

Abstract
The (formal) replacement of Co in cobalamin (Cbl) by NiII generates nibalamin (Nibl), a new transition‐metal analogue of vitamin B12. Described here is Nibl, synthesized by incorporation of a NiII ion into the metal‐free B12 ligand hydrogenobalamin (Hbl), itself prepared from hydrogenobyric acid (Hby). The related NiII corrin nibyric acid (Niby) was similarly synthesized from Hby, the metal‐free cobyric acid ligand. The solution structures of Hbl, and Niby and Nibl, were characterized by spectroscopic studies. Hbl features two inner protons bound at N2 and N4 of the corrin ligand, as discovered in Hby. X‐ray analysis of Niby shows the structural adaptation of the corrin ligand to NiII ions and the coordination behavior of NiII. The diamagnetic Niby and Nibl, and corresponding isoelectronic CoI corrins, were deduced to be isostructural. Nibl is a structural mimic of four‐coordinate base‐off Cbls, as verified by its ability to act as a strong inhibitor of bacterial adenosyltransferase.
Keywords: cobalamins, porphyrinoids, transition metals, crystal structures, vitamins
Nibalamin (Nibl), the novel NiII analogue of vitamin B12, was synthesized from the metal‐free B12 ligand hydrogenobalamin, prepared from biosynthetic hydrogenobyric acid (Hby). An X‐ray crystal analysis of nibyric acid, the NiII complex of Hby, revealed the first structural details of a nickel complex of a natural corrin ligand. Nibl is a structural mimic of the corresponding four‐coordinate cobalamins, which are of interest in biological investigations.

Introduction
Biologically active vitamin B12 derivatives exclusively utilize cobalt as their specific transition metal center, which is bound and activated exquisitely by a helical corrin macrocycle. [1] The metal‐free corrin ligand of vitamin B12, hydrogenobyric acid (Hby), has recently been made available as a consequence of engineered B12 biosynthesis in E. coli.[ 2 , 3 ] The availability of Hby has provided an unparalleled opportunity for the effective synthesis of metal‐free and transition metal analogues of the natural cobalt‐corrinoids a previously intractable challenge in bioinorganic and B12 chemistry. [4] We have recently used Hby for the synthesis of the corresponding zinc‐corrin zincobyric acid (Znby) and the Zn analogue of vitamin B12 zincobalamin (Znbl), of interest as luminescent structural B12 mimics. [5]
Herein, we report on the first nickel‐complexes of natural corrin ligands, including nibalamin (Nibl). We also describe the syntheses of crystalline nibyric acid (Niby), the novel NiII complex of Hby, [3] and hydrogenobalamin (Hbl), the metal‐free complete B12 ligand (see Scheme 1 and Scheme 2). Koppenhagen and co‐workers, back in the 1970’s, reported the isolation of Hbl from a Chromatium strain supplemented with 5,6‐dimethylbenz‐imidazole (DMB). They were able to characterize Hbl by UV/Vis‐spectroscopy and demonstrated that it could be converted into vitamin B12 by insertion of cobalt,[ 4e , 7 ] and later reported its mass spectrum. [8]
Scheme 1.
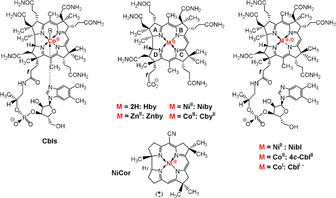
Structural formulae of cobalt, zinc, nickel and metal‐free corrinoids. Left: cobalamins with “base‐on” structures: vitamin B12 (R=CN, CNCbl), coenzyme B12 (R=5′‐deoxyadenosyl, AdoCbl), methylcobalamin (R=CH3, MeCbl), cob(II)alamin (R=e−, CblII). Center, top: the NiIIcorrin nibyrate (Niby), hydrogenobyric acid (Hby), CoII‐cobyric acid (CbyII) and zincobyric acid (Znby), where CoII and ZnII carry an unspecified axial ligand (e.g., solvent molecule). Center, bottom: Eschenmoser's synthetic racemic NiII‐corrin NiCor. [6] Right: nibalamin (Nibl), four‐coordinate “base off” cob(II)alamin (4 c ‐CblII) and cob(I)alamin (CblI−).
Scheme 2.

Preparation of the NiII‐corrins Niby and Nibl from Hby. i) 0.5 m Ni(OAc)2 pH 6, 1 h, 90 °C, Ar. ii) 3 equiv B12 nucleotide moiety,[ 1a , 12 ] HOBt, EDC*HCl, H2O, 0 °C, 4 d. iii) 0.5 m Ni(OAc)2 pH 6, 1 h, 90 °C, Ar (see the SI for details).
A NiII‐corrin, the NiCor (see Scheme 1), was prepared in the Eschenmoser labs as the first synthetic corrin, making use of the NiII ion as a “template” for the assembly of the corrin macro‐ring. [6] NiCor also became the object of the first X‐ray crystallographic investigation of the structure of a non‐cobalt corrin. [9] Four coordinate NiII complexes prefer to adopt a planar geometry and therefore are more structurally related to the corresponding CoI complexes. [10] Indeed, recently, there has been a resurgence in the quest for close Ni analogues of the B12 cofactors.[ 4c , 4f ] The planar ligand set of Nibl potentially represents a structural B12 mimic that is inert to the organometallic transformations typical of B12‐dependent enzymes, as suggested by its expected coordination chemistry and structural properties. Specific interest in Nibl, the NiII‐analogue of vitamin B12 and of other cobalamins (Cbls) (see Scheme 1), is thus a consequence not only of its chemistry, but also of its possible use as a molecular probe in B12 biology and biomedicine, helpful for the investigation of cobalamin‐dependent processes and their physiological effects. [11]
Results and Discussion
Nibyric acid (Niby) was prepared by dissolving 1.40 mg (1.6 μmol) of crystalline hydrogenobyric acid (Hby) [3] in 3.5 mL of deoxygenated 0.5 m aqueous NiIIacetate, pH 6, with stirring at 90 °C for 75 min. Separation on a short reverse phase column, evaporation and crystallization from aqueous acetonitrile yielded 0.90 mg (0.97 μmol, 61 %) of Niby, which was isolated as yellow crystals (see Scheme 2, Exptl. Part and Supporting Information (SI). The UV/Vis absorption spectrum of an aqueous solution of Niby displayed bands at 464 nm (shoulder), 448 nm and 334 nm (Figure 1), and exhibited similar gross features to those observed in an absorption spectrum of the Ni‐corrin NiCor (but with a slightly red‐shifted maxima).[ 6a , 6c ] The solution structure of the diamagnetic NiII‐corrin Niby (molecular formula C45H64N10O8Ni, for HR mass spectra see SI, Figure S3) was analyzed by NMR spectroscopy, providing assignment of all 52 non‐exchangeable H atoms and 44 C atoms (see SI, Figure S4 and Table S1). A 500 MHz 1H NMR spectrum of Niby in D2O displayed five high field singlets for the six methyl groups, a singlet of HC10 at 6.30 ppm, as well as several signals at intermediate field for HC19, HC3, HC8 and HC13 (see Figure 2). The data from homonuclear and heteronuclear correlations confirmed the stereostructure of Niby (see SI, Figure S5).
Figure 1.

Absorption spectra of aqueous solutions of metal‐free B12 ligands Hby and Hbl and of their NiII‐complexes Niby and Nibl at 298 K. Top: UV/Vis‐absorption spectra of Hby (c=31.5 μm, pH 5, black trace) and Niby (c=34.5 μm, unbuffered, red trace). Bottom: UV/Vis‐absorption of Hbl (pH 5, black trace) and of Nibl (unbuffered, red trace).
Figure 2.
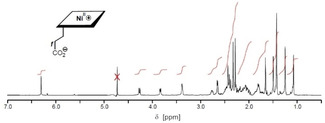
500 MHz 1H NMR spectrum of Niby in D2O (c=1.9 mm, 298 K); the water signal after presaturation is marked by an X.
The complete metal‐free ligand of the cobalamins, hydrogenobalamin (Hbl), was assembled by attaching the B12 nucleotide moiety[ 1a , 12 ] to the propionate moiety of Hby at 0 °C through application of the carbodiimide method (Scheme 2).[ 4d , 13 ] In brief, an aqueous solution of 9.12 mg (10.4 μmol) of Hby and of 14.71 mg (33.4 μmol) of the B12‐nucleotide was treated with 9.4 moleq of HOBt and degassed. To the frozen reaction mixture 4.4 moleq of EDC*HCl were added under Ar. Upon subsequent warm‐up of the reaction mixture to 0 °C, 16 moleq EDC*HCl were added and stirring was continued for 4 d (see SI). Work‐up, using RP18‐chromatographic purification, precipitation with MeCN and drying, furnished 11.3 mg (8.89 μmol, 85 % yield) of Hbl as an orange powder. An aqueous solution of Hbl at pH 5 exhibited UV/Vis [4e] and CD spectral features (SI, Figure S8 and S9) similar to those of Hby. [3] The UV/Vis absorption maximum at 525 nm of the α‐band of Hbl and the fluorescence emission maximum at 554 nm (SI, Figure S10) located the first excited singlet state of Hbl at E S near 221 kJ mol−1, marginally lower than for Hby. [3] The structure of Hbl (molecular formula C62H90N13O14P, see HR‐MS data in SI, Figure S11) in H2O was characterized by NMR spectroscopy (600 MHz 1H NMR spectrum in SI, Figure S12), providing assignment of 89 H atoms and of all 62 C atoms (see SI, Table S2). The two “inner” H atoms gave rise to singlets at δ=12.32 and δ=12.57 ppm, which were assigned to H(N4) and to H(N2), respectively, indicating a minor up‐field shift of both of them when compared to Hby. [3] The methyl group singlet of H3C1 at δ=0.81 ppm occurred at 0.47 ppm to higher field, compared to Hby, suggesting a temporary residence of the heteroaromatic DMB unit of Hbl near to its corrin moiety, a conclusion that was further supported by weak inter‐residual correlations in the 1H,1H ROESY spectra (see SI, Figure S13). However, the signals of the DMB moiety (HN2 at δ=8.35 ppm, HN4 at δ=7.31 ppm, HCN7 at δ=7.30 ppm) were found at similar chemical shift values to those of the free B12 nucleotide, [12] effectively incompatible with a time‐averaged positioning of the DMB part close to the corrin chromophore, as found for zincobalamin (Znbl) [5] and for typical “base‐on” CoIIICbls. [14]
The NiII‐corrin nibalamin (Nibl) was prepared by heating a deoxygenated aqueous solution of Hbl and Ni(OAc)2 for 1 h at 90 °C (Scheme 2), furnishing Nibl in 77 % yield as a yellow powder. An unbuffered aqueous solution of Nibl exhibited a UV/Vis spectrum that is incompatible with coordination by the DMB base and nearly indistinguishable (at >300 nm) from the spectrum of Niby, and similar to the spectrum of the NiII‐corrin NiCor[ 6a , 6c ] (see Figure 1). However, the absorption maxima of Nibl occurred at characteristically longer wavelengths when compared to the spectrum of the recently described vitamin B12‐derived 5,6‐dihydroxy‐5,6‐dihydronibalamin, which features an interrupted corrin π‐system. [4f] Lower pH values affected only the short wavelength part of the Nibl UV/Vis‐absorption spectrum, which was altered by DMB‐protonation, consistent with a pK a=4.35±0.06 for protonated Nibl‐H+ (see SI, Figure S20).
The structure of Nibl (molecular formula C62H88N13O14PNi, SI, Figure S17) was characterized in aqueous solution by heteronuclear NMR spectroscopy (see 500 MHz 1H NMR spectrum in Figure 3), providing assignment of all 73 non‐exchangeable H atoms and of all 62 C atoms (SI, Table S3). The positions of the singlets of H3C1A (δ=1.10 ppm), of HCN2 (δ=8.51 ppm), HN4 (δ=7.36 ppm) and HCN7 (δ=7.39 ppm) of the DMB moiety all indicate a base‐off form with a four‐coordinate NiII center. Hence, the UV/Vis and NMR spectral features of Nibl characterize it as an isoelectronic and, roughly, isostructural analogue of the diamagnetic cob(I)alamin (CblI), which is considered to feature a “base‐off” structure with a four‐coordinated CoI center.[ 15 , 16 ]
Figure 3.

500 MHz 1H NMR spectrum of Nibl in D2O (c=1.4 mm, 298 K); residual water signal after presaturation marked by an X.
The nickel corrin Niby was crystallized from aqueous acetonitrile, furnishing yellow single crystals (P 2 1 2 1 2 1) suitable for X‐ray analysis (see Figure 4). The incorporation of a NiII ion into the corrin macrocycle of the metal‐free Hby increased the effective symmetry of the corrin ligand as revealed by a comparison of the crystal structures of Niby and of Hby (see SI for details). Coordination of the NiII‐ion largely equalizes the lengths of the two diagonals, where the N2‐N4 diagonal of Niby exceeds its N1‐N3 counterpart by only Δd=0.047 Å, far less than in Hby (Δd=0.297 Å) [3] or in Znby (Δd=0.197 Å [5] ). The somewhat longer N2‐N4 diagonals in the metal corrins Niby and Znby appear to reflect the preferred mode of the conformational adaptation of the coordination hole of the flexible, unsymmetrical corrin ligand to bound metal ions. The radial size of the coordination hole also shrank upon NiII coordination as the average length of the N1‐N3 and N2‐N4 diagonals of Hby (d=3.82 Å) was reduced to d=3.71 Å in the complex Niby. Hence, the coordination of the NiII ion in Niby contracts the corrin ligand and makes it more symmetrical. This latter effect is also expressed by the regularly alternating bond lengths of the corrin π‐system in Niby, observations that are compatible with the model Ni‐corrin NiCor. [9]
Figure 4.
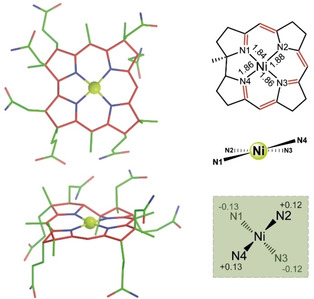
Crystal structure of Niby. Left. Crystallographic model of Niby in two projections; Right. Graphs representing the corrin core with display of N‐Ni bond lengths (top) and the coordination geometry around NiII center, highlighting the arrangement of the four inner corrin N atoms in a flattened tetrahedron around the NiII center (middle and bottom).
The four‐coordinate NiII ion sits very close to the plane of the four inner corrin N atoms, comparable to the situation in the NiII‐corrin NiCor, [9] and in typical CoIII‐corrins, [16a] but contrasting somewhat with the out of plane distance of 0.048 Å of the five‐coordinate CoII center of heptamethyl‐cob(II)yrinate perchlorate (CbinII) [17] (SI, Table S5). As expected,[ 9 , 18 ] the metal−N bonds in Niby (average Ni−N bond length=1.86 Å) are shorter than those found in the CoII analogue CbinII and in the CoIII‐corrin coenzyme B12 (AdoCbl), where average (CoII‐N) and (CoIII‐N) bond lengths of 1.89 Å [19] and of 1.90 Å, [20] respectively, were observed.
The coordination of the NiII ion barely affects the conformational properties of the metal‐free corrin ligand (Figures 5,6). Only a slight reduction of the helicity, h (Figure 7), of the inner corrin N atoms from h=12.9° in Hby to h=10.1° is seen in Niby. Indeed, the effect of the binding of the NiII ion on the corrin helicity is comparable to the situation in the enzyme‐bound four‐coordinate cob(II)alamin (4 c‐CblII(ACA)) of the adenosyltransferase ACA, [21] for which h=8°. [3] In contrast, in five‐coordinate CoII‐corrins the corrin helicity is significantly smaller, for example, h=6.1° in the CoII‐corrin CbinII, and in typical CoIII‐corrins planarization of the corrin ligand is still more pronounced, leading, for example, to h=3.5° in AdoCbl. [3]
Figure 5.
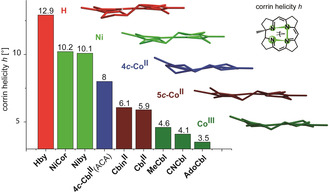
Structural adaptation of the helical corrin ligand to the coordinated metal ions. Comparison of the corrin helicity h in the structures of Hby, NiCor, Niby, of 4 c ‐CblII in adenosyltransferase (ACA), of five‐coordinate CbinII and CblII, and of six‐coordinate CoIII‐corrins MeCbl, CNCbl and AdoCbl (see text for details).
Figure 6.
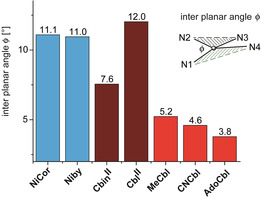
Adaptation of the coordination geometry around the corrin‐bound metal ions to the helical corrin ligand. Comparison of the interplanar angles φ in the structures of NiCor, Niby, 5‐coordinate CbinII and CblII, and the six‐coordinate CoIII corrins MeCbl, CNCbl and AdoCbl (see text for details).
Figure 7.
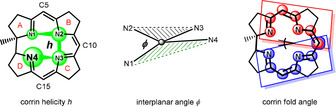
Geometrical parameters describing conformational effects in corrins. Left: corrin helicity (h=the dihedral angle N1‐N2‐N3‐N4 around a virtual bond between N2 and N3 of the corrin ligand), revealing the effect of bound metal‐ions on the corrin helix (Figure 5). [3] Center: interplanar angle (φ=angle between the two planes through the metal ion and corrin N′s N2/N4 or N1/N4, respectively) characterizing the deviation of the coordination environment at the metal center due to the helical corrin ligand (Figure 6). [3] Right: corrin fold (angle between the best two planes through the inner corrin atoms from N1 to C10 and from C10 to N4, respectively) describing the main conformational response of the corrin macrocycle to the binding of the DMB base in Cbls. [22] .
The observed lower drive of the four‐coordinate d 8 NiII ion to planarize the corrin macrocycle is similarly reflected by its own coordination geometry, which deviates strongly in Niby from the coplanar arrangement of the coordinating ligand atoms in typical four‐coordinate low‐spin NiII complexes.[ 9 , 10 , 18 ] In Niby a remarkably large interplanar angle φ (Figure 7) at NiII (φ=11.1°) results from extensive directional coordinative adaptation of the NiII‐center to the geometric requirements imposed by the helical corrin ligand (see Figure 6 and SI). φ is significantly larger in Niby than in CoIII‐corrins, which exhibit φ′s around 5° or less, [3] and is comparable to the situation in five‐coordinate CoII‐corrins CblII (φ=12°) and CbinII (φ=7.6°).
The corrin helicity h and the inter‐planar angle φ (Figure 7), were introduced recently as two complementary parameters characterizing inner conformational effects of the mutual structural adaptation of the corrin macrocycle and of the coordination geometry at the bound metal ion. [3] The so called corrin fold of the helical corrin macrocycle, [22] a classic parameter characterizing the nonplanar corrin ring in Cbls and in other “complete” cobamides (Figure 7), was not used in this current study. Conceived as a measure of the major conformational adaptation of the corrin ring to the cobalt coordination of the (bulky) DMB moiety in “base‐on” Cbls, it runs roughly along the Co‐C10 (east‐west) axis. [22] However, in four‐ and five‐coordinate metal‐corrins lacking the DMB unit, like CbinII, Niby and Znby, the calculated corrin‐fold is dominated by the effects of the corrin helicity and the intersection between the two relevant planes adheres to a north‐south direction (see SI Table S5 and Figure S22).
DFT calculations were carried out to test further the proposed close structural similarity between NiII‐corrins and their analogues with four‐coordinate cobalt centers (see Figure 8 and SI for further details). In order to minimize the relevance of peripheral H‐bonds between the amide functions in the implicit solvent calculations, the five‐coordinate lipophilic heptamethyl‐cob(II)yrinate perchlorate (CbinII) [17] was used as a starting model. Calculations of the structures of the related unknown corrins heptamethyl‐nibyrinate (Nibin), four‐coordinate heptamethyl‐cob(II)yrinate (4 c ‐CobIIin) and heptamethyl‐cob(I)yrinate (CobIin) were carried out. They indicate extensive structural similarities, but the equatorial metal−N bonds are shorter (by roughly 0.01–0.03 Å) in the CoI‐ and NiII‐corrins CobIin and Nibin in comparison to the four‐coordinate CoII‐corrin 4 c ‐CobIIin and 5‐coordinate CbinII. The N1‐N3 diagonal was calculated to be shorter than its N2‐N4 by roughly 0.01–0.02 Å, which is also in good qualitative agreement with the crystallographic data for Niby and CbinII. [17] All 4‐coordinate metal centers (NiII, CoI and CoII) were calculated to be located virtually in the best plane through the four corrin N‐atoms. The latter is arranged in a helix with a calculated value of h slightly decreasing from Nibin (7.6°) to CobIin to 4 c‐CobIIin (6.6°), and induced an interplanar angle φ that slightly decreased in the same order (from 8.4° to 7.5°). The calculations for five‐coordinate CbinII also reproduced, qualitatively, the still smaller value for h (5.4°), a larger value for φ (11.3°) and a significant displacement of the CoII center towards the axial β‐ligand (calculated as 0.112 Å).
Figure 8.
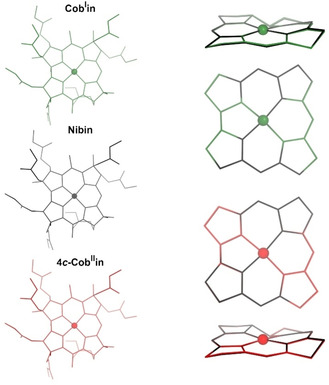
Calculated structures (left) of the four‐coordinate metal‐corrins Nibin (black), CobIin (green) and 4 c ‐CobIIin (orange) and structural superpositions (right) of the corrin cores of the cobalt complexes and Nibin, minimized at the four coordinating corrin N atoms.
The deduced utility of Nibl as a specific new structural mimic of four‐coordinate base‐off Cbls was initially tested in binding studies of Nibl to an adenosyltransferase enzyme (ATP:CoIrrinoid adenosyltransferase), which catalyzes the biosynthetic construction of AdoCbl by Coβ‐adenosylation of bound CblI−. Such bacterial [23] and human adenosyltransferases, [24] for example, BtuR, [23a] CobA, [23b] ACA, [21] and CblB, [24] have been shown to facilitate the adenosylation process by first inducing the corrin substrate to adopt a four‐coordinate structure, thus raising the redox potential of the CoII/CoI couple by around 250 mV,[ 23 , 24 ] thereby allowing the physiologically difficult reduction. We, thus, investigated the effect of Nibl on the adenosylation process by incubating the Brucella melitensis BtuR [23a] and the structural Cbl mimic Nibl in the presence of an excess of CblI (see SI, Figures S27–S29). As expected, Nibl itself was not a substrate for the enzyme and was not adenosylated. However, the presence of Nibl within the incubation did effectively inhibit the adenosyltransferase, reducing the activity of the enzyme by 38 % at a concentration of 1 μm, and by 60 % at 5 μm (Figure S29). Thus, the four‐coordinate NiII center of Nibl affords it the ability to bind within the active site of the adenosyltransferase and to prevent the productive binding of the natural substrate Cbl.
Conclusion
Herein, we have described the first NiII analogues of natural cobalt corrins vitamin B12 (CNCbl) and cobyric acid (Cby). The new Ni‐corrins Nibl and Niby display the same basic structural features and lack of coordinative activity as synthetic model NiII‐corrins, such as NiCor.[ 1a , 6a , 6c ] The NiII‐corrins are known to be exceptionally stable in regard to the chemical removal of their metal center, [25] and to exhibit no affinity for axially coordinating ligands.[ 1a , 6c , 7 ] This latter feature has been rationalized by the extraordinary stabilization of the low‐spin d8 configuration by the ligand field of the ring‐contracted corrin ligand.[ 1a , 9 ] As shown here, the natural corrin ligand undergoes only a small contraction of its coordination hole by about 0.03 to 0.04 Å to accommodate the low‐spin NiII ion. Indeed, the 15‐membered inner perimeter provided by the natural corrin macrocycle, selected for binding low‐spin cobalt ions, also binds NiII consistently in its low‐spin state. In contrast, in the porphyrinoid B12‐related nickel complex coenzyme F430[ 1a , 18 , 26 ] the 16‐membered porphyrinoid macrocycle is a key player in the active, specific adjustment of the spin state and coordinative activity of the nickel center to its function in the enzyme catalyzed methane formation. [27] Indeed, the discovery of coenzyme F430 provoked an entirely new look at the structural effect of the tetrapyrrolic macrocycle on the coordination chemistry of bound first‐row transition metals. [18]
A common feature of the valence shell of the low spin states of the transition metal ions NiII, CoIII, CoII and CoI is their unoccupied orbital, a key factor responsible for their strong sigma bonding interactions with the four inner corrin N atoms, leading to similar radial characteristics of their corrin complexes. A differing number of valence shell electrons in NiII‐, CoIII‐, CoII‐ and CoI‐corrins is transduced primarily into characteristically different reactivity of the metal centers in the axial direction, strongly affecting their potential binding sites there. Consequently, NiII‐corrins are to be considered particularly well‐suited structural mimics of corresponding isoelectronic CoI‐corrins, the critical intermediates in heterolytic organometallic transitions in B12‐dependent enzymes. [28] Crystallographic insights and DFT‐based structure calculations also indicate a structural similarity between NiII‐corrins and the exceptional four‐coordinate CoII‐corrins. This result contrasts strikingly with the mutually different structures of the typical five‐coordinate CoII‐corrins and their ZnII‐analogues [5] with similarly sized metal ions [29] that differ by the number of electrons in the valence shell.
The structural analysis of the Nibyrinates predicts that the constitutively robust Nibl would likely be an excellent redox‐resistant structural mimic for the elusive cob(I)alamin (CblI), a highly reactive redox‐active intermediate [30] that is found in B12‐dependent methyl group transferases, such as methionine synthase, [28a] as well as in the biosynthesis of AdoCbl from CblII (via CblI) by Cbl‐adenosyltransferases.[ 23 , 24 ] Nibl may, likewise, act as a good structural mimic of the recently described natural four‐coordinate CoII‐corrins, proposed as key intermediates in the enzymatic transformations catalyzed by the vitamin B12 tailoring enzyme CblC, [31] in corrinoid dehalogenases, [32] or as substrates for the reduction to CoI‐species in enzymatic cobalt alkylation.[ 14 , 33 ] Indeed, as verified here, Nibl is a very effective inhibitor of the bacterial Cbl‐adenosyltransferase BtuR.
We have developed a rational and direct synthetic path from hydrogenobyric acid (Hby) via hydrogenobalamin (Hbl) to nibalamin (Nibl), a novel transition‐metal analogue of the Cbls. Our recent studies with the RhIII analogue AdoRhbl of AdoCbl, [4d] with the ZnII analogue Znbl of CblII, [5] and now the NiII analogue Nibl of CblI, have furnished a valuable suite of cobalamin mimics for use in the study of B12‐dependent enzymatic processes,[ 16a , 28 , 30 , 34 , 35 ] and in B12‐dependent biological regulation. [36] Well‐characterized and adequately accessible transition metal analogues (Metbls) of the Cbls provide a promising small‐compound platform that may contribute significantly to the ongoing quest for innovative B12‐based biological and biomedical applications.[ 11 , 37 ] Along these lines, some Metbls may find applications as effective antivitamins B12.[ 4d , 11 ] The availability of selected Metbls and of related metal corrins (MetCors) will also allow more detailed experimental investigations into the chemical relevance of the coordination of transition metal ions by the uniquely skewed, strongly helical and unsymmetric natural corrin ligands. [3] Such studies will endow a more informed understanding of the specific evolutionary selection of cobalt rather than any other transition metal [1] for the task of complex organometallic catalysis achieved by the B12 cofactors.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by grants from the Austria Science Fund (FWF, P‐28892, P‐33059) to BK and (FWF M‐2005) to MP, and from the Biotechnology and Biological Sciences Research Council (BBSRC; BB/S014020/1, BB/K009249/1 and BB/S002197/1) and Royal Society (INF\R2\180062) to MJW. Part of the computational results presented was obtained using the HPC infrastructure LEO of the University of Innsbruck and the Vienna Scientific Cluster (VSC).
C. Kieninger, K. Wurst, M. Podewitz, M. Stanley, E. Deery, A. D. Lawrence, K. R. Liedl, M. J. Warren, B. Kräutler, Angew. Chem. Int. Ed. 2020, 59, 20129.
Dedicated to Professor Albert Eschenmoser on the occasion of his 95th birthday
Contributor Information
Prof. Dr. Martin J. Warren, Email: m.j.warren@kent.ac.uk.
Prof. Dr. Bernhard Kräutler, Email: bernhard.kraeutler@uibk.ac.at.
References
- 1.
- 1a. Eschenmoser A., Angew. Chem. Int. Ed. Engl. 1988, 27, 5–39; [Google Scholar]; Angew. Chem. 1988, 100, 5–40; [Google Scholar]
- 1b. Eschenmoser A., Angew. Chem. Int. Ed. 2011, 50, 12412–12472; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12618–12681; [Google Scholar]
- 1c. da Silva J. J. R. F., Williams R. J. P., The Biological Chemistry of the Elements, Clarendon Press, Oxford, 1991; [Google Scholar]
- 1d. Pratt J. M. in B12, Vol. I (Ed.: Dolphin D.), Wiley, New York, 1982, pp. 325–392. [Google Scholar]
- 2.
- 2a. Leeper F. J., Warren M. J., Kelly J. M., Lawrence A. D. in Handbook of Porphyrin Science, Vol. 25 (Ed.: Kadish K. M., Smith K. M., Guilard R.), World Scientific, Singapore, 2012, pp. 2–83; [Google Scholar]
- 2b. Deery E., Schroeder S., Lawrence A. D., Taylor S. L., Seyedarabi A., Waterman J., Wilson K. S., Brown D., Geeves M. A., Howard M. J., Pickersgill R. W., Warren M. J., Nat. Chem. Biol. 2012, 8, 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kieninger C., Deery E., Lawrence A. D., Podewitz M., Wurst K., Nemoto-Smith E., Widner F. J., Baker J. A., Jockusch S., Kreutz C. R., Liedl K. R., Gruber K., Warren M. J., Kräutler B., Angew. Chem. Int. Ed. 2019, 58, 10756–10760; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 10869–10873. [Google Scholar]
- 4.
- 4a. Blaser H. U., Winnacker E. L., Fischli A., Hardegger B., Bormann D., Hashimoto N., Schossig J., Keese R., Eschenmoser A., Helv. Chim. Acta 2015, 98, 1845–1920; [Google Scholar]
- 4b. Toohey J. I., Proc. Natl. Acad. Sci. USA 1965, 54, 934–942; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Holze G., Inhoffen H. H., Angew. Chem. 1985, 97, 887–888; [Google Scholar]
- 4d. Widner F. J., Lawrence A. D., Deery E., Heldt D., Frank S., Gruber K., Wurst K., Warren M. J., Kräutler B., Angew. Chem. Int. Ed. 2016, 55, 11281–11286; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11451–11456; [Google Scholar]
- 4e. Koppenhagen V. B. in B12, Vol. 2 (Ed.: Dolphin D.), Wiley, New York, 1982, pp. 105–150; [Google Scholar]
- 4f. Brenig C., Prieto L., Oetterli R., Zelder F., Angew. Chem. Int. Ed. 2018, 57, 16308–16312; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16546–16550. [Google Scholar]
- 5. Kieninger C., Baker J. A., Podewitz M., Wurst K., Jockusch S., Lawrence A. D., Deery E., Gruber K., Liedl K. R., Warren M. J., Kräutler B., Angew. Chem. Int. Ed. 2019, 58, 14568–14572; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 14710–14714. [Google Scholar]
- 6.
- 6a. Bertele E., Boos H., Dunitz J. D., Elsinger F., Eschenmoser A., Felner I., Gribi H. P., Gschwend H., Meyer E. F., Pesaro M., Scheffold R., Angew. Chem. Int. Ed. Engl. 1964, 3, 490–496; [Google Scholar]; Angew. Chem. 1964, 76, 393–399; [Google Scholar]
- 6b. Eschenmoser A., Pure Appl. Chem. 1969, 20, 1–14; [Google Scholar]
- 6c. Bertele E., Scheffold R., Gschwend H., Pesaro M., Fischli A., Roth M., Schossig J., Eschenmoser A., Helv. Chim. Acta 2015, 98, 1755–1844; [Google Scholar]
- 6d. Montforts F.-P., Osmers M., Leupold D. in Handbook of Porphyrin Science, Vol. 25 (Ed.: Kadish K. M., Smith K. M., Guilard R.), World Scientific, Singapore, 2012, pp. 266–308. [Google Scholar]
- 7. Koppenhagen V. B., Pfiffner J. J., J. Biol. Chem. 1971, 246, 3075–3077. [PubMed] [Google Scholar]
- 8. Grotjahn L., Koppenhagen V. B., Ernst L., Z. Naturforsch. B 1984, 39, 248–251. [Google Scholar]
- 9. Dunitz J. D., E. F. Meyer, Jr. , Helv. Chim. Acta 1971, 54, 77–89. [Google Scholar]
- 10.
- 10a. Lippard S. J., Berg J. M., Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, 1994; [Google Scholar]
- 10b. Kraatz H.-B., Metzler-Nolte N., Concepts and Models in Bioinorganic Chemistry, Wiley-VCH, Weinheim, 2006; [Google Scholar]
- 10c. Cotton F. A., Wilkinson G., Advanced Inorganic Chemistry, Interscience Publishers, New York, 1972. [Google Scholar]
- 11.
- 11a. Zelder F., Alberto R. in Handbook of Porphyrin Science, Vol. 25 (Ed.: Kadish K. M., Smith K. M., Guilard R.), World Scientific, Singapore, 2012, pp. 84–132; [Google Scholar]
- 11b. Zelder F., Chem. Commun. 2015, 51, 14004–14017; [DOI] [PubMed] [Google Scholar]
- 11c. Kräutler B., Chem. Eur. J. 2015, 21, 11280–11287; [DOI] [PubMed] [Google Scholar]
- 11d. Ruetz M., Gherasim C., Fedosov S. N., Gruber K., Banerjee R., Kräutler B., Angew. Chem. Int. Ed. 2013, 52, 2606–2610; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2668–2672; [Google Scholar]
- 11e. Mutti E., Ruetz M., Birn H., Kräutler B., Nexo E., Plos One 2013, 8, e75312; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11f. Ruetz M., Shanmuganathan A., Gherasim C., Karasik A., Salchner R., Kieninger C., Wurst K., Banerjee R., Koutmos M., Kräutler B., Angew. Chem. Int. Ed. 2017, 56, 7387–7392; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7493–7498. [Google Scholar]
- 12.F. Kreppelt, ETH Zürich, 10.3929/ethz-a-000626280 (Zürich), 1991. [DOI]
- 13. Widner F. J., Gstrein F., Kräutler B., Helv. Chim. Acta 2017, 100, e1700170. [Google Scholar]
- 14.
- 14a. Summers M. F., Marzilli L. G., Bax A., J. Am. Chem. Soc. 1986, 108, 4285–4294; [Google Scholar]
- 14b. Bax A., Marzilli L. G., Summers M. F., J. Am. Chem. Soc. 1987, 109, 566–574; [Google Scholar]
- 14c. Rossi M., Glusker J. P., Randaccio L., Summers M. F., Toscano P. J., Marzilli L. G., J. Am. Chem. Soc. 1985, 107, 1729–1738; [Google Scholar]
- 14d. Tollinger M., Konrat R., Kräutler B., Helv. Chim. Acta 1999, 82, 1596–1609. [Google Scholar]
- 15. Lexa D., Savéant J. M., Acc. Chem. Res. 1983, 16, 235–243. [Google Scholar]
- 16.
- 16a. Kräutler B., Puffer B. in Handbook of Porphyrin Science, Vol. 25 (Ed.: Kadish K. M., Smith K. M., Guilard R.), World Scientific, Singapore, 2012, pp. 133–265; [Google Scholar]
- 16b. Kumar M., Kozlowski P. M., Coord. Chem. Rev. 2017, 333, 71–81. [Google Scholar]
- 17. Kräutler B., Keller W., Hughes M., Caderas C., Kratky C., J. Chem. Soc. Chem. Commun. 1987, 1678–1680. [Google Scholar]
- 18. Kratky C., Waditschatka R., Angst C., Johansen J. E., Plaquevent J. C., Schreiber J., Eschenmoser A., Helv. Chim. Acta 1985, 68, 1312–1337. [Google Scholar]
- 19. Kräutler B., Keller W., Kratky C., J. Am. Chem. Soc. 1989, 111, 8936–8938. [Google Scholar]
- 20. Ouyang L., Rulis P., Ching W. Y., Nardin G., Randaccio L., Inorg. Chem. 2004, 43, 1235–1241. [DOI] [PubMed] [Google Scholar]
- 21. St. Maurice M. S., Mera P., Park K., Brunold T. C., Escalante-Semerena J. C., Rayment I., Biochemistry 2008, 47, 5755–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Pett V. B., Liebman M. N., Murray-Rust P., Prasad K., Glusker J. P., J. Am. Chem. Soc. 1987, 109, 3207–3215; [Google Scholar]
- 22b. Kratky C., Färber G., Gruber K., Wilson K., Dauter Z., Nolting H. F., Konrat R., Kräutler B., J. Am. Chem. Soc. 1995, 117, 4654–4670; [Google Scholar]
- 22c. Kratky C., Kräutler B. in Chemistry and Biochemistry of B12 (Ed.: Banerjee R.), Wiley, New York, 1999, pp. 9–41. [Google Scholar]
- 23.
- 23a. Costa F. G., Escalante-Semerena J. C., Biochemistry 2018, 57, 5076–5087; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Stich T. A., Buan N. R., Escalante-Semerena J. C., Brunold T. C., J. Am. Chem. Soc. 2005, 127, 8710–8719; [DOI] [PubMed] [Google Scholar]
- 23c. Park K., Mera P. E., Moore T. C., Escalante-Semerena J. C., Angew. Chem. Int. Ed. 2015, 54, 7158–7161; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7264–7267. [Google Scholar]
- 24.
- 24a. Stich T. A., Yamanishi M., Banerjee R., Brunold T. C., J. Am. Chem. Soc. 2005, 127, 7660–7661; [DOI] [PubMed] [Google Scholar]
- 24b. Gherasim C., Lofgren M., Banerjee R., J. Biol. Chem. 2013, 288, 13186–13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lewis N. J., Pfaltz A., Eschenmoser A., Angew. Chem. Int. Ed. Engl. 1983, 22, 735–736; [Google Scholar]; Angew. Chem. 1983, 95, 743–744. [Google Scholar]
- 26.
- 26a. Pfaltz A., Jaun B., Fassler A., Eschenmoser A., Jaenchen R., Gilles H. H., Diekert G., Thauer R. K., Helv. Chim. Acta 1982, 65, 828–865; [Google Scholar]
- 26b. Kratky C., Fässler A., Pfaltz A., Kräutler B., Jaun B., Eschenmoser A., J. Chem. Soc. Chem. Commun. 1984, 1368–1371. [Google Scholar]
- 27. Jaun B., Thauer R. K., Metal Ions in Life Sciences, Vol. 6 (Eds.: Sigel A., Sigel H., Sigel R. K. O.), deGruyter, Berlin, 2009, pp. 115–132. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Matthews R. G., Acc. Chem. Res. 2001, 34, 681–689; [DOI] [PubMed] [Google Scholar]
- 28b. Gruber K., Puffer B., Kräutler B., Chem. Soc. Rev. 2011, 40, 4346–4363; [DOI] [PubMed] [Google Scholar]
- 28c. Brunold T. C., Conrad K. S., Liptak M. D., Park K., Coord. Chem. Rev. 2009, 253, 779–794. [Google Scholar]
- 29.
- 29a. Cordero B., Gomez V., Platero-Prats A. E., Reves M., Echeverria J., Cremades E., Barragan F., Alvarez S., Dalton Trans. 2008, 2832–2838; [DOI] [PubMed] [Google Scholar]
- 29b. Shannon R. D., Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar]
- 30. Kräutler B. in Advances in Bioorganometallic Chemistry (Eds.: Hirao T., Moriuchi T.), Elsevier, Amsterdam, 2019, pp. 399–430. [Google Scholar]
- 31. Banerjee R., Gherasim C., Padovani D., Curr. Opin. Chem. Biol. 2009, 13, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.
- 32a. Bommer M., Kunze C., Fesseler J., Schubert T., Diekert G., Dobbek H., Science 2014, 346, 455–458; [DOI] [PubMed] [Google Scholar]
- 32b. Payne K. A. P., Quezada C. P., Fisher K., Dunstan M. S., Collins F. A., Sjuts H., Levy C., Hay S., Rigby S. E. J., Leys D., Nature 2015, 517, 513–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pallares I. G., Moore T. C., Escalante-Semerena J. C., Brunold T. C., Biochemistry 2014, 53, 7969–7982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.
- 34a. Banerjee R., Chem. Rev. 2003, 103, 2083–2094; [DOI] [PubMed] [Google Scholar]
- 34b. Toraya T., Chem. Rev. 2003, 103, 2095–2127; [DOI] [PubMed] [Google Scholar]
- 34c. Frey P. A., Hegeman A. D., Reed G. H., Chem. Rev. 2006, 106, 3302–3316; [DOI] [PubMed] [Google Scholar]
- 34d. Buckel W., Golding B. T., Annu. Rev. Microbiol. 2006, 60, 27–49; [DOI] [PubMed] [Google Scholar]
- 34e. Marsh E. N. G., Drennan C. L., Curr. Opin. Chem. Biol. 2001, 5, 499–505. [DOI] [PubMed] [Google Scholar]
- 35.
- 35a. Bridwell-Rabb J., Zhong A., Sun H. G., Drennan C. L., Liu H.-w., Nature 2017, 544, 322–326; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35b. Fujimori D. G., Curr. Opin. Chem. Biol. 2013, 17, 597–604; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35c. Zhang Q., van der Donk W., Liu W., Acc. Chem. Res. 2012, 45, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. Nahvi A., Barrick J. E., Breaker R. R., Nucleic Acids Res. 2004, 32, 143–150; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36b. Schaffner A., Li X. T., Gomez-Llorente Y., Leandrou E., Memou A., Clemente N., Yao C., Afsari F., Zhi L. T., Pan N. N., Morohashi K., Hua X. L., Zhou M. M., Wang C., Zhang H., Chen S. G., Elliott C. J., Rideout H., Ubarretxena-Belandia I., Yue Z. Y., Cell Res. 2019, 29, 313–329; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36c. Padmanabhan S., Perez-Castano R., Elias-Arnanz M., Curr. Opin. Struct. Biol. 2019, 57, 47–55. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Clardy S. M., Allis D. G., Fairchild T. J., Doyle R. P., Expert Opin. Drug Delivery 2011, 8, 127–140; [DOI] [PubMed] [Google Scholar]
- 37b. Hunger M., Mutti E., Rieder A., Enders B., Nexo E., Kräutler B., Chem. Eur. J. 2014, 20, 13103–13107; [DOI] [PubMed] [Google Scholar]
- 37c. Shell T. A., Lawrence D. S., Acc. Chem. Res. 2015, 48, 2866–2874; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37d. Lawrence A. D., Nemoto-Smith E., Deery E., Baker J. A., Schroeder S., Brown D. G., Tullet J. M. A., Howard M. J., Brown I. R., Smith A. G., Boshoff H. I., Barry C. E., Warren M. J., Cell Chem. Biol. 2018, 25, 941–951; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37e. Kainrath S., Stadler M., Reichhart E., Distel M., Janovjak H., Angew. Chem. Int. Ed. 2017, 56, 4608–4611; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4679–4682; [Google Scholar]
- 37f. Giedyk M., Jackowska A., Równicki M., Kolanowska M., Trylska J., Gryko D., Chem. Commun. 2019, 55, 763–766; [DOI] [PubMed] [Google Scholar]
- 37g. Braselmann E., Wierzba A. J., Polaski J. T., Chromiński M., Holmes Z. E., Hung S.-T., Batan D., Wheeler J. R., Parker R., Jimenez R., Gryko D., Batey R. T., Palmer A. E., Nat. Chem. Biol. 2018, 14, 964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary