

Abstract
Diels–Alder reactions have become established as one of the most effective ways to prepare stereochemically complex six‐membered rings. Different catalysis concepts have been reported, including dienophile activation by Lewis acids or H‐bond donors and diene activation by bases. Herein we report a new concept, in which an acidic prodiene is acidified by a Lewis acid to facilitate deprotonation by an imidazolium–aryloxide entity within a polyfunctional catalyst. A metal dienolate is thus formed, while an imidazolium–ArOH moiety probably forms hydrogen bonds with the dienophile. The catalyst type, readily prepared in few steps in high overall yield, was applied to 3‐hydroxy‐2‐pyrone and 3‐hydroxy‐2‐pyridone as well as cyclopentenone prodienes. Maleimide, maleic anhydride, and nitroolefin dienophiles were employed. Kinetic, spectroscopic, and control experiments support a cooperative mode of action. High enantioselectivity was observed even with unprecedented TONs of up to 3680.
Keywords: asymmetric catalysis, Diels–Alder cycloaddition, lactams, lactones, polyfunctional catalysts
In an efficient approach to catalytic asymmetric [4+2] cycloaddition, acidic prodienes were deprotonated by a betaine species to form a copper dienolate, and the dienophile was activated by hydrogen‐bond formation (see scheme). The polyfunctional catalyst promoted the transformation with high enantioselectivity as well as high turnover numbers.

Asymmetric Diels–Alder reactions (DAs) provide a very important tool to enantioselectively construct stereochemically complex six‐membered carbocycles. [1] Attractive features of this reaction type include stereospecificity regarding diene and dienophile components, wide applicability of different substrate types and the possibility to stereoselectively construct a number of stereogenic centers within the formed cyclohexene ring, thus creating structural complexity. [1] Various concepts for catalytic asymmetric DAs have been reported [1] including the use of chiral Lewis acids (LA), [2] primary or secondary amines/ammonium salts, [3] Brønsted acids, [4] hydrogen bond donors, [5] and organic Brønsted bases. [4]
With the traditional LA catalysts the dienophile's LUMO energy is decreased (Scheme 1). [2] More rarely, metal centers have also served as tethers to allow for quasi‐intramolecular reaction pathways. [6] In contrast, bases were described to increase the HOMO energy of acidic (pro)dienes.[ 1 , 3 , 4 ]
Scheme 1.
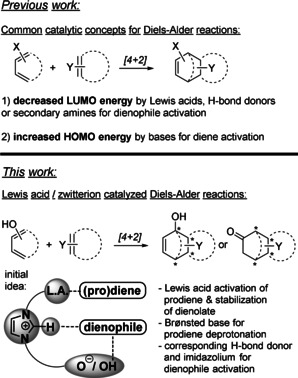
Comparison of previous Diels–Alder approaches with that presented.
In this article, we report the use of polyfunctional catalysts, in which the cooperation of a LA [7] and an aprotic imidazolium aryloxide moiety is utilized to generate the reactive diene. [8] The prodiene is acidified by coordination to the LA and then deprotonated by the aryloxide moiety. [9] The LA should stabilize the generated dienolate. Besides, hydrogen bond activation is crucial in this catalyst system, as control experiments show, most likely for activation of the dienophile.[ 10 , 11 ]
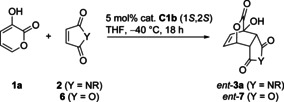
For our initial investigations, 3‐hydroxy‐2‐pyrones 1 were studied as diene component to form densely functionalized bicyclic lactones. Similar lactones have been described as valuable building blocks for the synthesis of a number of structurally complex, biologically active natural products such as taxol, [6b] and others.[ 12 , 13 ] Pyrones are usually quite reluctant to undergo DAs, [14] unless the electron density is increased by formation of a dienolate. [15] Consequently, chiral Brønsted bases—in particular cinchona alkaloid derivatives—have been reported for highly enantioselective activation. [16] Maleimides 2 were initially investigated as dienophiles, because succinimides not only represent an important structural motif in bioactive compounds, [17] but also because the previously reported methods using a combination of 1 and 2 suffered from moderate enantioselectivity and very limited scope.[ 16a , 16b , 16c ]
CuII/imidazolium/naphthoxide complex C1 a (5 mol %) [8] was found to catalyze the reaction of 1 a and 2A at room temperature with complete endo selectivity, yet low enantioselectivity (entry 1). At −40 °C, the ee could be significantly increased to 88 %, while an excellent yield was maintained (entry 2). Lower reaction temperatures gave a decreased enantioselectivity (not shown). Formally replacing the N‐Me substituent by the synthetically useful N‐benzyl protecting group caused a slightly improved enantioselectivity employing the same conditions (entry 3). Product 3 aB has been described as valuable precursor to antagonists of neurotransmitter SP, but was previously only accessible with moderate enantioselectivity. [18] A change in configuration of the iminosulfonamide moiety from (1S,2S) to (1R,2R) resulted in the formation of the opposite product enantiomer (ee=91 %, entry 4). This shows that the configurational outcome is dominated by the iminosulfonamide moiety, rather than the axially chiral part.
It was found that the choice of the sulfonamide unit heavily influences the enantioselectivity. Most residues R1 (Me, p‐tolyl, C6F5, 2‐ and 4‐nitrophenyl, 2‐naphthyl) resulted in poor to moderate enantioselectivity (for details see the SI). Nevertheless, with R1=1‐naphthyl in C1 b, which is accessible in an overall yield of 84 % starting from (R)‐BINAM (see SI), the enantiomeric excess increased to 96 % (entry 5). Adding 1 a by syringe pump further improved the ee to 98 % (entry 9).
To our surprise, with C1 b the optical antipode of 3 aB was formed, although the (1S,2S) iminosulfonamide configuration was used (compare entries 3 & 5). In contrast, the (1R,2R) configuration for C1 b resulted in no switch of the preferred enantiomer, but a significantly decreased ee value (entry 6). We suggest that the coordination of 1 at Cu might result in different coordination geometries depending on the sulfonamide residues. Lower loadings still resulted in good results (entries 7,8).
Different pyrones 1 were examined as prodienes under conditions of Table 1, entry 9, using catalysts C1 a and C1 b (Table 2). Next to 1 a, also the 4‐Me and 4‐halo substituted substrates were effective and only endo products were found (entries 4–6).
Table 1.
Development and optimization of the title reaction.

|
Entry |
C1 (config.) |
X [mol %] |
2 |
T [°C] |
Yield[a] [%] |
dr [b] |
ee [c] [%] |
|---|---|---|---|---|---|---|---|
|
1 |
C1 a (1S,2S) |
5 |
2A |
25 |
99 |
>98:2 |
30 |
|
2 |
C1 a (1S,2S) |
5 |
2A |
−40 |
97 |
>98:2 |
88 |
|
3 |
C1 a (1S,2S) |
5 |
2B |
−40 |
97 |
>98:2 |
90 |
|
4 |
C1 a (1R,2R) |
5 |
2B |
−40 |
95 |
>98:2 |
−91 |
|
5 |
C1 b (1S,2S) |
5 |
2B |
−40 |
98 |
>98:2 |
−96 |
|
6 |
C1 b (1R,2R) |
5 |
2B |
−40 |
96 |
>98:2 |
−40 |
|
7[d,e] |
C1 b (1S,2S) |
2.5 |
2B |
−40 |
98 |
>98:2 |
−96 |
|
8[d,e] |
C1 b (1S,2S) |
1 |
2B |
−40 |
95 |
>98:2 |
−90 |
|
9[e] |
C1 b (1S,2S) |
5 |
2B |
−40 |
98 |
>98:2 |
−98 |
[a] Yield of the isolated product. [b] Endo/exo ratios were determined by 1H NMR spectroscopy of the crude product. [c] The ee value of the endo isomer was determined by 1H NMR spectroscopy using saturated CDCl3 solutions of (R)‐(+)‐binaphthol. [16a] A minus sign indicates that the antipode of the enantiomer depicted was generated in excess. [d] Reaction time: 18 h. [e] Compound 1 a was added with a syringe pump.
Table 2.
Investigation of various 3‐hydroxy‐2‐pyrones and 3‐hydroxy‐2‐pyridones.

|
Entry |
C1 |
R |
1 or 4 |
3B or 5B |
Yield [%][a] |
dr [b] |
ee [%][c] |
|---|---|---|---|---|---|---|---|
|
1 |
C1 a |
H |
1 a |
3 aB |
97 |
>98:2 |
90 |
|
2 |
C1 b |
H |
1 a |
ent‐3 aB |
98 |
>98:2 |
−98 |
|
3[d] |
C1 b |
H |
1 a |
ent‐3 aB |
96 |
>98:2 |
−97 |
|
4 |
C1 a |
Me |
1 b |
3 bB |
93 |
>98:2 |
93 |
|
5 |
C1 a |
Cl |
1 c |
3 cB |
92 |
>98:2 |
95 |
|
6 |
C1 a |
Br |
1 d |
3 dB |
94 |
>98:2 |
93 |
|
7[e] |
C1 b |
H |
4 a |
ent‐5 aB |
97 |
>98:2 |
−94 |
|
8[e] |
C1 b |
allyl |
4 b |
ent‐5 bB |
98 |
>98:2 |
−96 |
|
9[e] |
C1 a |
allyl |
4 b |
5 bB |
95 |
>98:2 |
95 |
|
10[e] |
C1 b |
Cl |
4 c |
ent‐5 cB |
97 |
>98:2 |
−79 |
|
11[e,f] |
C1 b |
H |
4 a |
ent‐5 aC |
94 |
>98:2 |
−94 |
|
12[e,g] |
C1 b |
H |
4 a |
ent‐5 aF |
94 |
>98:2 |
−95 |
[a] Yield of the isolated product. [b] Endo/exo ratios were determined by 1H NMR spectroscopy of the crude product. [c] The ee value was determined by 1H NMR spectroscopy using saturated CDCl3 solutions of (R)‐(+)‐binaphthol. [16a] A minus sign indicates that the antipode of the enantiomer depicted was generated in excess. [d] The reaction was performed on a 5 mmol scale. [e] The reaction was performed at 25 °C. [f] 4‐O2N‐C6H4 was used for Bn in 2. [g] H was used for Bn in 2.
To showcase the practicality of this method, it was performed on a 5 mmol scale and provided excellent results (entry 3). In addition, catalyst recovery of C1 b was successful by silica gel filtration. [8] Employing the recovered catalyst provided ent‐3 aB in diastereopure form in 92 % yield and with 98 % ee for the second run and 92 % yield and with 96 % ee for the third run.
Besides pyrones 1, we also examined pyridones 4 (entries 7–12) which are valuable to prepare bioactive compounds like the antiviral drug Oseltamivir. [19] For catalytic asymmetric DAs of pyridones with 2, there is a single method available providing high enantioselectivity, which is currently limited though to N‐mesitylsulfonyl substituted substrates. [20] In our study 2‐nosyl (Ns) protected substrates 4 worked fine as well (entries 7–12). Again, only endo isomers were found and different enantiomers were available with catalysts C1 a and C1 b. [21] The 2‐Ns protecting group could be readily removed as shown for ent‐5 aB by PhSH/DBU at 25 °C in THF (yield: 94 %, ee 94 %).
Different maleimides 2 were also well tolerated (Table 3). Next to unbranched and branched N‐alkyl (entries 1,2), a Boc group (entry 3) was accepted which was removable from 3 aE in 92 % yield (ee=92 %) using TFA at 25 °C. Moreover, product formation was also achieved with unprotected maleimide (entry 4 and Table 2, entry 12). In addition, aromatic substituents with various electronic properties were tolerated (entry 5 and SI). The reaction could also be extended to maleic anhydride 6 providing product 7. [16j] All products were formed in high yields in diastereopure form with high enantioselectivity. [21]
Table 3.
Investigation of maleimide dienophiles 2 and maleic anhydride 6.
|
Entry |
2 or 6 |
Y |
(ent‐)3 a or 7 |
Yield [%][a] |
dr [b] |
ee [%][c] |
|---|---|---|---|---|---|---|
|
1 |
2A |
Me−N |
ent‐3 aA |
93 |
>98:2 |
93 |
|
2 |
2D |
cHex−N |
ent‐3 aD |
92 |
>98:2 |
96 |
|
3[d] |
2E |
Boc−N |
3 aE |
90 |
>98:2 |
−92 |
|
4[e] |
2F |
H−N |
ent‐ 3 aF |
89 |
>98:2 |
90 |
|
5 |
2G |
Ph−N |
ent‐3 aG |
94 |
>98:2 |
91 |
|
6[d,f] |
6 |
O |
7 |
92 |
>98:2 |
−84 |
[a] Yield of the isolated product. [b] Endo/exo ratios were determined by 1H NMR spectroscopy of the crude product. [c] The ee value was determined by 1H NMR spectroscopy using saturated CDCl3 solutions of (R)‐(+)‐binaphthol. [16a] A minus sign indicates that the antipode of the enantiomer depicted was generated in excess. [d] C1 a (1S,2S) was used as the catalyst. [e] The reaction was performed at −20 °C. [f] The reaction was performed at 0 °C.
Other types of C−H acidic prodienes can be applied as well as demonstrated for enone 8 (Scheme 2). With 2B catalyst C1 a produced bicycloheptane 9 exclusively as exo diastereomer featuring four contiguous stereocenters (98 % ee). [22] Surprisingly, this catalytic asymmetric DA is unknown, although compounds closely related to 9 have been reported as modulators of nuclear hormone receptor function. [23] The unexpected exo selectivity might possibly point to a stepwise process in that case.
Scheme 2.
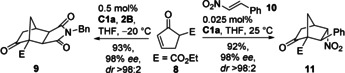
Extension to different substrate types (C1 a with 1S,2S configuration).
In addition, with nitroolefin 10 the cycloaddition proceeded very efficiently (TON 3680, 92 % yield) with just 0.025 mol % C1 a. The previously not available endo product was obtained with an ee of 98 %. Prior to this study, only the exo isomer of 11 was accessible with moderate dr by chiral amine catalysis with TONs <20.[ 24 , 25 ]
To learn more about the role of the aprotic betaine, experiments with several control catalysts were conducted (Table 4). Cs2CO3 (2.5 mol %) in the absence of a Cu complex formed racemic product 3 aB in 95 % yield as an endo/exo mixture (84:16).
Table 4.
Experiments with control catalyst systems.

|
|

[a] Yield of the isolated product. [b] Endo/exo ratios were determined by 1H NMR spectroscopy of the crude product. [c] The ee value was determined by 1H NMR spectroscopy using saturated CDCl3 solutions of (R)‐(+)‐binaphthol. [16a] [d] Without Cs2CO3.
With control catalysts C2–C4 (5 mol %) bearing either an alkyl moiety R, a simple imidazolium or one with an aliphatic alcohol moiety, the reaction outcome was almost identical as with only Cs2CO3. The experiments with C2 and C4 were also repeated in the absence of base providing yields of 5 % (ee n.d.) and 10 % (racemic), respectively. In contrast, using Cs2CO3 and C5 equipped with a neutral axially chiral residue bearing an aromatic OH group, stereoselectivity was considerably higher. However, nearly complete diastereoselectivity was only attained with control catalyst C6 containing an achiral zwitterionic imidazolium/aryloxide moiety. [26] In agreement with the observation that the configuration of the axially chiral binaphthol unit has a minor effect on stereocontrol, enantioselectivity was high with C6. Nevertheless, these results demonstrate that the presence of the zwitterionic moiety (achiral or chiral) is necessary to attain high control. [27]
Next to the control experiments, kinetic and spectroscopic experiments were performed (for details see SI). Blackmond's reaction progress kinetic analysis (RPKA) was done using 1H NMR spectroscopy. [28] By the “same excess” protocol, catalyst robustness and a possible product inhibition was assessed. [27] These experiments show that no significant catalyst decomposition and product inhibition occur in the model reaction.
The empirical rate law was determined by the variable time normalization analysis (VTNA) method introduced by Burés. [29] Again, the model reaction of 1 a and 2B was examined at −20 °C in [D8]THF using catalyst C1 b (1S,2S). Four reactions with different initial concentrations of C1 b, 1 a and 2B were used monitoring product ent‐3 aB. The best fit for the normalization of the time scale axis was achieved for the following rate law [Eq. 1]:
| (1) |
The reaction rate thus follows a second order kinetic dependence for C1 b, a first order for 2B, and zero order for 1 a. The latter might be explained by substrate saturation for 1 a. The second order dependence for C1 b indicates that two catalyst molecules are involved in the turnover‐limiting step. [30] There is also a significant positive nonlinear effect for the model reaction of 1 a and 2B catalyzed by C1 b (5 mol %) in THF at −40 °C. We suggest that a dimeric catalyst might be the active species, because a dimeric structure was also found for C6 by X‐ray analysis [31] (details are in the SI).
UV/Vis titrations were performed in which C1 b was treated with 1 a. Having added 1.0 equivalent of 1 a, the resulting spectrum is very similar to the one of the precatalyst, in which a neutral naphthol unit is present. With larger amounts of up to 10 equivalents of 1 a, the spectra did not significantly change anymore. This suggests that 1 a is deprotonated by the zwitterionic moiety. In combination with the zero order kinetic dependence for 1 a, we suggest that 1 a binds to the Cu center where it gets deprotonated resulting in substrate saturation. A copper dienolate could indeed be found by ESI HRMS (disodium salt: m/z 1128.2563, calculated: 1128.2564). The interpretation was validated by 1H NMR titrations, which were done in [D8]THF at −20 °C. Because the catalyst is paramagnetic, no well resolved signals were detected. By addition of 1 equivalent of 1 a, its signals almost disappeared caused by the paramagnetism. However, with an excess of 1 a the signals were appearing again, thus suggesting that the catalyst is already saturated with 1 equivalent.
In conclusion, we have reported an efficient way to catalyze [4+2] cycloadditions with acidic prodienes. They are activated by coordination to a LA and deprotonation by a zwitterionic moiety forming a metal dienolate. The acidic OH group generated during this proton transfer and the imidazolium unit are crucial for excellent diastereo‐ and enantioselectivity. In combination with the first order kinetic dependence for maleimide 2B, it is likely that the cycloaddition step is usually turnover‐limiting. The reaction was found to be applicable to different substrate types and TONs of up to 3680 were achieved. Moreover, the catalyst can be prepared in very high overall yield (C1 b: 84%), is stable during catalysis and can be readily recycled by a simple filtration protocol.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG, project 310990893). V.M.P. thanks the Landesgraduiertenförderung BW for a PhD scholarship. Svenja Bechtold is acknowledged for skillful experimental contributions during a research internship regarding the optimization of the ligand synthesis. Open access funding enabled and organized by Projekt DEAL.
V. Miskov-Pajic, F. Willig, D. M. Wanner, W. Frey, R. Peters, Angew. Chem. Int. Ed. 2020, 59, 19873.
References
- 1.
- 1a. Ishihara K., Sakakura A. in Science of Synthesis Stereoselective Synthesis, Vol. 3 (Eds.: De Vries J. G., Molander G. A., Evans P. A.), Thieme, Stuttgart, 2011, pp. 67–123; [Google Scholar]
- 1b. Du H., Ding K. in Handbook of Cyclization Reactions, Vol. 1, (Ed.: Ma S.), Wiley, Hoboken, 2010, pp. 1–57. [Google Scholar]
- 2. Mikami K., Aikawa K. in Catalytic Asymmetric Synthesis, 3rd ed. (Ed.: Ojima I.), Wiley, Hoboken, 2010, pp. 683–737. [Google Scholar]
- 3.
- 3a. Li J.-L., Liu T.-Y., Chen Y.-C., Acc. Chem. Res. 2012, 45, 1491–1500; [DOI] [PubMed] [Google Scholar]
- 3b. Klier L., Tur F., Poulsen P. H., Jørgensen K. A., Chem. Soc. Rev. 2017, 46, 1080. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Shen J., Tan C.-H., Org. Biomol. Chem. 2008, 6, 3229–3236; [DOI] [PubMed] [Google Scholar]
- 4b. Marcos V., Aleman J., Chem. Soc. Rev. 2016, 45, 6812–6832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rawal V. H., Thadani A. N. in Asymmetric Synthesis, 2nd ed. (Eds.: Christmann M., Bräse S.), Wiley-VCH, Weinheim, 2008, pp. 144–148. [Google Scholar]
- 6.
- 6a.For achiral stoichiometric Lewis acids, see: Narasaka K., Shimada S., Osoda K., Iwasawa N., Synthesis 1991, 1171–1172; [Google Scholar]
- 6b. Nicolaou K. C., Liu J. J., Hwang C.-K., Dai W.-M., Guy R. K., J. Chem. Soc. Chem. Commun. 1992, 1118–1119; [Google Scholar]
- 6c.for chiral stoichiometric Lewis acids, see: Ward D. E., Souweha M. S., Org. Lett. 2005, 7, 3533–3536; [DOI] [PubMed] [Google Scholar]
- 6d.for catalytic asymmetric methods, see: Ishihara J., Nakadachi S., Watanabe Y., Hatakeyama S., J. Org. Chem. 2015, 80, 2037–2041. [DOI] [PubMed] [Google Scholar]
- 7.For selected reviews on catalytic asymmetric Diels–Alder reactions, see:
- 7a. Reymond S., Cossy J., Chem. Rev. 2008, 108, 5359–5406; [DOI] [PubMed] [Google Scholar]
- 7b. Liu X., Zheng H., Xia Y., Lin L., Feng X., Acc. Chem. Res. 2017, 50, 2621–2631; [DOI] [PubMed] [Google Scholar]
- 7c. Du H., Ding K. in Comprehensive Enantioselective Organocatalysis, Vol. 3 (Ed.: Dalko P. I.), Wiley-VCH, Weinheim, 2013, pp. 1131–1162. [Google Scholar]
- 8.For Michael addition, see: Willig F., Lang J., Hans A. C., Ringenberg M. R., Pfeffer D., Frey W., Peters R., J. Am. Chem. Soc. 2019, 141, 12029–12043. [DOI] [PubMed] [Google Scholar]
- 9.“Lewis Acid–Brønsted Base Catalysis”: Shibasaki M., Kumagai N. in Cooperative Catalysis—Designing Efficient Catalysts for Synthesis (Ed.: Peters R.), Wiley-VCH, Weinheim, 2015. [Google Scholar]
- 10.For more cooperative polyfunctional LA/azolium/H-bond catalysts developed by our group, see:
- 10a. Mechler M., Peters R., Angew. Chem. Int. Ed. 2015, 54, 10303–10307; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10442–10446; [Google Scholar]
- 10b. Schmid J., Junge T., Lang J., Frey W., Peters R., Angew. Chem. Int. Ed. 2019, 58, 5447–5451; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5501–5505. [Google Scholar]
- 11.For related cooperative bi-/polyfunctional LA catalysts bearing cationic side arms developed by our group, see:
- 11a. Brodbeck D., Álvarez-Barcia S., Meisner J., Broghammer F., Klepp J., Garnier D., Frey W., Kästner J., Peters R., Chem. Eur. J. 2019, 25, 1515–1524; [DOI] [PubMed] [Google Scholar]
- 11b. Brodbeck D., Broghammer F., Meisner J., Klepp J., Garnier D., Frey W., Kästner J., Peters R., Angew. Chem. Int. Ed. 2017, 56, 4056–4060; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4115–4119; [Google Scholar]
- 11c. Broghammer F., Brodbeck D., Junge T., Peters R., Chem. Commun. 2017, 53, 1156–1159; [DOI] [PubMed] [Google Scholar]
- 11d. Meier P., Broghammer F., Latendorf K., Rauhut G., Peters R., Molecules 2012, 17, 7121–7150; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11e. Kull T., Cabrera J., Peters R., Chem. Eur. J. 2010, 16, 9132–9139; [DOI] [PubMed] [Google Scholar]
- 11f. Kull T., Peters R., Angew. Chem. Int. Ed. 2008, 47, 5461–5464; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 5541–5544; for related polymetallic catalysts, see, for example: [Google Scholar]
- 11g. Schmid J., Frey W., Peters R., Organometallics 2017, 36, 4313–4324; [Google Scholar]
- 11h. Mechler M., Frey W., Peters R., Organometallics 2014, 33, 5492–5508; [Google Scholar]
- 11i. Mechler M., Latendorf K., Frey W., Peters R., Organometallics 2013, 32, 112–130. [Google Scholar]
- 12. Suzuki T., Watanabe S., Kobayashi S., Tanino K., Org. Lett. 2017, 19, 922–925. [DOI] [PubMed] [Google Scholar]
- 13. Shimizu H., Okamura H., Yamashita N., Iwagawa T., Nakatani M., Tetrahedron Lett. 2001, 42, 8649–8651. [Google Scholar]
- 14. Afarinkia K., Vinader V., Nelson T. D., Posner G. H., Tetrahedron 1992, 48, 9111–9171. [Google Scholar]
- 15. Okamura H., Iwagawa T., Nakatani M., Tetrahedron Lett. 1995, 36, 5939–5942. [Google Scholar]
- 16.
- 16a.With maleimide: Okamura H., Nakamura Y., Iwagawa T., Nakatani M., Chem. Lett. 1996, 25, 193–194; [Google Scholar]
- 16b. Liu L., Cotelle Y., Bornhof A.-B., Besnard C., Sakai N., Matile S., Angew. Chem. Int. Ed. 2017, 56, 13066–13069; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13246–13249; [Google Scholar]
- 16c. Soh J. Y.-T., Tan C.-H., J. Am. Chem. Soc. 2009, 131, 6904–6905; [DOI] [PubMed] [Google Scholar]
- 16d.for the use of chiral N-acryloyloxazolidinones in combination with cinchona alkaloids, see: Ref. [13] and Okamura H., Morishige K., Iwagawa T., Nakatani M., Tetrahedron Lett. 1998, 39, 1211–1214; [Google Scholar]
- 16e.for the use of an α-chloroacrylate, see: Ref. [12];
- 16f.for the use of enones and α,β-unsaturated nitriles, see: Wang Y., Li H., Wang Y.-Q., Liu Y., Foxman B. M., Deng L., J. Am. Chem. Soc. 2007, 129, 6364–6365; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16g. Singh R. P., Bartelson K., Wang Y., Su H., Lu X., Deng L., J. Am. Chem. Soc. 2008, 130, 2422–2423; [DOI] [PubMed] [Google Scholar]
- 16h. Shi L.-M., Dong W.-W., Tao H.-Y., Dong X.-Q., Wang C.-J., Org. Lett. 2017, 19, 4532–4535; [DOI] [PubMed] [Google Scholar]
- 16i.for the use of nitroolefins, see: Bartelson K. J., Singh R. P., Foxman B. M., Deng L., Chem. Sci. 2011, 2, 1940–1944; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16j.for a non-enantioselective method with maleic anhydride, see: Corey E. J., Kozikowski A. P., Tetrahedron Lett. 1975, 16, 2389–2392. [Google Scholar]
- 17.For a review, see: Chauhan P., Kaur J., Chimni S. S., Chem. Asian J. 2013, 8, 328. [DOI] [PubMed] [Google Scholar]
- 18. Okamura H., Shimizu H., Nakamura Y., Iwagawa T., Nakatani M., Tetrahedron Lett. 2000, 41, 4147–4150. [Google Scholar]
- 19. Tienabe Kipassa N., Okamura H., Kina K., Hamada T., Iwagawa T., Org. Lett. 2008, 10, 815–816. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a.See Ref. [16c]; for early studies, see:
- 20b. Okamura H., Nagaike H., Iwagawa T., Nakatani M., Tetrahedron Lett. 2000, 41, 8317–8321; for additional studies, see: [Google Scholar]
- 20c. Takahashi T., Reddy U. V. S., Kohari Y., Seki C., Furuyama T., Kobayashi N., Okuyama Y., Kwon E., Uwai K., Tokiwa M., Takeshita M., Nakano H., Tetrahedron Lett. 2016, 57, 5771–5776; [Google Scholar]
- 20d. Chennapuram M., Reddy U. V. S., Seki C., Okuyama Y., Kwon E., Uwai K., Tokiwa M., Takeshita M., Nakano H., Eur. J. Org. Chem. 2017, 4633–4641. [Google Scholar]
- 21. Deposition Numbers 2003855 (for ent-5 aC (Table 2, entry 11)) and 1995951 (for ent-3 aC (see Table S2, entry 3)) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 22. Deposition Numbers 2002396 (for 9) and 1998669 (for 11a after reduction of the keto group (see the Supporting Information)) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 23. Salvati M. E., Attar R. M., Gottardis M. M., Balog J. A., Pickering D. A., Martinez R. L., Sun C., PCT Int. Appl. WO 2002067939 A1, 2002.
- 24. Fu J.-G., Shan Y.-F., Sun W.-B., Lin G.-Q., Sun B.-F., Org. Biomol. Chem. 2016, 14, 5229–5232. [DOI] [PubMed] [Google Scholar]
- 25.For the parent cyclopentenone, the endo isomer could be formed: Mose R., Jensen M. E., Preegel G., Jørgensen K. A., Angew. Chem. Int. Ed. 2015, 54, 13630–13634; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13834–13838. [Google Scholar]
- 26.For ammonium betaine organocatalysts, see: Uraguchi D., Ooi T., J. Synth. Org. Chem. Jpn. 2018, 76, 1144–1153. [Google Scholar]
- 27.In experiments using CuII complexes with common enantiomerically pure ligands, only poor to low enantioselectivity was attained (see the Supporting Information).
- 28.
- 28a. Baxter R. D., Sale D., Engle K. M., Yu J.-Q., Blackmond D. G., J. Am. Chem. Soc. 2012, 134, 4600–4606; [DOI] [PubMed] [Google Scholar]
- 28b. Blackmond D. G., J. Am. Chem. Soc. 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Burés J., Angew. Chem. Int. Ed. 2016, 55, 16084–16087; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16318–16321; [Google Scholar]
- 29b. Nielsen C. D.-T., Burés J., Chem. Sci. 2019, 10, 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.For cooperative bimetallic enantioselective catalysts, see, for example:
- 30a. Shibasaki M., Yoshikawa N., Chem. Rev. 2002, 102, 2187–2210; [DOI] [PubMed] [Google Scholar]
- 30b. Shibasaki M., Matsunaga S., Chem. Soc. Rev. 2006, 35, 269–279; [DOI] [PubMed] [Google Scholar]
- 30c. Shibasaki M., Kanai M., Matsunaga S., Kumagai N., Acc. Chem. Res. 2009, 42, 1117–1127; [DOI] [PubMed] [Google Scholar]
- 30d. Handa S., Gnanadesikan V., Matsunaga S., Shibasaki M., J. Am. Chem. Soc. 2007, 129, 4900–4901; [DOI] [PubMed] [Google Scholar]
- 30e. Park J., Hong S., Chem. Soc. Rev. 2012, 41, 6931–6943; [DOI] [PubMed] [Google Scholar]
- 30f. Mouri S., Chen Z., Mitsunuma H., Furutachi M., Matsunaga S., Shibasaki M., J. Am. Chem. Soc. 2010, 132, 1255–1257. [DOI] [PubMed] [Google Scholar]
- 31. Deposition Number 1995950 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary