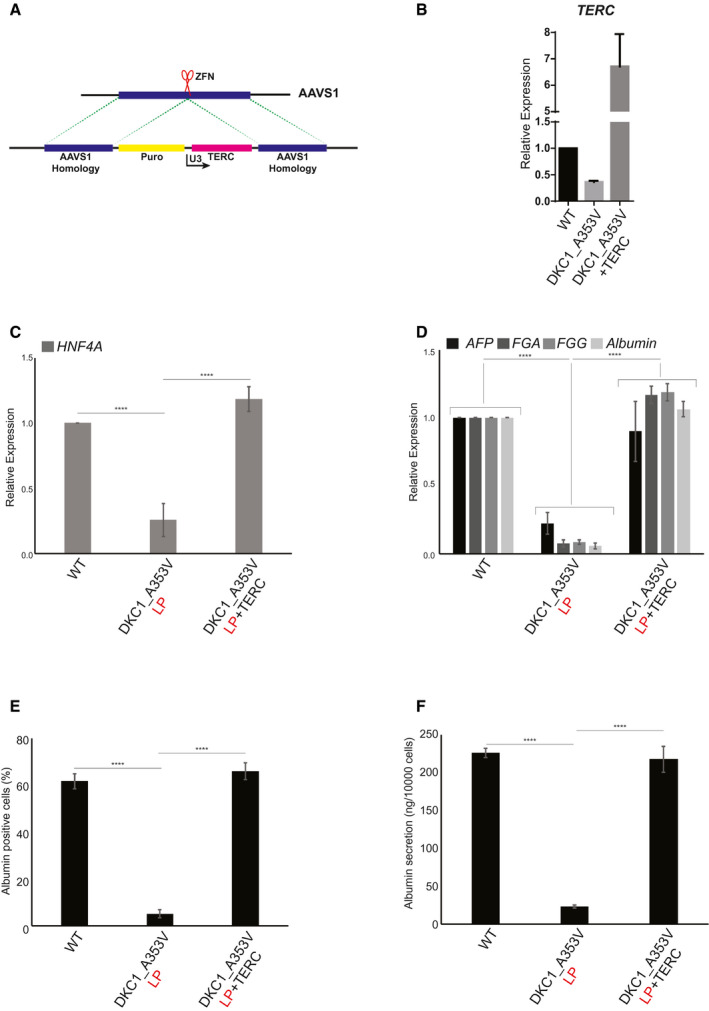
Reactivation of TERC rescues hepatocyte development from DKC1_A353V hESCs. (A) Model of AAVS1 targeting in DKC1_A353V hESCs. TERC is expressed under the control of a U3 promoter sequence in DKC1_A353V+TERC hESCs. (B) Quantification of TERC levels by real‐time quantitative PCR in WT, DKC1_A353V, and DKC1_A353V+TERC hESCs. (C) Quantification of HNF4α levels by quantitative RT‐PCR on day 11 of differentiation (hepatic endoderm stage) of WT, DKC1_A353V, and DKC1_A353V+TERC cells. (D) Relative expression of different hepatocyte markers (indicated in the figure) by real‐time quantitative PCR after 21 days of differentiation (mature hepatocyte stage) in WT, DKC1_A353V_LP, and DKC1_A353V+TERC cells. (E) Quantification of albumin‐positive cells by immunofluorescence after 21 days of differentiation in WT, DKC1_A353V_LP, and DKC1_A353V+TERC cells. (F) Quantification of albumin secretion by ELISA reading after 21 days of differentiation in WT, DKC1_A353V_LP, and DKC1_A353V+TERC cells. n = 3, mean ± SEM, ****P ≤ 0.0001. Statistical analysis was performed using one‐way ANOVA followed by Tukey’s post hoc test.
Stabilization of p53 Reduces Hepatocyte Differentiation from DKC1_A353V hESCs
Telomere shortening is a known inducer of senescence and cell death in mammalian cells,( 33 ) in a process that is regulated by p53 stabilization and activation of DNA damage response pathways.( 3 ) Therefore, we next decided to understand if p53 stabilization was involved in the failure to form hepatic endoderm observed in DKC1_A353V cells with short telomeres. We analyzed the levels of γH2AX, a marker of DNA damage signaling, and the stabilization of p53 during hepatic differentiation of WT, DKC1_A353V_EP, and DKC1_A353V_LP hESCs. It is clear that there is significant accrual of γH2AX specifically in DKC1_A353V_LP hESCs (Fig. 4A). Likewise, these cells showed clear stabilization of p53 levels (Fig. 4A), both of these being well‐established responses after telomere shortening and dysfunction in mammalian cells.
Fig. 4.
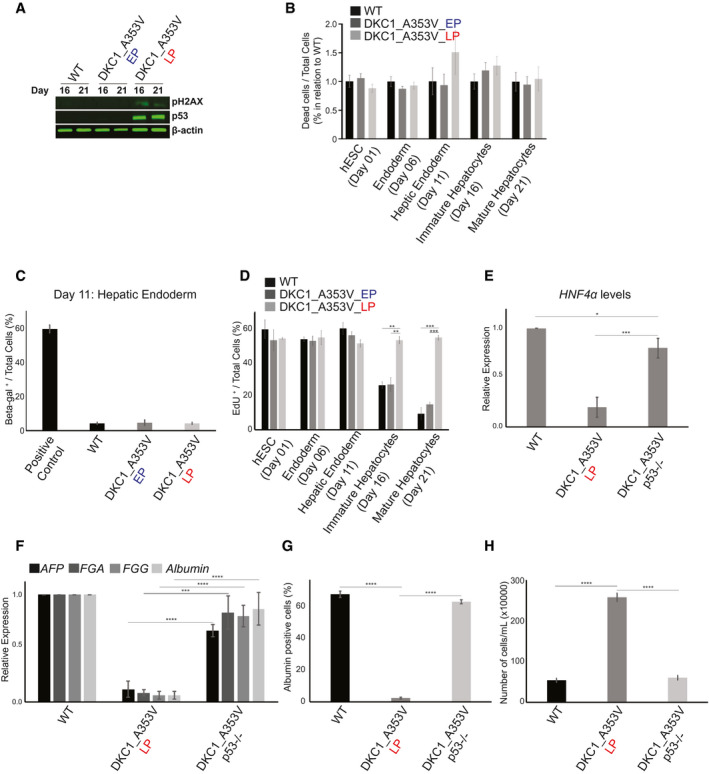
p53 stabilization impairs hepatocyte development from DKC1_A353V mutant hESCs. (A) Representative immunoblot analysis of γH2AX and p53 expression on days 16 and 21 of hepatic differentiation (immature and mature hepatocyte stages) of WT and DKC1_A353V hESCs (at early and late passages). β‐Actin is shown as loading control. (B) Quantification of dead cells by PI incorporation during different stages of hepatocyte development (indicated in the figure) in WT, DKC1_A353V_EP, and DKC1_A353V_LP cells. Fraction of PI‐positive cells presented over the total number of cells observed in each of the differentiation stages indicated. Results are presented in relation to WT cells for each stage. (C) Beta‐galactosidase‐positive cells were quantified (by light microscopy) in WT, DKC1_A353V_EP, and DKC1_A353V_LP cells on day 21 of differentiation (mature hepatocyte‐like stage). Positive control: senescent human primary fibroblasts (BJ cells). Fraction of beta‐galactosidase‐positive cells presented over the total number of cells observed in each condition. (D) Quantification of cellular proliferation by EdU incorporation during different stages of hepatocyte development (indicated in the figure) in WT, DKC1_A353V_EP, and DKC1_A353V_LP cells. Fraction of EdU‐positive cells presented over the total number of cells observed in each of the differentiation stages indicated. (E) Generation of hepatic endoderm population quantified by the relative gene expression (by real‐time quantitative PCR) of HNF4α in WT, DKC1_A353V_LP, and the p53‐ablated DKC1_A353V_p53–/– cells. (F) Relative expression of hepatocyte markers by real‐time quantitative PCR after 21 days of differentiation (mature hepatocyte stage) in WT, DKC1_A353V_LP, and DKC1_A353V_p53–/– cells. (G) Quantification of albumin‐positive cells by immunofluorescence after 21 days of differentiation in WT, DKC1_A353V_LP, and DKC1_A353V_p53–/– cells. (H) Total number of cells after hepatic differentiation of WT, DKC1_A353V_LP, and DKC1_A353V_p53–/– cells. Cells were collected on day 21, and the figure shows the total number of cells found in each population (total numbers quantified by cell counter). n = 3, mean ± SEM; *P ≤ 0.05, **P ≤ 0.0025, ***P ≤ 0.001, ****P ≤ 0.0001. Statistical analysis was performed using one‐way ANOVA followed by Tukey’s post hoc test.
As proliferation arrest and induction of cell death are common responses after p53 stabilization in cells with short telomeres,( 34 ) our next step was to verify if the observed low levels of hepatic endoderm formation in DKC1_A353V_LP cells were caused by reduced cellular viability. Surprisingly, however, and unlike the traditional cellular responses observed in terminally differentiated cells after telomere dysfunction, we did not observe increased cell death or senescence during the hepatic differentiation of cells with dysfunctional telomeres. DKC1_A353V_LP cells did not display increased cell death levels during any of the differentiation stages analyzed during hepatocyte development (Fig. 4B). Likewise, we did not detect activation of caspases 3 and 9 (which would be indicative of apoptosis) during differentiation of these cells (Supporting Fig. S4A). Similarly, we did not detect the induction of senescence in our cultures during hepatic endoderm formation (Fig. 4C), the developmental stage where DKC1_A353V_LP cells start to show reduced efficiency of differentiation. In fact, cellular proliferation was significantly increased in DKC1_A353V_LP cells when compared to WT and DKC1_A353V_EP cells during the later stages of differentiation, as quantified by EdU incorporation on days 1, 6, 11, 16, and 21 (Fig. 4D). This sustained proliferation capability led to significantly increased cell growth in DKC1_A353V_LP cells during the 21 days of hepatic differentiation when compared to WT and DKC1_A353V_EP cells (Supporting Fig. S4B). Combined, these data indicate that telomere shortening specifically impairs hepatocyte development through a mechanism that is independent from its well‐established role in cell death and/or cell cycle arrest.
These unexpected results prompted us to investigate in more detail the molecular regulation of hepatic differentiation in cells with short telomeres. For that, we initially decided to probe if the observed p53 up‐regulation during the hepatocyte differentiation of DKC1_A353V_LP hESCs cells (Fig. 4A) was a determining factor in the reduced efficiency of hepatocyte development observed in these cells. We ablated p53 (using CRISPR/Cas9 genome editing) in DKC1_A353V_LP hESCs, generating DKC1_A353V_p53–/– hESCs that retain short telomeres (as telomerase is still defective) but have no p53 stabilization (Supporting Fig. S5A,B). Surprisingly, our hepatic differentiation experiments showed that, despite retaining short telomeres, DKC1_A353V_p53–/– cells have normal hepatocyte development, with restored levels of HNF4α expression during the hepatic endoderm stage (day 11; Fig. 4E) and restored expression of hepatocyte markers at the end of the differentiation protocol (day 21; Fig. 4F). Likewise, there is a significant increase in the number of albumin‐positive cells at the mature hepatocyte‐like cell stage (Fig. 4G). Interestingly, despite ablation of p53, cellular growth is reduced to WT levels during hepatic differentiation of DKC1_A353V_p53–/– cells (Fig. 4H). Collectively, these data indicate that the activation of p53 by telomere shortening impairs hepatocyte development, through mechanisms that are independent from its well‐established role in triggering cellular senescence or death.
Restoring HNF4α Levels Successfully Rescues Hepatocyte Development and Function in Telomerase Mutant Cells
Without a direct role in cell death induction or growth arrest, we next focused on distinct roles of p53 activation during hepatocyte development. Intriguingly, the earliest phenotype observed during the hepatic differentiation of DKC1_A353V hESCs with short telomeres was reduced formation of hepatic endoderm, a developmental stage that is defined by the activation of HNF4α.( 27 , 35 ) Interestingly, it has been proposed that the stabilization of p53 in immortalized human hepatic cells suppresses the expression of HNF4α.( 36 ) We therefore hypothesized that the stabilization of p53 could similarly be preventing the activation of HNF4α during the formation of hepatic endoderm, and consequently blocking hepatocyte formation, as HNF4α regulates > 60% of genes expressed in human hepatocytes.( 37 ) Additionally, HNF4α is a key regulator of hepatocyte differentiation during embryonic development, and disruption of HNF4α in mature hepatocytes is linked to epithelial‐to‐mesenchymal transition( 38 , 39 ) and, similarly to what we observe in our in vitro system, increased cellular proliferation.( 37 , 40 )
We observed that in WT cells HNF4α expression starts on day 7 and increases continuously until day 11 of hepatocyte differentiation (Fig. 5A). To understand how essential this activation of HNF4α is during hepatocyte formation in vitro, we genetically silenced HNF4α in WT hESCs by constitutive expression of three different shRNA sequences from the AAVS1 safe harbor locus in WT cells (WT_shHNF4α; Supporting Fig. S5C). As expected, the suppression of HNF4α causes a significant reduction in hepatic differentiation (Fig. 5B) even in WT cells with long telomeres.
Fig. 5.
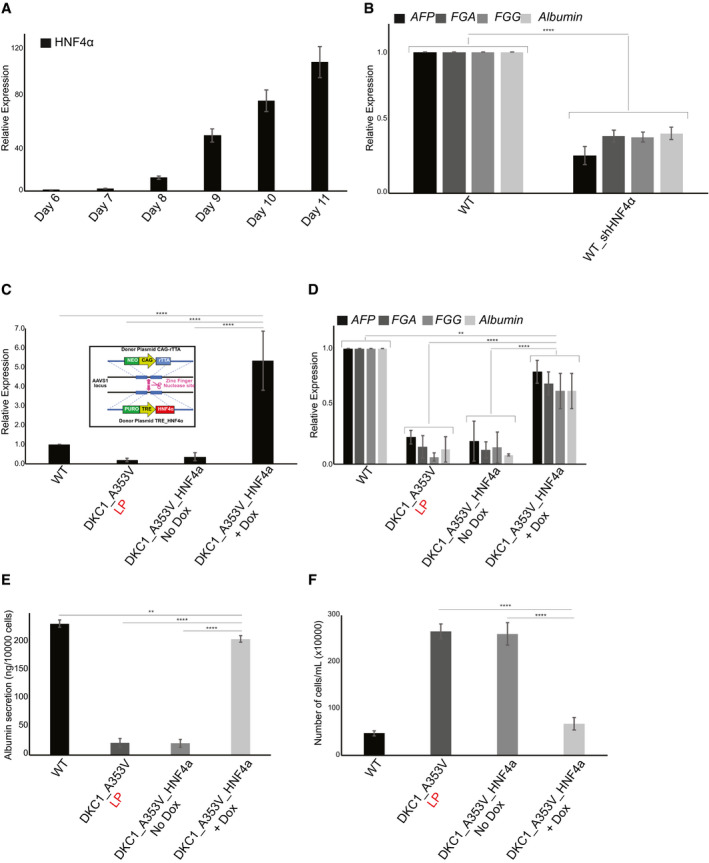
HNF4a expression rescues hepatocyte development in DKC1_A353V mutant cells that retain short telomeres. (A) HNF4α expression during hepatocyte derivation from WT hESCs. HNF4α expression increases until day 11 of differentiation (hepatic endoderm stage). (B) Relative expression of hepatocyte markers by real‐time quantitative PCR after 21 days of differentiation (mature hepatocyte stage) in WT and WT_sh HNF4α cells. (C) HNF4α expression in WT, DKC1_A353V_LP and conditional DKC1_A353V_ HNF4α cells with or without addition of DOX. Inset: Construction and cloning of conditional HNF4α cassette into the AAVS1 locus of DKC1_A353V hESCs. (D) Relative expression (by real‐time quantitative PCR) of hepatocyte markers after 21 days of differentiation in WT, DKC1_A353V_LP, and conditional DKC1_A353V_ HNF4α cells with or without addition of DOX. (E) Quantification of albumin secretion after 21 days of differentiation in WT, DKC1_A353V_LP and conditional DKC1_A353V_ HNF4α cells with or without addition of DOX. (F) Total number of cells after hepatic differentiation of WT, DKC1_A353V_LP, and conditional DKC1_A353V_ HNF4α cells with or without addition of DOX. Cells were collected on day 21, and the figure shows the total number of cells found in each population (total numbers quantified by cell counter). n = 3, mean ± SEM; **P ≤ 0.0025, ****P ≤ 0.0001. Statistical analysis was performed using one‐way ANOVA followed by Tukey’s post hoc test.
We therefore decided to assess if the reduced expression of HNF4α, caused by p53 stabilization, was directly related to the low levels of hepatocyte formation that we observed in DKC1_A353V cells with short telomeres. To rigorously investigate that, we again used genome engineering to conditionally express HNF4α from the AAVS1 safe harbor locus in late‐passage DKC1_A353V cells (in a doxycycline [DOX]–inducible system; Fig. 5C, inset). These DKC1_A353V‐TET‐ON_HNF4α cells retain short telomeres and p53 stabilization, but HNF4α expression can remain active, in a conditional, DOX‐dependent manner (Fig. 5C). We set out to perform hepatic differentiation in these cells. While early stages of differentiation remain unperturbed (endoderm, day 3; data not shown), it is clear that despite harboring short telomeres, activation of HNF4α expression in DKC1_A353V‐TET‐ON_HNF4α cells treated with DOX is able to rescue hepatocyte formation, as assessed by expression of different markers (Fig. 5D). Moreover, it also rescues hepatocyte function, as can be assessed by the expression and secretion of albumin (Fig. 5E). Even more interestingly, similarly to what was observed in DKC1_A353V cells with ablated p53, expression of HNF4α prevents the exacerbated cell growth observed in cells with dysfunctional telomeres (Fig. 5F), indicating that continuous cellular growth is associated with the loss of HNF4α expression and a possible change in cellular identity under these settings.
Taken together, our data indicate that restoring HNF4α levels successfully overcomes the deleterious consequences of telomere dysfunction observed during hepatocyte development in telomerase mutant cells.
Discussion
In the present study we demonstrate that progressive telomere shortening leads to repression of HNF4α activation during hepatocyte development. This repression of HNF4α is linked to the activation of p53 following accumulation of DNA damage, leading to a reduced efficiency to generate functional, hepatocyte‐like cells from telomerase mutants. Interestingly, the activation of p53 and suppression of HNF4α caused a significant increase in cellular proliferation during hepatocyte development and was not associated with increased cell death or senescence.
Telomere syndromes are a group of heterogenous diseases that are characterized, molecularly, by mutations in telomere biology genes.( 4 , 5 , 6 , 7 ) These mutations lead to widespread tissue defects and reduced life span in humans. Telomere length is the primary determinant of disease onset and progression in these patients, with disease‐associated phenotypes appearing at an earlier age, and with more severe presentation, with each successive generation as the telomere length progressively shortens. This genetic anticipation phenomenon is also observed in telomerase‐null mice, which develop worsening phenotypes with successive breeding( 41 ) and eventually die at prereproductive ages, therefore limiting the genetic lineage.( 42 ) Interestingly, telomerase‐deficient mice do not fully recapitulate the spectrum of phenotypes observed in patients, significantly reducing their use for the understanding of the underlying causes of disease and for the development of therapies against specific phenotypes.
Hepatic involvement is common in patients and can present in diverse ways (including elevated liver enzymes as well as histopathologic and imaging abnormalities), and liver disease has important implications for morbidity and mortality in patients with telomere disease.( 43 ) Moreover, even in patients with liver disease and cirrhosis with no detectable mutations in telomerase, there is significant telomere shortening observed in hepatic cells.( 44 ) This progressive shortening of telomeres is likely associated with impaired liver regeneration, potentially leading to a faster disease progression.( 43 ) Accordingly, it has recently been described that a rare subpopulation of hepatocytes retains TERT expression and is responsible for regeneration after injury in the adult mouse liver.( 15 ) While these indicate a clear connection between telomerase biology, telomere homeostasis, and hepatocyte function, the mechanisms leading to liver disease in settings of mutant telomerase remain obscure.
Here, we used the targeted differentiation of hESCs as a model to decipher mechanisms that lead to hepatocyte failure in settings of dysfunctional telomeres, using cells with a DKC1_A353V mutation that has been associated with liver disease.( 43 ) We observed a significant induction of p53 expression during hepatocyte development in settings of short telomeres. Higher p53 levels are also observed in samples from liver disease patients,( 45 ) indicating that under settings of high stress, liver cells can activate the p53 pathway as a protective response to injury. However, p53 activation can also act as a potentiator of liver disease and be involved in its pathogenesis, by reducing the regenerative capacity of liver cells.( 46 )
Interestingly, our results show that activation of p53 significantly reduces hepatocyte formation as DKC1_A353V cells that retained short telomeres but had genetically ablated p53 show normal hepatocyte development. We demonstrate that this reduction was not related to senescence or induction of cell death; instead, failure to generate hepatocytes in DKC1_A353V cells was associated with increased cellular proliferation. Ablation of p53 not only rescued hepatocyte formation but also reduced cellular growth, indicating that, in these settings, p53 is acting independently from its well‐established role in proliferation arrest. Our data show that the first phenotype during hepatocyte development in DKC1_A353V with short telomeres, the inability of these cells to up‐regulate HNF4α, was directly related to p53 stabilization. Accordingly, p53 has been shown to down‐regulate HNF4α through its proximal P1 promoter,( 36 ) indicating that p53 stabilization in DKC1_A353V cells with short telomeres could prevent hepatocyte formation by preventing HNF4α expression. We confirmed this hypothesis using DKC1_A353V cells with short telomeres and efficient p53 response but with conditional expression of HNF4α. In these cells, hepatic endoderm develops normally, and hepatocytes are formed with a similar efficiency when compared to WT cells. Interestingly, conditional mouse models of HNF4α ablation show that silencing of this transcription factor leads to up‐regulation of genes associated with proliferation and cell cycle control, causing increased cellular proliferation, loss of hepatocyte identity, and reduced hepatocyte formation,( 37 ) similar to what we observed in our cells with short telomeres and low HNF4α levels. We show that the conditional reexpression of HNF4α is able to reduce cellular growth during development of hepatocyte‐like cells in DKC1_A353V_LP cells.
Taken together, our data suggest that the loss of HNF4α is a determining factor in the reduced ability of hESCs with short telomeres to form hepatocytes in vitro from hESCs. While our system does not directly recapitulate the in vivo environment of liver disease in patients with telomere syndromes, our findings do suggest that HNF4α is a major target of telomere dysfunction. HNF4α is a key regulator of hepatic differentiation, maintenance of liver homeostasis, and liver metabolism.( 31 ) It also acts as a suppressor of liver fibrosis and cirrhosis.( 47 , 48 , 49 ) Recent genetic evidence has established telomere maintenance and telomerase modulation as major aspects of liver disease and carcinogenesis, with a high number of human hepatocellular carcinomas (HCCs) showing mutations in the TERT promoter( 28 ) that lead to its reactivation.( 50 ) Similar to our findings, HCCs show telomere shortening, up‐regulation of p53, loss of HNF4α, and increased proliferation, leading to a change in cellular identity.( 28 ) While our current work does not directly address the mechanisms leading to the progression of liver disease and HCC in patients, we believe that the many similarities between our data and what is observed in vivo demonstrate the relevance of proper telomere maintenance in hepatic cells. While still in its infancy, the reproducibly, robustness, and ease of genetic manipulation transform the in vitro differentiation of hESCs into an efficient model to study the development of liver disease, as has been shown by other groups.( 51 , 52 , 53 ) Future experiments using this system, coupled with more traditional mouse models and clinical samples, will be essential for us to determine to what extent the restoration of HNF4α can normalize human hepatocytes in telomere syndrome patients suffering from liver disease.
Author Contributions
M.M., E.L.N. and L.F.Z.B. designed the experiments and analyzed the data; M.M., E.L.N., W.C.F., A.T.V., H.J., K.A.B. and L.F.Z.B. performed the experiments; M.M., E.L.N. and L.F.Z.B. wrote the manuscript.
Supporting information
Supplementary Material
Supported by CNPq, Brazil (E.L.N.); the Phillip Majerus Postdoctoral Fellowship (A.T.V.); the National Heart, Lung, and Blood Institute’s T32 Training Grant in Molecular Hematology (HL007088‐41, to M.M. and W.C.F.); the National Fellowship Foundation (K.A.B.); the National Heart, Lung, and Blood Institute (1R01HL137793‐01, to L.F.Z.B.); the Department of Defense (BM160054); and grants from the Siteman Cancer Center at Washington University in St. Louis, the American Cancer Society, the V Foundation for Cancer Research, the Edward Mallinckrodt Jr. Foundation, the Concern Foundation, the American Federation for Aging Research, and the Longer Life Foundation. This project was also supported by a pilot grant from the Washington University DDRCC program (NIDDK P30 DK052574, to L.F.Z.B.).
Potential conflict of interest: Nothing to report.
See Editorial on Page 1166
References
Author names in bold designate shared co‐first authorship.
- 1. Smith EM, Pendlebury DF, Nandakumar J. Structural biology of telomeres and telomerase. Cell Mol Life Sci 2020;77:61‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet 2019;20:299‐309. [DOI] [PubMed] [Google Scholar]
- 3. Lazzerini‐Denchi E, Sfeir A. Stop pulling my strings—what telomeres taught us about the DNA damage response. Nat Rev Mol Cell Biol 2016;17:364‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet 2012;13:693‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Savage SA. Beginning at the ends: telomeres and human disease. F1000Res 2018;7:524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bertuch AA. The molecular genetics of the telomere biology disorders. RNA Biol 2016;13:696‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Armanios M. Syndromes of telomere shortening. Annu Rev Genomics Hum Genet 2009;10:45‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walne AJ, Dokal I. Advances in the understanding of dyskeratosis congenita. Br J Haematol 2009;145:164‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alder JK, Hanumanthu VS, Strong MA, DeZern AE, Stanley SE, Takemoto CM, et al. Diagnostic utility of telomere length testing in a hospital‐based setting. Proc Natl Acad Sci USA 2018;115:E2358‐E2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wu RA, Upton HE, Vogan JM, Collins K. Telomerase mechanism of telomere synthesis. Annu Rev Biochem 2017;86:439‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutat Res 2012;730:52‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Niewisch MR, Savage SA. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol 2019;12:1037‐1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rudolph KL, Chang S, Millard M, Schreiber‐Agus N, DePinho RA. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 2000;287:1253‐1258. [DOI] [PubMed] [Google Scholar]
- 14. Sirma H, Kumar M, Meena JK, Witt B, Weise JM, Lechel A, et al. The promoter of human telomerase reverse transcriptase is activated during liver regeneration and hepatocyte proliferation. Gastroenterology 2011;141:326‐337, 337e1‐3. [DOI] [PubMed] [Google Scholar]
- 15. Lin S, Nascimento EM, Gajera CR, Chen L, Neuhofer P, Garbuzov A, et al. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature 2018;556:244‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carulli L, Anzivino C. Telomere and telomerase in chronic liver disease and hepatocarcinoma. World J Gastroenterol 2014;20:6287‐6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Donati B, Valenti L. Telomeres, NAFLD and chronic liver disease. Int J Mol Sci 2016;17:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Calado RT, Brudno J, Mehta P, Kovacs JJ, Wu C, Zago MA, et al. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology 2011;53:1600‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hartmann D, Srivastava U, Thaler M, Kleinhans KN, N'Kontchou G, Scheffold A, et al. Telomerase gene mutations are associated with cirrhosis formation. Hepatology 2011;53:1608‐1617. [DOI] [PubMed] [Google Scholar]
- 20. Lazzerini Denchi E, Celli G, de Lange T. Hepatocytes with extensive telomere deprotection and fusion remain viable and regenerate liver mass through endoreduplication. Genes Dev 2006;20:2648‐2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fok WC, Niero ELO, Dege C, Brenner KA, Sturgeon CM, Batista LFZ. p53 mediates failure of human definitive hematopoiesis in dyskeratosis congenita. Stem Cell Reports 2017;9:409‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Batista LF, Pech MF, Zhong FL, Nguyen HN, Xie KT, Zaug AJ, et al. Telomere shortening and loss of self‐renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 2011;474:399‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Agarwal S, Loh YH, McLoughlin EM, Huang J, Park IH, Miller JD, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature 2010;464:292‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winkler T, Hong SG, Decker JE, Morgan MJ, Wu C, Hughes WM 5th, et al. Defective telomere elongation and hematopoiesis from telomerase‐mutant aplastic anemia iPSCs. J Clin Invest 2013;123:1952‐1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fok WC, Shukla S, Vessoni AT, Brenner KA, Parker R, Sturgeon CM, et al. Posttranscriptional modulation of TERC by PAPD5 inhibition rescues hematopoietic development in dyskeratosis congenita. Blood 2019;133:1308‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Donaires FS, Alves‐Paiva RM, Gutierrez‐Rodrigues F, da Silva FB, Tellechea MF, Moreira LF, et al. Telomere dynamics and hematopoietic differentiation of human DKC1‐mutant induced pluripotent stem cells. Stem Cell Res 2019;40:101540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mallanna SK, Duncan SA. Differentiation of hepatocytes from pluripotent stem cells. Curr Protoc Stem Cell Biol 2013;26:Unit 1G 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nault JC, Ningarhari M, Rebouissou S, Zucman‐Rossi J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat Rev Gastroenterol Hepatol 2019;16:544‐558. [DOI] [PubMed] [Google Scholar]
- 29. Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 1999;402:551‐555. [DOI] [PubMed] [Google Scholar]
- 30. Watt AJ, Garrison WD, Duncan SA. HNF4: a central regulator of hepatocyte differentiation and function. Hepatology 2003;37:1249‐1253. [DOI] [PubMed] [Google Scholar]
- 31. Lau HH, Ng NHJ, Loo LSW, Jasmen JB, Teo AKK. The molecular functions of hepatocyte nuclear factors—in and beyond the liver. J Hepatol 2018;68:1033‐1048. [DOI] [PubMed] [Google Scholar]
- 32. Sim X, Cardenas‐Diaz FL, French DL, Gadue P. A doxycycline‐inducible system for genetic correction of iPSC disease models. Methods Mol Biol 2016;1353:13‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet 2019;20:299‐309. [DOI] [PubMed] [Google Scholar]
- 34. Laptenko O, Prives C. p53: master of life, death, and the epigenome. Genes Dev 2017;31:955‐956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hanawa M, Takayama K, Sakurai F, Tachibana M, Mizuguchi H. Hepatocyte nuclear factor 4 alpha promotes definitive endoderm differentiation from human induced pluripotent stem cells. Stem Cell Rev Rep 2017;13:542‐551. [DOI] [PubMed] [Google Scholar]
- 36. Maeda Y, Hwang‐Verslues WW, Wei G, Fukazawa T, Durbin ML, Owen LB, et al. Tumour suppressor p53 down‐regulates the expression of the human hepatocyte nuclear factor 4α (HNF4α) gene. Biochem J 2006;400:303‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bonzo JA, Ferry CH, Matsubara T, Kim JH, Gonzalez FJ. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4α in adult mice. J Biol Chem 2012;287:7345‐7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Santangelo L, Marchetti A, Cicchini C, Conigliaro A, Conti B, Mancone C, et al. The stable repression of mesenchymal program is required for hepatocyte identity: a novel role for hepatocyte nuclear factor 4alpha. Hepatology 2011;53:2063‐2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ning BF, Ding J, Yin C, Zhong W, Wu K, Zeng X, et al. Hepatocyte nuclear factor 4 alpha suppresses the development of hepatocellular carcinoma. Cancer Res 2010;70:7640‐7651. [DOI] [PubMed] [Google Scholar]
- 40. Walesky C, Apte U. Role of hepatocyte nuclear factor 4α (HNF4α) in cell proliferation and cancer. Gene Expr 2015;16:101‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hao LY, Armanios M, Strong MA, Karim B, Feldser DM, Huso D, et al. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell 2005;123:1121‐1131. [DOI] [PubMed] [Google Scholar]
- 42. Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 1997;91:25‐34. [DOI] [PubMed] [Google Scholar]
- 43. Kapuria D, Ben‐Yakov G, Ortolano R, Cho MH, Kalchiem‐Dekel O, Takyar V, et al. The spectrum of hepatic involvement in patients with telomere disease. Hepatology 2019;69:2579‐2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wiemann SU, Satyanarayana A, Tsahuridu M, Tillmann HL, Zender L, Klempnauer J, et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J 2002;16:935‐942. [DOI] [PubMed] [Google Scholar]
- 45. Krstic J, Galhuber M, Schulz TJ, Schupp M, Prokesch A. p53 as a dichotomous regulator of liver disease: the dose makes the medicine. Int J Mol Sci 2018;19:921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yan Z, Miao X, Zhang B, Xie J. p53 as a double‐edged sword in the progression of non‐alcoholic fatty liver disease. Life Sci 2018;215:64‐72. [DOI] [PubMed] [Google Scholar]
- 47. Yue HY, Yin C, Hou JL, Zeng X, Chen YX, Zhong W, et al. Hepatocyte nuclear factor 4alpha attenuates hepatic fibrosis in rats. Gut 2010;59:236‐246. [DOI] [PubMed] [Google Scholar]
- 48. Babeu JP, Boudreau F. Hepatocyte nuclear factor 4‐alpha involvement in liver and intestinal inflammatory networks. World J Gastroenterol 2014;20:22‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lazarevich NL, Shavochkina DA, Fleishman DI, Kustova IF, Morozova OV, Chuchuev ES, et al. Deregulation of hepatocyte nuclear factor 4 (HNF4) as a marker of epithelial tumors progression. Exp Oncol 2010;32:167‐171. [PubMed] [Google Scholar]
- 50. Chiba K, Lorbeer FK, Shain AH, McSwiggen DT, Schruf E, Oh A, et al. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two‐step mechanism. Science 2017;357:1416‐1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Takayama K, Akita N, Mimura N, Akahira R, Taniguchi Y, Ikeda M, et al. Generation of safe and therapeutically effective human induced pluripotent stem cell‐derived hepatocyte‐like cells for regenerative medicine. Hepatol Commun 2017;1:1058‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Corbett JL, Duncan SA. iPSC‐derived hepatocytes as a platform for disease modeling and drug discovery. Front Med (Lausanne) 2019;6:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jing R, Corbett JL, Cai J, Beeson GC, Beeson CC, Chan SS, et al. A screen using iPSC‐derived hepatocytes reveals NAD+ as a potential treatment for mtDNA depletion syndrome. Cell Rep 2018;25:1469‐1484.e1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material