

Abstract
Inspired by the dynamic morphology control of molecular assemblies in biological systems, we have developed pH‐responsive transformable peptide‐based nanoparticles for photodynamic therapy (PDT) with prolonged tumor retention times. The self‐assembled peptide–porphyrin nanoparticles transformed into nanofibers when exposed to the acidic tumor microenvironment, which was mainly driven by enhanced intermolecular hydrogen bond formation between the protonated molecules. The nanoparticle transformation into fibrils improved their singlet oxygen generation ability and enabled high accumulation and long‐term retention at tumor sites. Strong fluorescent signals of these nanomaterials were detected in tumor tissue up to 7 days after administration. Moreover, the peptide assemblies exhibited excellent anti‐tumor efficacy via PDT in vivo. This in situ fibrillar transformation strategy could be utilized to design effective stimuli‐responsive biomaterials for long‐term imaging and therapy.
Keywords: fibrillar transformation, peptides, photodynamic therapy, photosensitizers, self-assembly
Fibrillar‐transformable peptide‐porphyrin (PWG) nanoparticles activated by the acidic inter‐ and intracellular tumor microenvironment were developed for photodynamic therapy (PDT). The transformation of nanoparticles into nanofibers improved their singlet oxygen generation ability and enabled high accumulation and long‐term retention at tumor sites, resulting in excellent anti‐tumor efficacy via PDT in vivo.
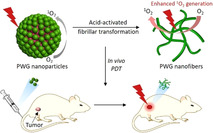
Introduction
Phototherapy is regarded as a powerful method for cancer treatment due to its safety and selectivity. [1] Photodynamic therapy (PDT) requires the combination of three components, namely a photosensitizer, light, and oxygen, which separately are intrinsically nontoxic, but together enable the generation of reactive oxygen species or more particularly singlet oxygen (1O2), leading to cell apoptosis and necrosis in targeted tissue. [2] Dual selectivity is a distinctive hallmark of PDT, which results from the selective uptake of the photosensitizer by tumor tissues and the ability to restrict photosensitizer light activation to a specific area, sparing healthy tissue and leading to irreversible cytotoxicity of the tumor tissue only. [3] The selection of the type of photosensitizer is a critical part for effective PDT. The porphyrin family includes several excellent photosensitizers because of their high quantum yield in 1O2 generation, and their chemical diversity, allowing to absorb light throughout the whole visible electromagnetic spectrum. [2c] Some porphyrin formulations, including Photofrin and Visudyne, have already proceeded to clinical trials and have been approved for the treatment of various cancers. [4] However, the application of porphyrins in PDT faces some challenges which mainly result from the hydrophobicity of many porphyrins, leading to their tendency to aggregate in aqueous solution. This does not only induce a decrease of their photodynamic yield due to self‐quenching, but also causes inadequate pharmacokinetics and insufficient selectivity towards malignant tissues. [5] Nanotechnology brings inspiration to overcome photosensitizer solubility issues; employing nanoparticles allows selective delivery of photosensitizers into tumors presumably via the enhanced permeation and retention (EPR) effect. [6] Several non‐covalent formulations of photosensitizers into organic nanostructures, such as liposomes, micelles, hydrogels, polymeric nanoparticles and solid lipid nanoparticles have been reported. [7] However, the high local concentration of the individual photosensitizers in some of these nanomaterials still induces self‐quenching, along with a reduced PDT activity. [8] Thus, it is of great interest to design nanoparticles of which the photosensitizing capacity can be activated by specific cancer‐associated events to permit a high therapeutic efficacy in PDT. [9]
Peptide self‐assembled nanomaterials have been widely applied in PDT based on their remarkable biocompatibility, biodegradability, structural and functional versatility. [10] Several peptides possess specific properties, such as their response to pH, redox reactions and enzymes, making it possible to design stimuli‐responsive nanomaterials displaying high anti‐tumor efficiency. [11] Peptides have been co‐assembled with photosensitizers through multiple weak intermolecular interactions to construct self‐assembled nanostructures.[ 2b , 12 ] In addition, peptide‐photosensitizer conjugates have been synthesized to create multifunctional nanostructures for PDT. [13] Once released in the tumor site, low molecular weight photosensitizers tend to quickly diffuse away and get cleared from tumor tissues, causing limited tumor accumulation and short retention times, which strongly limit the application of PDT in clinical trials for cancer therapy. [14] Thus, it is urgent to develop nanomaterials to overcome this issue. Wang, Lam and co‐workers developed several stimuli‐responsive in situ self‐assembled and transformable peptide‐based nanoparticles which underwent a transformation to fibrillar structures in the tumor microenvironment; these structures performed excellently in tumor imaging and therapy in vivo by blocking for example HER2 dimerization.[ 7b , 15 ] Compared to traditional nanoparticles, these fibril‐transformable nanoparticles possessed prolonged retention time in tumors and thus caused an enhanced photoacoustic imaging signal, selective cytotoxicity against cancer cells and remarkable tumor suppression efficacy in vivo. [15] However, until now, transmorphic peptide nanoparticles have been rarely utilized to improve the tumor retention time of photosensitizers for PDT. A specific feature of most tumor microenvironments is that they are more acidic (pH around 6.5) than blood and extracellular fluid in healthy tissue. [16] In addition, the acidity is increased further in subcellular compartments of cancer cells, such as the lysosomes (pH 4.5–5.0). Based on the endogenous acidity differences, pH‐responsive nanostructures are a useful design option for PDT.
Herein, we present, to the best of our knowledge, for the first time an acid‐activatable peptide‐based transmorphic nanosystem for PDT. A peptide‐porphyrin conjugate was designed by coupling the pH‐responsive dipeptide tryptophan‐glycine (WG) to a hydrophobic porphyrin (P) core via amidation. The dipeptide was composed of tryptophan to provide delocalized π electrons for fluorescence, and glycine to supply carboxyl groups serving as the acid‐sensitive unit. [17] This synthesized peptide‐porphyrin conjugate (PWG) self‐assembled into nanoparticles under physiological conditions (Scheme 1). When the nanoparticles reached the tumor tissue, the protonation of PWG owing to the increased acidity facilitated the formation of intermolecular hydrogen bonds, inducing the transformation of nanoparticles into nanofibers. The mechanism of this fibrillar transformation was studied at the molecular level via all‐atom molecular dynamic simulations. Based on this transformable nanosystem, we achieved an excellent tumor imaging effect and high photodynamic therapeutic efficacy. Furthermore, the peptide–porphyrin nanoparticles displayed a remarkably long‐term fluorescence after intravenous injection. This peptide‐porphyrin platform can not only be explored for the development of highly efficient treatment modalities against cancer, but also offers a basis to attain a better fundamental understanding of the effect of morphology of nanomaterials on their biological activity.
Scheme 1.
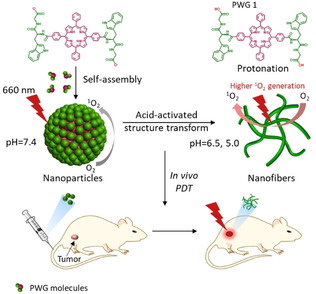
A schematic illustration of self‐assembly and fibrillar transformation of the acid‐activated peptide‐porphyrin (PWG) nanoparticles and their application in PDT. Protonation of PWG (PWG 1) in the acidic microenvironment of tumor tissue and lysosomes induces the transformation of the nanoparticles into aggregated nanofibers, which has a beneficial effect on singlet oxygen generation.
Results and Discussion
The peptide‐porphyrin conjugate was synthesized through solid phase peptide synthesis. In Figure S1, S2, S3 and S4, the MALDI‐TOF MS, LC‐MS, 1H NMR and 13C NMR results indicated high‐purity conjugates were obtained after purification by HPLC. Dynamic light scattering (DLS) showed that the PWG molecules self‐assembled into PWG nanoparticles with an average diameter of approximately 160 nm (Figure 1 a) and a low polydispersity index (PDI) of 0.173 in aqueous solution. Nanoparticles in the sub‐200 nm size range have great potential in preferentially accumulating in tumor tissue based on the EPR effect. [18] Compared to molecularly dissolved PWG molecules in ethanol, a bathochromic shift and a broadening of the Soret and Q bands appeared in the absorption spectra of the nanoparticles, as a result of the self‐assembly process (Figure S5). In Figure 1 b, the DLS results showed no obvious change in size and PDI of these nanoparticles after incubating them in phosphate buffer solution at pH 7.4 for 32 h, confirming these assembled PWG nanoparticles were stable at neutral pH. Moreover, the stability of the PWG nanoparticles was investigated in Dulbecco's modified eagle medium (DMEM) containing 10 % FBS (pH 7.4). No significant changes were observed in the size and PDI for 32 h (Figure S6), which showed the high stability of PWG nanoparticles under normal physiological conditions.
Figure 1.
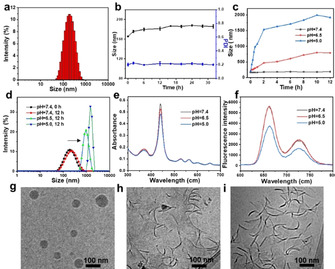
Self‐assembly and fibrillar transformation of the acid‐activatable PWG nanoparticles. a) DLS size of self‐assembled PWG nanoparticles in water. b) Size and PDI of nanoparticles in phosphate buffer solution at pH 7.4 as a function of time measured by DLS. c) Size of PWG nanoparticles in phosphate buffer solution at pH 7.4, 6.5 and 5.0 as a function of time, as measured by DLS. d) Size of the nanoparticles in phosphate buffer solution at pH 7.4, 6.5 and 5.0 measured by DLS after 12 h incubation. e) Changes in UV/Vis absorption and f) fluorescence of PWG nanostructures in phosphate buffer solution at pH 7.4, 6.5 and 5.0. g–i) Cryo‐TEM images of PWG nanostructures in phosphate buffer solution at pH 7.4, 6.5, and 5.0 respectively, after 12 h incubation.
The pH‐responsive behavior of the PWG nanoparticles was tested by exposing them to phosphate buffer solutions at lower pH (pH 6.5 and 5.0). As shown in Figure 1 c and Figure S7, the size and PDI of the nanoparticles increased over time at pH 6.5 and reached 788 nm and 0.263 after 12 h incubation. With a further decrease of pH, the size and PDI of these nanoparticles increased even further, up to 2 μm and 0.63 at pH 5.0 after 12 h incubation (Figure 1 d and Figure S7). The increase in acidity of the phosphate buffer solution led to a gradual decrease in absorption at 440 nm, reflecting possible changes in PWG self‐assembly (Figure 1 e). Concomitantly, the fluorescence intensity of the nanostructures was similar at pH 7.4 and pH 6.5 but was found to decrease dramatically at pH 5.0 (Figure 1 f). This fluorescence quenching was probably caused by the pH‐induced aggregation at pH 5.0. [19] Cryo‐TEM images depicted in Figure 1 g showed that PWG nanoparticles were solid spheres with an average diameter of 100 nm at neutral pH, slightly smaller than their hydrodynamic size measured by DLS (Figure 1 a). After adding them to phosphate buffer solution of pH 6.5, most of the nanoparticles transformed into aggregated PWG nanofibers with a length around 100 nm (Figure 1 h). At lower pH (pH 5.0), nearly all nanoparticles transformed into fibrous structures with more severe aggregation (Figure 1 i). These results indicated that the peptide nanoparticles were pH‐responsive and that acidic conditions (pH 6.5 and 5.0) induced their transformation from solid spheres to fibrillar structures.
The influence of acidity on the photoactivity of the PWG nanostructures was evaluated by investigating their singlet oxygen generation (1O2) capacity using 9,10‐anthracenediylbis‐ (methylene)dimalonic acid (ABDA) as indicator at various pH values. As shown in Figure 2 a, the bleaching of ABDA by PWG nanostructures was 16 % at pH 7.4, increased to 34 % at pH 6.5, and increased further to 51 % at pH 5.0 upon 15 min illumination with a 660 nm laser (Figure 2 a and b). The 1O2 generating capacity of PWG nanofibers at pH 6.5 was twofold and at pH 5.0 even threefold higher than that of the PWG nanoparticles at pH 7.4, indicating that the fibrillar transformation at acidic conditions enhances the production of 1O2 of PWG nanostructures. This can be explained by enhanced intersystem crossing through fibrillar transformation. [20] The photosensitizing process to generate 1O2, is triggered when the photosensitizer absorbs a photon, followed by the promotion of an electron from the ground state to an electronically excited singlet state. [21] This singlet excited state decays back to an excited triplet state via a variety of competitive relaxation processes, such as emission of a photon via fluorescence, nonradiative relaxation (heat generation), and intersystem crossing, leading to energy transfer from the photosensitizer triplet directly to molecular oxygen resulting in the generation of singlet oxygen (1O2). Thus, enhancing the intersystem crossing process will facilitate the production of 1O2. [21] We compared the fluorescence and heat generation of nanoparticles and nanofibers at different pH. As shown in Figure 1 c, the fluorescence of aggregated PWG nanofibers at pH 5.0 was lower than that of PWG nanostructures at pH 7.4 and 6.5. Meanwhile, the heat generation of PWG nanostructures decreased as the pH decreased from 7.4 to 5.0 (Figure S8). As a result, the intersystem crossing mechanism was a more preferred pathway after this fibrillar transformation, which as a consequence led to the improvement of the 1O2 generation.
Figure 2.
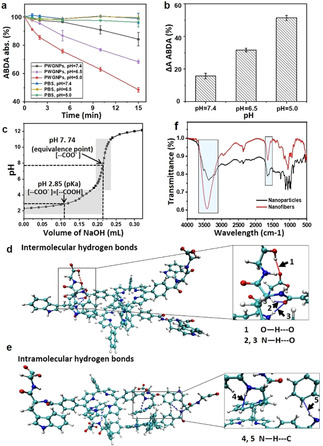
Characteristics of fibrillar‐transformable PWG nanoparticles at different pH. a) Rate of oxidation of ABDA sensitized by irradiating PWG nanostructures at 660 nm in phosphate buffer solutions at pH 7.4, 6.5 and 5.0, measured by the decrease in the absorption of ABDA at 378 nm as a function of time. The pure phosphate buffer solutions at pH 7.4, 6.5 and 5.0 were used as control groups. All samples were processed under irradiation (660 nm, 0.2 W cm−2) for 15 min. b) The rate of decay of ABDA absorbance at pH 7.4, 6.5 and 5.0 after 15 min irradiation in presence of PWG nanostructures. c) The pH titration curve of PWG. d) Typical conformation of protonated PWG 1 dimers obtained with AAMD simulations showing the dominant intermolecular hydrogen bonding interactions. The arrows represent intermolecular hydrogen bonds. Arrow 1=O−H⋅⋅⋅O, arrows 2 and 3=N−H⋅⋅⋅O. e) Typical conformation of deprotonated PWG dimers obtained with AAMD simulations displaying the predominant intramolecular hydrogen bonding interactions. The arrows represent intramolecular hydrogen bonds. Arrows 4 and 5=N−H⋅⋅⋅C. f) FTIR spectra of PWG nanoparticles at pH 7.4 and nanofibers at pH 5.0. The rectangle represents the peak area indicative for hydrogen bonding.
The ability of the peptide‐porphyrin conjugate to arrange itself in different assemblies is a result of an intricate interplay between van der Waals interactions, hydrophobic effects, electrostatic interactions, π‐π stacking interactions, as well as hydrogen‐bonding, which are affected at different pH values. [22] Due to the presence of the carboxyl and amino groups in PWG, the self‐assembly was pH sensitive. [23] To explore the mechanism of acid‐activated structural transformation of these assemblies, the noncovalent bond interactions of the peptide‐porphyrin conjugates at different pH were investigated. In Figure 2 c, the equivalence point and pK a value of the carboxyl groups of PWG were determined to be 7.74 and 2.85 respectively via acid‐base titration in aqueous solution. Based on the near‐neutral equivalence point (7.74), it could be assumed that almost all carboxyl groups of PWG exist in deprotonated form (‐COO−) at pH 7.4. Meanwhile, lower pH resulted in increasing protonation of the carboxyl groups (‐COOH) of PWG. [24]
All‐atom molecular dynamics (AAMD) simulations were performed for the self‐assembly of the protonated PWG (PWG 1) and deprotonated PWG dimers. Figure 2 d showed that the intermolecular hydrogen bonds between the adjacent carboxyl groups (O−H⋅⋅⋅O), as well as between the NH and carbonyl groups (N−H⋅⋅⋅O) were formed in the protonated PWG 1 dimer. However, in Figure 2 e, intramolecular hydrogen bonds between the ‐NH and the carbon atom of the phenyl ring (N−H⋅⋅⋅C) were readily formed in the deprotonated PWG dimer. The weak intramolecular hydrogen bonding as well as a higher level of repulsion between the deprotonated PWG, favored the formation of nanoparticles at pH 7.4. [25] As the pH decreased, the carboxyl groups of PWG became progressively protonated and stronger intermolecular hydrogen bonding occurred, becoming the dominant force to stabilize protonated PWG 1 dimers, thereby facilitating the formation of PWG nanofibers with long‐range order.[ 24 , 26 ] Thus, the changes of type and strength of hydrogen bonds at lower pH induced the fibrillar transformation of PWG nanoparticles to nanofibers. The differences in hydrogen bonds between PWG nanoparticles and nanofibers were confirmed by Fourier transform infrared spectroscopy (FTIR) (Figure 2 f). Compared to the sharp signals for “free” ‐OH groups in the spectrum of ‐COOH at 3470 cm−1 and “free” ‐NH groups at 3447 cm−1, a broad peak at 3418 cm−1, showing a red shift, was observed in the self‐assembled nanofibers, which was indicative of intermolecular hydrogen bonding (O−H⋅⋅⋅O) and (N−H⋅⋅⋅O) occurring in the self‐assembled nanofibers due to the interactions between ‐COOH groups, as well as between the ‐COOH and ‐NH.[ 23 , 27 ] Meanwhile, a broad vibrational band appeared at 3373 cm−1 in the self‐assembled nanoparticles, which, when compared to “free” ‐NH, corresponded to the intramolecular hydrogen‐bonded NH stretching vibration between the ‐NH and the carbon atom of the phenyl ring (N−H⋅⋅⋅C) which was much weaker than those with the ‐COOH group. [27b] Moreover, the peak at 1661 cm−1 of ‐C=O in nanoparticles showed a red shift of about 20 nm to 1640 cm−1 in the nanofibers, also indicating that hydrogen bonding interactions of ‐COOH groups occurred in the nanofibers. [23] These results provide a deeper understanding of the interaction mechanism between peptide‐porphyrin conjugates at the molecular level, which could be significant in guiding the design of smart peptide‐based nanomaterials and improving their application prospects in photodynamic therapy.
Next, cellular uptake and intracellular localization of the PWG nanoparticles were investigated by confocal laser scanning microscopy (CLSM) after incubating breast cancer cells (MCF‐7 cells) with the nanoparticles at 37 °C for 12 h. LysoTracker Green DND‐26 was used to mark acidic lysosomes to determine the cellular distribution. In Figure 3 a and b, the CLSM images and their magnified images of MCF‐7 cells showed that red fluorescence from PWG colocalized well with the green fluorescence from LysoTracker Green DND‐26, indicating that most of the PWG nanostructures were internalized into lysosomes (pH 5.0) in MCF‐7 cells. Intracellular photoactivity of the PWG nanoparticles was examined by CLSM using 2,7‐ dichlorofluorescein diacetate (DCFH‐DA) for the detection of 1O2.
Figure 3.
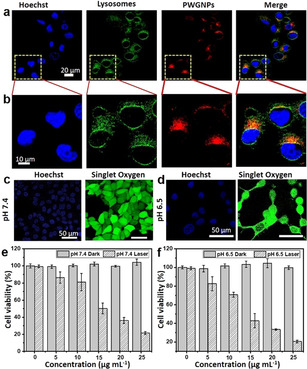
In vitro PDT evaluation of fibrillar‐transformable PWG nanoparticles on MCF‐7 cells. a) Colocalization of PWG nanostructures in MCF‐7 cancer cells. CLSM images indicating the nuclei (stained with Hoechst), lysosomes (LysoTracker Green DND‐26), and PWG nanostructures (red), including the overlay. b) Magnified CLSM images of (a). The scale bar is the same for all images. c) CLSM images of intracellular 1O2 generation in MCF‐7 cells treated with PWG nanoparticles at pH 7.4. d) CLSM images of intracellular 1O2 generation in MCF‐7 cells treated with PWG nanoparticles at pH 6.5. e) Dark cytotoxicity and phototoxicity of PWG nanoparticles in MCF‐7 cells at pH 7.4 determined by MTT assay. f) Dark cytotoxicity and phototoxicity of PWG nanoparticles in MCF‐7 cells at pH 6.5 determined by MTT assay.
The intracellular 1O2 generation in MCF‐7 cells treated with PWG nanoparticles at pH 7.4 and 6.5 was investigated by CLSM. In Figure 3 c, bright green fluorescence was observed in MCF‐7 cells treated with PWG nanoparticles at pH 7.4 after irradiation by a 660 nm laser (0.12 W cm−2) for 10 min, showing notable 1O2 generation. Compared with the experiment at pH 7.4, stronger green fluorescence was observed in MCF‐7 cells treated with PWG nanoparticles at pH 6.5 (Figure 3 d), indicating enhanced 1O2 generation. Meanwhile, hardly any fluorescence was detected from the control group, in which MCF‐7 cells treated with PWG nanoparticles at pH 7.4 and 6.5 were not illuminated (Figure S9). Furthermore, the dark cytotoxicity and phototoxicity of the PWG nanoparticles toward cancer cells treated at pH 7.4 and 6.5 were evaluated by MTT assays. As shown in Figure 3 e and f, no obvious dark cytotoxicity was observed in MCF‐7 cells treated with 25 μg mL−1 PWG nanoparticles at pH 7.4 and 6.5, showing their good biocompatibility. Under irradiation for 10 min by a 660 nm laser (0.12 W cm−2), the viability of MCF‐7 cells decreased with the increase of the PWG nanoparticle concentration. The IC50 value for cancer cells at pH 6.5 (12.5 μg mL−1) was lower than the IC50 value at pH 7.4 (15 μg mL−1), indicating that these self‐assembled PWG nanostructures showed higher phototoxicity for cancer cells in the acidic tumor microenvironment (pH 6.5) for PDT than under normal physiological conditions (pH 7.4).
An in vivo imaging system was used to investigate the biodistribution in tumor‐bearing mice. As shown in Figure 4 a, red fluorescence started to appear at the tumor site at 8 h post injection. Further high accumulation of PWG nanoparticles was observed via the increased fluorescence at the tumor site with time. The tumor targeting effect presumably benefited from the enhanced permeability and retention (EPR) effect of small PWG nanoparticles in tumor tissue. [28] Remarkably, the fluorescence signal in the tumor increased over prolonged periods of time, reached a maximum at 72 h and remained strong up to 168 h (Figure 4 a). The average fluorescence intensity in the tumor site was quantified to investigate the accumulation of the PWG nanostructures at different time points. As shown in Figure 4 b, more than 64 % of the maximum average fluorescence intensity remained in the tumor even 168 h (7 days) after intravenous administration. The ex vivo fluorescence imaging of the tumor and main organs provided additional insight into the tumor targeting efficiency and retention behavior of these PWG nanostructures. As shown in Figure 4 c, similar results were obtained as for the in vivo imaging studies. The fluorescence signal in the tumor was quite strong at 72 h post injection and even could be detected at 360 h post injection (Figure 4 c). These findings confirmed that PWG nanostructures selectively accumulated and possessed superior retention behavior at tumor sites. Meanwhile, the fluorescence signal of PWG nanostructures in the major organs, including liver, heart, spleen, lung and kidney decreased over time and was nearly eliminated after 360 h, ensuring the biosafety of these materials.
Figure 4.
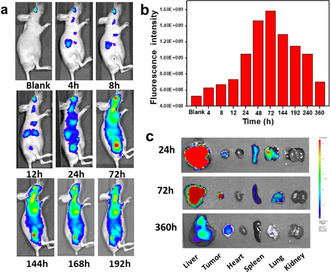
In vivo biodistribution of the fibrillar‐transformable PWG nanoparticles. a) Fluorescence imaging of tumor‐bearing mice at varying time points after intravenous injection of PWG nanoparticles over a period of 192 h. b) Quantified relative fluorescence intensity in the tumor region of (a). c) Ex vivo fluorescence images of main organs of tumor‐bearing mice at 24, 72 and 360 h post injection of PWG nanoparticles.
The above results showed that these PWG nanostructures exhibited high accumulation and ultralong tumor retention, especially when compared to similar peptide‐porphyrin nanoparticles without pH‐activatable features. [8] This phenomenon might mainly result from the enhanced accumulation and retention effect of the transformation to fibrils of the PWG nanoparticles at the tumor site. [7b] Furthermore, the aggregated nanofibers sedimented at the tumor interstitium increased the uptake of nanofibers by cancer cells because of the direct interaction between nanofibers and cells. Additionally, the intracellular uptake of nanofibers further enhanced the retention effect. [22a] The enhanced accumulation and retention effect of this fibrillar transition for nanofibers at tumor sites will not only make the PWG nanostructure become a novel fluorescent probe useful for tumor long‐term imaging, but also ensures its high therapeutic PDT efficacy.
Next, tumor‐bearing mice were used to evaluate the efficacy of PWG nanoparticles for in vivo PDT. Twenty mice were divided into four groups, which were treated with PWG nanoparticles with irradiation (PWGNPs+Laser), 5 % glucose aqueous solution (Control), PWG nanoparticles without irradiation (PWGNPs), and irradiation only (Laser), respectively. At 24 h post‐injection, the mice of the PWGNPs + Laser and Laser groups were irradiated by a 660 nm laser for 20 min (0.3 W cm−2), and the other two groups were treated without laser. As shown in the tumor growth profiles, the PWGNPs + Laser group displayed strong antitumor efficiency (Figure 5 a) and all tumors of the mice were completely eradicated after 21 days treatment (Figure 5 b). In sharp contrast, the tumor volume (Figure 5 c) increased to over 6‐fold compared to the original one in the other three groups (Control, PWGNPs and Laser groups). These results showed that the PWG nanostructures display highly effective PDT against tumors, which might result from the large amount of tumor accumulation of the PWG nanofibers possessing high 1O2 generation capacity. [14] Figure 5 d showed that the body weights of the mice in all groups were not significantly different and showed a continued increase during treatment, indicating the high biocompatibility of this PDT system. In Figure S10, the hematoxylin and eosin (H&E) sections of the organs from the mice of the four groups did not show obvious pathological changes, which confirmed that we developed a biocompatible treatment for tumor ablation. These self‐assembled PWG nanostructures could be a biocompatible and safe material that facilitates both tumor imaging and PDT.
Figure 5.
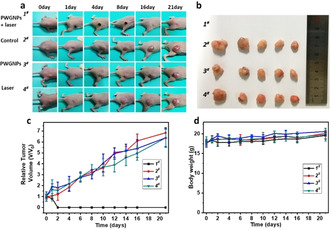
In vivo therapeutic efficacy of PDT. a) Photographs of tumor‐bearing mice at various time points. b) Tumors taken at the end of the antitumor studies (21st day) of the mice in different groups treated with PWGNPs + Laser (1#), Control (2#), PWGNPs (3#), Laser (4#). Mean: s.d., n=5. c) The relative tumor volume growth profiles and d) Body weight change of the mice in the different groups.
Conclusion
In summary, we have developed acid‐activatable transmorphic peptide‐based nanoparticles for PDT. The self‐assembled peptide‐porphyrin nanoparticles transformed into nanofibers in the acidic environment of tumor tissues and lysosomes, exhibiting enhanced singlet oxygen generation. Molecular dynamics simulations were performed to study the mechanism of structure transformation of peptide‐porphyrin nanoparticles, which showed that the fibrillar transformation was mainly driven by the enhanced hydrogen bonding between protonated peptide‐porphyrin dimers at lower pH. These acid‐activatable nanoparticles possessed remarkable phototoxicity against cancer cells in vitro. In vivo fluorescence imaging showed that the peptide‐porphyrin nanostructures displayed remarkable tumor targeting specificity, high accumulation and long‐term retention. Moreover, these PWG nanostructures showed excellent in vivo anti‐tumor activity when being employed for PDT, without off‐target side effects. Our study provides new insight into the design of transformable peptide‐based nanostructures and demonstrates their utility in tumor targeting, long‐term imaging and anti‐tumor therapy.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge financial support from the Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035) and the Eurotech Postdoc Programme, under the Marie Skłodowska‐Curie grant agreement No 754462 as well as the National Natural Science Fund BRICS STI Framework Programme of China (No. 51861145304), Innovation Research Community Science Fund of National Natural Science Foundation of China (No. 21821005), and the Key Research Program of Frontier Sciences of the Chinese Academy of Sciences (CAS, Grant No. QYZDB‐SSW‐JSC034). All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee in compliance with Chinese law for experimental animals with an approval number of IPEAECA2019106.
B. Sun, R. Chang, S. Cao, C. Yuan, L. Zhao, H. Yang, J. Li, X. Yan, J. C. M. van Hest, Angew. Chem. Int. Ed. 2020, 59, 20582.
Contributor Information
Prof. Xuehai Yan, Email: yanxh@ipe.ac.cn.
Prof. Jan C. M. van Hest, Email: J.C.M.v.Hest@tue.nl.
References
- 1.
- 1a. Agostinis P., Berg K., Cengel K. A., Foster T. H., Girotti A. W., Gollnick S. O., Hahn S. M., Hamblin M. R., Juzeniene A., Kessel D., Korbelik M., Moan J., Mroz P., Nowis D., Piette J., Wilson B. C., Golab J., Ca-Cancer J. Clin. 2011, 61, 250–281; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Castano A. P., Mroz P., Hamblin M. R., Nat. Rev. Cancer 2006, 6, 535–545; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Jiang Y., Zhao X., Huang J., Li J., Upputuri P. K., Sun H., Han X., Pramanik M., Miao Y., Duan H., Nat. Commun. 2020, 11, 1857; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Li J. C., Cui D., Jiang Y. Y., Huang J. G., Cheng P. H., Pu K. Y., Adv. Mater. 2019, 31, 1905091. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Qi S., Kwon N., Yim Y., Nguyen V.-N., Yoon J., Chem. Sci. 2020, 10.1039/D0SC01171A; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Liu K., Xing R. R., Zou Q. L., Ma G. H., Mohwald H., Yan X. H., Angew. Chem. Int. Ed. 2016, 55, 3036–3039; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3088–3091; [Google Scholar]
- 2c. Ethirajan M., Chen Y. H., Joshi P., Pandey R. K., Chem. Soc. Rev. 2011, 40, 340–362; [DOI] [PubMed] [Google Scholar]
- 2d. Li J., Cui D., Huang J., He S., Yang Z., Zhang Y., Luo Y., Pu K., Angew. Chem. Int. Ed. 2019, 58, 12680–12687; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 12810–12817. [Google Scholar]
- 3. Cao H. Q., Wang L., Yang Y., Li J., Qi Y. F., Li Y., Li Y., Wang H., Li J. B., Angew. Chem. Int. Ed. 2018, 57, 7759–7763; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7885–7889. [Google Scholar]
- 4.
- 4a. Detty M. R., Gibson S. L., Wagner S. J., J. Med. Chem. 2004, 47, 3897–3915; [DOI] [PubMed] [Google Scholar]
- 4b. Juarranz A., Jaen P., Sanz-Rodriguez F., Cuevas J., Gonzalez S., Clin. Transl. Oncol. 2008, 10, 148–154; [DOI] [PubMed] [Google Scholar]
- 4c. Plaetzer K., Krammer B., Berlanda J., Berr F., Kiesslich T., Lasers Med. Sci. 2009, 24, 259–268. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Jin J. G., Zhu Y. C., Zhang Z. H., Zhang W. A., Angew. Chem. Int. Ed. 2018, 57, 16354–16358; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16592–16596; [Google Scholar]
- 5b. Escudero C., Crusats J., Diez-Perez I., El-Hachemi Z., Ribo J. M., Angew. Chem. Int. Ed. 2006, 45, 8032–8035; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 8200–8203. [Google Scholar]
- 6.
- 6a. Lucky S. S., Soo K. C., Zhang Y., Chem. Rev. 2015, 115, 1990–2042; [DOI] [PubMed] [Google Scholar]
- 6b. Li Y. P., Lin T. Y., Luo Y., Liu Q. Q., Xiao W. W., Guo W. C., Lac D., Zhang H. Y., Feng C. H., Wachsmann-Hogiu S., Walton J. H., Cherry S. R., Rowland D. J., Kukis D., Pan C. X., Lam K. S., Nat. Commun. 2014, 5, 4712; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Kim J., Cho H. R., Jeon H., Kim D., Song C., Lee N., Choi S. H., Hyeon T., J. Am. Chem. Soc. 2017, 139, 10992–10995. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Calixto G. M. F., Bernegossi J., de Freitas L. M., Fontana C. R., Chorilli M., Molecules 2016, 21, 342; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. He P. P., Li X. D., Wang L., Wang H., Acc. Chem. Res. 2019, 52, 367–378; [DOI] [PubMed] [Google Scholar]
- 7c. Pille J., van Lith S. A. M., van Hest J. C. M., Leenders W. P. J., Biomacromolecules 2017, 18, 1302–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zou Q. L., Abbas M., Zhao L. Y., Li S. K., Shen G. Z., Yan X. H., J. Am. Chem. Soc. 2017, 139, 1921–1927. [DOI] [PubMed] [Google Scholar]
- 9. Li S. K., Zou Q. L., Li Y. X., Yuan C. Q., Xing R. R., Yan X. H., J. Am. Chem. Soc. 2018, 140, 10794–10802. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Sun B. B., Tao K., Jia Y., Yan X. H., Zou Q. L., Gazit E., Li J. B., Chem. Soc. Rev. 2019, 48, 4387–4400; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Wang Y., Cheetham A. G., Angacian G., Su H., Xie L. S., Cui H. G., Adv. Drug Delivery Rev. 2017, 110, 112–126; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Laor D., Sade D., Shaham-Niv S., Zaguri D., Gartner M., Basavalingappa V., Raveh A., Pichinuk E., Engei H., Lwasaki K., Yamamoto T., Noothalapati H., Gazit E., Nat. Commun. 2019, 10, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shah A., Malik M. S., Khan G. S., Nosheen E., Iftikhar F. J., Khan F. A., Shukla S. S., Akhter M. S., Kraatz H. B., Aminabhavi T. M., Chem. Eng. J. 2018, 353, 559–583. [Google Scholar]
- 12. Sun B. B., Wang L., Li Q., He P. P., Liu H. L., Wang H., Yang Y., Li J. B., Biomacromolecules 2017, 18, 3506–3513. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Chen J., Stefflova K., Niedre M. J., Wilson B. C., Chance B., Glickson J. D., Zheng G., J. Am. Chem. Soc. 2004, 126, 11450–11451; [DOI] [PubMed] [Google Scholar]
- 13b. Overchuk M., Zheng M., Rajora M. A., Charron D. M., Chen J., Zheng G., ACS Nano 2019, 13, 4560–4571. [DOI] [PubMed] [Google Scholar]
- 14. Zheng N., Zhang Z. Y., Kuang J., Wang C. S., Zheng Y. B., Lu Q., Bai Y. G., Li Y., Wang A. G., Song W. Z., ACS Appl. Mater. Interfaces 2019, 11, 18224–18232. [DOI] [PubMed] [Google Scholar]
- 15. Zhang L., Jing D., Jiang N., Rojalin T., Baehr C. M., Zhang D. L., Xiao W. W., Wu Y., Cong Z. Q., Li J. J., Li Y. P., Wang L., Lam K. S., Nat. Nanotechnol. 2020, 15, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kneipp J., Kneipp H., Wittig B., Kneipp K., J. Phys. Chem. C 2010, 114, 7421–7426. [Google Scholar]
- 17. Fan Z., Chang Y., Cui C. C., Sun L. M., Wang D. H., Pan Z., Zhang M. J., Nat. Commun. 2018, 9, 2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bae K. H., Chung H. J., Park T. G., Mol. Cells 2011, 31, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang Y. Z., Xiao N., Liu S. G., Han L., Li N. B., Luo H. Q., Mater. Sci. Eng. C 2020, 108, 110401. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Jeong H. G., Choi M. S., Isr. J. Chem. 2016, 56, 110–118; [Google Scholar]
- 20b. Mitra S., Foster T. H., Photochem. Photobiol. 2005, 81, 849–859. [DOI] [PubMed] [Google Scholar]
- 21. Zhao L. Y., Liu Y. M., Chang R., Xing R. R., Yan X. H., Adv. Funct. Mater. 2019, 29, 1806877. [Google Scholar]
- 22.
- 22a. Liu X. S., Chen Y. J., Li H., Huang N., Jin Q., Ren K. F., Ji J., ACS Nano 2013, 7, 6244–6257; [DOI] [PubMed] [Google Scholar]
- 22b. Pandeeswar M., Govindaraju T., Mol. Syst. Des. Eng. 2016, 1, 202–207. [Google Scholar]
- 23. Barnaby S. N., Frayne S. H., Smoak E. M., Banerjee I. A., Mater. Sci. Eng. C 2011, 31, 620–628. [Google Scholar]
- 24. Wash P. L., Maverick E., Chiefari J., Lightner D. A., J. Am. Chem. Soc. 1997, 119, 3802–3806. [Google Scholar]
- 25.
- 25a. Liu J. H., Li J. S., Jiang Y., Yang S., Tan W. H., Yang R. H., Chem. Commun. 2011, 47, 11321–11323; [DOI] [PubMed] [Google Scholar]
- 25b. Zhu Z. G., Xu N., Yu Q. P., Guo L., Cao H., Lu X. H., Cai Y. L., Macromol. Rapid Commun. 2015, 36, 1521–1527. [DOI] [PubMed] [Google Scholar]
- 26. Song B., Wei H., Wang Z. Q., Zhang X., Smet M., Dehaen W., Adv. Mater. 2007, 19, 416–420. [Google Scholar]
- 27.
- 27a. Mohamed M. G., Hsiao C. H., Hsu K. C., Lu F. H., Shih H. K., Kuo S. W., RSC Adv. 2015, 5, 12763–12772; [Google Scholar]
- 27b. Wang F. C., Feve M., Lam T. M., Pascault J. P., J. Polym. Sci. Part B 1994, 32, 1315–1320. [Google Scholar]
- 28.
- 28a. Fang J., Nakamura H., Maeda H., Adv. Drug Delivery Rev. 2011, 63, 136–151; [DOI] [PubMed] [Google Scholar]
- 28b. Torchilin V., Adv. Drug Delivery Rev. 2011, 63, 131–135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary